

Crystal Structure, Hirshfeld Analysis, and DFT Calculations of Three Trinuclear Cu(II) Polymorphs
 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis
2.3. X-ray Crystallography and Data Collection
2.4. DFT Calculations
2.5. Software
3. Results and Discussion
3.1. Crystal Structure Descriptions
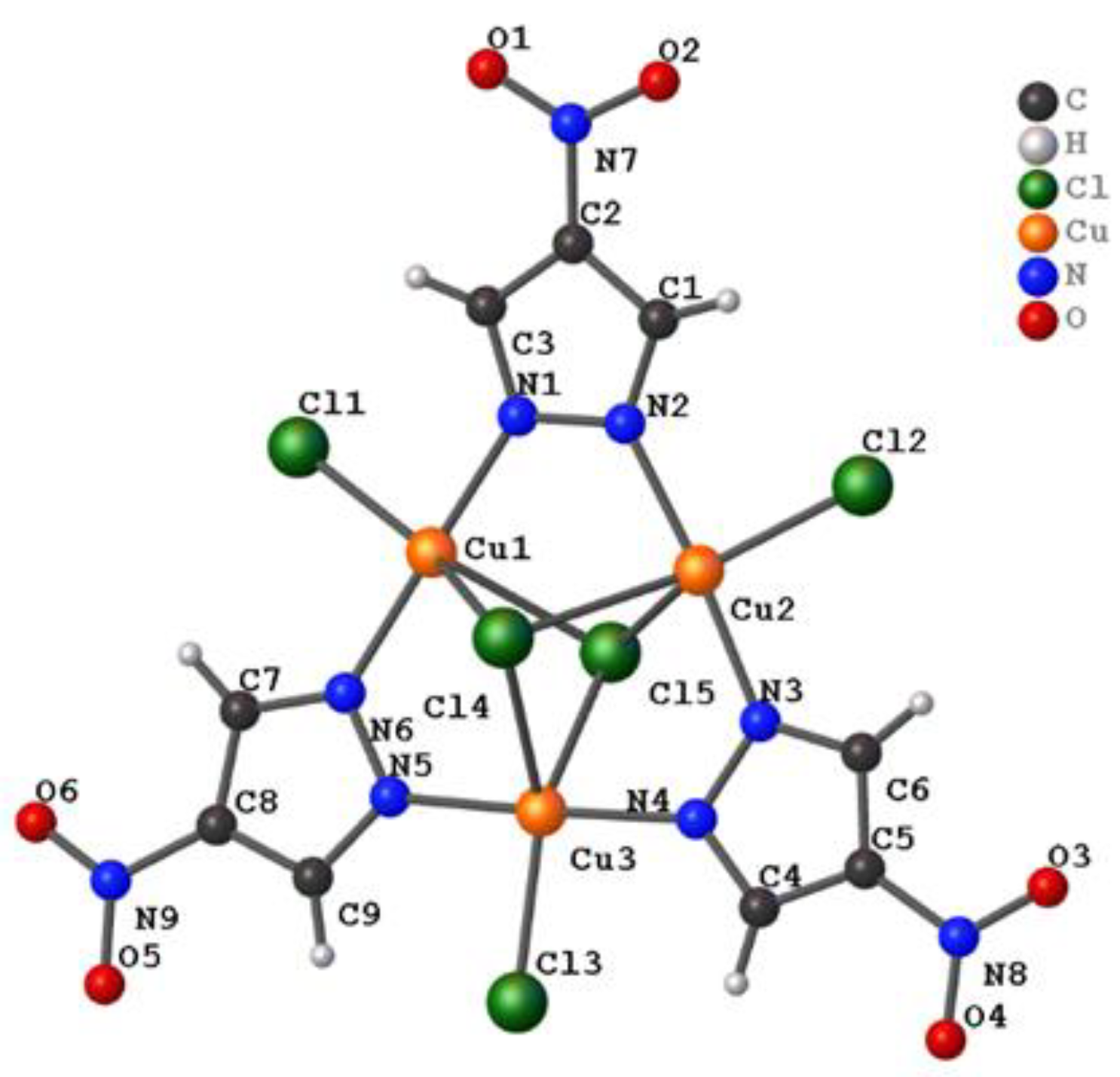
3.1.1. General Structure Description
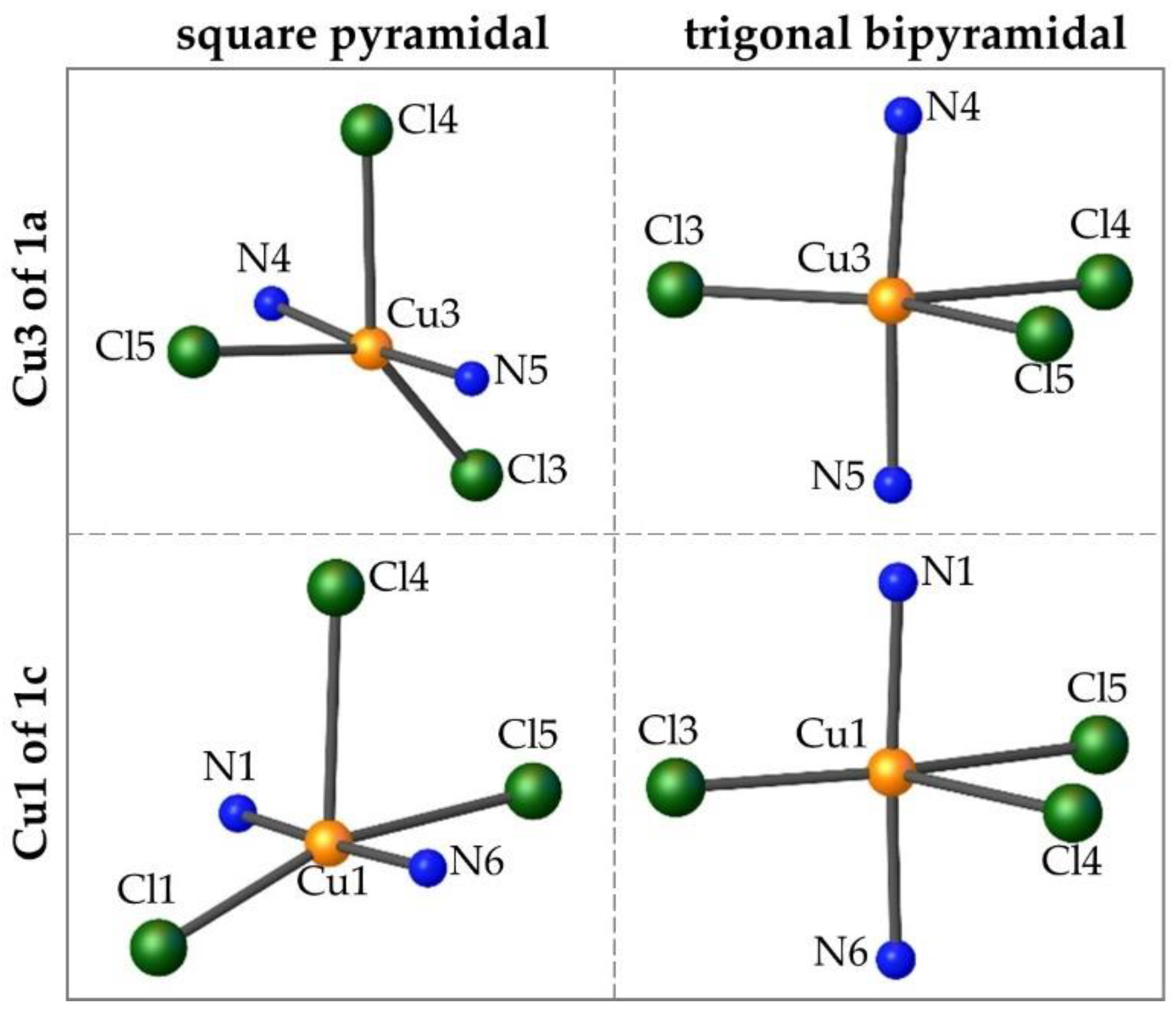
3.1.2. Tau Parameter Determination
3.1.3. Ligand Position in Relation to the Cu3 Plane
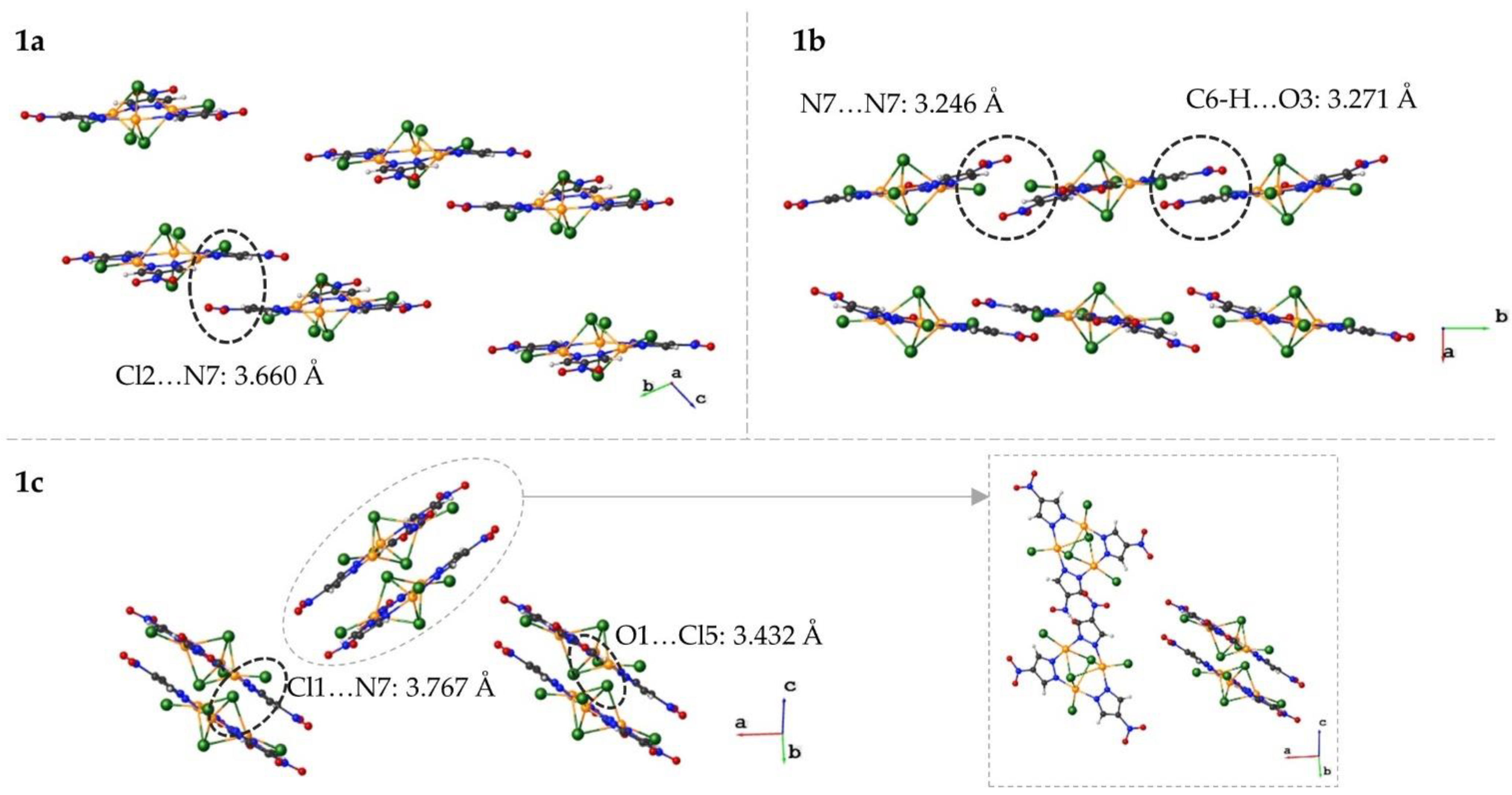
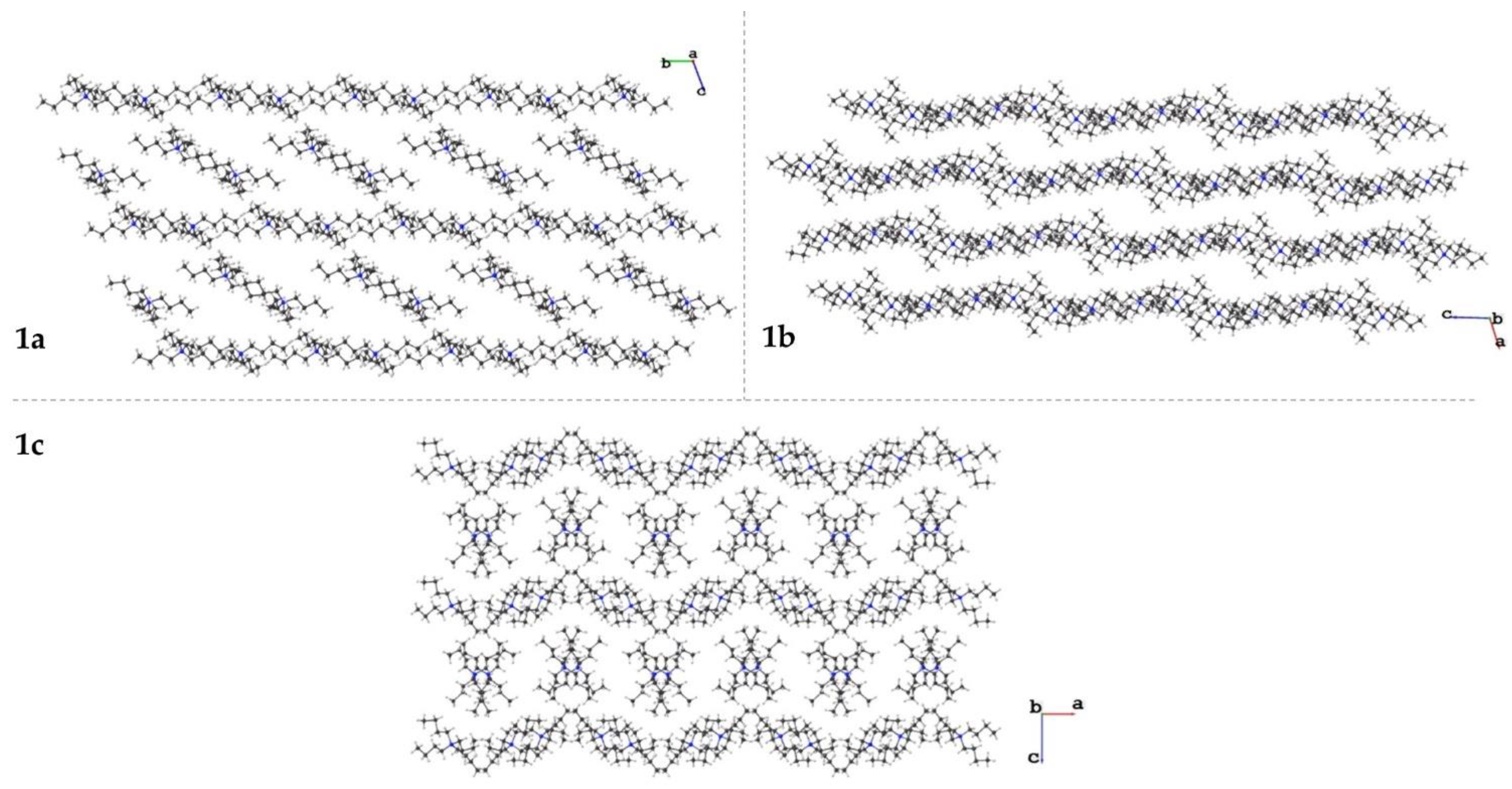
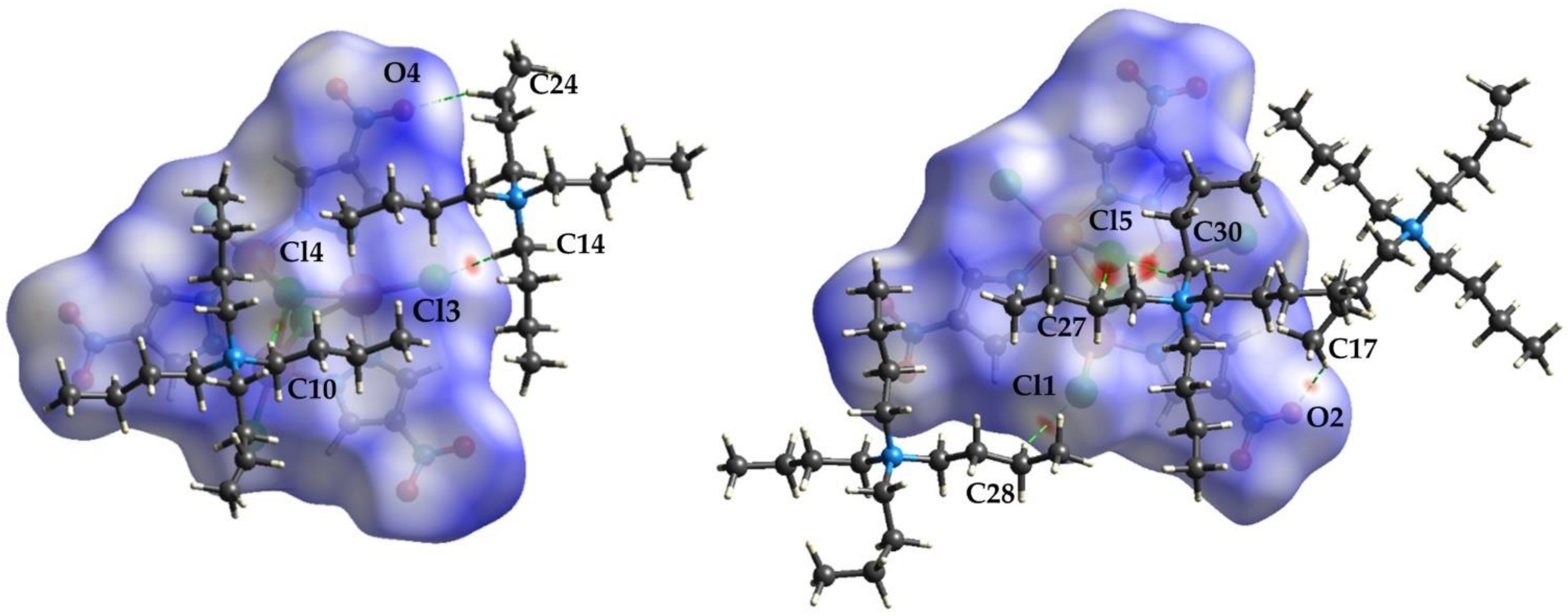
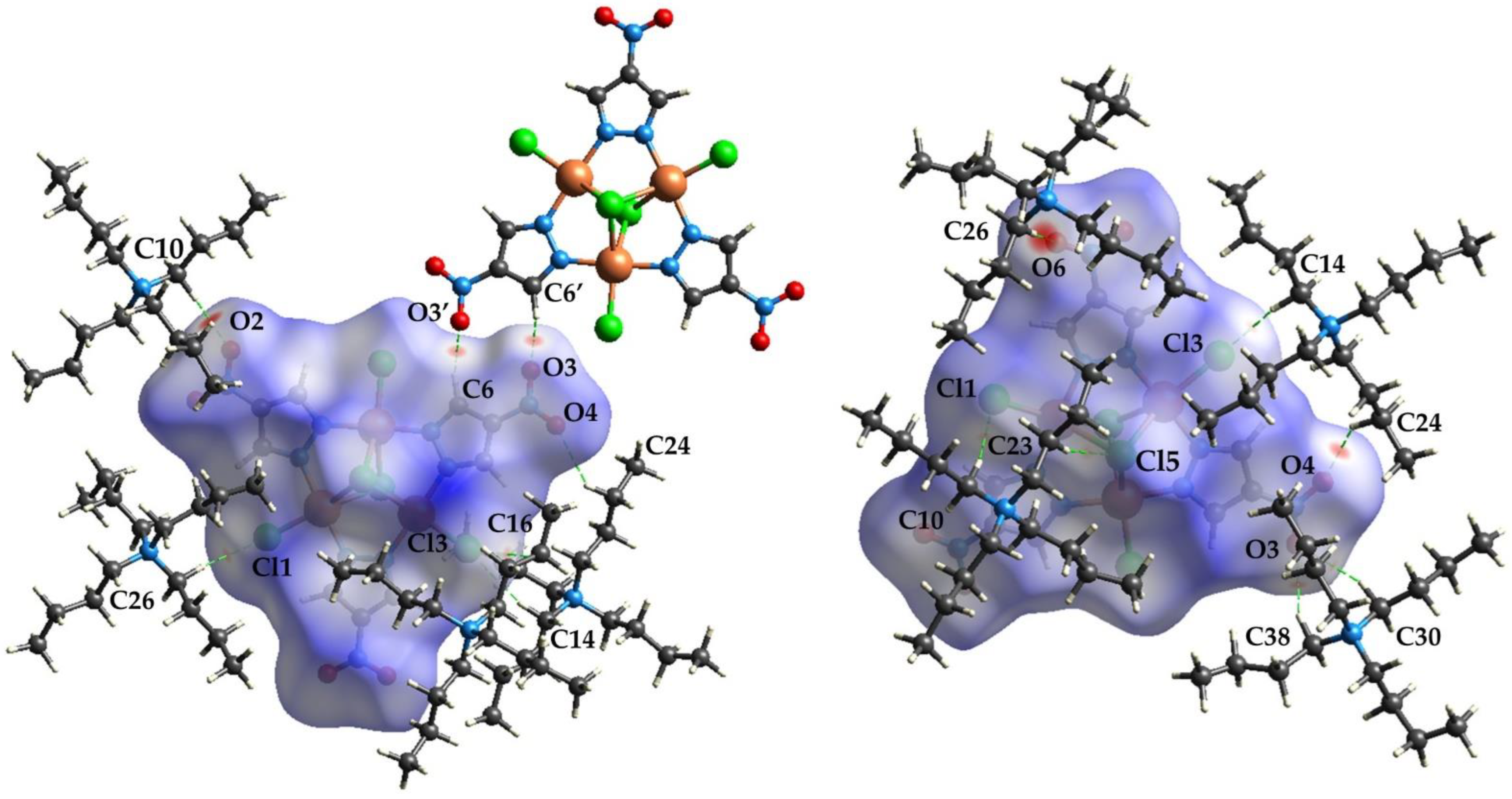
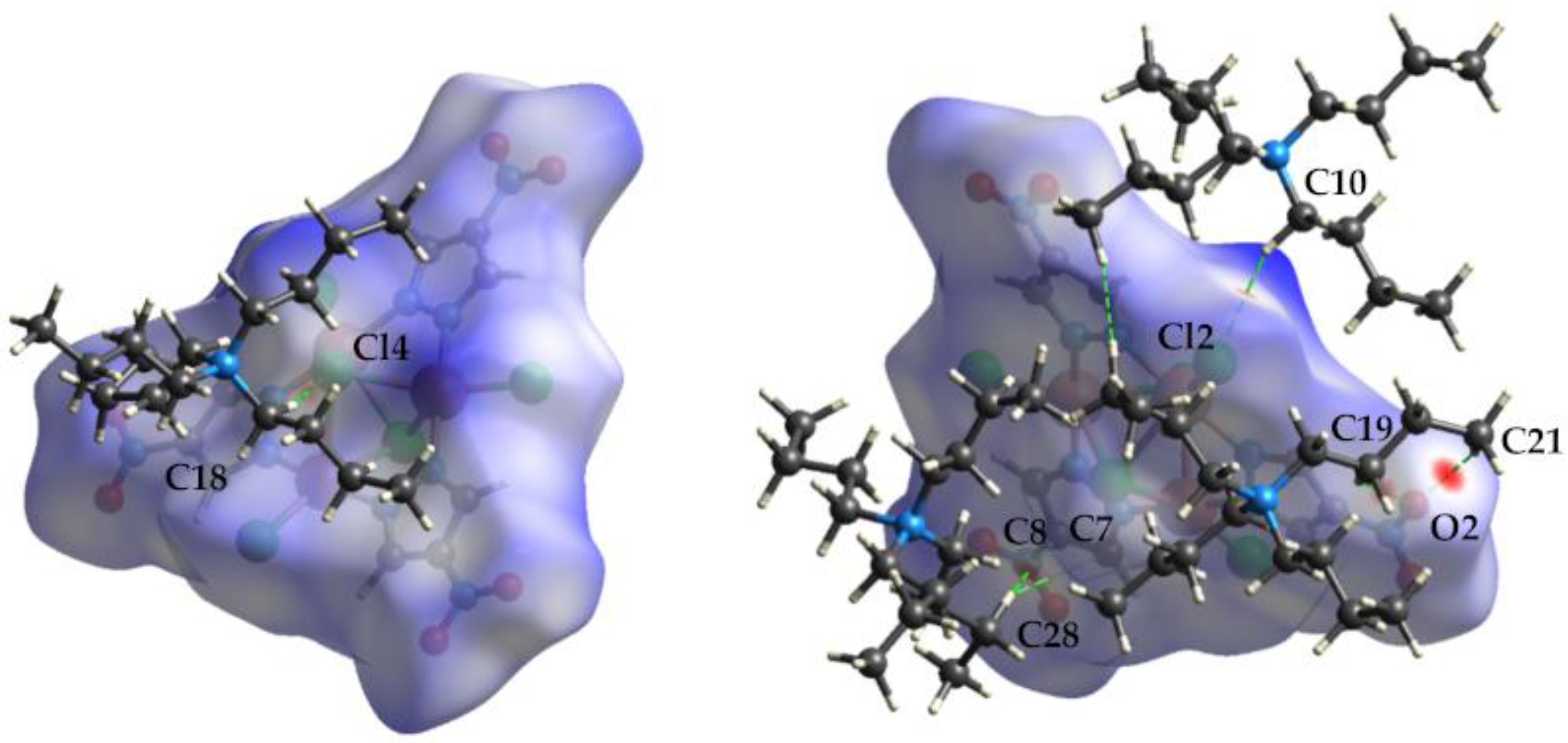
3.1.4. Crystal Packing
3.2. DFT Calculations
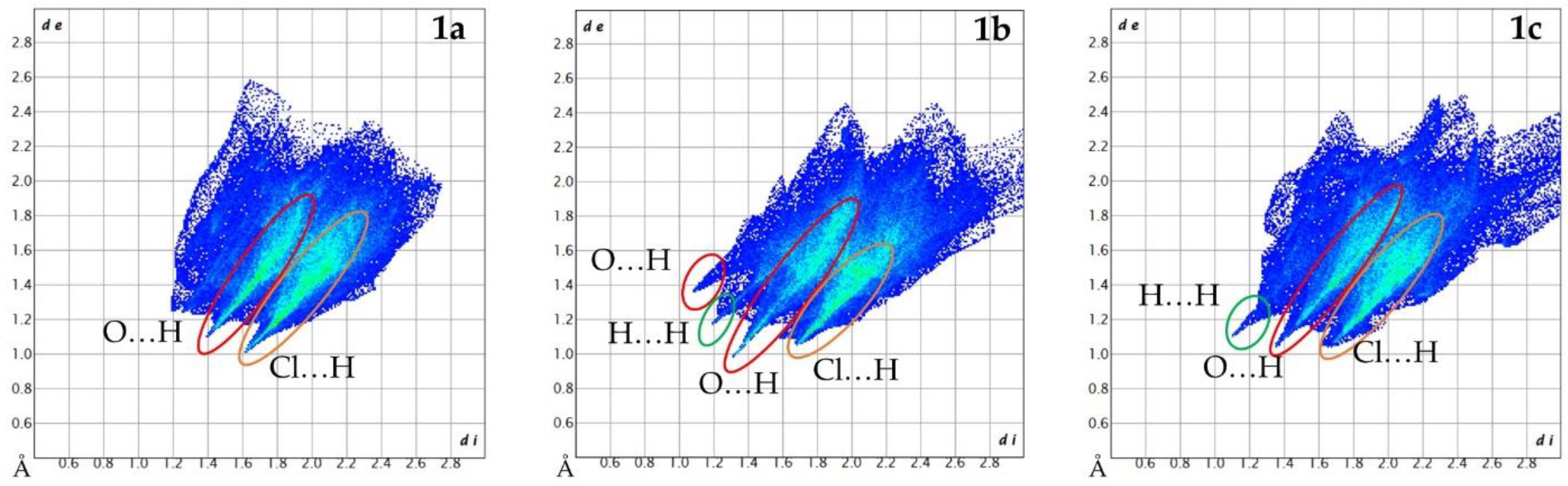
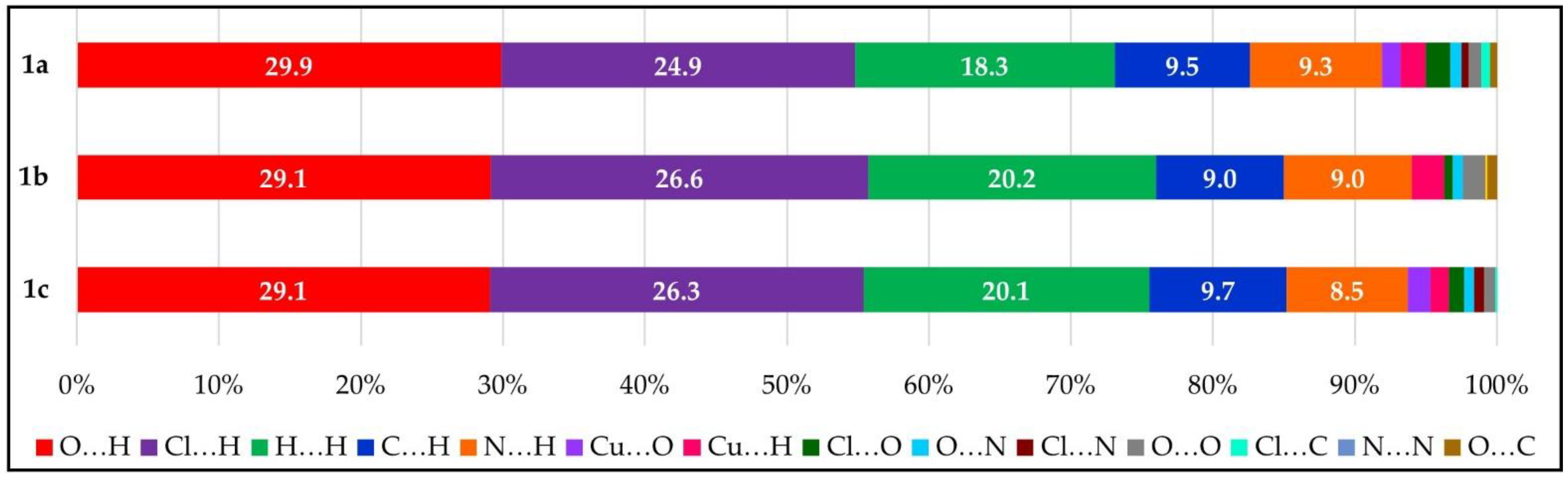
3.3. Hirshfeld Surface Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bernstein, J. Polymorphism in Molecular Crystals; International Union of Crystallography Monographs on Crystallography; Oxford University Press: Oxford, UK, 2007. [Google Scholar] [CrossRef]
- Stahly, G.P. Diversity in Single- and Multiple-Component Crystals. The Search for and Prevalence of Polymorphs and Cocrystals. Cryst. Grow. Des. 2007, 7, 1007–1026. [Google Scholar] [CrossRef] [Green Version]
- Hilfiker, R. Polymorphism: In the Pharmaceutical Industry; Wiley-VCH: Weinheim, Germany, 2006. [Google Scholar]
- Saha, B.K.; Nath, N.K.; Thakuria, R. Polymorphs with Remarkably Distinct Physical and/or Chemical Properties. Chem. Rec. 2002, e202200173. [Google Scholar] [CrossRef] [PubMed]
- Benito, Q.; Le Goff, X.F.; Maron, S.; Fargues, A.; Garcia, A.; Martineau, C.; Taulelle, F.; Kahlal, S.; Gacoin, T.; Boilot, J.-P.; et al. Polymorphic Copper Iodide Clusters: Insights into the Mechanochromic Luminescence Properties. J. Am. Chem. Soc. 2014, 136, 11311–11320. [Google Scholar] [CrossRef]
- Dave, S.; Sahu, R.; Tripathy, B.C. Electrochemical Applications of Metal-Organic Frameworks; Elsevier: Amsterdam, The Netherlands, 2022; pp. 17–35. [Google Scholar]
- Chai, W.; Hong, M.; Song, L.; Jia, G.; Shi, H.; Guo, J.; Shu, K.; Guo, B.; Zhang, Y.; You, W.; et al. Three Reversible Polymorphic Copper(I) Complexes Triggered by Ligand Conformation: Insights into Polymorphic Crystal Habit and Luminescent Properties. Inorg. Chem. 2015, 54, 4200–4207. [Google Scholar] [CrossRef]
- Muthukumar, P.; Pannipara, M.; Al-Sehemi, A.G.; Moon, D.; Anthony, S.P. Polymorphs of a Copper Coordination Compound: Interlinking Active Sites Enhance the Electrocatalytic Activity of the Coordination Polymer Compared to the Coordination Complex. CrystEngComm 2020, 22, 425–429. [Google Scholar] [CrossRef]
- Wöhlert, S.; Runčevski, T.; Dinnebier, R.E.; Ebbinghaus, S.G.; Näther, C. Synthesis, Structures, Polymorphism, and Magnetic Properties of Transition Metal Thiocyanato Coordination Compounds. Cryst. Grow. Des. 2014, 14, 1902–1913. [Google Scholar] [CrossRef]
- Fromm, K.M.; Doimeadios, J.L.S.; Robin, A.Y. Concomitant Crystallization of Two Polymorphs—A Ring and a Helix: Concentration Effect on Supramolecular Isomerism. Chem. Commun. 2005, 36, 4548–4550. [Google Scholar] [CrossRef]
- White-Morris, R.L.; Olmstead, M.M.; Attar, S.; Balch, A.L. Intermolecular Interactions in Polymorphs of Trinuclear Gold(I) Complexes: Insight into the Solvoluminescence of AuI3(MeNCOMe)3. Inorg. Chem. 2005, 44, 5021–5029. [Google Scholar] [CrossRef]
- Brog, J.-P.; Chanez, C.-L.; Crochet, A.; Fromm, K.M. Polymorphism, What It Is and How to Identify It: A Systematic Review. RSC Adv. 2013, 3, 16905. [Google Scholar] [CrossRef]
- Angaridis, P.A.; Baran, P.; Boča, R.; Cervantes-Lee, F.; Haase, W.; Mezei, G.; Raptis, R.G.; Werner, R. Synthesis and Structural Characterization of Trinuclear CuII −Pyrazolato Complexes Containing μ3 -OH, μ3 -O, and μ3 -Cl Ligands. Magnetic Susceptibility Study of [PPN]2[(μ3-O)Cu3(μ-pz)3Cl3]. Inorg. Chem. 2002, 41, 2219–2228. [Google Scholar] [CrossRef]
- Boudalis, A.K.; Rogez, G.; Heinrich, B.; Raptis, R.G.; Turek, P. Towards Ionic Liquids with Tailored Magnetic Properties: Bmim+ Salts of Ferro- and Antiferromagnetic CuII3 Triangles. Dalton Trans. 2017, 46, 12263–12273. [Google Scholar] [CrossRef] [Green Version]
- Kreiger, D.I.; Mathivathanan, L.; Raptis, R.G. Coordination Polymers Based on Pyrazole-4-Carboxaldehyde-Containing Cu3N6 Metallacycles as Building Units. CrystEngComm 2019, 21, 3047–3055. [Google Scholar] [CrossRef]
- Mathivathanan, L.; Boudalis, A.K.; Turek, P.; Pissas, M.; Sanakis, Y.; Raptis, R.G. Interactions between H-Bonded [CuII3(μ3-OH)] Triangles; a Combined Magnetic Susceptibility and EPR Study. Phys. Chem. Chem. Phys. 2018, 20, 17234–17244. [Google Scholar] [CrossRef] [PubMed]
- Mathivathanan, L.; Rogez, G.; Ben Amor, N.; Robert, V.; Raptis, R.G.; Boudalis, A.K. Origin of Ferromagnetism and Magnetic Anisotropy in a Family of Copper(II) Triangles. Chem. Eur. J. 2020, 26, 12769–12784. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Carrillo, M.; Chakraborty, I.; Mezei, G.; Webster, R.D.; Raptis, R.G. Tuning of the [Cu3(μ-O)]4+/5+ Redox Couple: Spectroscopic Evidence of Charge Delocalization in the Mixed-Valent [Cu3(μ-O)]5+ Species. Inorg. Chem. 2008, 47, 7644–7650. [Google Scholar] [CrossRef] [PubMed]
- Mezei, G.; Raptis, R.G. Effect of Pyrazole-Substitution on the Structure and Nuclearity of Cu(II)-Pyrazolato Complexes. Inorg. Chim. Acta 2004, 357, 3279–3288. [Google Scholar] [CrossRef]
- Mezei, G.; Rivera-Carrillo, M.; Raptis, R.G. Effect of Copper-Substitution on the Structure and Nuclearity of Cu(II)-Pyrazolates: From Trinuclear to Tetra-, Hexa- and Polynuclear Complexes. Inorg. Chim. Acta 2004, 357, 3721–3732. [Google Scholar] [CrossRef]
- Boča, R.; Dlháň, L.; Mezei, G.; Ortiz-Pérez, T.; Raptis, R.G.; Telser, J. Triangular, Ferromagnetically-Coupled CuII3−Pyrazolato Complexes as Possible Models of Particulate Methane Monooxygenase (PMMO). Inorg. Chem. 2003, 42, 5801–5803. [Google Scholar] [CrossRef]
- Mezei, G.; Raptis, R.G.; Telser, J. Trinuclear, Antiferromagnetically Coupled CuII Complex with an EPR Spectrum of Mononuclear CuII: Effect of Alcoholic Solvents. Inorg. Chem. 2006, 45, 8841–8843. [Google Scholar] [CrossRef]
- Maresca, K.P.; Rose, D.J.; Zubieta, J. Synthesis and Characterization of a Binuclear Rhenium Nitropyrazole Complex [Re2O3Cl2(PPh3)2(C3H2N3O2)2]. Inorg. Chim. Acta 1997, 260, 83–88. [Google Scholar] [CrossRef]
- Perrin, D.D.; Armarego, W.L.F.; Perrin, D.R. Purification of Laboratory Chemicals; Pergamon Press: Oxford, UK, 1980. [Google Scholar]
- Data Collection: SMART-NT Software Reference Manual; Version 5.0; Bruker AXS, Inc.: Madison, WI, USA, 1998.
- Data Reduction: SAINT-NT Software Reference Manual; Version 4.0; Bruker AXS, Inc.: Madison, WI, USA, 1996.
- Sheldrick, G.M. SHELXTL-NT; Version 5.1; Bruker AXS, Inc.: Madison, WI, USA, 1999. [Google Scholar]
- Becke, A.D. Density-functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hariharan, P.C.; Pople, J.A. The Influence of Polarization Functions on Molecular Orbital Hydrogenation Energies. Theoret. Chim. Acta 1973, 28, 213–222. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From Visualization to Analysis, Design and Prediction. J. Appl. Cryst. 2020, 53, 226–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spackman, P.R.; Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer: A Program for Hirshfeld Surface Analysis, Visualization and Quantitative Analysis of Molecular Crystals. J. Appl. Cryst. 2021, 54, 1006–1011. [Google Scholar] [CrossRef]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld Surface Analysis. CrystEngComm 2009, 11, 19–32. [Google Scholar] [CrossRef]
- Sundareswaran, S.; Karuppannan, S. Hirshfeld Surface Analysis of Stable and Metastable Polymorphs of Vanillin. Crys. Res. Technol. 2020, 55, 2000083. [Google Scholar] [CrossRef]
- McKinnon, J.J.; Fabbiani, F.P.A.; Spackman, M.A. Comparison of Polymorphic Molecular Crystal Structures through Hirshfeld Surface Analysis. Cryst. Grow. Des. 2007, 7, 755–769. [Google Scholar] [CrossRef]
- Spackman, M.A.; McKinnon, J.J. Fingerprinting Intermolecular Interactions in Molecular Crystals. CrystEngComm 2002, 4, 378–392. [Google Scholar] [CrossRef]
- Addison, A.W.; Rao, T.N.; Reedijk, J.; van Rijn, J.; Verschoor, G.C. Synthesis, Structure, and Spectroscopic Properties of Copper(II) Compounds Containing Nitrogen–Sulphur Donor Ligands; the Crystal and Molecular Structure of Aqua[1,7-bis(N-methylbenzimidazol-2′-yl)-2,6-dithiaheptane]copper(II) perchlorate. J. Chem. Soc. Dalton Trans. 1984, 7, 1349–1356. [Google Scholar] [CrossRef]
- Baker, T.M.; Howard, K.M.; Brennessel, W.W.; Neidig, M.L. Crystal Structure of a Third Polymorph of Tris (acetyl acetonato-κ2O,O′)iron(III). Acta Cryst. 2015, E71, m228–m229. [Google Scholar] [CrossRef]
- Morosin, B. The Crystal Structure of Trisacetylacetonatochromium(III). Acta Cryst. 1965, 19, 131–137. [Google Scholar] [CrossRef]
- Geremia, S.; Demitri, N. Crystallographic Study of Manganese(III) Acetylacetonate: An Advanced Undergraduate Project with Unexpected Challenges. J. Chem. Educ. 2005, 82, 460. [Google Scholar] [CrossRef]
- Vrdoljak, V.; Prugovečki, B.; Matković-Čalogović, D.; Hrenar, T.; Dreos, R.; Siega, P. Three Polymorphic Forms of a Monomeric Mo(VI) Complex: Building Blocks for Two Metal–Organic Supramolecular Isomers. Intermolecular Interactions and Ligand Substituent Effects. Cryst. Grow. Des. 2013, 13, 3773–3784. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1a [22] | 1b | 1c | |
|---|---|---|---|
| Formula | C41H78Cl5Cu3N11O6 | C41H78Cl5Cu3N11O6 | C41H78Cl5Cu3N11O6 |
| Formula Weight | 1189.01 | 1189.01 | 1189.01 |
| Temperature (K) | 299(2) | 298(2) | 298(2) |
| Crystal System | Triclinic | Monoclinic | Orthorhombic |
| Space Group | P-1 (No. 2) | P21/n (No. 14) | Pbca (No. 61) |
| a/Å | 13.121(1) | 15.526(2) | 21.944(2) |
| b/Å | 15.183(1) | 20.938(3) | 15.783(2) |
| c/Å | 15.625(1) | 18.904(3) | 33.886(4) |
| α/° | 108.778(2) | 90 | 90 |
| β/° | 102.082(2) | 108.746(2) | 90 |
| γ/° | 95.916(2) | 90 | 90 |
| V/Å3 | 2832.5(5) | 5819(1) | 11,736(2) |
| Z | 2 | 4 | 8 |
| Dcalc/g cm−3 | 1.394 | 1.357 | 1.346 |
| μ/mm−1 | 1.402 | 1.365 | 1.354 |
| Refl. Collected | 12,635 | 30,317 | 62,961 |
| Unique Refl. | 8156 | 10,274 | 10,358 |
| Obs. Refl. (I > 2σ(I)) | 4654 | 6065 | 4571 |
| θ range/° | 1.42–23.30 | 1.50–25.03 | 1.20–25.04 |
| Data | 8156 | 10,274 | 10,358 |
| Restraints/param. | 0/603 | 0/603 | 6/603 |
| R(F); Rw(F) (I > 2σ(I)) | 0.0401; 0.0914 | 0.0476; 0.1294 | 0.0795; 0.2176 |
| GooF | 0.914 | 1.000 | 1.003 |
| 1a | 1b | 1c | |
|---|---|---|---|
| Cu1–Cl1 | 2.234(2) | 2.275(1) | 2.256(2) |
| Cu1–Cl4 | 2.501(2) | 2.659(1) | 2.680(3) |
| Cu1–Cl5 | 2.628(1) | 2.510(1) | 2.480(2) |
| Cu1–N1 | 1.939(4) | 1.948(4) | 1.950(6) |
| Cu1–N6 | 1.950(4) | 1.944(3) | 1.942(6) |
| Cu2–Cl2 | 2.255(2) | 2.232(1) | 2.267(2) |
| Cu2–Cl4 | 2.558(1) | 2.594(1) | 2.679(2) |
| Cu2–Cl5 | 2.614(2) | 2.560(1) | 2.490(2) |
| Cu2–N2 | 1.947(4) | 1.950(4) | 1.937(6) |
| Cu2–N3 | 1.946(4) | 1.951(3) | 1.926(7) |
| Cu3–Cl3 | 2.276(2) | 2.271(1) | 2.255(3) |
| Cu3–Cl4 | 2.584(2) | 2.513(1) | 2.465(2) |
| Cu3–Cl5 | 2.501(1) | 2.615(1) | 2.642(2) |
| Cu3–N4 | 1.960(4) | 1.945(3) | 1.964(7) |
| Cu3–N5 | 1.966(4) | 1.953(3) | 1.950(7) |
| Cu1…Cu2 | 3.381(1) | 3.4194(9) | 3.420(2) |
| Cu1…Cu3 | 3.389(1) | 3.4042(8) | 3.386(2) |
| Cu2…Cu3 | 3.433(1) | 3.397(1) | 3.423(2) |
| 1a | 1b | 1c | |||||
|---|---|---|---|---|---|---|---|
| Cu1 | ∠1 | Cl1-Cu1-Cl5 | 119.56 | Cl1-Cu1-Cl5 | 132.23 | Cl1-Cu1-Cl5 | 163.74 |
| ∠2 | Cl1-Cu1-Cl4 | 160.39 | Cl1-Cu1-Cl4 | 147.74 | Cl1-Cu1-Cl4 | 116.35 | |
| ∠3 | Cl4-Cu1-Cl5 | 79.99 | Cl4-Cu1-Cl5 | 80.03 | Cl4-Cu1-Cl5 | 79.89 | |
| ∠4 | N1-Cu1-N6 | 173.43 | N1-Cu1-N6 | 172.60 | N1-Cu1-N6 | 176.43 | |
| τ | (∠4 − ∠2)/60 | 0.22 | (∠4 − ∠2)/60 | 0.41 | (∠4 − ∠1)/60 | 0.21 | |
| Cu2 | ∠1 | Cl2-Cu2-Cl5 | 120.58 | Cl1-Cu1-Cl5 | 132.23 | Cl2-Cu2-Cl5 | 133.70 |
| ∠2 | Cl2-Cu2-Cl4 | 160.14 | Cl1-Cu1-Cl4 | 147.74 | Cl2-Cu2-Cl4 | 146.56 | |
| ∠3 | Cl4-Cu2-Cl5 | 79.24 | Cl4-Cu1-Cl5 | 80.03 | Cl4-Cu2-Cl5 | 79.73 | |
| ∠4 | N2-Cu2-N3 | 173.32 | N1-Cu1-N6 | 172.60 | N2-Cu2-N3 | 172.86 | |
| τ | (∠4 − ∠2)/60 | 0.22 | (∠4 − ∠2)/60 | 0.54 | (∠4 − ∠2)/60 | 0.44 | |
| Cu3 | ∠1 | Cl3-Cu3-Cl5 | 138.45 | Cl1-Cu1-Cl5 | 132.23 | Cl3-Cu3-Cl5 | 130.39 |
| ∠2 | Cl3-Cu3-Cl4 | 140.69 | Cl1-Cu1-Cl4 | 147.74 | Cl3-Cu3-Cl4 | 148.69 | |
| ∠3 | Cl4-Cu3-Cl5 | 80.85 | Cl4-Cu1-Cl5 | 80.03 | Cl4-Cu3-Cl5 | 80.90 | |
| ∠4 | N4-Cu3-N5 | 174.94 | N1-Cu1-N6 | 172.60 | N4-Cu3-N5 | 174.36 | |
| τ | (∠4 − ∠2)/60 | 0.57 | (∠4 − ∠1)/60 | 0.51 | (∠4 − ∠2)/60 | 0.43 | |
| Στ | 1.01 | 1.46 | 1.08 | ||||
| Cl1 | Cl2 | Cl3 | pz1 | pz2 | pz3 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1a | 0.799 | 0.758 | −0.017 | 1.540 | 1.574 | −0.278 | −0.133 | −0.075 | 0.486 | 0.486 |
| 1b | −0.275 | −0.116 | 0.150 | 0.241 | 0.541 | 0.427 | −0.145 | 0.013 | 0.295 | 0.585 |
| 1c | 0.912 | −0.248 | −0.486 | 0.178 | 1.646 | 0.276 | 0.018 | −0.302 | 0.008 | 0.596 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rue, K.L.; Mathivathanan, L.; Mezei, G.; Mebel, A.M.; Raptis, R.G. Crystal Structure, Hirshfeld Analysis, and DFT Calculations of Three Trinuclear Cu(II) Polymorphs. Crystals 2022, 12, 1611. https://doi.org/10.3390/cryst12111611
Rue KL, Mathivathanan L, Mezei G, Mebel AM, Raptis RG. Crystal Structure, Hirshfeld Analysis, and DFT Calculations of Three Trinuclear Cu(II) Polymorphs. Crystals. 2022; 12(11):1611. https://doi.org/10.3390/cryst12111611
Chicago/Turabian StyleRue, Kelly L., Logesh Mathivathanan, Gellert Mezei, Alexander M. Mebel, and Raphael G. Raptis. 2022. "Crystal Structure, Hirshfeld Analysis, and DFT Calculations of Three Trinuclear Cu(II) Polymorphs" Crystals 12, no. 11: 1611. https://doi.org/10.3390/cryst12111611