
First Principles Study on the Adsorption of Hydrogen Atoms on the Surface of Plutonium-Aluminum Systems

Abstract
:1. Introduction
2. Materials and Methods
2.1. Calculation Method
2.2. Calculation Model
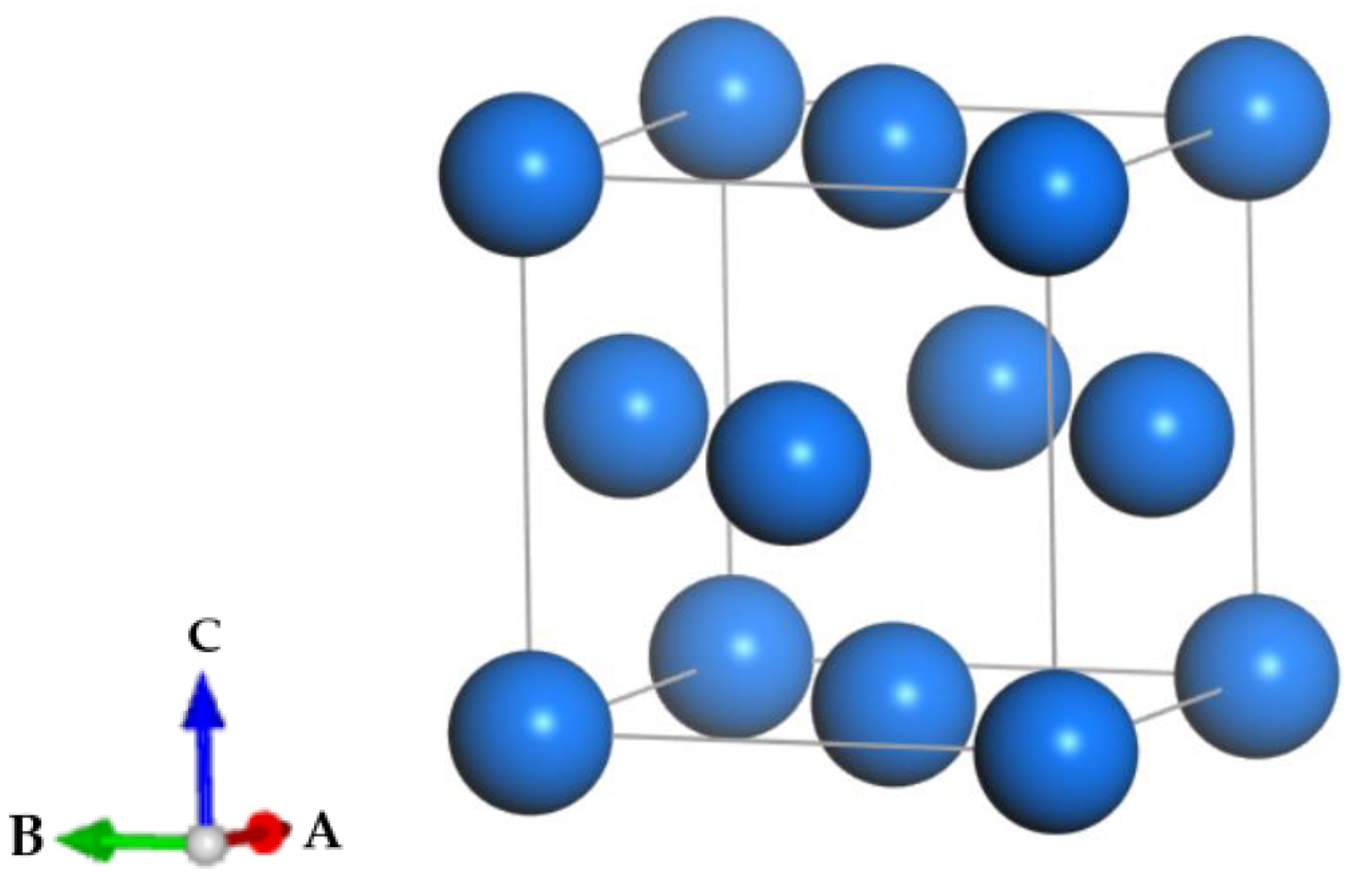
2.2.1. δ-Pu Lattice Optimization
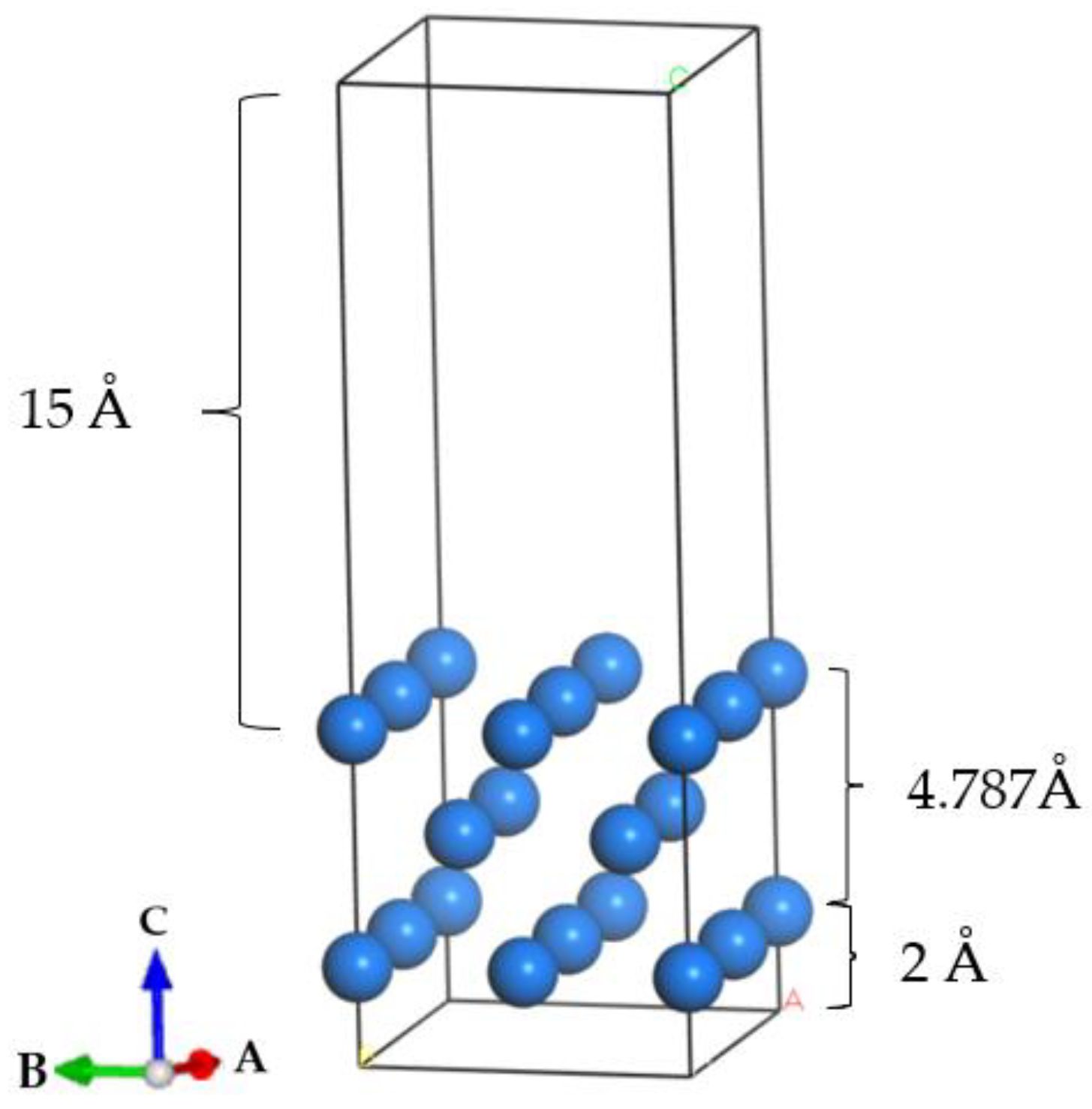
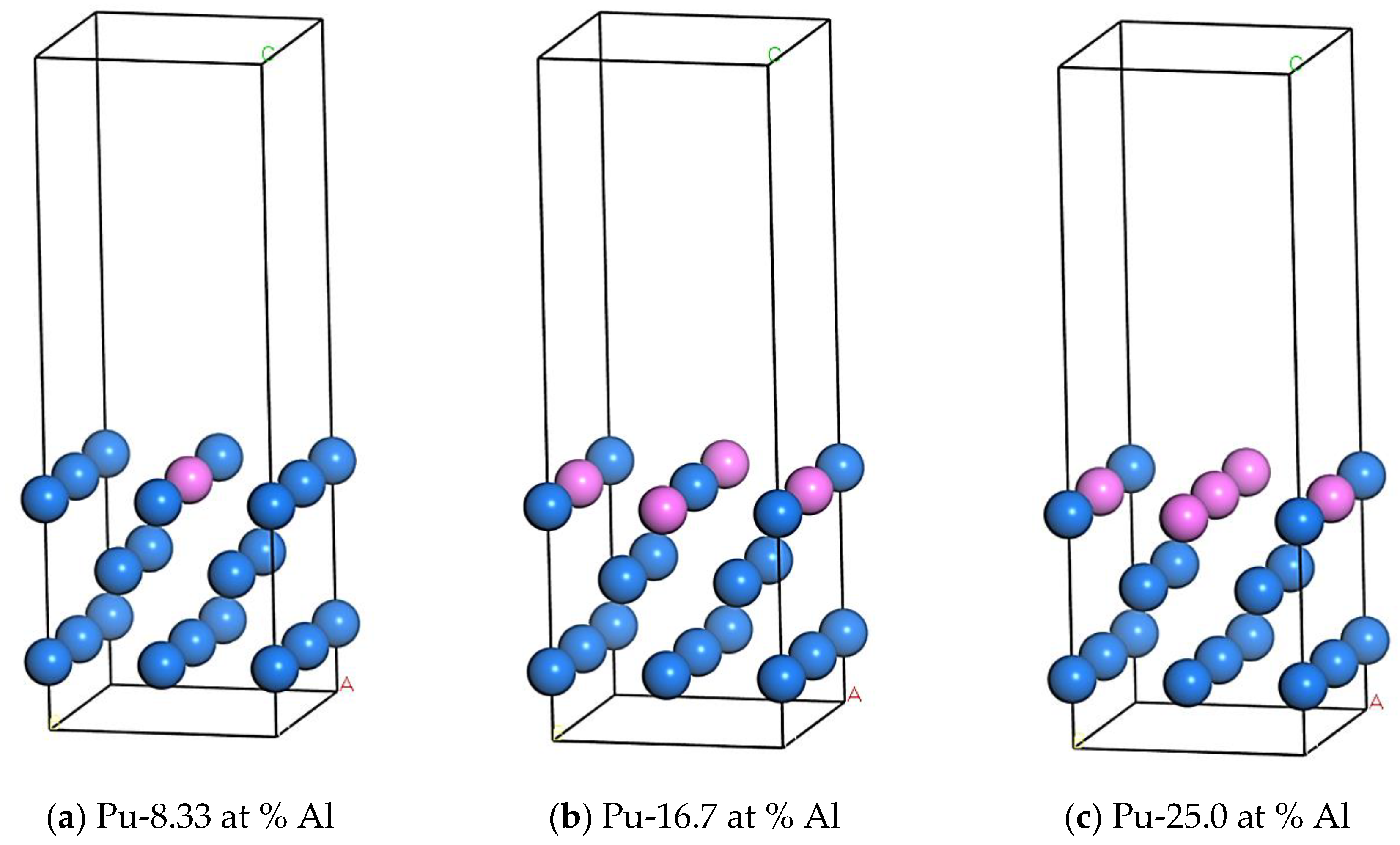
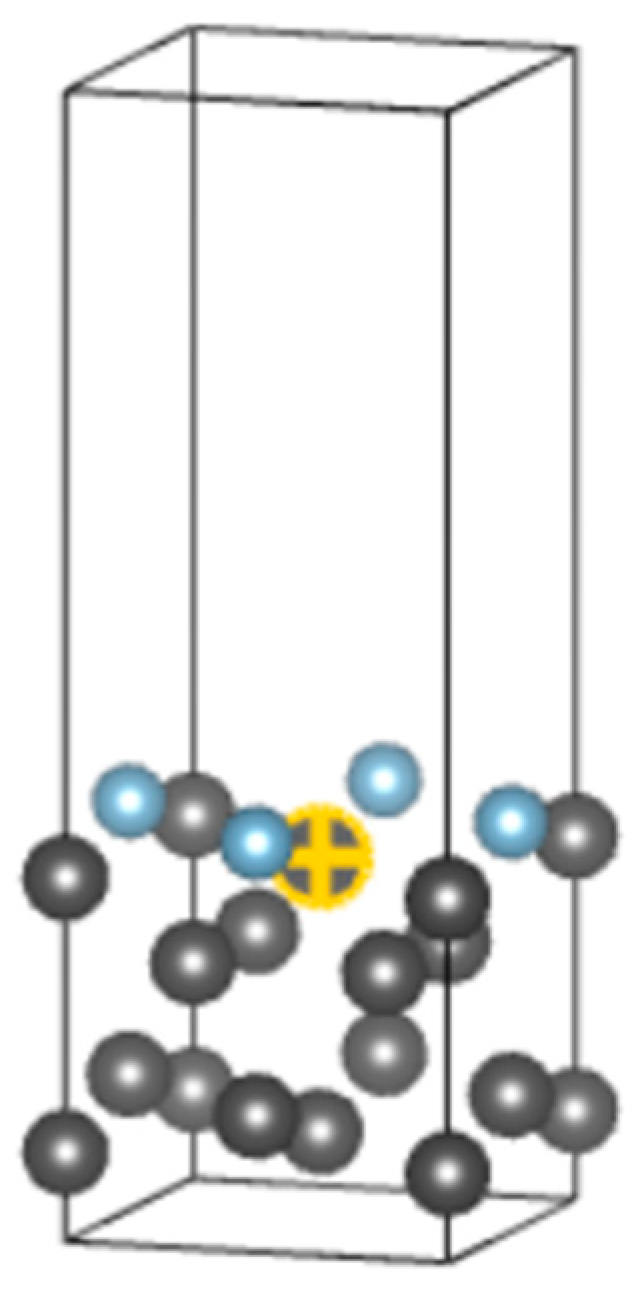
2.2.2. Establishment of Doping System Model
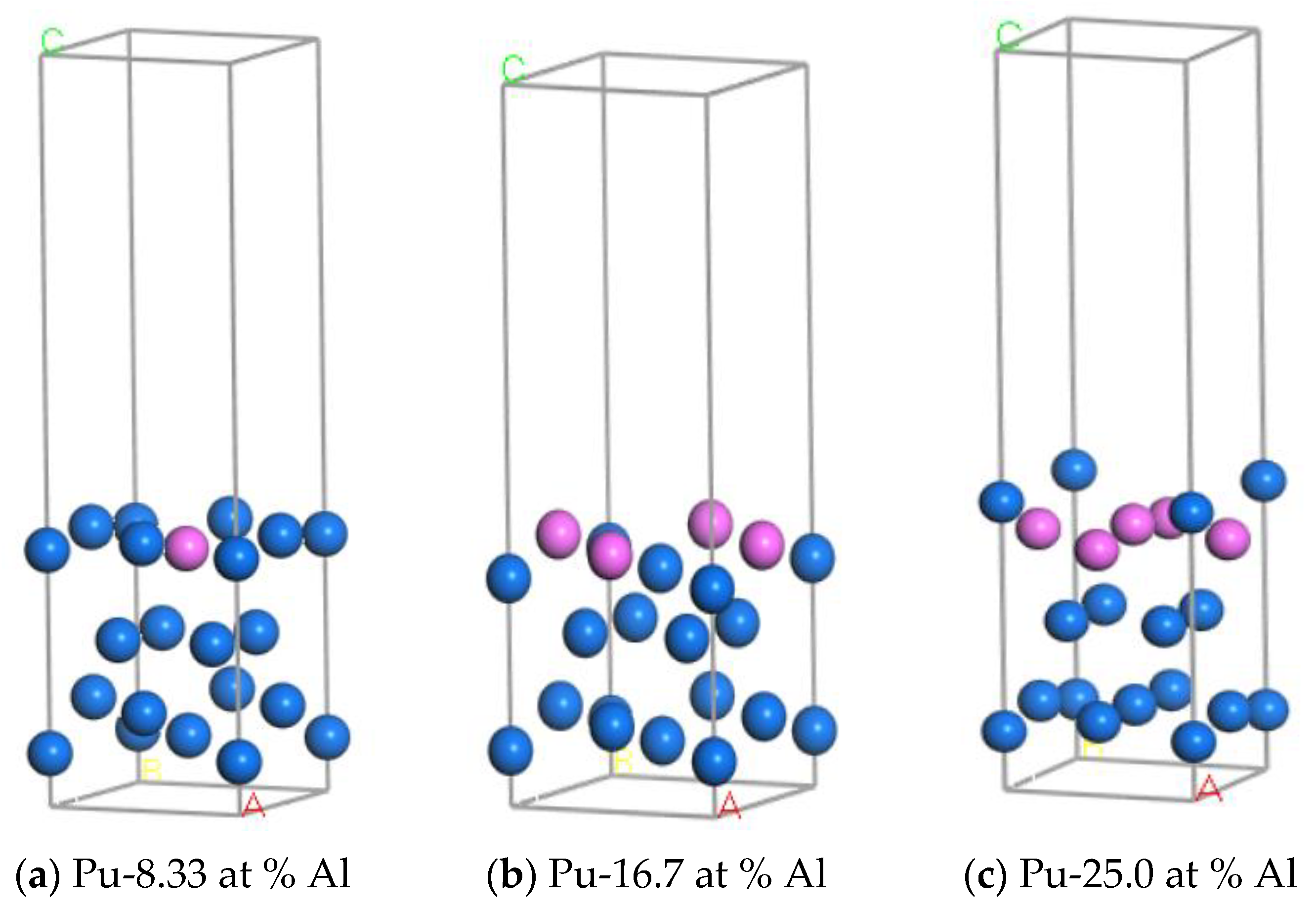
2.2.3. Structure Optimization and Surface Energy Calculation of Doped System
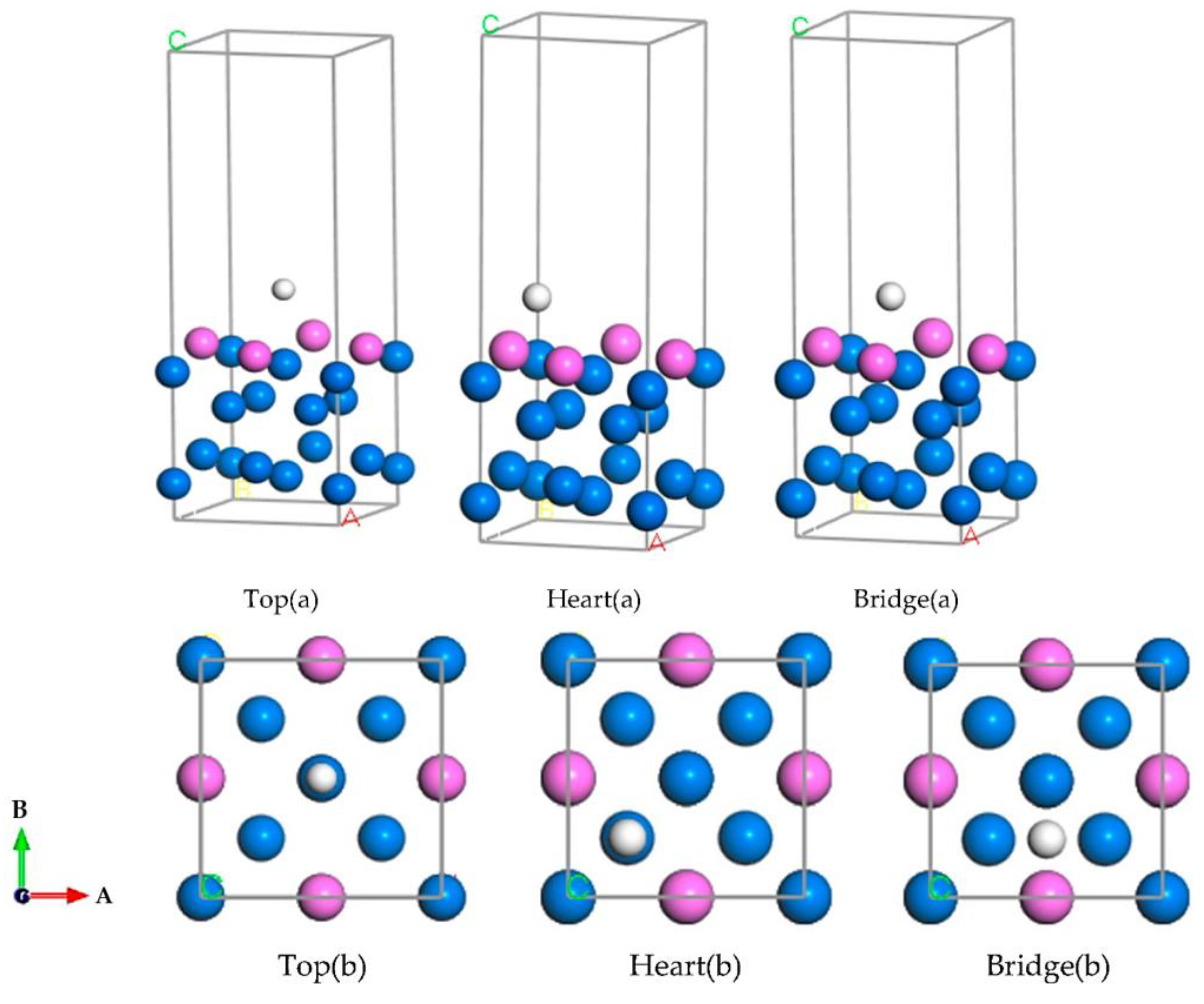
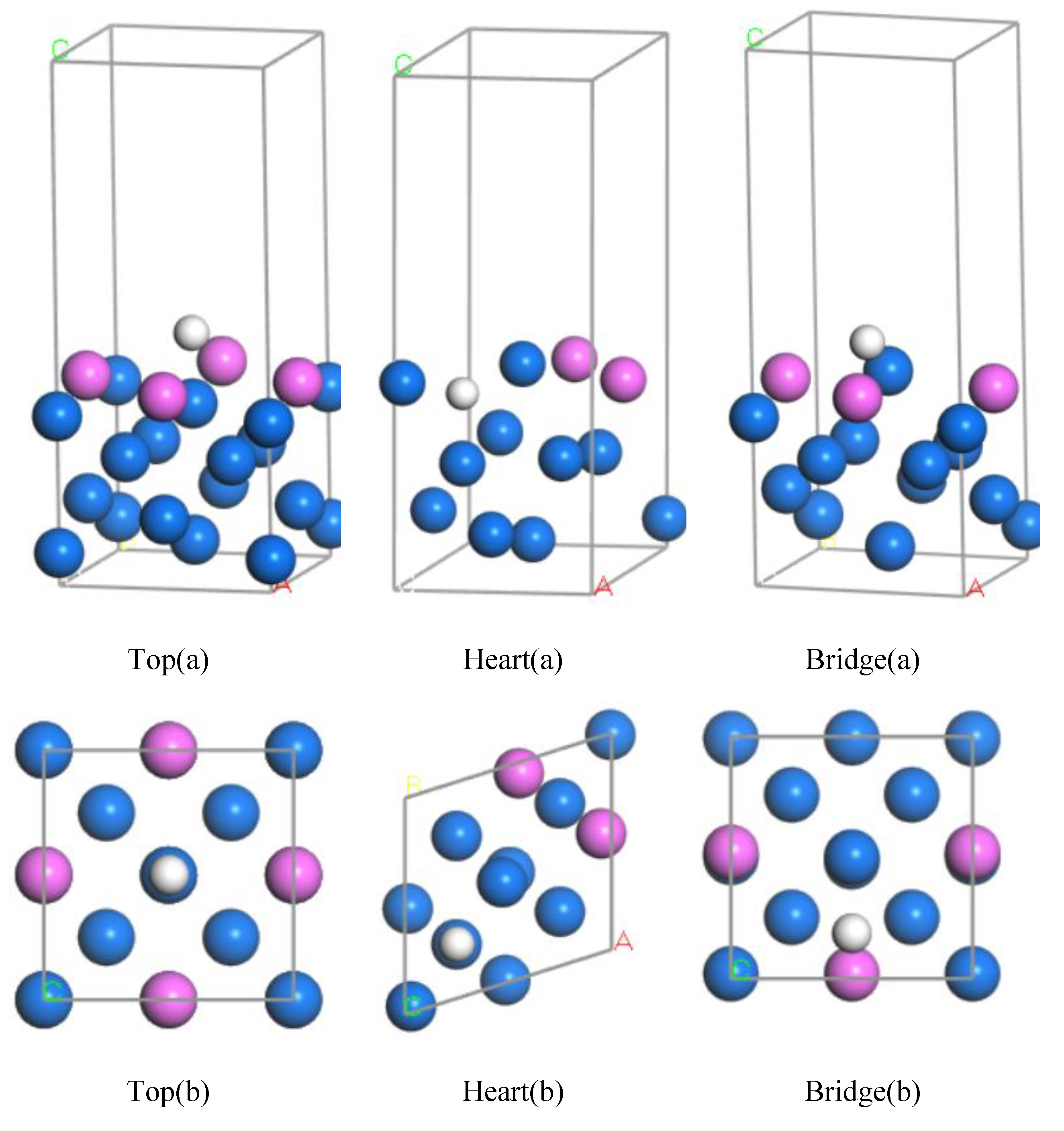
2.2.4. Adsorption Model and Calculation of H Atom
3. Results
3.1. Calculation of Adsorption Energy
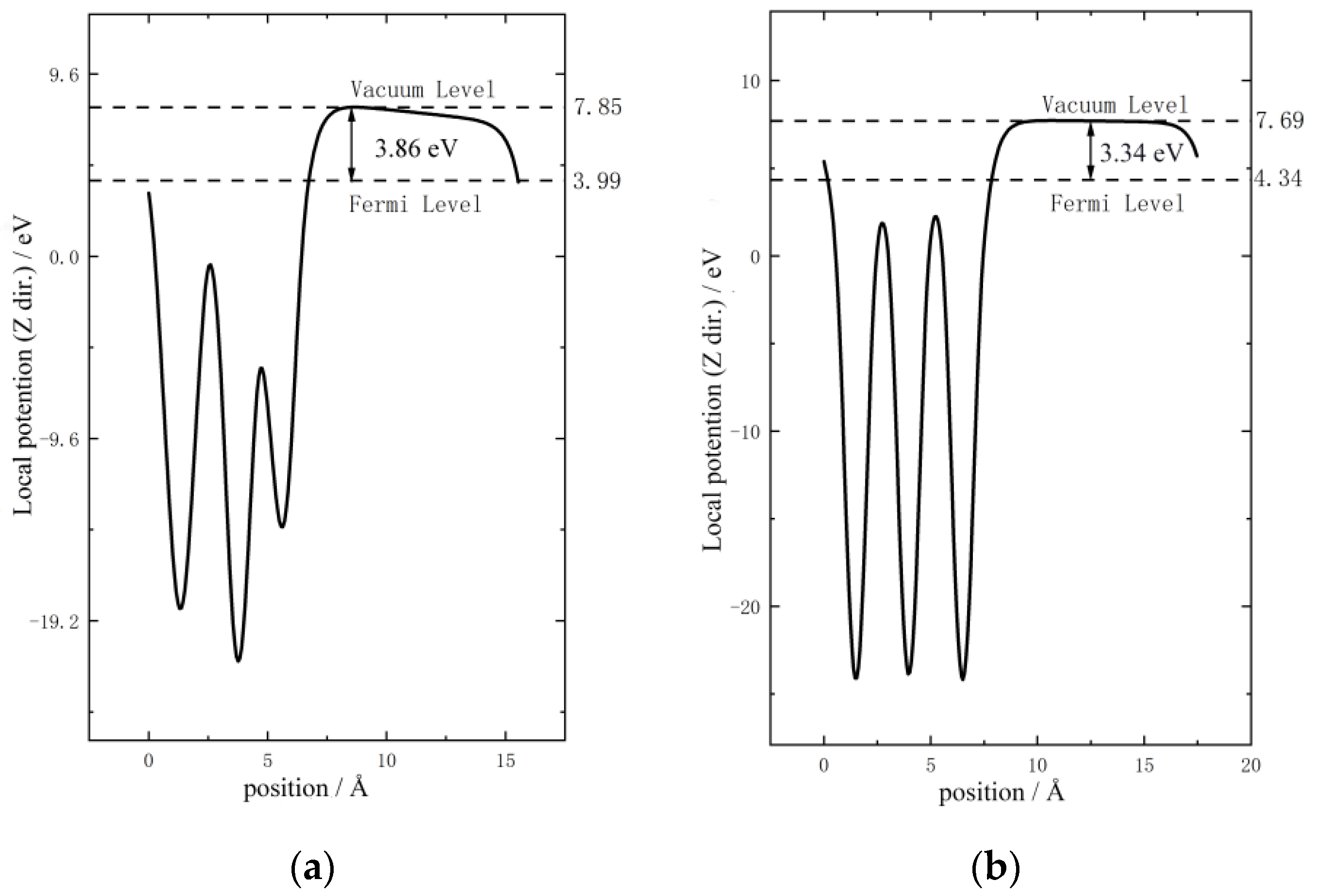
3.2. Surface Work Function Analysis
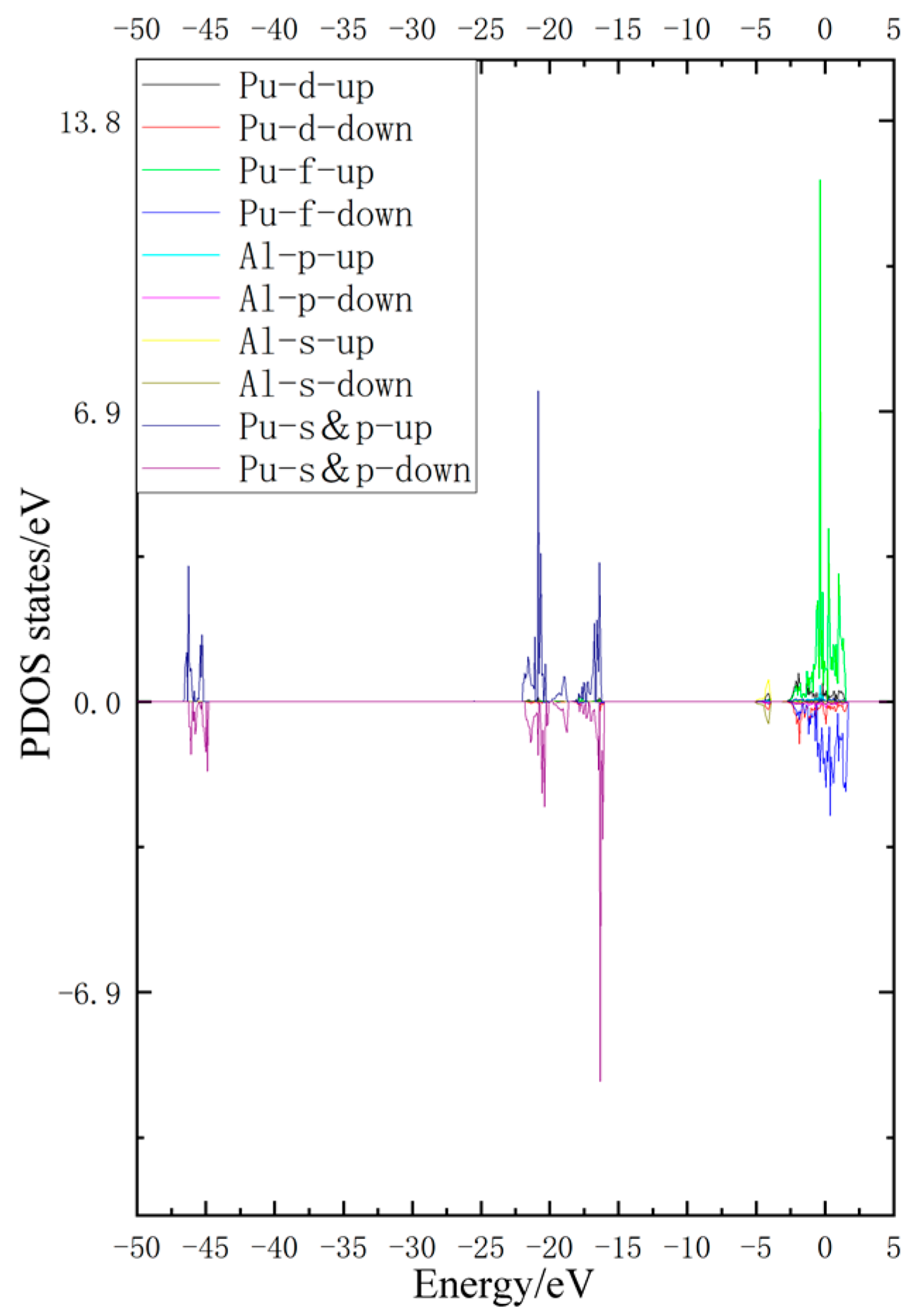
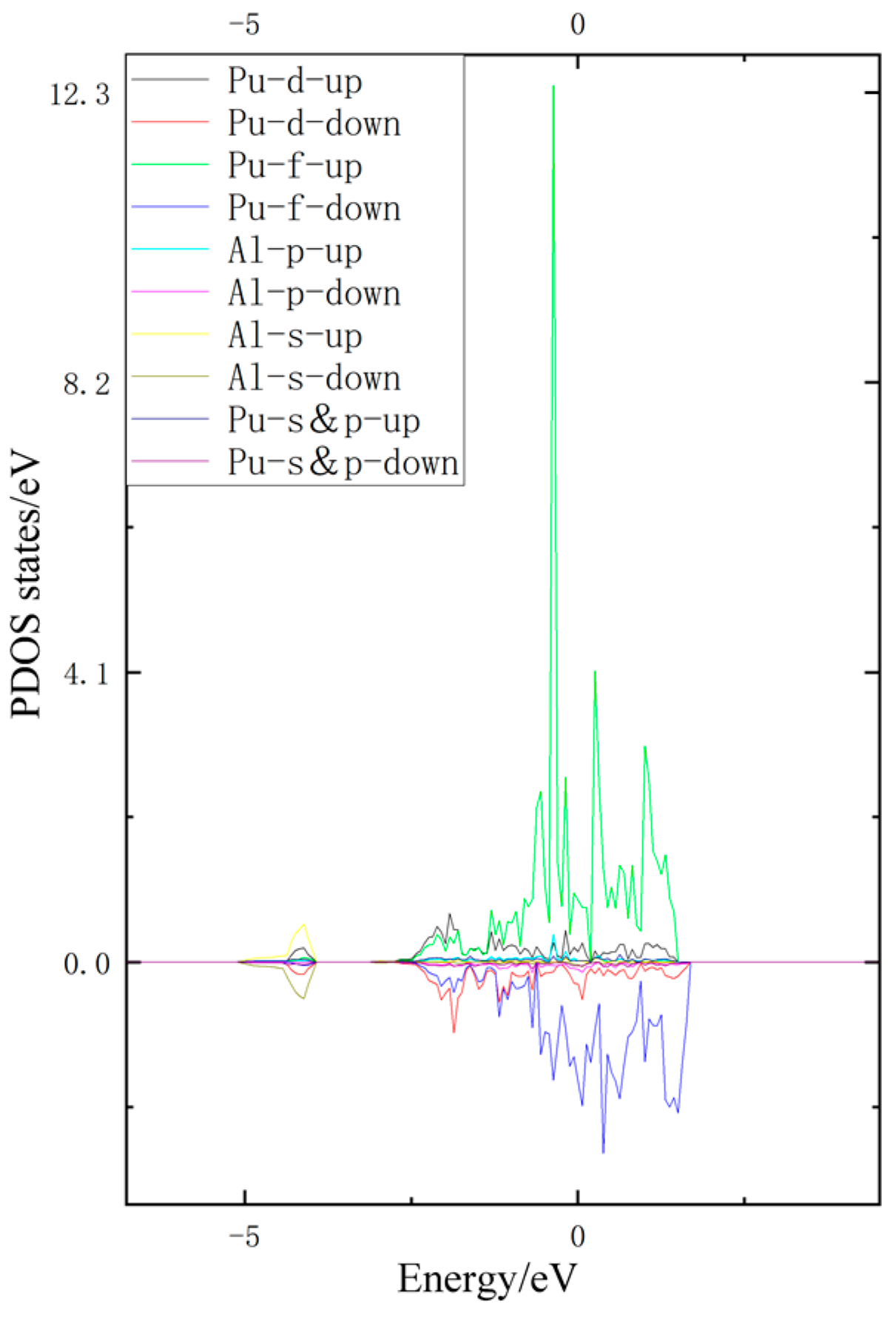
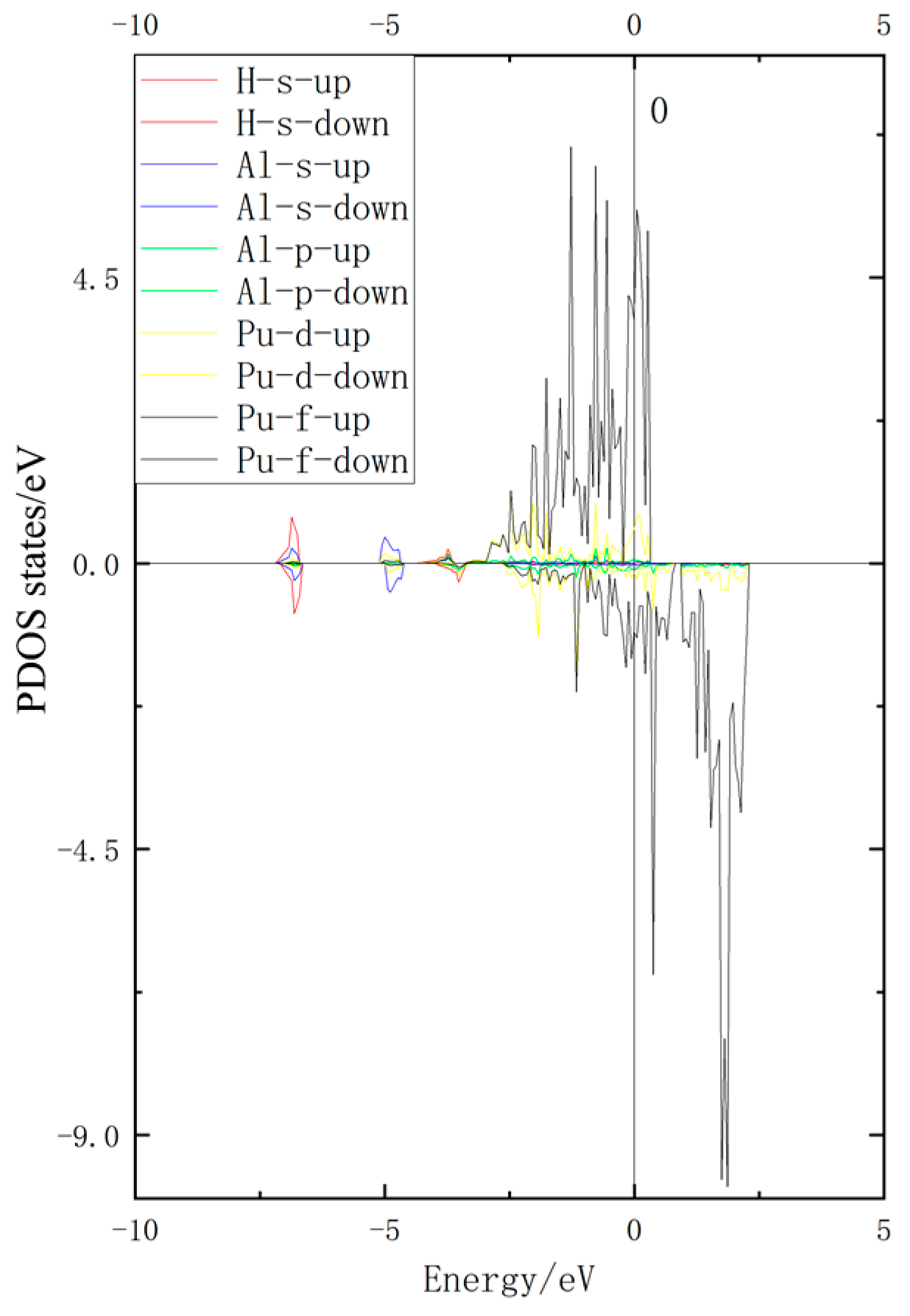
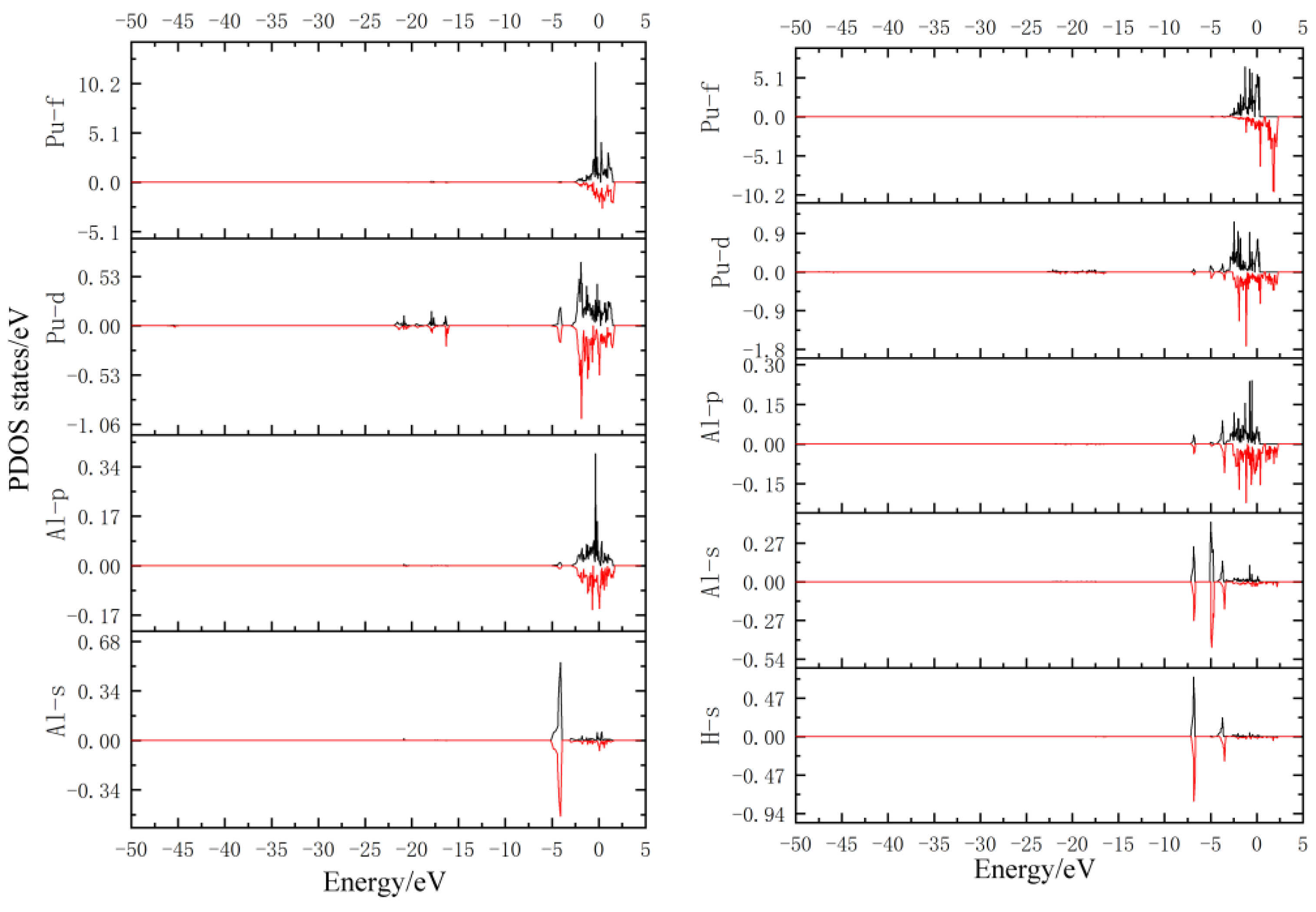
3.3. Density of States Analysis
3.4. Bader Charge Analysis
4. Conclusions
- The surface energy of the Pu-16.7 at % Al doping model is the lowest, 0.040925 eV, and the structure is the most stable when two aluminum atoms replace the intermittent position on the plutonium (100) surface.
- By DOS analysis of the unabsorbed doping model, the Pu/5f and Pu/6d electrons interact strongly with the Al/3p electrons, forming a stable structure. At −4 eV away from the Fermi level, the Al/3s and the Pu/6d are also hybridized to a lesser extent.
- According to the adsorption energy analysis of the adsorption model, among the three adsorption positions of the H atoms, the central position has the largest adsorption energy and the most stable adsorption. The energy of the whole system is the lowest, and the H atoms are chemisorbed.
- According to the density of states analysis, the H/1s hybridizes with the Al/3s and the Pu/6d, and the H atom interacts with both atoms.
- According to the surface work function analysis, the Pu-Al doped system is less likely to lose electrons than the δ-Pu system after co-adsorption of the H atoms, thus slowing down further chemical corrosion.
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Hecker, S.S. Plutonium—An Element Never at Equilibrium. Metall. Mater. Trans. A 2008, 39, 1585–1592. [Google Scholar] [CrossRef]
- Chen, Q.; Cao, K.; Ao, B.; Lai, X. First-principles Investigation of Adsorption of Rare Gas Atoms on Pu(100) Surface. At. Energy Sci. Technol. 2013, 47, 1931–1936. [Google Scholar]
- Boring, A.M.; Smith, J.L. Plutonium condensed-matter physics—A survey of theory and experiment. Los Alamos Sci. 2000, 26, 90–127. [Google Scholar]
- Jvan Ek, J.; Sterne, P.A.; Gonis, A. Phase stability of plutonium. Phys. Rev. B Condens. Matter 1993, 48, 16280. [Google Scholar] [CrossRef]
- Haschke, J.M.; Allen, T.H.; Stakebake, J.L. Reaction kinetics of plutonium with oxygen, water and humid air: Moisture enhancement of the corrosion rate. J. Alloys Compd. 1997, 28, 23–35. [Google Scholar] [CrossRef]
- Smith, C.S. Properties of Plutonium Metal. Phys. Rev. 1954, 94, 233–239. [Google Scholar] [CrossRef]
- Meng, D.; Luo, W.; Gan, L.; Chen, H. Density functional study of CO2 adsorption on Pu(100) surface. Acta Phys. Sin. 2009, 58, 8224–8229. [Google Scholar] [CrossRef]
- Qi, C.; Wang, T.; Wang, J.; Tao, S.; Qin, M. Study on Adsorption Behavior of CO2 on the δ-Pu(100) Surface Based on First-principles. Rare Met. Mater. Eng. 2021, 50, 2728–2737. [Google Scholar]
- Wei, H.; Hu, R.; Xiong, X.; Wang, G.; Song, H.; Luo, S. A first principle theory study of molecular hydrogen adsorption on δ-Pu(100) surface. J. Mol. Sci. 2010, 26, 37–41. [Google Scholar]
- Xiong, X.; Wei, H.; Luo, S.; Liu, G. A periodical density functional theory study of CO adsorption on δ-Pu(111) surface. J. Mol. Sci. 2009, 25, 50–54. [Google Scholar]
- Luo, W.; Wang, Q.; Ruan, W.; Xie, A.; Zhang, D.; Wang, X.; Gao, T. Study on Reaction Mechanism and Electron Density of Plutonium-hydrogen Interaction. At. Energy Sci. Technol. 2018, 52, 1345–1352. [Google Scholar]
- Hohenberg, P.; Kohn, W. Inhomogneous electron gas. Phys. Rev. 1964, 136, B864. [Google Scholar] [CrossRef] [Green Version]
- Kohn, W.; Sham, L.J. Quantum Density Oscillations in an Inhomogeneous Electron Gas. Phys. Rev. 1965, 137, 1697–1705. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projected augmented-wave method. Phys. Rev. B Condens. Matter 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Perdew, J.; Burke, P.K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1998, 77, 3865–3868. [Google Scholar] [CrossRef]
- Perdew, J.P.; Chevary, J.A.; Vosko, S.H.; Jackson, K.A.; Pederson, M.R.; Singh, D.J.; Fiolhais, C. Erratum: Atoms, molecules, solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation. Phys. Rev. B Condens. Matter 1993, 46, 6671–6687. [Google Scholar] [CrossRef]
- Huda, M.N.; Ray, A.K. Electronic structures and bonding of oxygen on plutonium layers. Eur. Phys. J. B Condens. Matter Complex Syst. 2004, 40, 337–346. [Google Scholar] [CrossRef]
- Li, G.; Lai, X.; Sun, Y. An All-electron FLAPW Study of Geometric and Electronic Structures for δ-Pu Monolayer. Acta Phys.-Chim. Sin. 2005, 21, 686–689. [Google Scholar]
- Li, L.; Zhu, M.; Zheng, G.; Li, Y.; Yang, Y.; Liu, Y.; Su, H. First-Principles Study on the Adsorption Behavior of O2 on the Surface of Plutonium Gallium System. Materials 2022, 15, 5035. [Google Scholar] [CrossRef]
- Wei, H. Study of Adsorption, Dissociation and Diffusion Processes of Atoms and Molecules on δ-Pu. Ph.D. Thesis, China Academy of Engineering Physics, Beijing, China, 2010. [Google Scholar]
- Eriksson, O.; Cox, L.E.; Cooper, B.R.; Wills, J.M.; Fernando, G.W.; Hao, Y.G.; Boring, A.M. Possibility of a δ-like surface for α-Pu: Theory. Phys. Rev. B Condens. Matter 1992, 46, 13576–13583. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Q.; Wang, J.; Hang, G.; Yu, W.; Wang, T. First-principles study on δ-Pu surface stability. AIP Adv. 2022, 12, 065001. [Google Scholar] [CrossRef]
- Bader, R.; Matta, C.F. Atomic charges are measurable quantum expectation values: A rebuttal of criticisms of QTAIM charges. J. Phys. Chem. A 2004, 108, 8385–8394. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Surface | Eslab (eV) | A (a2) | NAl | EAl (eV) | NPu | EPu (eV) | Esur (eV) |
|---|---|---|---|---|---|---|---|
| a | −148.683 | 45.8397 | 1 | −3.74957 | 11 | −13.683632 | 0.060935 |
| b | −141.323 | 36.8048 | 2 | −3.74957 | 10 | −13.683632 | 0.040925 |
| c | −129.265 | 28.9142 | 3 | −3.74957 | 9 | −13.683632 | 0.088821 |
| Site | Ebefore (eV) | Eafter (eV) | Eads (eV) |
|---|---|---|---|
| Top | −141.323357 | −145.244679 | 3.921322 |
| Heart | −141.323357 | −145.982763 | 4.659406 |
| Bridge | −141.323357 | −145.512543 | 4.189186 |
| Num. | Type | X | Y | Z | Charge | Transfer |
|---|---|---|---|---|---|---|
| 1 | Pu | 0 | 0 | 1.354808 | 16.40103 | 0.401032 |
| 2 | Pu | 0 | 0 | 5.548894 | 16.12665 | 0.12665 |
| 3 | Pu | 3.033303 | 3.033303 | 1.354808 | 16.40103 | 0.401032 |
| 4 | Pu | 3.033303 | 3.033303 | 5.548894 | 16.12665 | 0.12665 |
| 5 | Pu | 1.516652 | 1.516652 | 4.09589 | 15.92393 | −0.07607 |
| 6 | Pu | 4.549955 | 4.549955 | 4.09589 | 15.92393 | −0.07607 |
| 7 | Pu | 1.516652 | 4.549955 | 4.09589 | 15.92393 | −0.07607 |
| 8 | Pu | 4.549955 | 1.516652 | 4.09589 | 15.92393 | −0.07607 |
| 9 | Pu | 0 | 3.033303 | 2.071963 | 15.71913 | −0.28087 |
| 10 | Pu | 3.033303 | 0 | 2.071963 | 15.71913 | −0.28087 |
| 11 | Al | 0 | 3.033303 | 6.240134 | 2.905343 | −0.09466 |
| 12 | Al | 3.033303 | 0 | 6.240134 | 2.905343 | −0.09466 |
| Num. | Type | X | Y | Z | Charge | Transfer |
|---|---|---|---|---|---|---|
| 1 | Pu | 0.0128 | 0.0128 | 1.020247 | 15.66798 | −0.33202 |
| 2 | Pu | 6.174373 | 6.174373 | 5.424792 | 15.75725 | −0.24275 |
| 3 | Pu | 3.141484 | 3.141484 | 1.020247 | 15.66798 | −0.33202 |
| 4 | Pu | 3.28848 | 3.28848 | 5.424792 | 15.75725 | −0.24276 |
| 5 | Pu | 1.577142 | 1.577142 | 3.424649 | 16.03702 | 0.037016 |
| 6 | Pu | 4.731427 | 4.731427 | 3.803609 | 16.12979 | 0.129787 |
| 7 | Pu | 1.67714 | 4.631429 | 3.892134 | 16.19174 | 0.191736 |
| 8 | Pu | 4.631429 | 1.67714 | 3.892134 | 16.19174 | 0.191736 |
| 9 | Pu | 6.121118 | 3.341735 | 1.657537 | 16.1954 | 0.195396 |
| 10 | Pu | 3.341735 | 6.121118 | 1.657537 | 16.1954 | 0.195396 |
| 11 | Al | 0.204273 | 2.950012 | 5.869963 | 2.728273 | −0.27173 |
| 12 | Al | 2.950012 | 0.204273 | 5.869963 | 2.728273 | −0.27173 |
| 13 | H | 1.577142 | 1.577142 | 6.083913 | 1.751932 | 0.751932 |
| Num. | Type | X | Y | Z | Charge | Transfer |
|---|---|---|---|---|---|---|
| 1 | Pu | 5.979441 | 5.979441 | 1.503701 | 15.95619 | −0.04381 |
| 2 | Pu | 0.028456 | 0.028456 | 6.493678 | 15.81387 | −0.18613 |
| 3 | Pu | 2.993552 | 2.993552 | 1.503701 | 15.95967 | −0.04033 |
| 4 | Pu | 2.962541 | 2.962541 | 6.493678 | 15.81493 | −0.18507 |
| 5 | Pu | 1.495499 | 1.495499 | 3.945217 | 16.20650 | 0.206497 |
| 6 | Pu | 4.486496 | 4.486496 | 4.006787 | 16.16924 | 0.16924 |
| 7 | Pu | 1.495499 | 4.486496 | 3.968255 | 15.99755 | −0.00245 |
| 8 | Pu | 4.486496 | 1.495499 | 3.968255 | 15.99755 | −0.00245 |
| 9 | Pu | 5.979441 | 2.993552 | 1.503701 | 15.95793 | −0.04207 |
| 10 | Pu | 0.028456 | 2.962541 | 6.493678 | 15.81441 | −0.18559 |
| 11 | Pu | 2.993552 | 5.979441 | 1.503701 | 15.95793 | −0.04207 |
| 12 | Pu | 2.962541 | 0.028456 | 6.493678 | 15.81441 | −0.18559 |
| 13 | H | 1.495499 | 1.495499 | 7.648295 | 1.53980 | 0.53980 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Su, H.; Zhu, M.; Li, L.; Huang, H.; Yang, Y. First Principles Study on the Adsorption of Hydrogen Atoms on the Surface of Plutonium-Aluminum Systems. Crystals 2022, 12, 1592. https://doi.org/10.3390/cryst12111592
Su H, Zhu M, Li L, Huang H, Yang Y. First Principles Study on the Adsorption of Hydrogen Atoms on the Surface of Plutonium-Aluminum Systems. Crystals. 2022; 12(11):1592. https://doi.org/10.3390/cryst12111592
Chicago/Turabian StyleSu, Huan, Min Zhu, Longxian Li, Huang Huang, and Yang Yang. 2022. "First Principles Study on the Adsorption of Hydrogen Atoms on the Surface of Plutonium-Aluminum Systems" Crystals 12, no. 11: 1592. https://doi.org/10.3390/cryst12111592