
Structure and Properties of Cubic PuH2 and PuH3: A Density Functional Theory Study
 , and
, and
Abstract
:1. Introduction
2. Computational Methodology
3. Results and Discussion
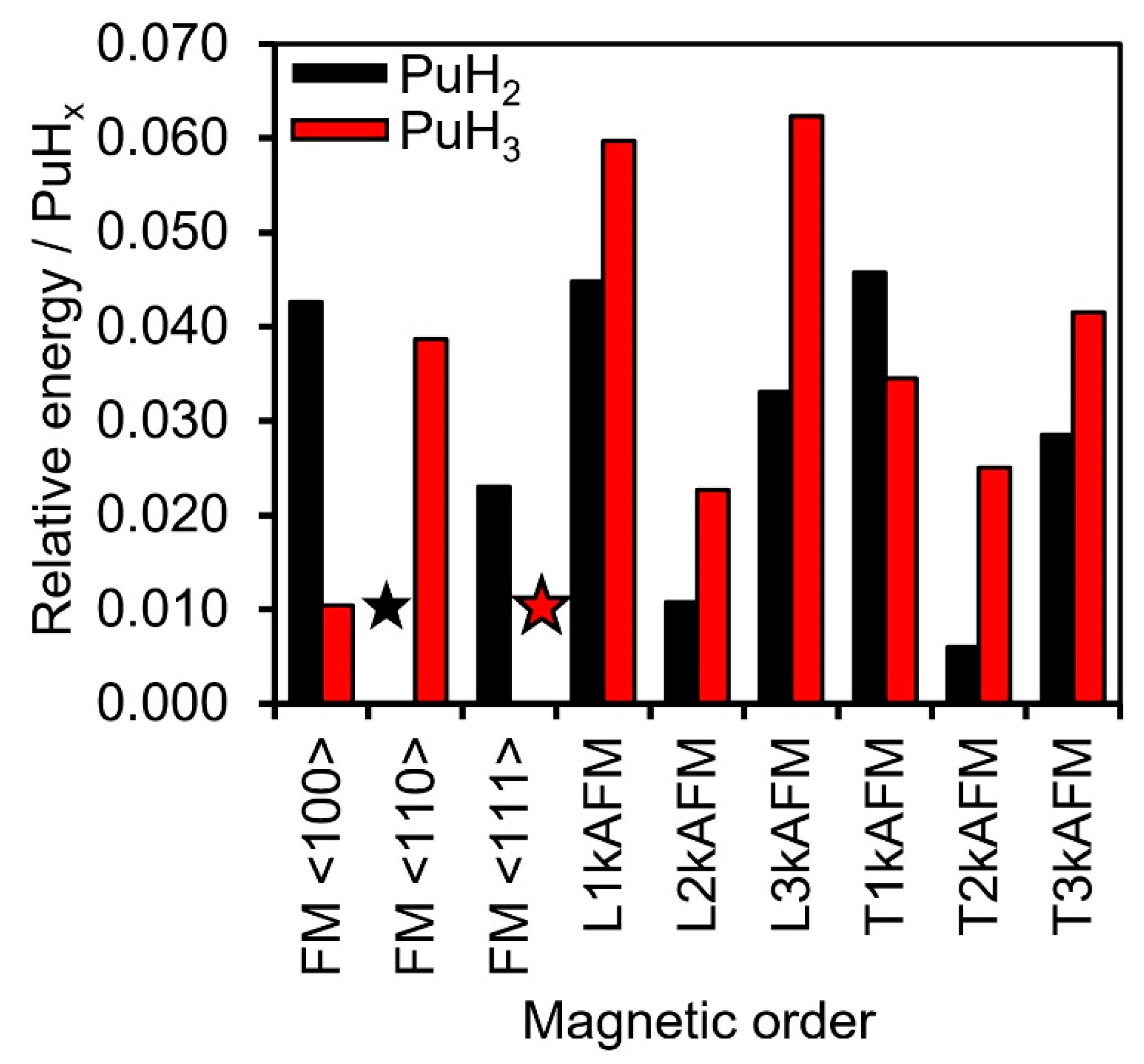
3.1. Thermodynamic Stability
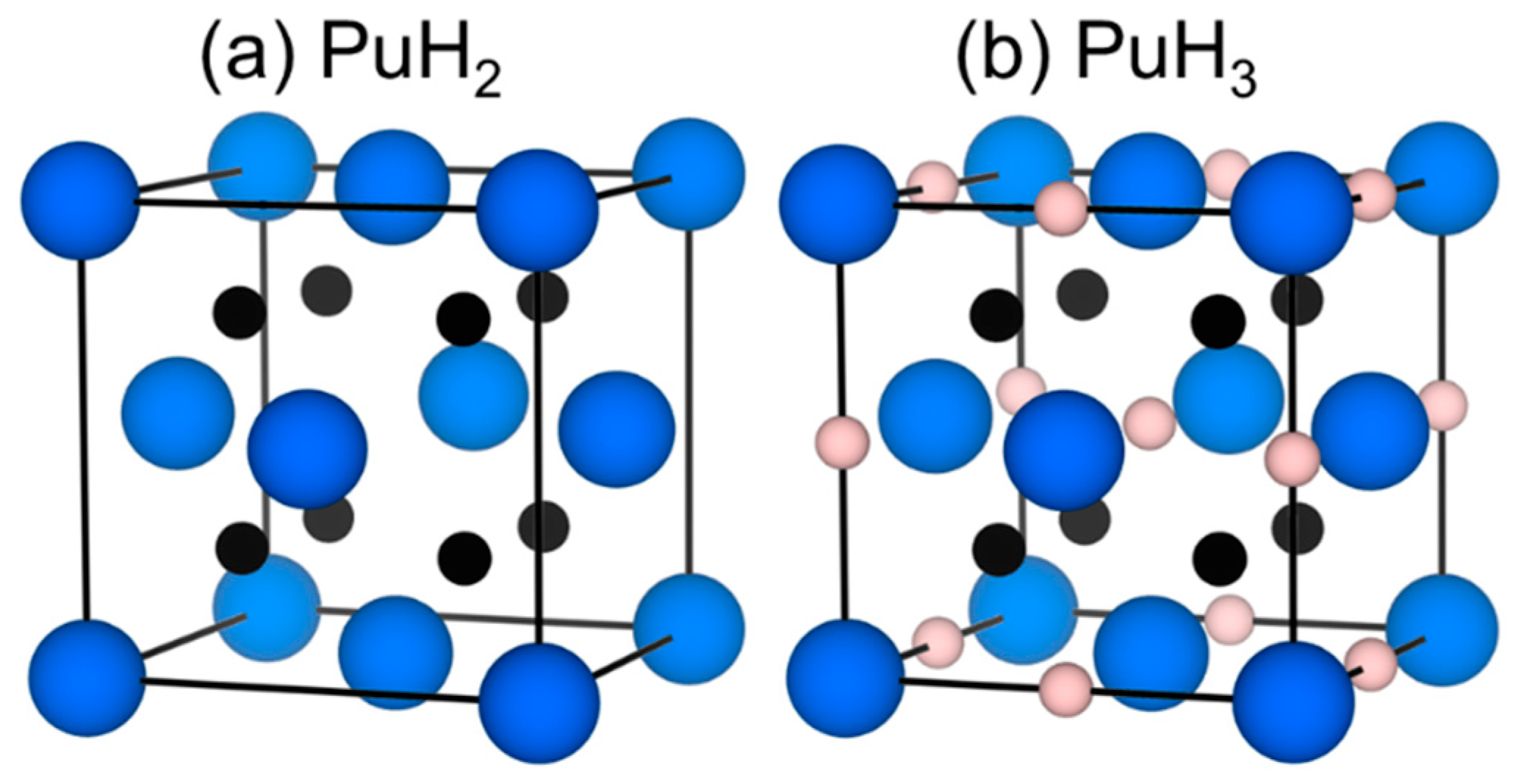
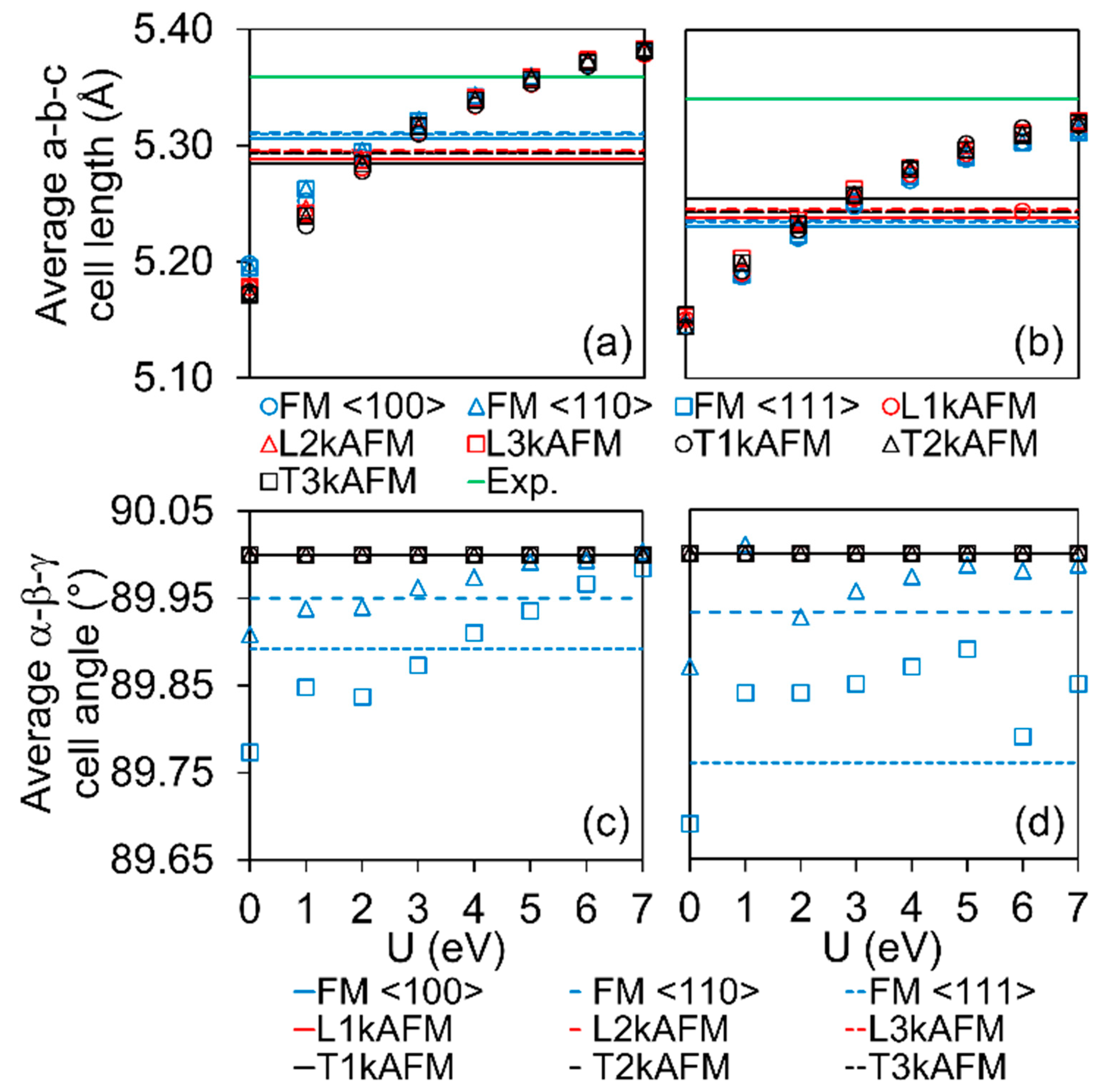
3.2. Symmetry and Structure
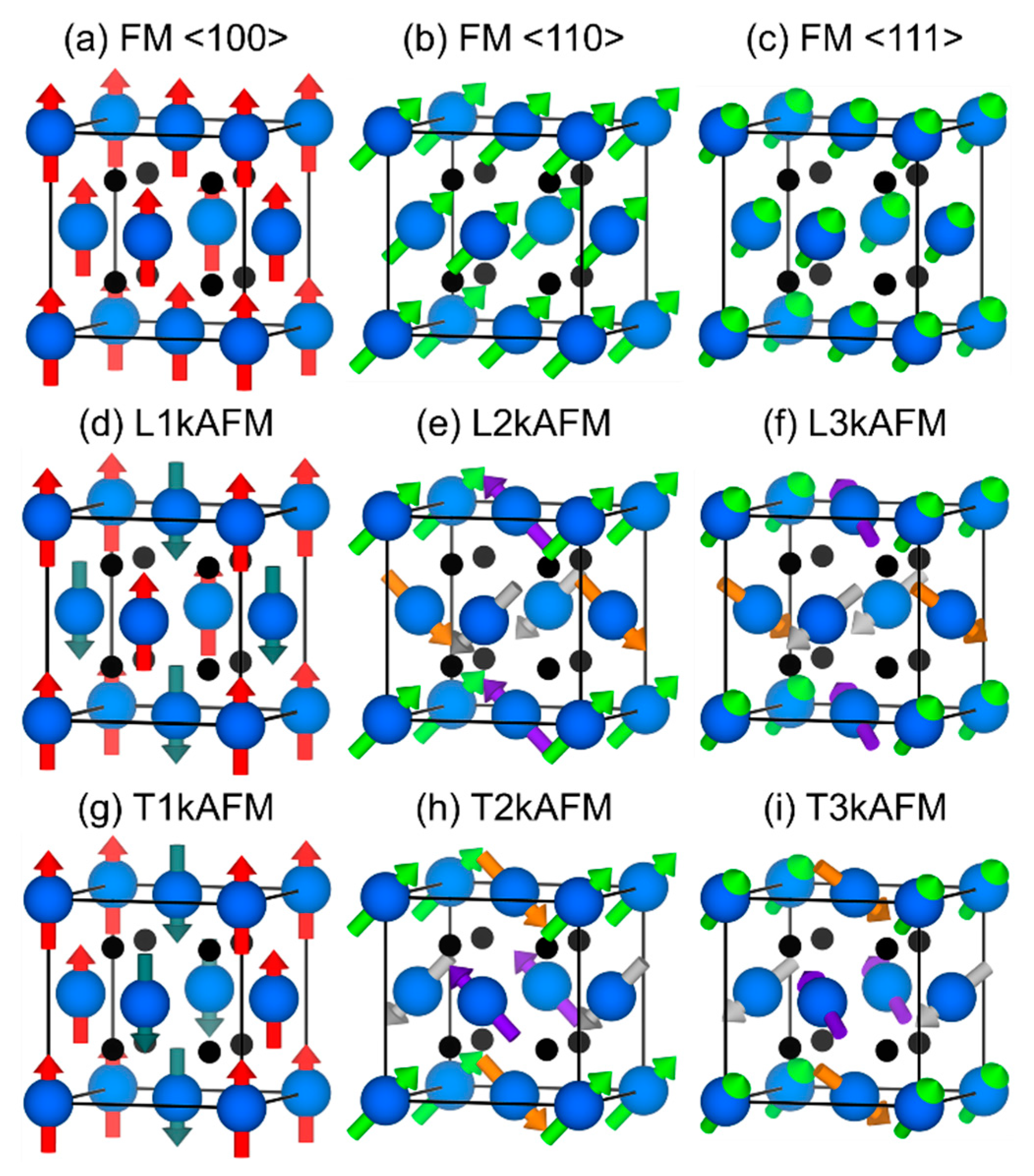
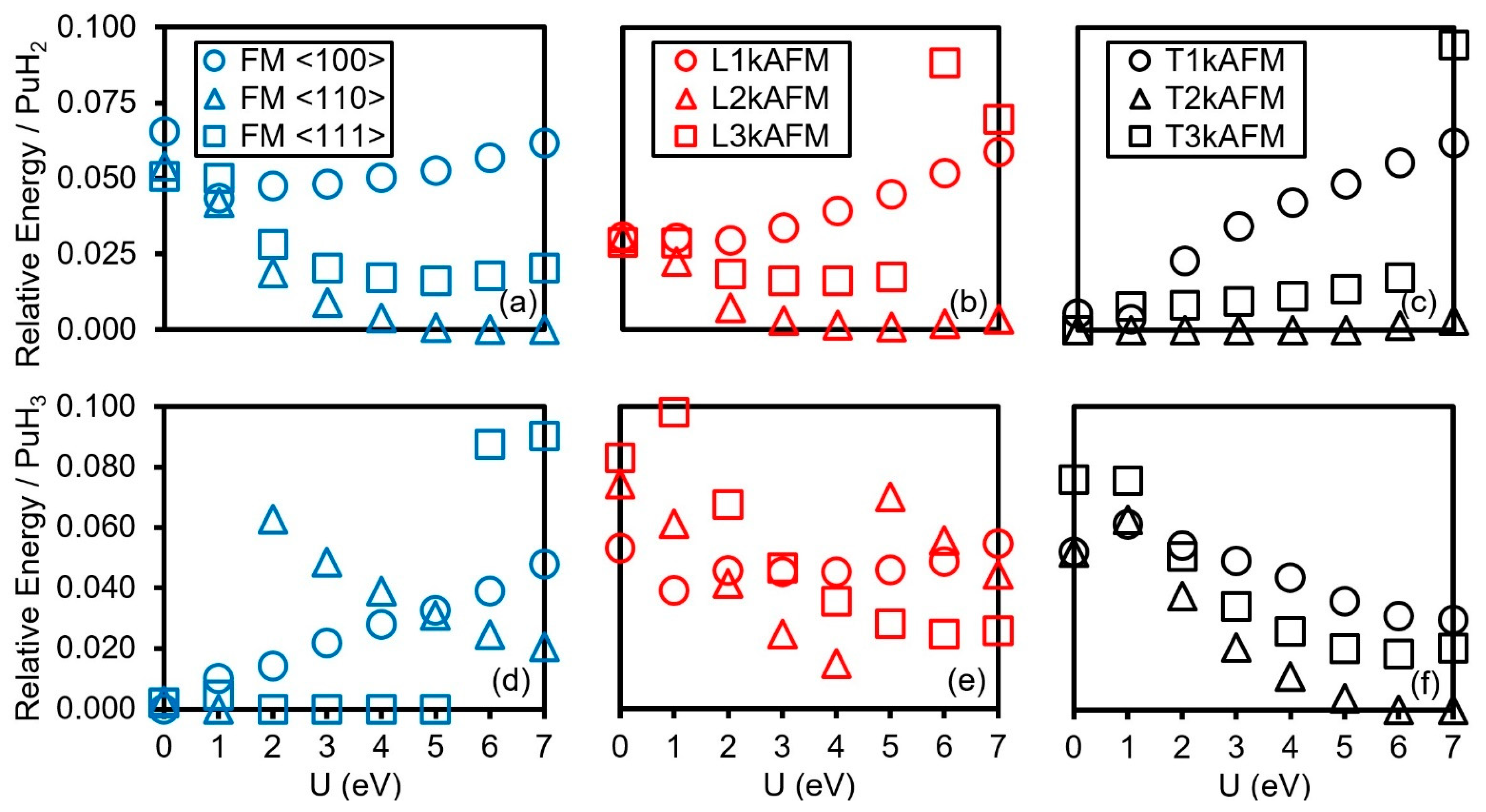
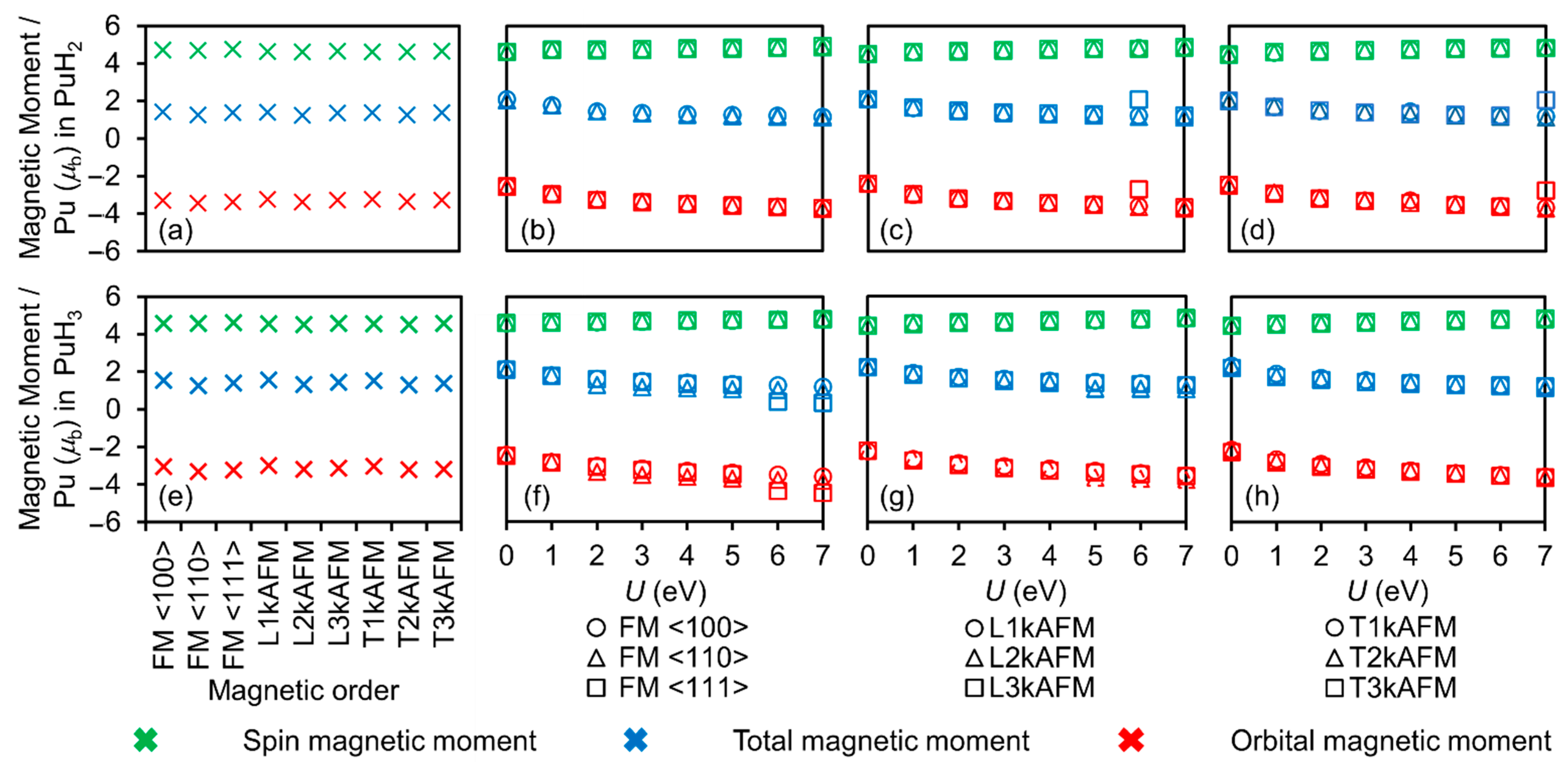
3.3. Magnetic Moments
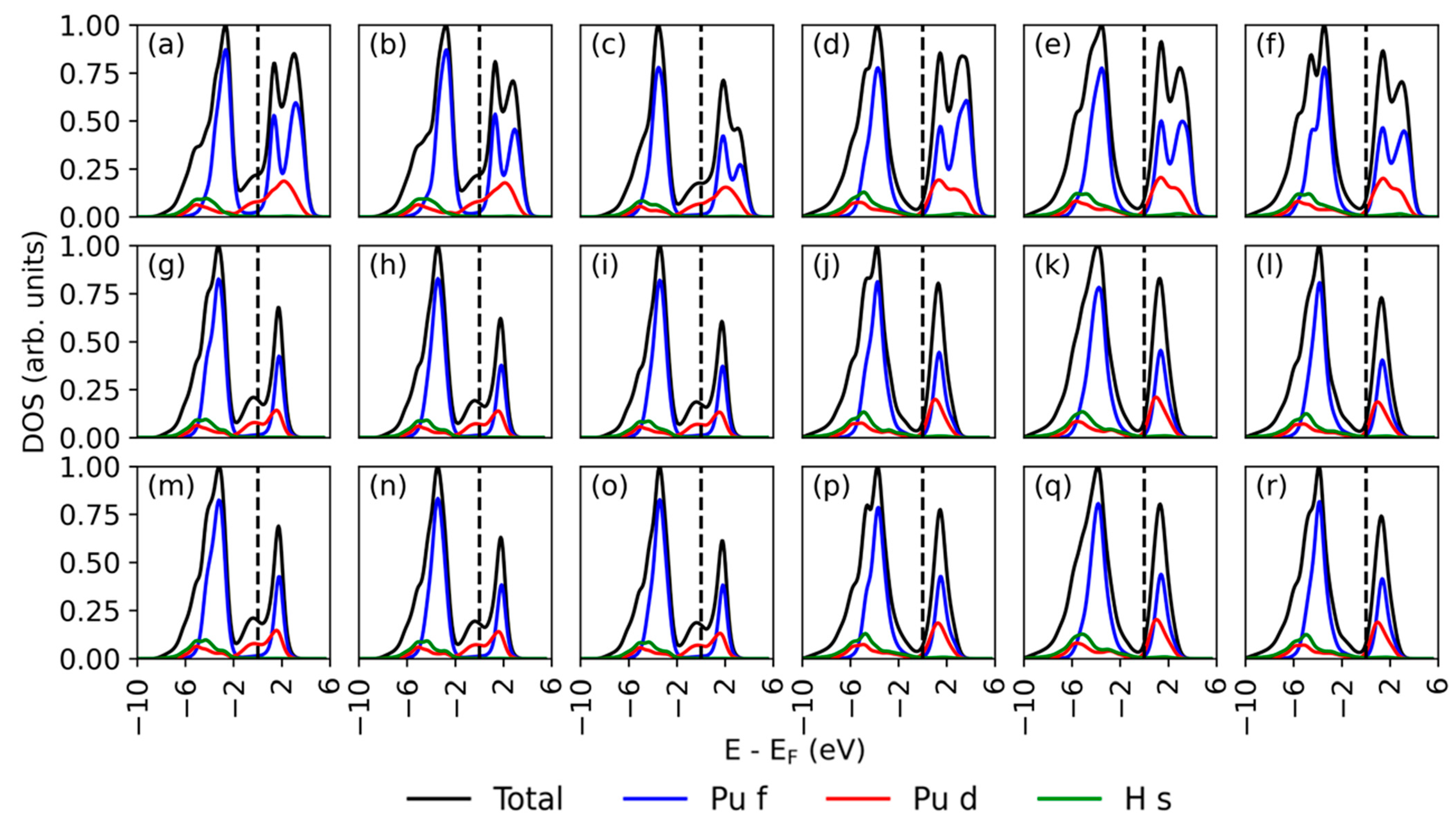
3.4. Electronic Structure
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Patterson, W. Fifty years of hopes and fears. Nature 2007, 449, 664. [Google Scholar] [CrossRef]
- Hyatt, N.C. Safe management of the UK separated plutonium inventory: A challenge of materials degradation. NPJ Mater. Degrad. 2020, 4, 1. [Google Scholar] [CrossRef]
- Nuclear Decommissioning Authority. Progress on Plutonium Consolidation, Storage and Disposition; Nuclear Decommissioning Authority: Moor Row, UK, 2019.
- Oetting, F.L. The Chemical Thermodynamic Properties of Plutonium Compounds. Chem. Rev. 1967, 67, 261. [Google Scholar] [CrossRef]
- Haschke, J.M.; Hodges, A.E.; Lucas, R.L. Equilibrium and structural properties of the PuH system. J. Less-Common Met. 1987, 133, 155. [Google Scholar] [CrossRef]
- Allen, T.H.; Haschke, J.M. Hydride-Catalyzed Corrosion of Plutonium by Air: Initiation by Plutonium Monoxide Monohydride; LA-13462-MS; Los Alamos National Lab.: Los Alamos, NM, USA, 1998. [Google Scholar] [CrossRef] [Green Version]
- Haschke, J.M.; Allen, T.H. Plutonium hydride, sesquioxide and monoxide monohydride: Pyrophoricity and catalysis of plutonium corrosion. J. Alloys Compd. 2001, 320, 58. [Google Scholar] [CrossRef]
- Hecker, S.S.; Martz, J.C. Plutonium Aging: From Mystery to Enigma. In Ageing Studies and Lifetime Extension of Materials; Springer: Boston, MA, USA, 2001; pp. 23–52. [Google Scholar]
- Ganda, F.; Greenspan, E. Incineration of Plutonium in PWR Using Hydride Fuel. In Proceedings of the 2005 International Conference on Advances in Nuclear Power Plants, ARWIF-2005, Oak Ridge, TN, USA, 6–18 February 2005. [Google Scholar]
- Ganda, F.; Greenspan, E. Plutonium recycling in hydride fueled PWR cores. Nucl. Eng. Des. 2009, 239, 1489. [Google Scholar] [CrossRef]
- Wicke, O.J. (Ed.) Plutonium Handbook: A Guide to the Technology; American Nuclear Society: La Grange Park, IL, USA, 1992. [Google Scholar]
- Flotow, H.E.; Haschke, J.M.; Yamauchi, S. Part 9—The Actinide Hydrides. In The Chemical Thermodynamics of Actinide Elements and Compounds; IAEA: Vienna, Austria, 1984; p. 115. [Google Scholar]
- Haschke, J.M.; Stakebake, J.L. Handling, Storage, and Disposition of Plutonium and Uranium. In The Chemistry of the Actinide and Transactinide Elements; Springer: Dordrecht, The Netherlands, 2007; pp. 3199–3272. [Google Scholar]
- Stakebake, J.L. The storage behavior of plutonium metal, alloys, and oxide: A review. J. Nucl. Mater. 1971, 38, 241. [Google Scholar] [CrossRef]
- Hecker, S.S. Plutonium—An element never at equilibrium. Metall. Mater. Trans. A Phys. Metall. Mater. Sci. 2008, 39, 1585. [Google Scholar] [CrossRef]
- Ai, J.; Liu, T.; Gao, T.; Ao, B. First-principles study of electronic structure and metal-insulator transition of plutonium dihydride and trihydride. Comput. Mater. Sci. 2012, 51, 127. [Google Scholar] [CrossRef]
- Ao, B.Y.; Wang, X.L.; Shi, P.; Chen, P.H.; Ye, X.Q.; Lai, X.C.; Gao, T. First-principles LDA + U calculations investigating the lattice contraction of face-centered cubic Pu hydrides. J. Nucl. Mater. 2012, 424, 183. [Google Scholar] [CrossRef]
- Li, S.; Ao, B.; Ye, X.; Qiu, R.; Gao, T. New Insights into the Crystal Structures of Plutonium Hydrides from First-Principles Calculations. J. Phys. Chem. C 2018, 122, 10103. [Google Scholar] [CrossRef]
- Ao, B.Y.; Ai, J.J.; Gao, T.; Wang, X.L.; Shi, P.; Chen, P.H.; Ye, X.Q. Metal-insulator transition of plutonium hydrides: DFT + U calculations in the FPLAPW basis. Chin. Phys. Lett. 2012, 29, 017102. [Google Scholar] [CrossRef]
- Ao, B.Y.; Shi, P.; Guo, Y.; Gao, T. The abnormal lattice contraction of plutonium hydrides studied by first-principles calculations. Chin. Phys. B 2013, 22, 037103. [Google Scholar] [CrossRef]
- Guo, Y.; Ai, J.J.; Gao, T.; Ao, B.Y. Structural, magnetic, electronic, and elastic properties of face-centered cubic PuHx (x = 2, 3): GGA (LSDA) + U + so. Chin. Phys. B 2013, 22, 057103. [Google Scholar] [CrossRef]
- Sudhapriyanga, G.; Santhosh, M.; Rajeswarapalanichamy, R.; Iyakutti, K. First Principles Study of Pressure Induced Phase Transition, Electronic and magnetic Properties of Plutonium trihydride. Int. J. Sci. Eng. Res. 2014, 5, 114. [Google Scholar]
- Zhang, C.; Yang, Y.; Zhang, P. Thermodynamics Study of Dehydriding Reaction on the PuO2(110) Surface from First Principles. J. Phys. Chem. C 2018, 122, 7790. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, P. Hydriding and dehydriding energies of PuHx from ab initio calculations. Phys. Lett. A 2015, 379, 1649. [Google Scholar] [CrossRef]
- Zheng, J.J.; Li, W.D.; Li, S.N.; Zhang, P.; Wang, B.T. Mechanical and thermodynamic properties of plutonium dihydride. J. Alloys Compd. 2018, 750, 258. [Google Scholar] [CrossRef]
- Zheng, J.J.; Wang, B.T.; Di Marco, I.; Li, W.D. Electronic structure and phase stability of plutonium hydrides: Role of Coulomb repulsion and spin-orbital coupling. Int. J. Hydrogen Energy 2014, 39, 13255. [Google Scholar] [CrossRef]
- Li, R.S.; Lu, X.; Wang, J.T.; Xin, D.Q.; Yao, X.G. A many-body perspective on dual 5f states in two plutonium hydrides. Chem. Phys. Lett. 2020, 740, 137079. [Google Scholar] [CrossRef]
- Li, S.; Guo, Y.; Ye, X.; Gao, T.; Ao, B. Structural, magnetic, and dynamic properties of PuH2+x (x = 0, 0.25, 0.5, 0.75, 1): A hybrid density functional study. Int. J. Hydrogen Energy 2017, 42, 30727–30737. [Google Scholar] [CrossRef]
- Muromura, T.; Yahata, T.; Ouchi, K.; Iseki, M. The variation of lattice parameter with hydrogen content of non-stoichiometric plutonium dihydride. J. Inorg. Nucl. Chem. 1972, 34, 171. [Google Scholar] [CrossRef]
- Willis, J.O.; Ward, J.W.; Smith, J.L.; Kosiewicz, S.T.; Haschke, J.M.; Hodges, A.E. Electronic properties and structure of the plutonium-hydrogen system. Phys. B C 1985, 130, 527. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef] [PubMed]
- Heyd, J.; Scuseria, G.E.; Ernzerhof, M. Hybrid functionals based on a screened Coulomb potential. J. Chem. Phys. 2003, 118, 8207. [Google Scholar] [CrossRef] [Green Version]
- Steiner, S.; Khmelevskyi, S.; Marsmann, M.; Kresse, G. Calculation of the magnetic anisotropy with projected-augmented-wave methodology and the case study of disordered Fe1-xCox alloys. Phys. Rev. B 2016, 93, 224425. [Google Scholar] [CrossRef]
- Csonka, G.I.; Perdew, J.P.; Ruzsinszky, A.; Philipsen, P.H.T.; Lebègue, S.; Paier, J.; Vydrov, O.A.; Ángyán, J.G. Assessing the performance of recent density functionals for bulk solids. Phys. Rev. B 2009, 79, 155107. [Google Scholar] [CrossRef]
- Schimka, L.; Harl, J.; Kresse, G. Improved hybrid functional for solids: The HSEsol functional. J. Chem. Phys. 2011, 134, 024116. [Google Scholar] [CrossRef]
- Liechtenstein, A.I.; Anisimov, V.I.; Zaanen, J. Density-functional theory and strong interactions: Orbital ordering in Mott-Hubbard insulators. Phys. Rev. B 1995, 52, R5467. [Google Scholar] [CrossRef] [Green Version]
- Dudarev, S.; Botton, G. Electron-energy-loss spectra and the structural stability of nickel oxide: An LSDA + U study. Phys. Rev. B 1998, 57, 1505. [Google Scholar] [CrossRef]
- Moxon, S.; Symington, A.R.; Tse, J.S.; Dawson, J.; Flitcroft, J.M.; Parker, S.C.; Cooke, D.J.; Harker, R.M.; Molinari, M. The energetics of carbonated PuO2 surfaces affects nanoparticle morphology: A DFT+: U study. Phys. Chem. Chem. Phys. 2020, 22, 7728. [Google Scholar] [CrossRef] [PubMed]
- Pegg, J.T.; Shields, A.E.; Storr, M.T.; Wills, A.S.; Scanlon, D.O.; de Leeuw, N.H. Hidden magnetic order in plutonium dioxide nuclear fuel. Phys. Chem. Chem. Phys. 2018, 20, 20943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pegg, J.T.; Aparicio-Anglès, X.; Storr, M.; de Leeuw, N.H. DFT + U study of the structures and properties of the actinide dioxides. J. Nucl. Mater. 2017, 492, 269. [Google Scholar] [CrossRef]
- Desgranges, L.; Ma, Y.; Garcia, P.; Baldinozzi, G.; Siméone, D.; Fischer, H.E. What Is the Actual Local Crystalline Structure of Uranium Dioxide, UO2? A New Perspective for the Most Used Nuclear Fuel. Inorg. Chem. 2017, 56, 321. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Aldred, A.T.; Cinader, G.; Lam, D.J.; Weber, L.W. Magnetic properties of neptunium and plutonium hydrides. Phys. Rev. B 1979, 19, 300. [Google Scholar] [CrossRef]
- Kim, J.W.; Mun, E.D.; Baiardo, J.P.; Smith, A.I.; Richmond, S.; Mitchell, J.; Schwartz, D.; Zapf, V.S.; Mielke, C.H. Detecting low concentrations of plutonium hydride with magnetization measurements. J. Appl. Phys. 2015, 117, 053905. [Google Scholar] [CrossRef]
- Cinader, G.; Zamir, D.; Hadari, Z. NMR study of the plutonium hydride system. Phys. Rev. B 1976, 14, 912. [Google Scholar] [CrossRef]
- Yang, X.; Yang, Y.; Lu, Y.; Sun, Z.; Hussain, S.; Zhang, P. First-principles GGA + U calculation investigating the hydriding and diffusion properties of hydrogen in PuH2+x, 0 ≤ x ≤ 1. Int. J. Hydrogen Energy 2018, 43, 13632. [Google Scholar] [CrossRef]
- Dorado, B.; Amadon, B.; Freyss, M.; Bertolus, M. DFT + U calculations of the ground state and metastable states of uranium dioxide. Phys. Rev. B 2009, 79, 235125. [Google Scholar] [CrossRef]
- Meredig, B.; Thompson, A.; Hansen, H.A.; Wolverton, C.; Van De Walle, A. Method for locating low-energy solutions within DFT + U. Phys. Rev. B 2010, 82, 195128. [Google Scholar] [CrossRef] [Green Version]
- Mulford, R.N.R.; Sturdy, G.E. The Plutonium-Hydrogen System. I. Plutonium Dihydride and Dideuteride. J. Am. Chem. Soc. 1955, 77, 3449. [Google Scholar] [CrossRef]
- Mulford, R.N.R.; Sturdy, G.E. The Plutonium-Hydrogen System. II. Solid Solution of Hydrogen in Plutonium Dihydride. J. Am. Chem. Soc. 1956, 78, 3897. [Google Scholar] [CrossRef]
- Ward, J.W. Electronic structure and bonding in transuranics: Comparison with lanthanides. J. Less-Common Met. 1983, 93, 279. [Google Scholar] [CrossRef] [Green Version]
- Haschke, J.M.; Hodges, A.E.; Smith, C.M.; Oetting, F.L. Equilibria and thermodynamic properties of the plutonium-hydrogen system. J. Less-Common Met. 1980, 73, 41. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Magnetic Order | PBEsol+U+SOC—U (eV) | HSE06sol +SOC | |||||||
|---|---|---|---|---|---|---|---|---|---|
| 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | ||
| FM <100> | I4/mmm | I4/mmm | I4/mmm | I4/mmm | I4/mmm | I4/mmm | I4/mmm | I4/mmm | I4/mmm |
| FM <110> | P | C2/m | C2/m | C2/m | C2/m | C2/m | C2/m | C2/m | Immm (C2/m) |
| FM <111> | Rm | Rm | Rm | Rm | Rm | Rm | Rm | Rm | Rm |
| L1kAFM | I4/mmm | I4/mmm | I4/mmm | I4/mmm | I4/mmm | I4/mmm | I4/mmm | I4/mmm | I4/mmm |
| L2kAFM | I4/mmm | I4/mmm | I4/mmm | I4/mmm | I4/mmm | I4/mmm | I4/mmm | I4/mmm | I4/mmm |
| L3kAFM | Fmm | Fmm | Fmm | Fmm | Fmm | Fmm | Fmm | Fmm | Fmm |
| T1kAFM | Fmmm | Fmmm | Fmmm | Fmmm | Fmmm | Fmmm | Fmmm | Fmmm | Fmmm |
| T2kAFM | Cmca | Cmca | Cmca (Pbca) | Cmca | Cmca | Cmca | Cmca | Cmca | Pbca (Fmmm) |
| T3kAFM | Pa | Pa | Pa | Pa | Pa | Pa | Pa | Pa | Pa |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smith, T.; Moxon, S.; Cooke, D.J.; Gillie, L.J.; Harker, R.M.; Storr, M.T.; da Silva, E.L.; Molinari, M. Structure and Properties of Cubic PuH2 and PuH3: A Density Functional Theory Study. Crystals 2022, 12, 1499. https://doi.org/10.3390/cryst12101499
Smith T, Moxon S, Cooke DJ, Gillie LJ, Harker RM, Storr MT, da Silva EL, Molinari M. Structure and Properties of Cubic PuH2 and PuH3: A Density Functional Theory Study. Crystals. 2022; 12(10):1499. https://doi.org/10.3390/cryst12101499
Chicago/Turabian StyleSmith, Thomas, Samuel Moxon, David J. Cooke, Lisa J. Gillie, Robert M. Harker, Mark T. Storr, Estelina Lora da Silva, and Marco Molinari. 2022. "Structure and Properties of Cubic PuH2 and PuH3: A Density Functional Theory Study" Crystals 12, no. 10: 1499. https://doi.org/10.3390/cryst12101499