

A Lection in Humbleness: Crystallization of Chiral and Zwitterionic APIs Baclofen and Phenibut

Abstract
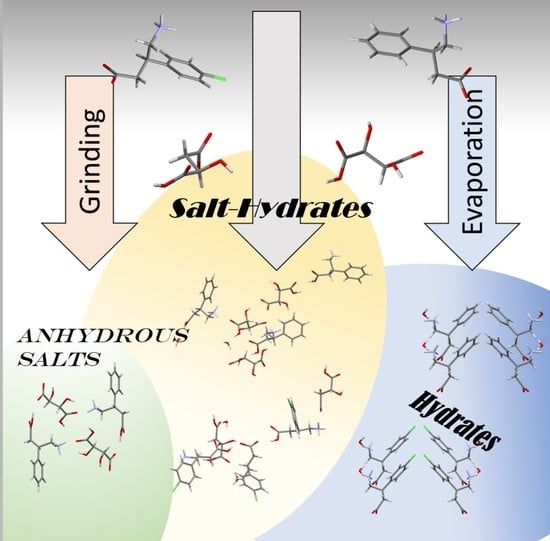
:
1. Introduction
2. Materials and Methods
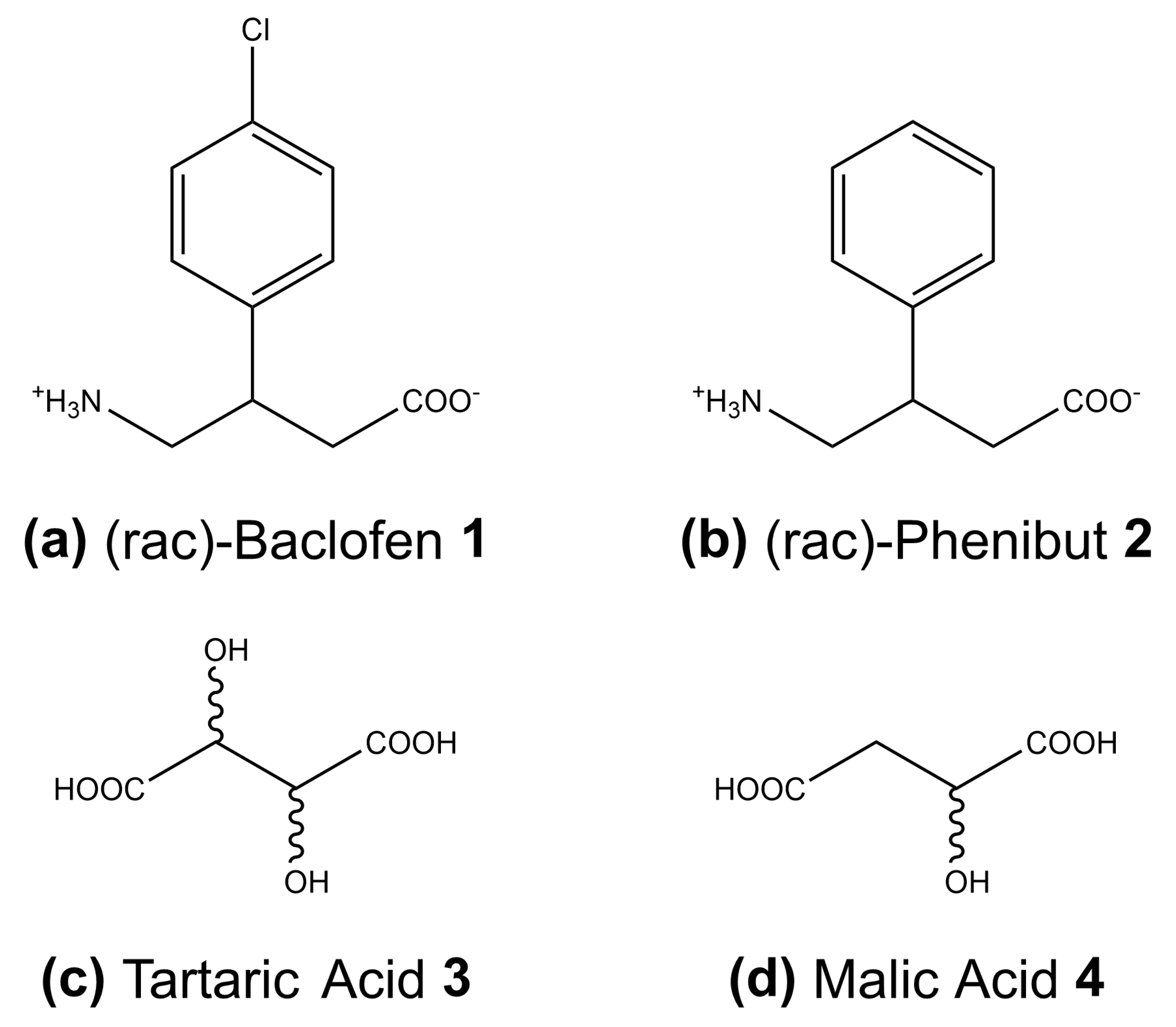
2.1. Chemicals
2.2. Sample Preparation
2.3. Powder X-ray Diffraction (PXRD)
2.4. Single-Crystal X-ray Diffraction (SC-XRD)
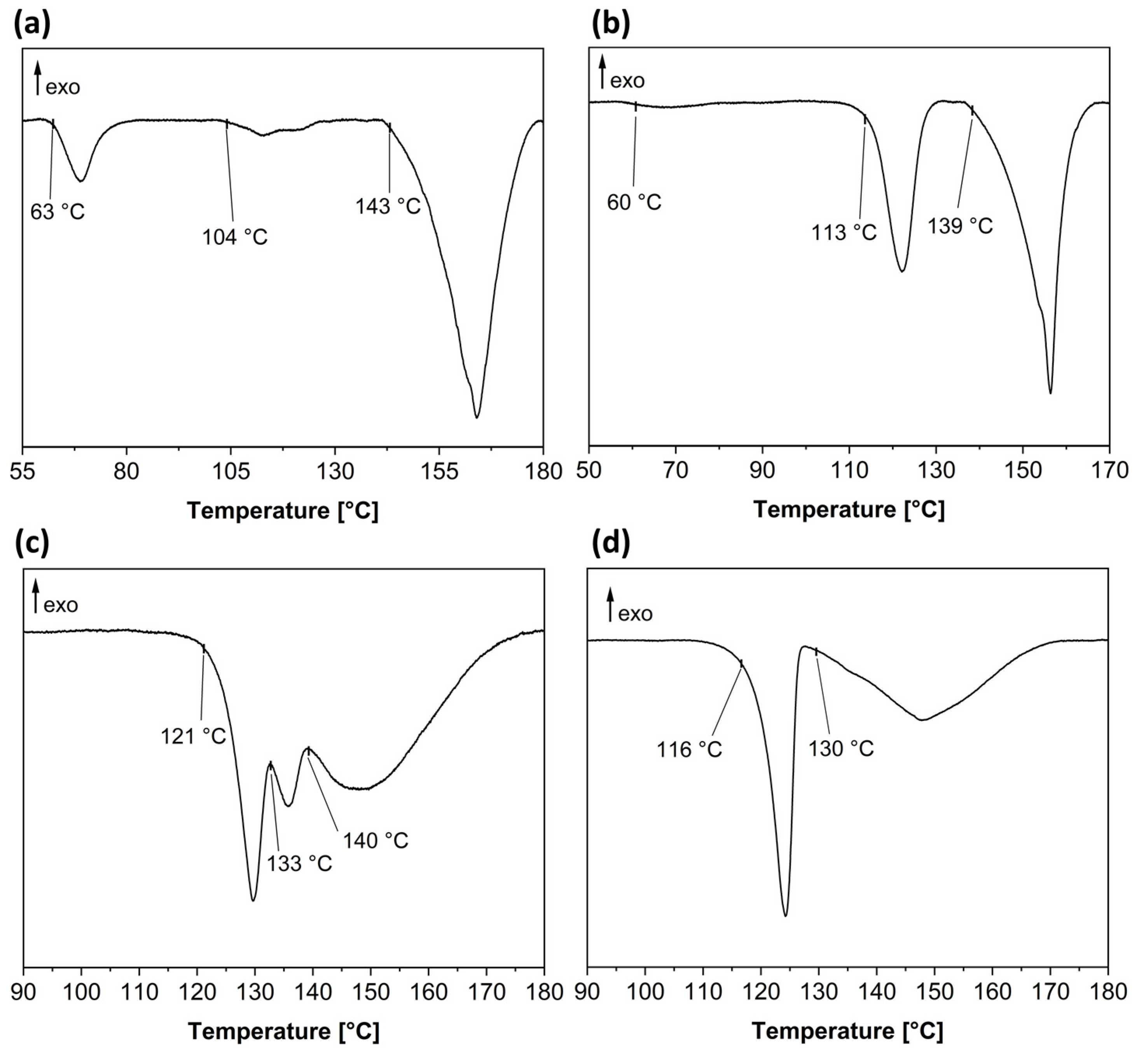
2.5. Thermal Analysis
2.6. IR-Spectra
3. Results
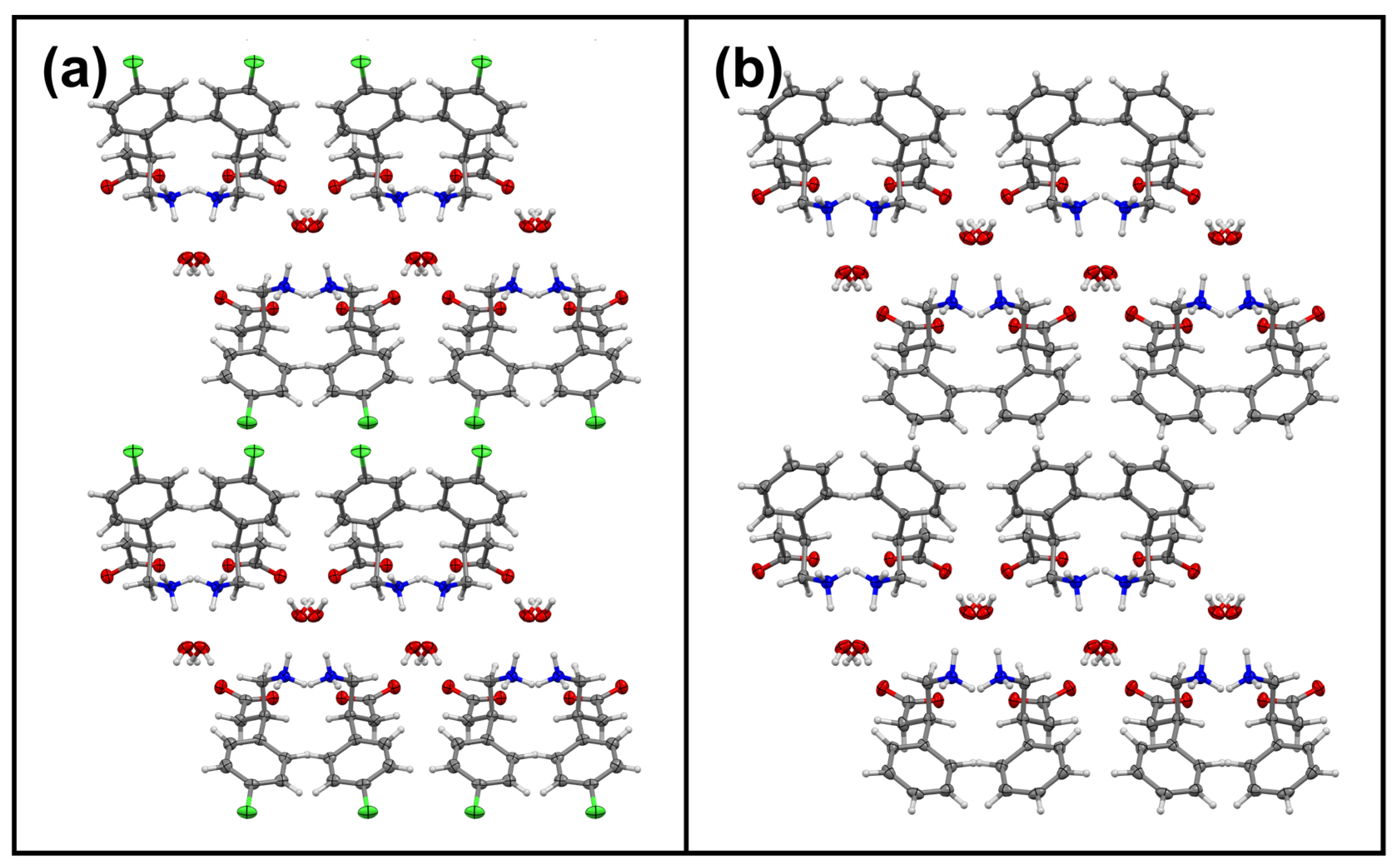
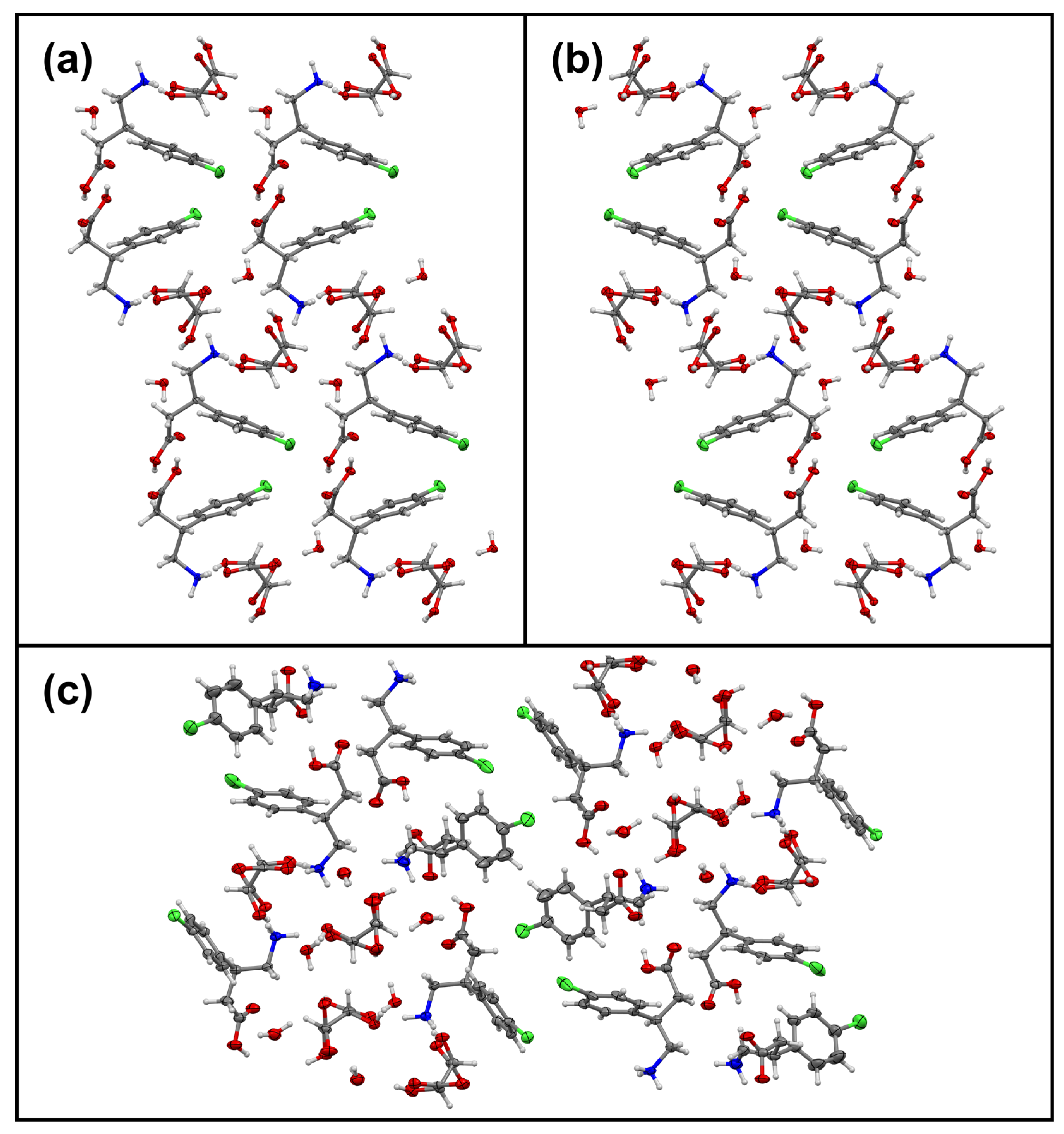
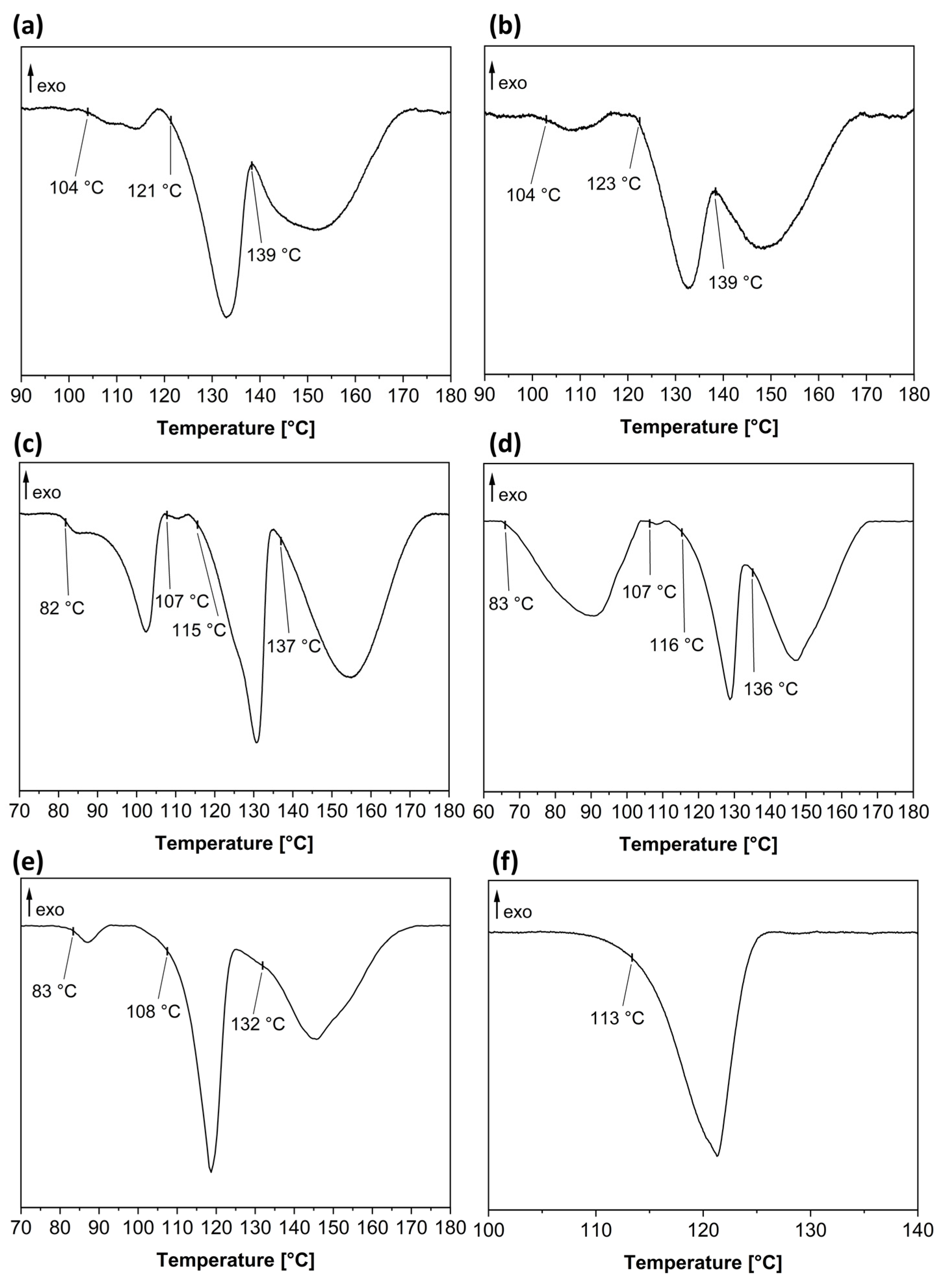
3.1. Overview of All Received Single Crystals and Pure Hydrates of Baclofen and Phenibut
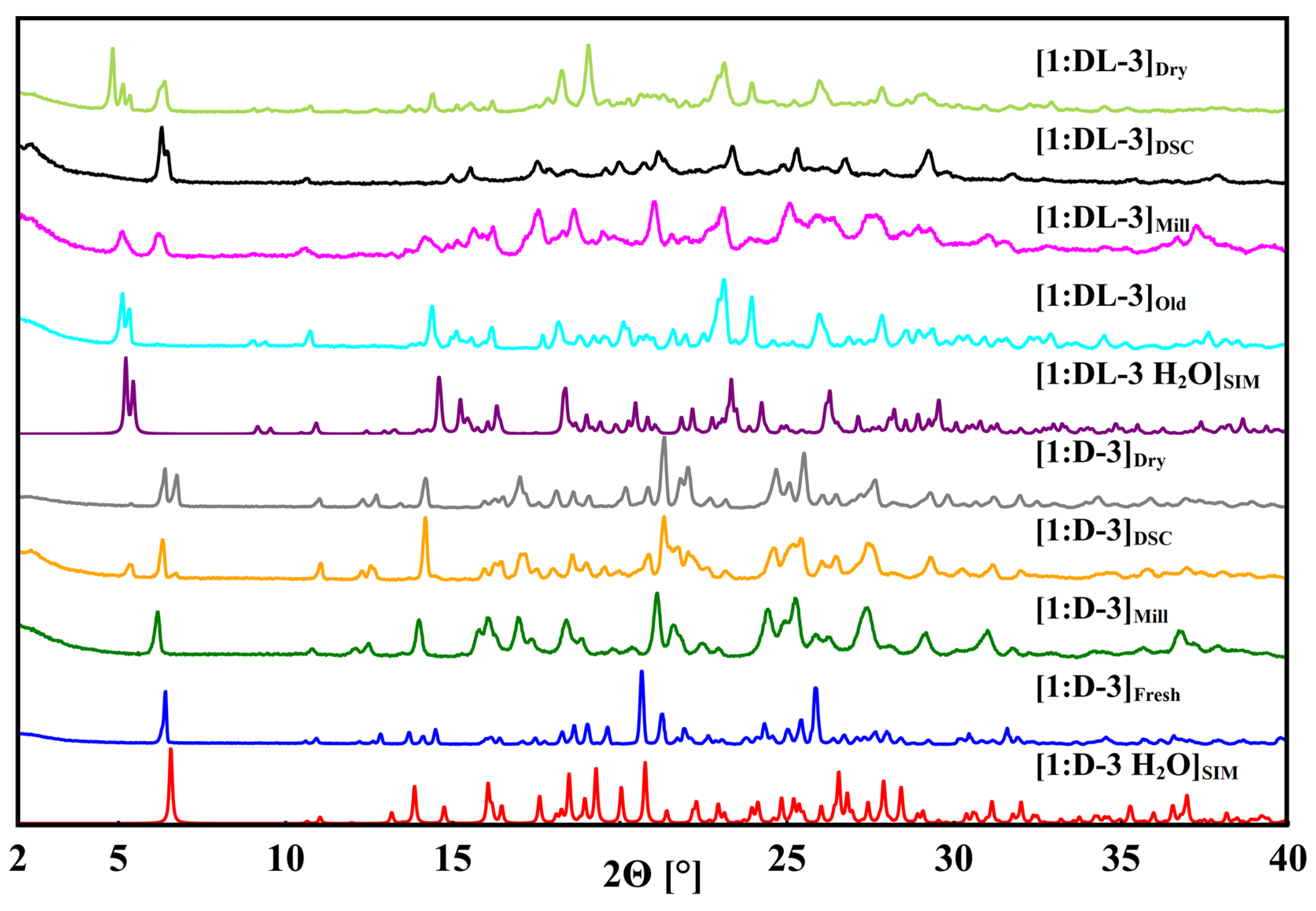
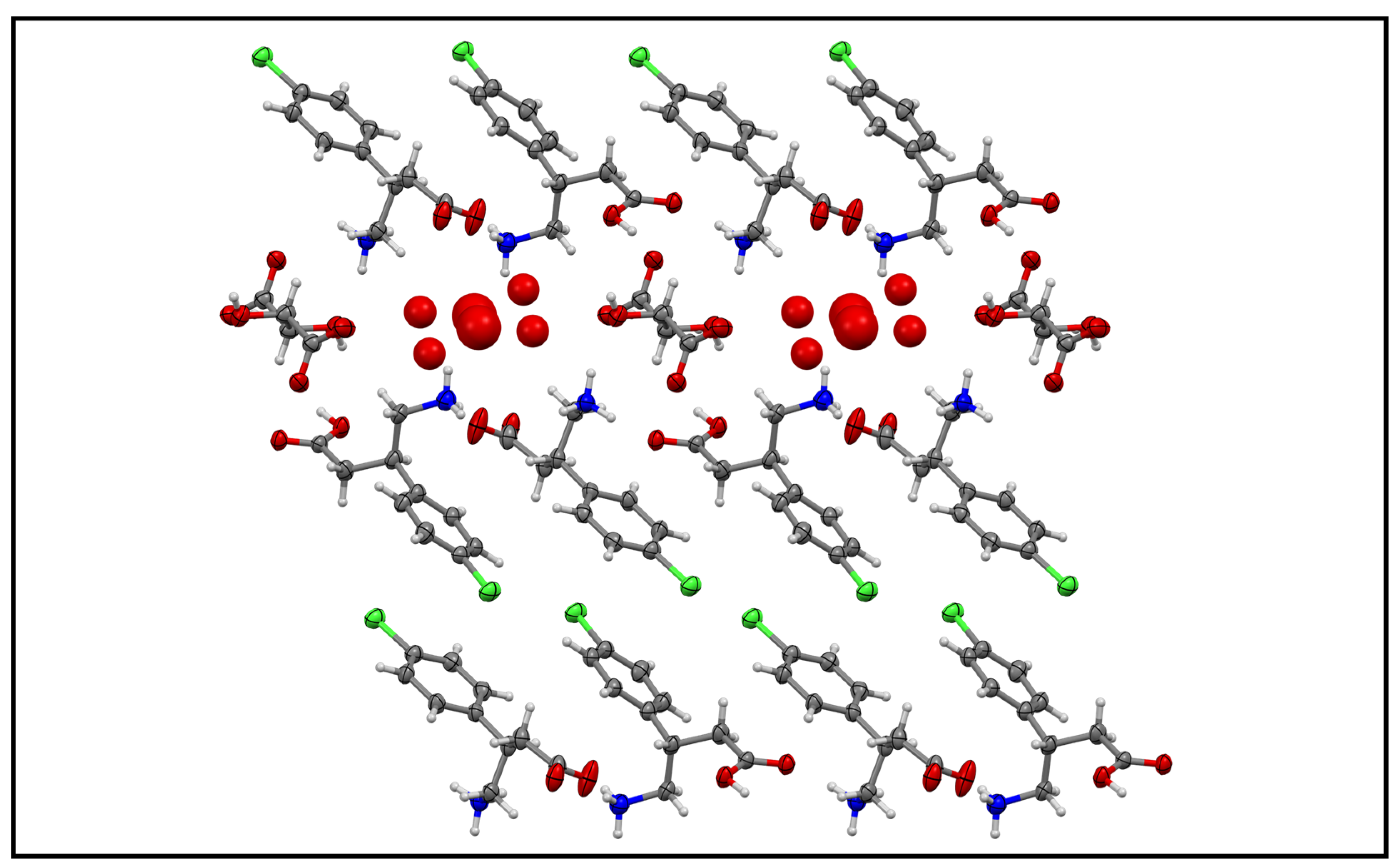
3.2. Baclofen and Tartaric Acid Species
3.3. Baclofen and Malic Acid Species
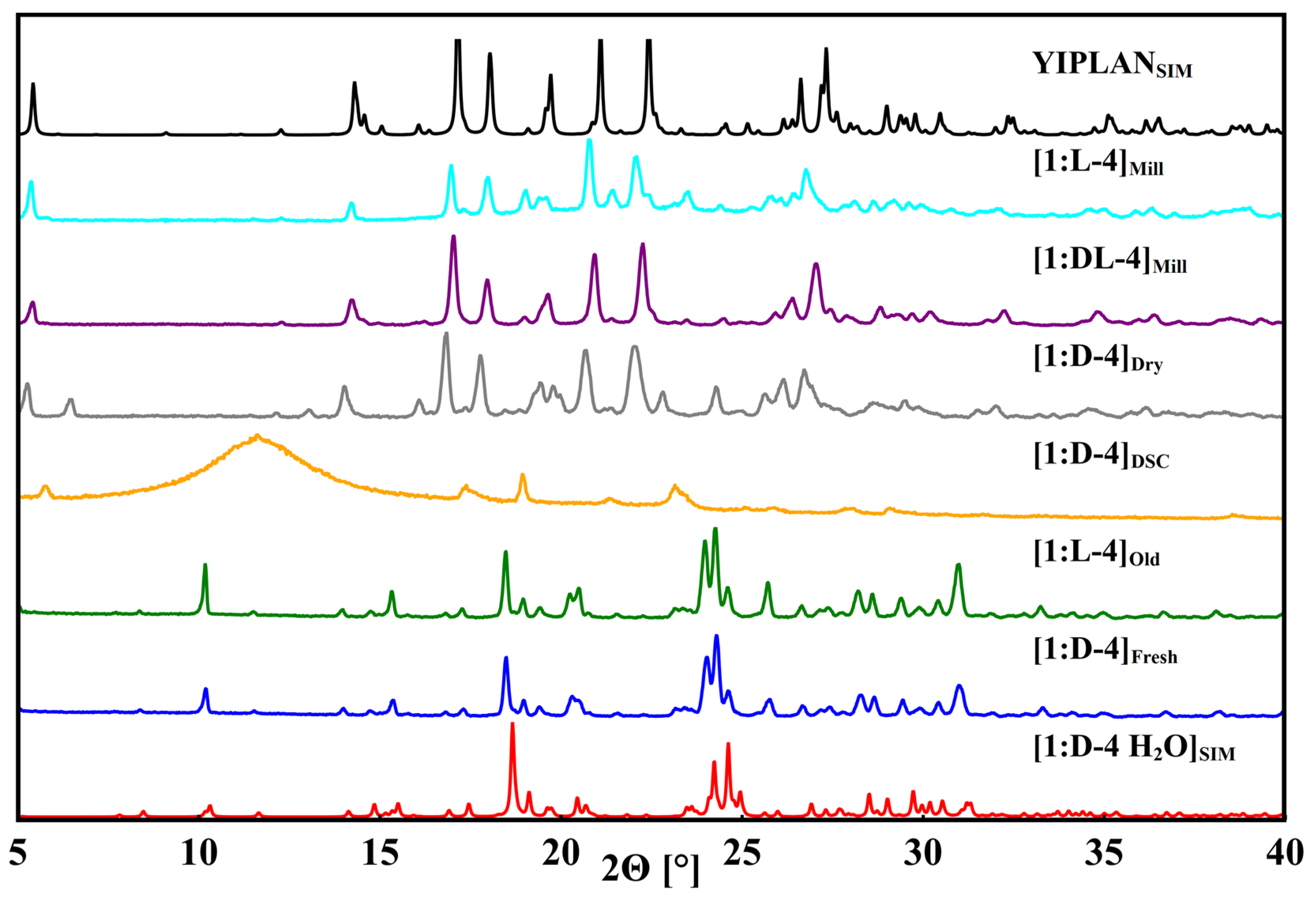
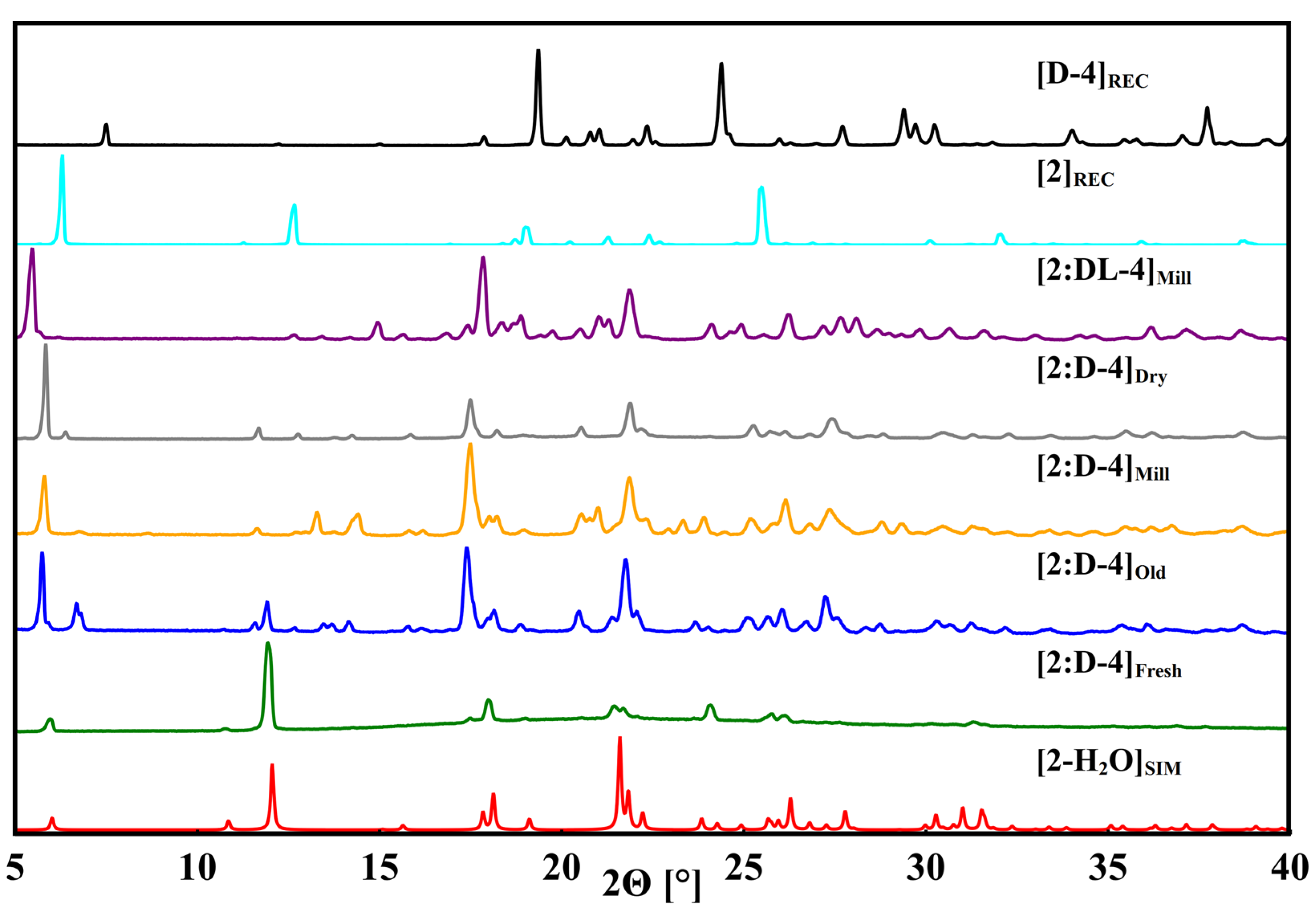
3.4. Phenibut and Tartaric Acid Species
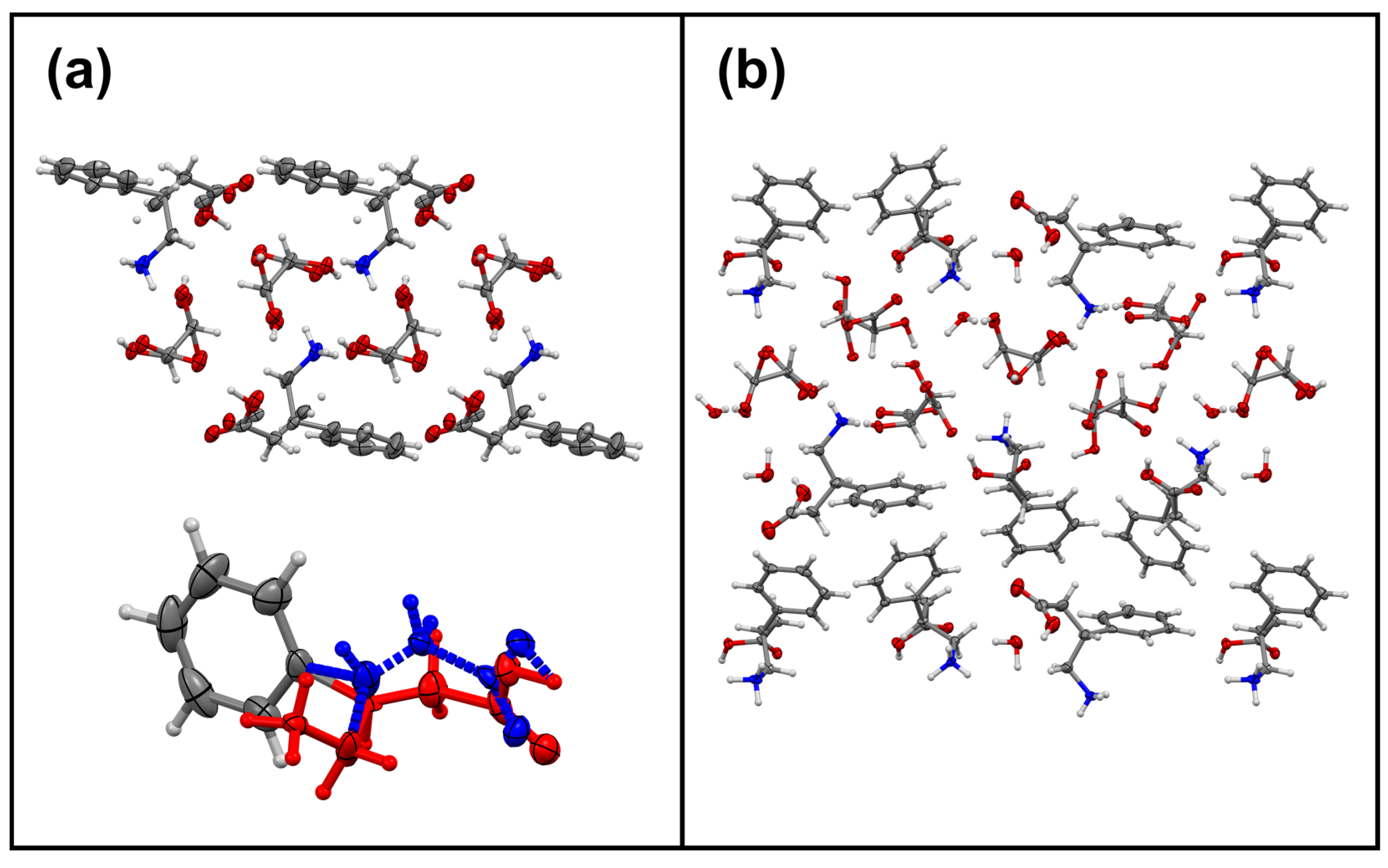
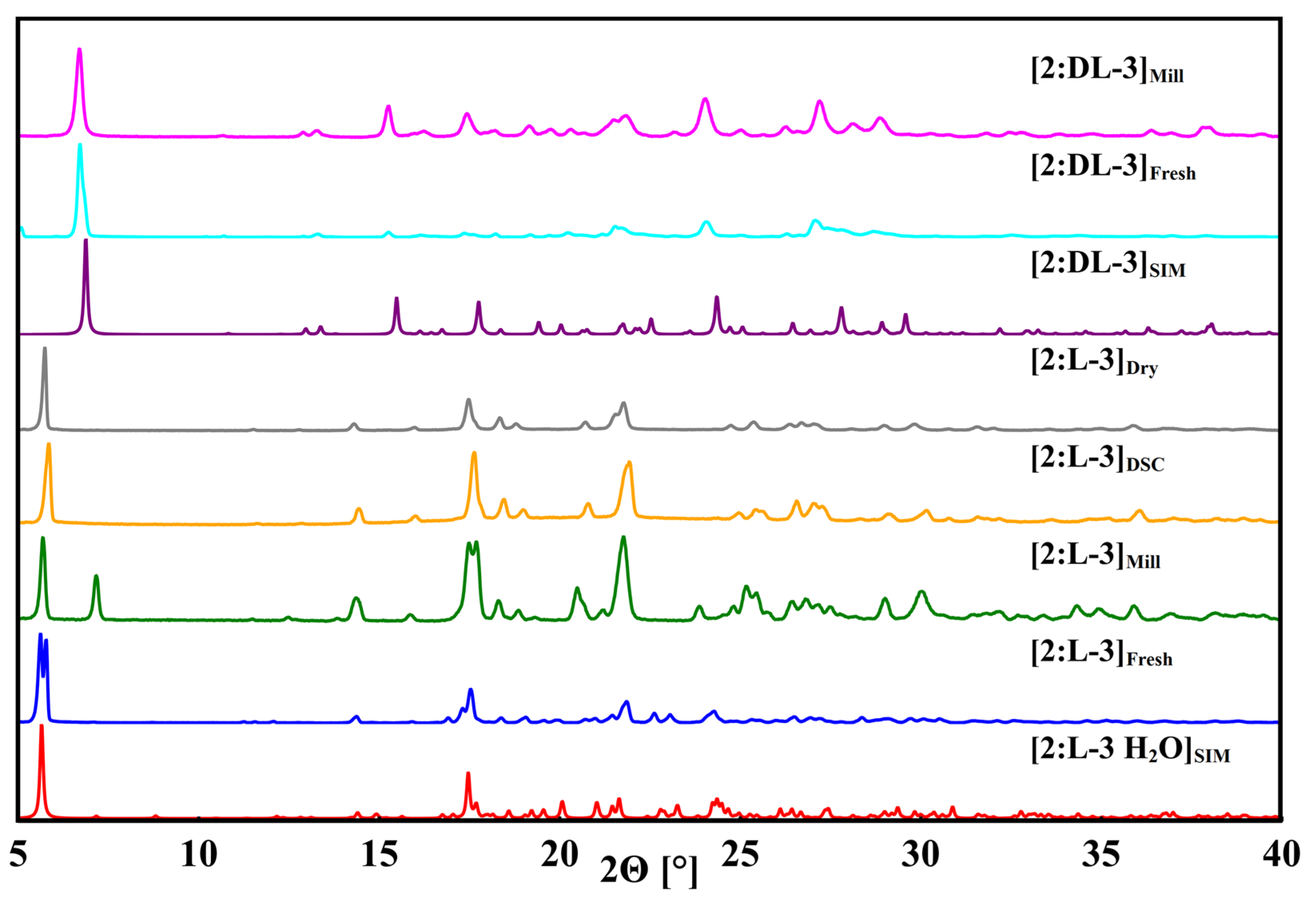
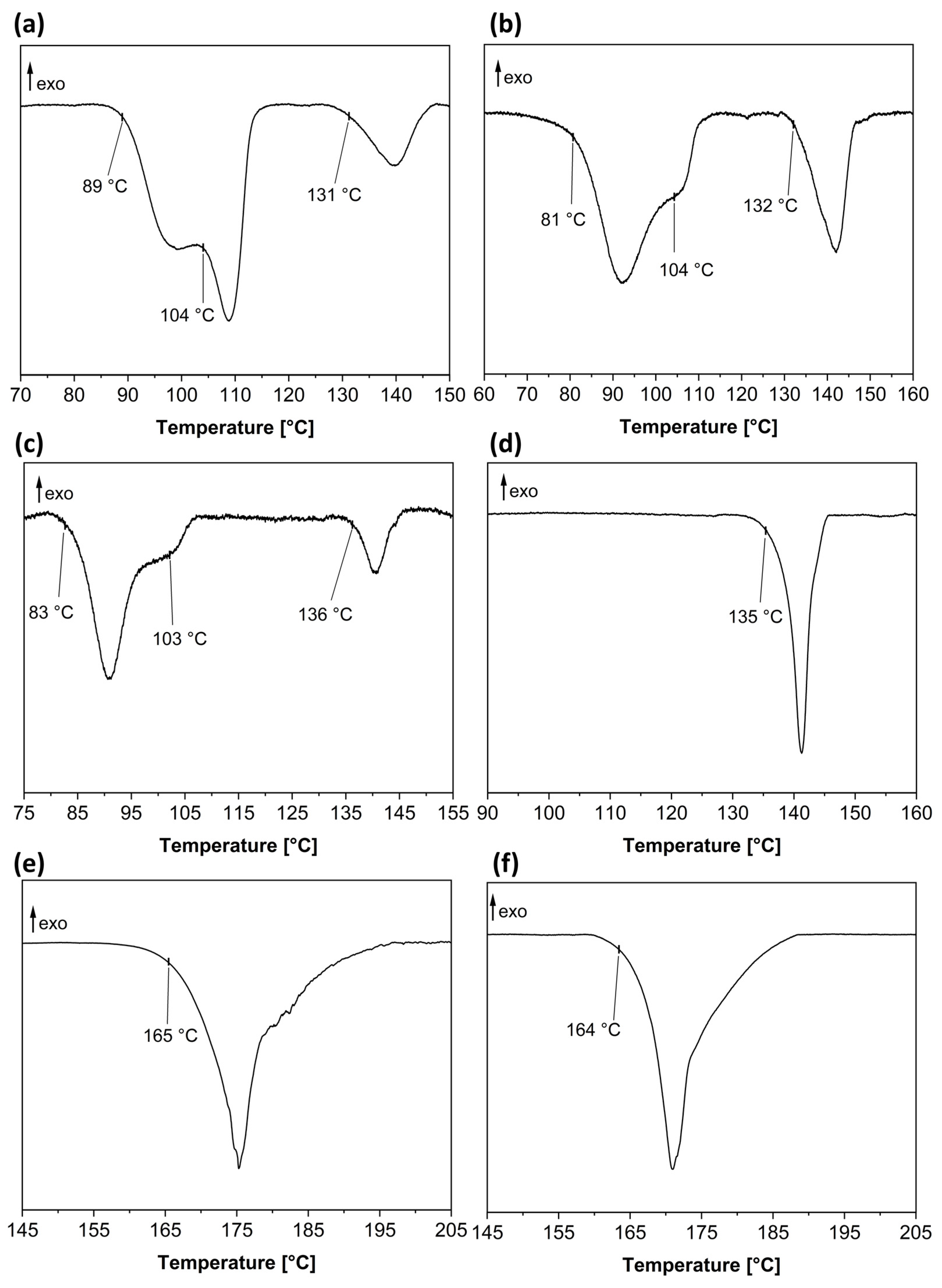
3.5. Phenibut and Malic Acid Species
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Morrison, G.; zur Loye, H.-C. Expanding the Chemistry of Salt-Inclusion Materials: Utilizing the Titanyl Ion as a Structure Directing Agent for the Targeted Synthesis of Salt-Inclusion Titanium Silicates. Cryst. Growth Des. 2020, 20, 8071–8078. [Google Scholar] [CrossRef]
- Hou, Y.; Sun, X.; Dou, M.; Lu, C.; Liu, J.; Rao, W. Cellulose Nanocrystals Facilitate Needle-like Ice Crystal Growth and Modulate Molecular Targeted Ice Crystal Nucleation. Nano Lett. 2021, 21, 4868–4877. [Google Scholar] [CrossRef]
- Uehara, T. Simulation of Polyhedral Crystal Growth Based on the Estimated Surface Energy of Crystallographic Planes. MSA 2021, 12, 519–533. [Google Scholar] [CrossRef]
- Wu, C.-S.; Ikeyama, J.; Nakabayashi, S.; Sugiyama, T.; Yoshikawa, H.Y. Growth Promotion of Targeted Crystal Face by Nanoprocessing via Laser Ablation. J. Phys. Chem. C 2019, 123, 24919–24926. [Google Scholar] [CrossRef]
- Jain, R.; Mallette, A.J.; Rimer, J.D. Controlling Nucleation Pathways in Zeolite Crystallization: Seeding Conceptual Methodologies for Advanced Materials Design. J. Am. Chem. Soc. 2021, 143, 21446–21460. [Google Scholar] [CrossRef]
- Orehek, J.; Teslić, D.; Likozar, B. Continuous Crystallization Processes in Pharmaceutical Manufacturing: A Review. Org. Process Res. Dev. 2021, 25, 16–42. [Google Scholar] [CrossRef]
- Tappan, B.A.; Brutchey, R.L. Polymorphic Metastability in Colloidal Semiconductor Nanocrystals. ChemNanoMat 2020, 6, 1567–1588. [Google Scholar] [CrossRef]
- Selekman, J.A.; Roberts, D.; Rosso, V.; Qiu, J.; Nolfo, J.; Gao, Q.; Janey, J. Development of a Highly Automated Workflow for Investigating Polymorphism and Assessing Risk of Forming Undesired Crystal Forms within a Crystallization Design Space. Org. Process Res. Dev. 2016, 20, 70–75. [Google Scholar] [CrossRef]
- Ostwald, W. Studien über die Bildung und Umwandlung fester Körper. Z. Phys. Chem. 1897, 22U, 289–330. [Google Scholar] [CrossRef]
- Ahn, B.; Bosetti, L.; Mazzotti, M. Accounting for the Presence of Molecular Clusters in Modeling and Interpreting Nucleation and Growth. Cryst. Growth Des. 2022, 22, 661–672. [Google Scholar] [CrossRef]
- Fu, H.; Gao, X.; Zhang, X.; Ling, L. Recent Advances in Nonclassical Crystallization: Fundamentals, Applications, and Challenges. Cryst. Growth Des. 2022, 22, 1476–1499. [Google Scholar] [CrossRef]
- Maurya, R.S.; Jayanthi, S.; Chandaluri, C.G.; Radhakrishnan, T.P. Monitoring Molecular Microparticles through the Amorphous-to-Crystalline Transformation and Fluorescence Enhancement/Tuning. Chem. Mater. 2022, 34, 244–253. [Google Scholar] [CrossRef]
- Weber, J.; Bracco, J.N.; Yuan, K.; Starchenko, V.; Stack, A.G. Studies of Mineral Nucleation and Growth Across Multiple Scales: Review of the Current State of Research Using the Example of Barite (BaSO4). ACS Earth Space Chem. 2021, 5, 3338–3361. [Google Scholar] [CrossRef]
- Rizvi, A.K.; Roberts, K.J.; Izumi, T. The Influence of Supersaturation and the Presence of Biuret on the Nucleation, Growth and Morphology of Urea Crystallised from Ethanolic Solutions. Isr. J. Chem. 2021, 61, 727–742. [Google Scholar] [CrossRef]
- Peng, H.; Tian, N.; Yu, C.; Gao, Y.; Li, K.; Yan, H.; Zhao, P.; Wu, S.; Chen, M.; Gong, J. Insights into the Role of Dipentaerythritol in the Thermodynamics and Nucleation Behavior of a Pentaerythritol–Water System. Cryst. Growth Des. 2022, 22, 449–460. [Google Scholar] [CrossRef]
- Ouyang, J.; Xing, X.; Chen, J.; Zhou, L.; Liu, Z.; Heng, J.Y. Effects of solvent, supersaturation ratio and silica template on morphology and polymorph evolution of vanillin during swift cooling crystallization. Particuology 2022, 65, 93–104. [Google Scholar] [CrossRef]
- Chen, Y.-M.; Gao, J. Synthesis and characterization of ZnO nanoparticles in n-hexanol solution. Mater. Lett. X 2021, 12, 100106. [Google Scholar] [CrossRef]
- Arzig, M.; Künecke, U.; Salamon, M.; Uhlmann, N.; Wellmann, P.J. Influence of the growth conditions on the formation of macro-steps on the growth interface of SiC-Crystals. J. Cryst. Growth 2021, 576, 126361. [Google Scholar] [CrossRef]
- Zhang, L.; Xu, Y.; Lou, B.; Qin, X.; Zhang, L.; Liu, X.; Yuan, H.; Zhang, Y.; Rohani, S.; Lu, J. Effect of Additives on Preferential Crystallization for the Chiral Resolution of Citrulline: Experimental, Statistical, and Molecular Dynamics Simulation Studies. Cryst. Growth Des. 2022, 22, 2392–2406. [Google Scholar] [CrossRef]
- Hoffmann, J.; Flannigan, J.; Cashmore, A.; Briuglia, M.L.; Steendam, R.R.E.; Gerard, C.J.J.; Haw, M.D.; Sefcik, J.; Horst, J.H. ter. The unexpected dominance of secondary over primary nucleation. Faraday Discuss. 2022, 235, 109–131. [Google Scholar] [CrossRef]
- Carpenter, J.E.; Grünwald, M. Pre-Nucleation Clusters Predict Crystal Structures in Models of Chiral Molecules. J. Am. Chem. Soc. 2021, 143, 21580–21593. [Google Scholar] [CrossRef] [PubMed]
- Bučar, D.-K.; Lancaster, R.W.; Bernstein, J. Disappearing polymorphs revisited. Angew. Chem. Int. Ed. 2015, 54, 6972–6993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Gabriele, B.; Davey, R.J.; Cruz-Cabeza, A.J. Concerning Elusive Crystal Forms: The Case of Paracetamol. J. Am. Chem. Soc. 2020, 142, 6682–6689. [Google Scholar] [CrossRef] [PubMed]
- Malec, L.M.; Gryl, M.; Oszajca, M.T.; Brela, M.Z.; Stadnicka, K.M. Chasing the Co-crystal Disappearing Polymorph with Ab Initio Methods. Cryst. Growth Des. 2021, 21, 6902–6912. [Google Scholar] [CrossRef]
- Lapin, I. Phenibut (beta-phenyl-GABA): A tranquilizer and nootropic drug. CNS Drug Rev. 2001, 7, 471–481. [Google Scholar] [CrossRef]
- Komisarek, D.; Pallaske, M.; Vasylyeva, V. Crystal Structure and Thermal Properties of Phenibut, Phenibut H2O and Phenibut HCl: A Case for Phase Stability Based on Structural Considerations. Z. Anorg. Allg. Chem. 2021, 647, 984–991. [Google Scholar] [CrossRef]
- Belov, F.; Villinger, A.; Langermann, J. von. (R)-Baclofen (R)-4-amino-3-(4-chloro-phen-yl)butanoic acid. Acta Crystallogr. E Crystallogr. Commun. 2022, 78 Pt 1, 33–35. [Google Scholar] [CrossRef]
- Gendron, F.-X.; Mahieux, J.; Sanselme, M.; Coquerel, G. Resolution of Baclofenium Hydrogenomaleate By Using Preferential Crystallization. A First Case of Complete Solid Solution at High Temperature and a Large Miscibility Gap in the Solid State. Cryst. Growth Des. 2019, 19, 4793–4801. [Google Scholar] [CrossRef]
- Takagi, Y.; Yamada, H.; Ebara, H.; Hayashi, H.; Kidani, S.; Toyooka, K.; Ishino, Y.; Kitano, Y.; Nakanami, A.; Kagechika, K.; et al. Intrathecal baclofen therapy for severe spasticity in an adult with tethered cord syndrome: A case report. J. Med. Case Rep. 2021, 15, 442. [Google Scholar] [CrossRef]
- Romito, J.W.; Turner, E.R.; Rosener, J.A.; Coldiron, L.; Udipi, A.; Nohrn, L.; Tausiani, J.; Romito, B.T. Baclofen therapeutics, toxicity, and withdrawal: A narrative review. SAGE Open Med. 2021, 9, 20503121211022197. [Google Scholar] [CrossRef]
- Kent, C.N.; Park, C.; Lindsley, C.W. Classics in Chemical Neuroscience: Baclofen. ACS Chem. Neurosci. 2020, 11, 1740–1755. [Google Scholar] [CrossRef] [PubMed]
- Agabio, R.; Baldwin, D.S.; Amaro, H.; Leggio, L.; Sinclair, J.M.A. The influence of anxiety symptoms on clinical outcomes during baclofen treatment of alcohol use disorder: A systematic review and meta-analysis. Neurosci. Biobehav. Rev. 2021, 125, 296–313. [Google Scholar] [CrossRef] [PubMed]
- Garbutt, J.C.; Kampov-Polevoy, A.B.; Pedersen, C.; Stansbury, M.; Jordan, R.; Willing, L.; Gallop, R.J. Efficacy and tolerability of baclofen in a U.S. community population with alcohol use disorder: A dose-response, randomized, controlled trial. Neuropsychopharmacology 2021, 46, 2250–2256. [Google Scholar] [CrossRef] [PubMed]
- Leggio, L.; Litten, R.Z. The GABA-B receptor agonist baclofen helps patients with alcohol use disorder: Why these findings matter. Neuropsychopharmacology 2021, 46, 2228–2229. [Google Scholar] [CrossRef]
- Levine, M.; Lovecchio, F. New Designer Drugs. Emerg. Med. Clin. N. Am. 2021, 39, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Jouney, E.A. Phenibut (β-Phenyl-γ-Aminobutyric Acid): An Easily Obtainable “Dietary Supplement” with Propensities for Physical Dependence and Addiction. Curr. Psychiatry Rep. 2019, 21, 23. [Google Scholar] [CrossRef] [PubMed]
- Doyno, C.R.; White, C.M. Sedative-Hypnotic Agents That Impact Gamma-Aminobutyric Acid Receptors: Focus on Flunitrazepam, Gamma-Hydroxybutyric Acid, Phenibut, and Selank. J. Clin. Pharmacol. 2021, 61 (Suppl. S2), S114–S128. [Google Scholar] [CrossRef]
- Kupats, E.; Vrublevska, J.; Zvejniece, B.; Vavers, E.; Stelfa, G.; Zvejniece, L.; Dambrova, M. Safety and Tolerability of the Anxiolytic and Nootropic Drug Phenibut: A Systematic Review of Clinical Trials and Case Reports. Pharmacopsychiatry 2020, 53, 201–208. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, Y.; Huang, X.; Li, X.; Zong, S.; Wang, N.; Hao, H. Conformational Selectivity and Evolution Affected by the Desolvation Process. Cryst. Growth Des. 2022, 22, 1283–1291. [Google Scholar] [CrossRef]
- Venu, N.; Vishweshwar, P.; Ram, T.; Surya, D.; Apurba, B. (S)-3-(Ammoniomethyl)-5-methylhexanoate (pregabalin). Acta Crystallogr. C Struct. Chem. 2007, 63 Pt 5, o306–o308. [Google Scholar] [CrossRef]
- CrysAlisPRO; Oxford Diffraction/Agilent Technologies UK Ltd.: Yarnton, England, 2014.
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 2015, C71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. A short history of SHELX. Acta. Crystallogr. Sect. A Found. Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Crystallogr. 2020, 53 Pt 1, 226–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Ibers, J.A. Gabapentin and gabapentin monohydrate. Acta Crystallogr. C Struct. Chem. 2001, 57 Pt 5, 641–643. [Google Scholar] [CrossRef]
- Khandavilli, U.B.R.; Lusi, M.; Frawley, P.J. Plasticity in zwitterionic drugs: The bending properties of Pregabalin and Gabapentin and their hydrates. IUCrJ 2019, 6 Pt 4, 630–634. [Google Scholar] [CrossRef] [Green Version]
- Vasudev, P.G.; Aravinda, S.; Ananda, K.; Veena, S.D.; Nagarajan, K.; Shamala, N.; Balaram, P. Crystal structures of a new polymorphic form of gabapentin monohydrate and the e and z isomers of 4-tertiarybutylgabapentin. Chem. Biol. Drug Des. 2009, 73, 83–96. [Google Scholar] [CrossRef]
- Wang, Y.; Du, S.; Wu, S.; Li, L.; Zhang, D.; Yu, B.; Zhou, L.; Bekele, H.K.; Gong, J. Thermodynamic and molecular investigation into the solubility, stability and self-assembly of gabapentin anhydrate and hydrate. J. Chem. Thermodyn. 2017, 113, 132–143. [Google Scholar] [CrossRef]
- Córdova-Villanueva, E.N.; Rodríguez-Ruiz, C.; Sánchez-Guadarrama, O.; Rivera-Islas, J.; Herrera-Ruiz, D.; Morales-Rojas, H.; Höpfl, H. Diastereomeric Salt Formation by the γ-Amino Acid RS -Baclofen and L -Malic Acid: Stabilization by Strong Heterosynthons Based on Hydrogen Bonds between RNH 3+ and COOH/COO– Groups. Cryst. Growth Des. 2018, 18, 7356–7367. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | 1 • H2O | 2 • H2O |
|---|---|---|
| Formula | C10H12ClNO2, H2O | C10H13NO2, H2O |
| Formula Weight [gmol−1] | 231.67 | 197.23 |
| T [K] | 100 | 100 |
| Crystal System | orthorhombic | orthorhombic |
| Space Group | Aea 2 | Aea 2 |
| a [Å] | 9.5543 (2) | 9.7521 (3) |
| b [Å] | 32.6466 (6) | 29.3294 (11) |
| c [Å] | 7.1995 (1) | 7.1397 (2) |
| β [°] | 90 | 90 |
| V [Å3] | 2245.64 (7) | 2042.12 (11) |
| Z, Z’ | 8, 1 | 8, 1 |
| R1, wR2 [%] | 4.56, 12.11 | 4.42, 10.4 |
| Hooft | 0.003 (16) | −0.18 (16) |
| Source | Cu Kα (λ = 1.54184) | Cu Kα (λ = 1.54184) |
| Quick Phase Transition ≤ 3 d | Phase Transition ≥ 3 d or in DSC | Remains Stable, DSC Melting Only |
|---|---|---|
| 1 • H2O, Aea2, only stable in mother liquid | 1:D−3 • H2O, P1, transition in about 1 week | 2:DL−3, P, disorder along GABA subunit |
| 1:L−3 • H2O, P1, transition in about 1 week | ||
| 1:DL−3 • H2O, P21/n, DSC transition, no decomposition over time | ||
| 2 • H2O, Aea2, transition upon drying—slower than 1 • H2O | 1:D−4 • H2O, P, strongly disordered water molecules; no decomposition over time | |
| 2:L−3 • H2O, P21, DSC transition, no decomposition over time |
| Parameters | 1:L−3 • H2O | 1:D−3 • H2O | 1:DL−3 • H2O |
|---|---|---|---|
| Formula | C10H13ClNO2, C4H5O6, H2O | C10H13ClNO2, C4H5O6, H2O | (C10H13ClNO2)2, C10H12ClNO2, (C4H5O6)2, (H2O)3 |
| Formula Weight [gmol−1] | 381.76 | 381.76 | 995.19 |
| T [K] | 100 | 100 | 100 |
| Crystal System | triclinic | triclinic | monoclinic |
| Space Group | P1 | P1 | P21/n |
| a [Å] | 7.3819 (1) | 7.3781 (1) | 19.2882 (4) |
| b [Å] | 8.5625 (1) | 8.5661 (1) | 7.2929 (3) |
| c [Å] | 13.8601 (2) | 13.8636 (1) | 32.3679 (10) |
| α [°] | 75.551 (1) | 75.546 (1) | 90 |
| β [°] | 88.406 (1) | 88.339 (1) | 92.378 (2) |
| γ [°] | 89.616 (1) | 89.529 (19) | 90 |
| V [Å3] | 848.02 (2) | 848.111 (17) | 4549.2 (3) |
| Z, Z’ | 2, 2 | 2, 2 | 4, 1 |
| R1, wR2 [%] | 2.56, 6.50 | 2.76, 7.26 | 6.56, 18.38 |
| Hooft | −0.003 (6) | −0.003 (7) | - |
| Source | Cu Kα (λ = 1.54184) | Cu Kα (λ = 1.54184) | Cu Kα (λ = 1.54184) |
| Parameters | 1:D−4 • H2O |
|---|---|
| Formula | C10H12ClNO2, C10H13ClNO2, (C10H13ClNO2)0.5, (H2O)2 |
| Formula Weight [gmol−1] | 1051.91 |
| T [K] | 100 |
| Crystal System | triclinic |
| Space Group | |
| a [Å] | 6.1571 (1) |
| b [Å] | 11.8661 (2) |
| c [Å] | 17.6166 (5) |
| α [°] | 77.292 (2) |
| β [°] | 82.763 (2) |
| γ [°] | 76.139 (2) |
| V [Å3] | 1215.34 (5) |
| Z, Z’ | 4, 2 |
| R1, wR2 [%] | 8.31, 21.28 |
| Hooft | - |
| Source | Cu Kα (λ = 1.54184) |
| Parameters | 2:L−3 • H2O | 2:DL−3 |
|---|---|---|
| Formula | (C10H14NO2)3, (C4H5O6)3, H2O | C10H14NO2, C4H5O6 |
| Formula Weight [gmol−1] | 1023.93 | 329.20 |
| T [K] | 100 | 100 |
| Crystal System | monoclinic | triclinic |
| Space Group | P21 | |
| a [Å] | 7.4642 (2) | 7.2070 (0) |
| b [Å] | 20.0354 (6) | 8.2646 (0) |
| c [Å] | 15.8509 (5) | 13.2223 (3) |
| α [°] | 90 | 93.570 (2) |
| β [°] | 100.400 (3) | 103.006 (2) |
| γ [°] | 90 | 97.319 (2) |
| V [Å3] | 2331.53 (12) | 757.73 (2) |
| Z, Z’ | 2, 1 | 2, 1 |
| R1, wR2 [%] | 5.89, 13.85 | 5.01, 12.72 |
| Hooft | 0.1 (2) | - |
| Source | Cu Kα (λ = 1.54184) | Cu Kα (λ = 1.54184) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Herbst, M.; Komisarek, D.; Strothmann, T.; Vasylyeva, V. A Lection in Humbleness: Crystallization of Chiral and Zwitterionic APIs Baclofen and Phenibut. Crystals 2022, 12, 1393. https://doi.org/10.3390/cryst12101393
Herbst M, Komisarek D, Strothmann T, Vasylyeva V. A Lection in Humbleness: Crystallization of Chiral and Zwitterionic APIs Baclofen and Phenibut. Crystals. 2022; 12(10):1393. https://doi.org/10.3390/cryst12101393
Chicago/Turabian StyleHerbst, Marco, Daniel Komisarek, Till Strothmann, and Vera Vasylyeva. 2022. "A Lection in Humbleness: Crystallization of Chiral and Zwitterionic APIs Baclofen and Phenibut" Crystals 12, no. 10: 1393. https://doi.org/10.3390/cryst12101393