
Application of Fundamental Techniques for Physicochemical Characterizations to Understand Post-Formulation Performance of Pharmaceutical Nanocrystalline Materials



Abstract
:1. Introduction
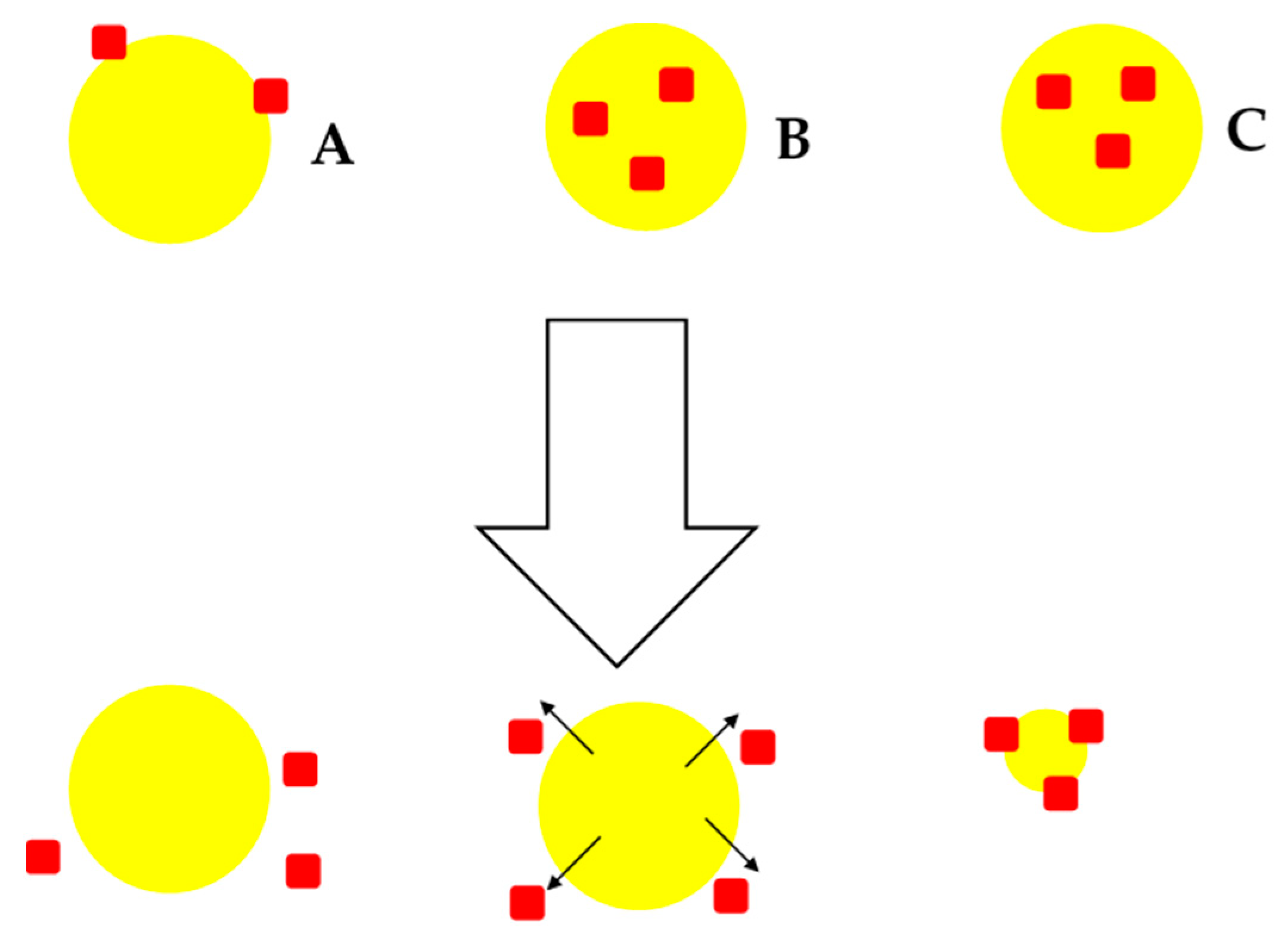
2. Formulation and Morphology of NCM
2.1. Top-Down Production Approaches
2.1.1. HPH (IDD-P®, DissoCubes®, and Nanopure®)
2.1.2. MM (NanoCrystals®)
2.1.3. Bottom-Up Production Approaches
3. Nucleation and Crystal Growth
3.1. Supramolecular Processes in Crystal Growth
3.2. Crystal Habit, Morphology, and Growth
4. Techniques used in the Physicochemical Characterisations of NCM
- Specific surface area
- Particle size
- Particle size distribution or polydispersity index
- Crystallinity
- Surface Reactivity
- Solubility
- Agglomeration
- Surface charge
- Elemental/molecular composition
- Surface chemistry
4.1. Dynamic Light Scattering (DLS)
4.1.1. Particles Size (PS)
4.1.2. Surface Charge
4.2. Thermal Analysis
4.2.1. Thermal Gravimetric Analysis (TGA)
4.2.2. Differential Scanning Calorimetry (DSC)
4.3. X-Ray Techniques
4.3.1. Powder X-ray Diffraction (PXRD)
4.3.2. Small Angle X-ray Scattering (SAXS)
4.3.3. X-Ray Photon Spectroscopy (XPS)
4.4. Vibrational Spectroscopy
4.4.1. Fourier Transform Infrared (FTIR) Spectroscopy
4.4.2. Raman Spectroscopy (RS)
4.5. Microscopy Techniques
4.5.1. Scanning Electron Microscopy (SEM)
4.5.2. Scanning Electron Microscopy Energy Dispersive X-ray Spectroscopy (SEM-EDX)
4.5.3. Transmission Electron Microscopy (TEM)
4.6. Quantitative Techniques
4.6.1. (Ultra) High-Performance Liquid Chromatography ((U)HPLC)
4.6.2. Ultraviolet-Visible Spectrophotometry (UV-Vis Spec)
5. Applications of Characterization Techniques on Prediction of NCM Performance
5.1. Prediction of Physical and Chemical Stability of NCM
5.1.1. Suspension Stability
5.1.2. Phase Behaviour
5.1.3. Determination of Critical Aggregation or Micelle Concentration
5.1.4. Chemical Degradation Analysis
5.2. In Vitro Dissolution and Kinetics of Drug Release
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Tiwari, G.; Tiwari, R.; Bannerjee, S.; Bhati, L.; Pandey, S.; Pandey, P.; Sriwastawa, B. Drug delivery systems: An updated review. Int. J. Pharm. Investig. 2012, 2, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farokhzad, O.C.; Langer, R. Impact of Nanotechnology on Drug Discovery & Development Pharmanext. ACS Nano 2009, 3, 16–20. [Google Scholar] [PubMed]
- Junghanns, J.U.A.H.; Müller, R.H. Nanocrystal technology, drug delivery and clinical applications. Int. J. Nanomed. 2008, 3, 295–309. [Google Scholar] [CrossRef] [Green Version]
- Müller, R.H. Lipid nanoparticles: Recent advances. Adv. Drug Deliv. Rev. 2007, 59, 375–376. [Google Scholar] [CrossRef]
- Muller, R.H.; Keck, C.M. Challenges and solutions for the delivery of biotech drugs—A review of drug nanocrystal technology and lipid nanoparticles. J. Biotechnol. 2004, 113, 151–170. [Google Scholar] [CrossRef]
- Blagden, N.; de Matas, M.; Gavan, P.T.; York, P. Crystal engineering of active pharmaceutical ingredients to improve solubility and dissolution rates. Adv. Drug Deliv. Rev. 2007, 59, 617–630. [Google Scholar] [CrossRef]
- Subramanian, S.; Zaworotko, M.J. Manifestations of noncovalent bonding in the solid state. 6. [H 4 (cyclam)] 4+ (cyclam = 1,4,8,11-tetraazacyclotetradecane) as a template for crystal engineering of network hydrogen-bonded solids. Can. J. Chem. 1995, 73, 414–424. [Google Scholar] [CrossRef]
- Desiraju, G.R. Crystal Engineering: The design of Organic Solids. J. Appl. Crystallogr. 1991, 24, 265. [Google Scholar] [CrossRef] [Green Version]
- Desiraju, G.R. Supramolecular Synthons in Crystal Engineering. Angew. Chem. Int. Ed. 1995, 34, 2311–2327. [Google Scholar] [CrossRef]
- Aakeroy, C.B.; Aakeroy, A.; Sinha, A.S. Co-Crystals: Introduction and Scope; Royal Society of Chemistry: London, UK, 2018; Volume 11. [Google Scholar]
- Bolton, O.; Matzger, A.J. Improved stability and smart-material functionality realized in an energetic cocrystal. Angew. Chem. Int. Ed. 2011, 50, 8960–8963. [Google Scholar] [CrossRef]
- Brittain, H.G. Pharmaceutical cocrystals: The coming wave of new drug substances. J. Pharm. Sci. 2013, 102, 311–317. [Google Scholar] [CrossRef]
- Brittain, H.G. Cocrystal Systems of Pharmaceutical Interest: 2010. Cryst. Growth Des. 2011, 36, 361–381. [Google Scholar] [CrossRef]
- Sekhon, B. Pharmaceutical co-crystals—A review. ARS Pharm. 2009, 150, 99–117. [Google Scholar]
- Merisko-Liversidge, E.; Liversidge, G.G. Nanosizing for oral and parenteral drug delivery: A perspective on formulating poorly-water soluble compounds using wet media milling technology. Adv. Drug Deliv. Rev. 2011, 63, 427–440. [Google Scholar] [CrossRef] [PubMed]
- Merisko-Liversidge, E.; Liversidge, G.G.; Cooper, E.R. Nanosizing: A formulation approach for poorly-water-soluble compounds. Eur. J. Pharm. Sci. 2003, 18, 113–120. [Google Scholar] [CrossRef]
- Sinha, B.; Muller, R.H.; Moschwitzer, J.P. Bottom-up approaches for preparing drug nanocrystals: Formulations and factors affecting particle size. Int. J. Pharm. 2013, 8, 384–392. [Google Scholar] [CrossRef] [PubMed]
- De Waard, H.; Frijlink, H.W.; Hinrichs, W.L.J. Bottom-up preparation techniques for nanocrystals of lipophilic drugs. Pharm. Res. 2011, 28, 1220–1223. [Google Scholar] [CrossRef] [Green Version]
- Witika, B.A.; Smith, V.J.; Walker, R.B. A comparative study of the effect of different stabilizers on the critical quality attributes of self-assembling nano co-crystals. Pharmaceutics 2020, 12, 182. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.Y.; Yoo, J.Y.; Kwak, H.S.; Nam, B.U.; Lee, J. Role of polymeric stabilizers for drug nanocrystal dispersions. Curr. Appl. Phys. 2005, 5, 472–474. [Google Scholar] [CrossRef]
- Ghosh, I.; Bose, S.; Vippagunta, R.; Harmon, F. Pharmaceutical Nanotechnology Nanosuspension for improving the bioavailability of a poorly soluble drug and screening of stabilizing agents to inhibit crystal growth. Int. J. Pharm. 2011, 409, 260–268. [Google Scholar] [CrossRef]
- Raghava Srivalli, K.M.; Mishra, B. Drug nanocrystals: A way toward scale-up. Saudi Pharm. J. 2016, 24, 386–404. [Google Scholar] [CrossRef] [Green Version]
- Al Shaal, L.; Müller, R.H.; Keck, C.M. Preserving hesperetin nanosuspensions for dermal application. Pharmazie 2010, 65, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Piao, H.; Kamiya, N.; Cui, F.; Goto, M. Preparation of a solid-in-oil nanosuspension containing l-ascorbic acid as a novel long-term stable topical formulation. Int. J. Pharm. 2011, 420, 156–160. [Google Scholar] [CrossRef]
- Patel, D.A.; Patel, M.R.; Patel, K.R.; Patel, N.M. Buccal mucosa as a route for systemic drug delivery: A review. Int. J. Drug Dev. Res. 2012, 1, 15–30. [Google Scholar]
- Dressman, J.B.; Reppas, C. In vitro–in vivo correlations for lipophilic, poorly water-soluble drugs. Eur. J. Pharm. Sci. 2000, 11, S73–S80. [Google Scholar] [CrossRef]
- Kesisoglou, F.; Panmai, S.; Wu, Y. Nanosizing—Oral formulation development and biopharmaceutical evaluation. Adv. Drug Deliv. Rev. 2007, 59, 631–644. [Google Scholar] [CrossRef]
- Wu, Y.; Loper, A.; Landis, E.; Hettrick, L.; Novak, L.; Lynn, K.; Chen, C.; Thompson, K.; Higgins, R.; Batra, U.; et al. The role of biopharmaceutics in the development of a clinical nanoparticle formulation of MK-0869: A Beagle dog model predicts improved bioavailability and diminished food effect on absorption in human. Int. J. Pharm. 2004, 285, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Edelhauser, H.F.; Rowe-Rendleman, C.L.; Robinson, M.R.; Dawson, D.G.; Chader, G.J.; Grossniklaus, H.E.; Rittenhouse, K.D.; Wilson, C.G.; Weber, D.A.; Kuppermann, B.D.; et al. Ophthalmic drug delivery systems for the treatment of retinal diseases: Basic research to clinical applications. Investig. Ophthalmol. Vis. Sci. 2010, 51, 5403–5420. [Google Scholar] [CrossRef]
- Makoni, P.A.; Khamanga, S.M.; Walker, R.B. Muco-adhesive clarithromycin-loaded nanostructured lipid carriers for ocular delivery: Formulation, characterization, cytotoxicity and stability. J. Drug Deliv. Sci. Technol. 2020, 61, 102171. [Google Scholar] [CrossRef]
- Kassem, M.A.; Abdel Rahman, A.A.; Ghorab, M.M.; Ahmed, M.B.; Khalil, R.M. Nanosuspension as an ophthalmic delivery system for certain glucocorticoid drugs. Int. J. Pharm. 2007, 340, 126–133. [Google Scholar] [CrossRef]
- Baba, K.; Nishida, K. Steroid nanocrystals prepared using the nano spray dryer B-90. Pharmaceutics 2013, 5, 107–114. [Google Scholar] [CrossRef] [Green Version]
- Ali, H.S.M.; York, P.; Ali, A.M.A.; Blagden, N. Hydrocortisone nanosuspensions for ophthalmic delivery: A comparative study between microfluidic nanoprecipitation and wet milling. J. Control Release 2011, 149, 175–181. [Google Scholar] [CrossRef]
- Ganta, S.; Paxton, J.W.; Baguley, B.C.; Garg, S. Formulation and pharmacokinetic evaluation of an asulacrine nanocrystalline suspension for intravenous delivery. Int. J. Pharm. 2009, 367, 179–186. [Google Scholar] [CrossRef]
- Ben Zirar, S.; Astier, A.; Muchow, M.; Gibaud, S. Comparison of nanosuspensions and hydroxypropyl-β-cyclodextrin complex of melarsoprol: Pharmacokinetics and tissue distribution in mice. Eur. J. Pharm. Biopharm. 2008, 70, 649–656. [Google Scholar] [CrossRef]
- Zhang, D.; Chen, M.; Zheng, T.; Wang, S. Preparation and Characterization of an Oridonin Nanosuspension for Solubility and Dissolution Velocity Enhancement AU—Gao, Lei. Drug Dev. Ind. Pharm. 2007, 33, 1332–1339. [Google Scholar] [CrossRef]
- Rabinow, B.; Kipp, J.; Papadopoulos, P.; Wong, J.; Glosson, J.; Gass, J.; Sun, C.S.; Wielgos, T.; White, R.; Cook, C.; et al. Itraconazole IV nanosuspension enhances efficacy through altered pharmacokinetics in the rat. Int. J. Pharm. 2007, 339, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Sun, M.; Guo, C.; Yu, A.; Xi, Y.; Cui, J.; Lou, H.; Zhai, G. Preparation and characterization of intravenously injectable curcumin nanosuspension AU—Gao, Yan. Drug Deliv. 2011, 18, 131–142. [Google Scholar] [CrossRef]
- Lu, Y.; Wang, Z.H.; Li, T.; McNally, H.; Park, K.; Sturek, M. Development and evaluation of transferrin-stabilized paclitaxel nanocrystal formulation. J. Control Release 2014, 176, 76–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarnes, A.; Kovalainen, M.; Häkkinen, M.R.; Laaksonen, T.; Laru, J.; Kiesvaara, J.; Ilkka, J.; Oksala, O.; Rönkkö, S.; Järvinen, K.; et al. Nanocrystal-based per-oral itraconazole delivery: Superior in vitro dissolution enhancement versus Sporanox® is not realized in in vivo drug absorption. J. Control Release 2014, 180, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Müller, R.H.; Jacobs, C. Buparvaquone mucoadhesive nanosuspension: Preparation, optimisation and long-term stability. Int. J. Pharm. 2002, 237, 151–161. [Google Scholar] [CrossRef]
- Witika, B.A.; Smith, V.J.; Walker, R.B. Quality by Design Optimization of Cold Sonochemical Synthesis of Zidovudine-Lamivudine Nanosuspensions. Pharmaceutics 2020, 12, 367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aucamp, M.; Milne, M. The physical stability of drugs linked to quality-by-design (QbD) and in-process technology (PAT) perspectives. Eur. J. Pharm. Sci. 2019, 139, 105057. [Google Scholar] [CrossRef]
- Shegokar, R.; Müller, R.H. Nanocrystals: Industrially feasible multifunctional formulation technology for poorly soluble actives. Int. J. Pharm. 2010, 399, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Rabinow, B.E. Nanosuspensions in drug delivery. Nat. Rev. Drug Discov. 2004, 3, 785. [Google Scholar] [CrossRef]
- Muller, R.H.; Jacobs, C.; Kayser, O. Nanosuspensions as particulate drug formulations in therapy: Rationale for development and what we can expect for the future. Adv. Drug Deliv. Rev. 2001, 47, 3–19. [Google Scholar] [CrossRef]
- Keck, C.M.; Müller, R.H. Drug nanocrystals of poorly soluble drugs produced by high pressure homogenisation. Eur. J. Pharm. Biopharm. 2006, 62, 3–16. [Google Scholar] [CrossRef]
- Li, X.; Zhou, L.; Ma, J.; Gao, L.; Wang, X.; Liu, G. Drug nanocrystals: In vivo performances. J. Control Release 2012, 160, 418–430. [Google Scholar] [CrossRef]
- Lu, Y.; Li, Y.; Wu, W. Injected nanocrystals for targeted drug delivery. Acta Pharm. Sin. B 2016, 6, 106–113. [Google Scholar] [CrossRef] [Green Version]
- Juhnke, M.; Martin, D.; John, E. Generation of wear during the production of drug nanosuspensions by wet media milling. Eur. J. Pharm. Biopharm. 2012, 81, 214–222. [Google Scholar] [CrossRef]
- Van Eerdenbrugh, B.; Van den Mooter, G.; Augustijns, P. Top-down production of drug nanocrystals: Nanosuspension stabilization, miniaturization and transformation into solid products. Int. J. Pharm. 2008, 364, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Karashima, M.; Kimoto, K.; Yamamoto, K.; Kojima, T.; Ikeda, Y. A novel solubilization technique for poorly soluble drugs through the integration of nanocrystal and cocrystal technologies. Eur. J. Pharm. Biopharm. 2016, 107, 142–150. [Google Scholar] [CrossRef]
- De Smet, L.; Saerens, L.; De Beer, T.; Carleer, R.; Adriaensens, P.; Van Bocxlaer, J.; Vervaet, C.; Remon, J.P. Formulation of itraconazole nanococrystals and evaluation of their bioavailability in dogs. Eur. J. Pharm. Biopharm. 2014, 87, 107–113. [Google Scholar] [CrossRef] [Green Version]
- Witika, B.A.; Smith, V.J.; Walker, R.B. Top-Down Synthesis of a Lamivudine-Zidovudine Nano Co-Crystal. Crystals 2021, 11, 33. [Google Scholar] [CrossRef]
- Xia, D.; Cui, Y.G. and F. Application of Precipitation Methods for the Production of Water-insoluble Drug Nanocrystals: Production Techniques and Stability of Nanocrystals. Curr. Pharm. Des. 2014, 20, 408–435. [Google Scholar] [CrossRef] [PubMed]
- Dalvi, S.V.; Yadav, M.D. Effect of ultrasound and stabilizers on nucleation kinetics of curcumin during liquid antisolvent precipitation. Ultrason. Sonochem. 2015, 24, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Sander, J.R.G.; Bučar, D.K.; Henry, R.F.; Zhang, G.G.Z.; MacGillivray, L.R. Pharmaceutical nano-cocrystals: Sonochemical synthesis by solvent selection and use of a surfactant. Angew. Chem. Int. Ed. 2010, 49, 7284–7288. [Google Scholar] [CrossRef]
- Huang, Y.; Li, J.-M.; Lai, Z.-H.; Wu, J.; Lu, T.-B.; Chen, J.-M. Phenazopyridine-phthalimide nano-cocrystal: Release rate and oral bioavailability enhancement. Eur. J. Pharm. Sci. 2017. [Google Scholar] [CrossRef]
- Hong, C.; Xie, Y.; Yao, Y.; Li, G.; Yuan, X.; Shen, H. A Novel strategy for pharmaceutical cocrystal generation without knowledge of stoichiometric ratio: Myricetin cocrystals and a ternary phase diagram. Pharm. Res. 2015, 32, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Hong, C.; Li, G.; Ma, P.; Xie, Y. The generation of myricetin-nicotinamide nanococrystals by top down and bottom up technologies. Nanotechnology 2016, 27. [Google Scholar] [CrossRef] [PubMed]
- Allen, F.H. The Cambridge Structural Database: A quarter of a million crystal structures and rising. Acta Crystallogr. Sect. B Struct. Sci. 2002, 58, 380–388. [Google Scholar] [CrossRef] [PubMed]
- Bruno, I.J.; Cole, J.C.; Edgington, P.R.; Kessler, M.; Macrae, C.F.; McCabe, P.; Pearson, J.; Taylor, R. New software for searching the Cambridge Structural Database and visualizing crystal structures. Acta Crystallogr. B 2002, 58, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Vance, E.J. Growth and Perfection of Crystals. J. Am. Chem. Soc. 1959, 81, 3489–3490. [Google Scholar] [CrossRef]
- Doremus, R.; Roberts, B.; Turnbull, D. Growth and Perfection of Crystals. J. Polym. Sci. 1959, 38, 2053–2054. [Google Scholar] [CrossRef]
- Gagniere, E.; Mangin, D.; Veesler, S.; Puel, F. Co-Crystallization in Solution and Scale-up Issues; Royal Society of Chemistry: London, UK, 2012; ISBN 9781849733502. [Google Scholar]
- Cho, E.J.; Holback, H.; Liu, K.C.; Abouelmagd, S.A.; Park, J.; Yeo, Y. Nanoparticle characterization: State of the art, challenges, and emerging technologies. Mol. Pharm. 2013, 10, 2093–2110. [Google Scholar] [CrossRef] [Green Version]
- Stefaniak, A.B.; Hackley, V.A.; Roebben, G.; Ehara, K.; Hankin, S.; Postek, M.T.; Lynch, I.; Fu, W.-E.; Linsinger, T.P.J.; Thünemann, A.F. Nanoscale reference materials for environmental, health and safety measurements: Needs, gaps and opportunities. Nanotoxicology 2013, 7, 1325–1337. [Google Scholar] [CrossRef]
- Lewinski, N.; Colvin, V.; Drezek, R. Cytotoxicity of nanopartides. Small 2008, 4, 26–49. [Google Scholar] [CrossRef] [PubMed]
- Almeida, J.P.M.; Chen, A.L.; Foster, A.; Drezek, R. In vivo biodistribution of nanoparticles. Nanomedicine 2011, 6, 815–835. [Google Scholar] [CrossRef]
- Malvern Instruments Ltd. Surfactant micelle characterization using dynamic light scattering. Malvern Instrum. 2006, MRK809-01, 1–5. [Google Scholar]
- Malvern Instruments White Paper: Dynamic Light Scattering, Common terms defined; Malvern Instruments Limited: Worcestershire, UK, 2011; pp. 1–6.
- Lu, X.-Y.; Wu, D.-C.; Li, Z.-J.; Chen, G.-Q. Polymer nanoparticles. Prog. Mol. Biol. Transl. Sci. 2011, 104, 299–323. [Google Scholar] [CrossRef] [PubMed]
- Hoo, C.M.; Starostin, N.; West, P.; Mecartney, M.L. A comparison of atomic force microscopy (AFM) and dynamic light scattering (DLS) methods to characterize nanoparticle size distributions. J. Nanopart. Res. 2008, 10, 89–96. [Google Scholar] [CrossRef]
- Boyd, R.D.; Pichaimuthu, S.K.; Cuenat, A. New approach to inter-technique comparisons for nanoparticle size measurements; using atomic force microscopy, nanoparticle tracking analysis and dynamic light scattering. Colloids Surfaces A Physicochem. Eng. Asp. 2011, 387, 35–42. [Google Scholar] [CrossRef]
- Mahl, D.; Diendorf, J.; Meyer-Zaika, W.; Epple, M. Possibilities and limitations of different analytical methods for the size determination of a bimodal dispersion of metallic nanoparticles. Colloids Surfaces A Physicochem. Eng. Asp. 2011, 377, 386–392. [Google Scholar] [CrossRef]
- Murdock, R.C.; Braydich-Stolle, L.; Schrand, A.M.; Schlager, J.J.; Hussain, S.M. Characterization of nanomaterial dispersion in solution prior to in vitro exposure using dynamic light scattering technique. Toxicol. Sci. 2008, 101, 239–253. [Google Scholar] [CrossRef] [Green Version]
- Kirby, B.J.; Hasselbrink, E.F. Zeta potential of microfluidic substrates: 2. Data for polymers. Electrophoresis 2004, 25, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Kirby, B.J.; Hasselbrink, E.F. Zeta potential of microfluidic substrates: 1. Theory, experimental techniques, and effects on separations This. Electrophoresis 2004, 25, 187–202. [Google Scholar] [CrossRef]
- Malamatari, M.; Somavarapu, S.; Taylor, K.M.; Buckton, G. Solidification of nanosuspensions for the production of solid oral dosage forms and inhalable dry powders Solidification of nanosuspensions for the production of solid oral dosage forms and inhalable dry powders. Expert Opin. Drug Deliv. 2016. [Google Scholar] [CrossRef]
- Peng, H.R.; Gong, M.M.; Chen, Y.Z.; Liu, F. Thermal stability of nanocrystalline materials: Thermodynamics and kinetics. Int. Mater. Rev. 2017, 62, 303–333. [Google Scholar] [CrossRef]
- Verdonck, E.; Schaap, K.; Thomas, L.C. A discussion of the principles and applications of Modulated Temperature DSC (MTDSC). Int. J. Pharm. 1999, 192, 3–20. [Google Scholar] [CrossRef]
- Chogale, M.M.; Ghodake, V.N.; Patravale, V.B. Performance parameters and characterizations of nanocrystals: A brief review. Pharmaceutics 2016, 8, 26. [Google Scholar] [CrossRef]
- Kocbek, P.; Baumgartner, S.; Kristl, J. Preparation and evaluation of nanosuspensions for enhancing the dissolution of poorly soluble drugs. Int. J. Pharm. 2006, 312, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Valo, H.; Kovalainen, M.; Laaksonen, P.; Häkkinen, M.; Auriola, S.; Peltonen, L.; Linder, M.; Järvinen, K.; Hirvonen, J.; Laaksonen, T. Immobilization of protein-coated drug nanoparticles in nanofibrillar cellulose matrices-Enhanced stability and release. J. Control Release 2011, 156, 390–397. [Google Scholar] [CrossRef]
- Clas, S.; Dalton, C.; Hancock, B. Differential scanning calorimetry: Applications in drug development. Pharm. Sci. Technol. Today 1999, 2, 311–320. [Google Scholar] [CrossRef]
- Mourdikoudis, S.; Pallares, R.M.; Thanh, N.T.K. Characterization techniques for nanoparticles: Comparison and complementarity upon studying nanoparticle properties. Nanoscale 2018, 10, 12871–12934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingham, B.; Toney, M.F. X-Ray Diffraction for Characterizing Metallic Films. In Metallic Films for Electronic, Optical and Magnetic Applications: Structure, Processing and Properties; Elsevier Ltd.: Amsterdam, The Netherlands, 2013; pp. 3–38. ISBN 9780857090577. [Google Scholar]
- Müllertz, A.; Perrie, Y.; Rades, T. Advances in Delivery Science and Technology: Analytical Techniques in the Pharmaceutical Sciences; Rathbone, M.J., Ed.; Springer Science and Business Media LLC: New York, NY, USA, 2016; ISBN 9781493940271. [Google Scholar]
- Engelhard, M.H.; Droubay, T.C.; Du, Y. X-ray Photoelectron Spectroscopy Applications. In Encyclopedia of Spectroscopy and Spectrometry; Elsevier: Amsterdam, The Netherlands, 2017; pp. 716–724. ISBN 9780128032244. [Google Scholar]
- Chauhan, A. Powder XRD Technique and its Applications in Science and Technology. J. Anal. Bioanal. Tech. 2014, 5. [Google Scholar] [CrossRef] [Green Version]
- Kirtansinh, G.; Piyushbhai, P.; Natubhai, P. Application of Analytical Techniques in Preformulation Study: A Review. Int. J. Pharm. Biol. Arch. 2011, 2, 1319–1326. [Google Scholar]
- Hirsch, P.B. Elements of X-Ray Diffraction, 2nd ed.; Wesley-Addion Publishing Company: Reading, MA, USA, 1957; Volume 8, ISBN 0201610914. [Google Scholar]
- Upadhyay, S.; Parekh, K.; Pandey, B. Influence of crystallite size on the magnetic properties of Fe3O4 nanoparticles. J. Alloys Compd. 2016, 678, 478–485. [Google Scholar] [CrossRef]
- Li, W.; Zamani, R.; Rivera Gil, P.; Pelaz, B.; Ibáñez, M.; Cadavid, D.; Shavel, A.; Alvarez-Puebla, R.A.; Parak, W.J.; Arbiol, J.; et al. CuTe nanocrystals: Shape and size control, plasmonic properties, and use as SERS probes and photothermal agents. J. Am. Chem. Soc. 2013, 135, 7098–7101. [Google Scholar] [CrossRef]
- Yan, W.; Mahurin, S.M.; Overbury, S.H.; Dai, S. Nanoengineering catalyst supports via layer-by-layer surface functionalization. Top. Catal. 2006, 39, 199–212. [Google Scholar] [CrossRef]
- Wang, W.; Chen, X.; Cai, Q.; Mo, G.; Jiang, L.S.; Zhang, K.; Chen, Z.J.; Wu, Z.H.; Pan, W. In situ SAXS study on size changes of platinum nanoparticles with temperature. Eur. Phys. J. B 2008, 65, 57–64. [Google Scholar] [CrossRef]
- Cipolla, D.; Wu, H.; Salentinig, S.; Boyd, B.; Rades, T.; Vanhecke, D.; Petri-Fink, A.; Rothin-Rutishauser, B.; Eastman, S.; Redelmeier, T.; et al. Formation of drug nanocrystals under nanoconfinement afforded by liposomes. RSC Adv. 2016, 6, 6223–6233. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Mudie, S.; Cipolla, D.; Rades, T.; Boyd, B.J. Solid State Characterization of Ciprofloxacin Liposome Nanocrystals. Mol. Pharm. 2019, 16, 184–194. [Google Scholar] [CrossRef] [PubMed]
- Schilt, Y.; Berman, T.; Wei, X.; Barenholz, Y.; Raviv, U. Using solution X-ray scattering to determine the high-resolution structure and morphology of PEGylated liposomal doxorubicin nanodrugs. Biochim. Biophys. Acta 2016, 1860, 108–119. [Google Scholar] [CrossRef]
- Li, X.; Hirsh, D.J.; Cabral-Lilly, D.; Zirkel, A.; Gruner, S.M.; Janoff, A.S.; Perkins, W.R. Doxorubicin physical state in solution and inside liposomes loaded via a pH gradient. Biochim. Biophys. Acta 1998, 1415, 23–40. [Google Scholar] [CrossRef] [Green Version]
- Matthew, J. Surface Analysis by AUGER and X-Ray Photoelectron Spectroscopy; Briggs, D., Grant, J.T., Eds.; John Wiley & Sons, Ltd.: Chichester, UK, 2004; Volume 36, ISBN 1-901019-04-7. [Google Scholar]
- Ray, S.; Shard, A.G. Quantitative Analysis of Adsorbed Proteins by X-ray Photoelectron Spectroscopy. Anal. Chem. 2011, 83, 8659–8666. [Google Scholar] [CrossRef]
- Beloqui Redondo, A.; Jordan, I.; Ziazadeh, I.; Kleibert, A.; Giorgi, J.B.; Wörner, H.J.; May, S.; Abbas, Z.; Brown, M.A. Nanoparticle-Induced Charge Redistribution of the Air–Water Interface. J. Phys. Chem. C 2015, 119, 2661–2668. [Google Scholar] [CrossRef]
- Qiu, L.; Liu, F.; Zhao, L.; Ma, Y.; Yao, J. Comparative XPS study of surface reduction for nanocrystalline and microcrystalline ceria powder. Appl. Surf. Sci. 2006, 252, 4931–4935. [Google Scholar] [CrossRef]
- Chow, S.F.; Wan, K.Y.; Cheng, K.K.; Wong, K.W.; Sun, C.C.; Baum, L.; Chow, A.H.L. Development of highly stabilized curcumin nanoparticles by flash nanoprecipitation and lyophilization. Eur. J. Pharm. Biopharm. 2015, 94, 436–449. [Google Scholar] [CrossRef]
- Dong, Y.; Feng, S.S. Methoxy poly(ethylene glycol)-poly(lactide) (MPEG-PLA) nanoparticles for controlled delivery of anticancer drugs. Biomaterials 2004, 25, 2843–2849. [Google Scholar] [CrossRef]
- Mu, L.; Feng, S.S. A novel controlled release formulation for the anticancer drug paclitaxel (Taxol®): PLGA nanoparticles containing vitamin E TPGS. J. Control Release 2003, 86, 33–48. [Google Scholar] [CrossRef]
- El-Hagrasy, A.S.; Morris, H.R.; D’Amico, F.; Lodder, R.A.; Drennen, J.K. Near-infrared spectroscopy and imaging for the monitoring of powder blend homogeneity. J. Pharm. Sci. 2001, 90, 1298–1307. [Google Scholar] [CrossRef]
- Robert, M.S.; Webster, F.X.; Kiemle, D.J.; Bryce, D.L. Spectrometric Identification of Organic Compounds, 3rd ed.; Wiley: Hoboken, NJ, USA, 1976; Volume 30, ISBN 0471393622. [Google Scholar]
- Lai, F.; Pini, E.; Corrias, F.; Perricci, J.; Manconi, M.; Fadda, A.M.; Sinico, C. Formulation strategy and evaluation of nanocrystal piroxicam orally disintegrating tablets manufacturing by freeze-drying. Int. J. Pharm. 2014, 467, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Witika, B.A. Formulation Development, Manufacture and Evaluation of a Lamivudine-Zidovudine Nano Co-Crystal Thermo-Responsive Suspension. Ph.D. Thesis, Rhodes University, Makhanda, South Africa, 2020. [Google Scholar]
- Sharma, P.; Zujovic, Z.D.; Bowmaker, G.A.; Denny, W.A.; Garg, S. Evaluation of a crystalline nanosuspension: Polymorphism, process induced transformation and in vivo studies. Int. J. Pharm. 2011, 408, 138–151. [Google Scholar] [CrossRef] [PubMed]
- Loridant, S.; Lucazeau, G.; Le Bihan, T. A high-pressure Raman and X-ray diffraction study of the perovskite SrCeO3. J. Phys. Chem. Solids 2002, 63, 1983–1992. [Google Scholar] [CrossRef]
- Colomban, P. ReviewRaman Studies of Inorganic Gels and of Their Sol-to-Gel, Gel-to-Glass and Glass-to-Ceramics Transformation. J. Raman Spectrosc. 1996, 27, 747–758. [Google Scholar] [CrossRef]
- Durán, P.; Capel, F.; Tartaj, J.; Gutierrez, D.; Moure, C. Heating-rate effect on the BaTiO3 formation by thermal decomposition of metal citrate polymeric precursors. Solid State Ion. 2001, 141–142, 529–539. [Google Scholar] [CrossRef]
- Parayanthal, P.; Pollak, F.H. Raman Scattering in Alloy Semiconductors: “Spatial Correlation” Model. Phys. Rev. Lett. 1984, 52, 1822–1825. [Google Scholar] [CrossRef]
- Ager, J.W.; Veirs, D.K.; Rosenblatt, G.M. Spatially resolved Raman studies of diamond films grown by chemical vapor deposition. Phys. Rev. B 1991, 43, 6491–6499. [Google Scholar] [CrossRef]
- Critchley, L. Is Raman Spectroscopy Useful in Nanomaterial Analysis? Available online: https://www.azonano.com/article.aspx?ArticleID=5273 (accessed on 21 March 2021).
- Mu, S.; Li, M.; Guo, M.; Yang, W.; Wang, Y.; Li, J.; Fu, Q.; He, Z. Spironolactone nanocrystals for oral administration: Different pharmacokinetic performances induced by stabilizers. Colloids Surfaces B Biointerfaces 2016, 147, 73–80. [Google Scholar] [CrossRef]
- De Waard, H.; De Beer, T.; Hinrichs, W.L.J.; Vervaet, C.; Remon, J.P.; Frijlink, H.W. Controlled crystallization of the lipophilic drug fenofibrate during freeze-drying: Elucidation of the mechanism by in-line raman spectroscopy. AAPS J. 2010, 12, 569–575. [Google Scholar] [CrossRef] [PubMed]
- Swarbick, J. Encyclopedia of Pharmaceutical Technology, 3rd ed.; CRC Press: Boca Raton, FL, USA, 2018; ISBN 0824725387. [Google Scholar]
- Klienebudde, P. The Crystallite-Gel-Model for microcrystalline Celluslose in Wet Granulation, Extrusion and Spheronization. Pharm. Res. 1997, 14, 804–809. [Google Scholar] [CrossRef] [PubMed]
- Pitchayajittipong, C.; Price, R.; Shur, J.; Kaerger, J.S.; Edge, S. Characterisation and functionality of inhalation anhydrous lactose. Int. J. Pharm. 2010, 390, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Crisp, J.L.; Dann, S.E.; Blatchford, C.G. Antisolvent crystallization of pharmaceutical excipients from aqueous solutions and the use of preferred orientation in phase identification by powder X-ray diffraction. Eur. J. Pharm. Sci. 2011, 42, 568–577. [Google Scholar] [CrossRef]
- Ho, R.; Naderi, M.; Heng, J.Y.Y.; Williams, D.R.; Thielmann, F.; Bouza, P.; Keith, A.R.; Thiele, G.; Burnett, D.J. Effect of milling on particle shape and surface energy heterogeneity of needle-Shaped crystals. Pharm. Res. 2012, 29, 2806–2816. [Google Scholar] [CrossRef] [PubMed]
- Kubavat, H.A.; Shur, J.; Ruecroft, G.; Hipkiss, D.; Price, R. Investigation into the influence of primary crystallization conditions on the mechanical properties and secondary processing behaviour of fluticasone propionate for carrier based dry powder inhaler formulations. Pharm. Res. 2012, 29, 994–1006. [Google Scholar] [CrossRef]
- Otte, A.; Teresa, C. Assessment of Milling-Induced Disorder of Two Pharmaceutical Compounds. J. Pharm. Sci. 2012, 101, 322–332. [Google Scholar] [CrossRef]
- Park, M.H.; Kim, J.H.; Jeon, J.W.; Park, J.K.; Lee, B.J.; Suh, G.H.; Cho, C.W. Preformulation studies of bee venom for the preparation of bee venom-loaded PLGA particles. Molecules 2015, 20, 15072–15083. [Google Scholar] [CrossRef] [Green Version]
- Patel, P.; Ahir, K.; Patel, V.; Manani, L.; Patel, C. Drug-Excipient compatibility studies: First step for dosage form development. Pharma Innov. J. 2015, 4, 14–20. [Google Scholar]
- Ruska, E.; Knoll, M.; Ruska, E. Das Elektronenmikroskop. Zeitschrift Phys. 1932, 78, 318–339. [Google Scholar] [CrossRef]
- Kuntsche, J.; Horst, J.C.; Bunjes, H. Cryogenic transmission electron microscopy (cryo-TEM) for studying the morphology of colloidal drug delivery systems. Int. J. Pharm. 2011, 417, 120–137. [Google Scholar] [CrossRef]
- Klang, V.; Matsko, N.B. Electron microscopy of pharmaceutical systems. Adv. Imaging Electron. Phys. 2014, 181, 125–208. [Google Scholar] [CrossRef]
- Swartz, M.E. Ultra performance liquid chromatography (UPLC): An introduction. Sep. Sci. Re-Defin. 2005, 586, 8–14. [Google Scholar]
- Hamilton, R.J.; Sewell, P.A. Introduction to High Performance Liquid Chromatography; Hamilton, R.J., Sewell, P.A., Eds.; Springer: Dordrecht, The Netherlands, 1982; Volume 15, ISBN 978-94-009-5938-5. [Google Scholar]
- Davankov, V.A. Separation of enantiomeric compounds using chiral HPLC systems. A brief review of general principles, advances, and development trends. Chromatographia 1989, 27, 475–482. [Google Scholar] [CrossRef]
- Aygün, Ş.F.; Özcimder, M. A comparison of normal(-CN) and reversed (C-18) phase chromatographic behaviour of polycyclic aromatic hydrocarbons. Turk. J. Chem. 1996, 20, 269–275. [Google Scholar]
- de Villiers, A.; Lestremau, F.; Szucs, R.; Gélébart, S.; David, F.; Sandra, P. Evaluation of ultra performance liquid chromatography. Part I. Possibilities and limitations. J. Chromatogr. A 2006, 1127, 60–69. [Google Scholar] [CrossRef]
- Vervoort, R.J.M.; Debets, A.J.J.; Claessens, H.A.; Cramers, C.A.; De Jong, G.J. Optimisation and characterisation of silica-based reversed-phase liquid chromatographic systems for the analysis of basic pharmaceuticals. J. Chromatogr. A 2000, 897, 1–22. [Google Scholar] [CrossRef]
- Snyder, R.J.; Kirkland, J.; Glajch, J.L. Practical HPLC Method Development, 2nd ed.; John Wiley and Sons: New York, NY, USA, 1997; Volume 41. [Google Scholar]
- Simpson, C. Practical HPLC; The Whitefriars Press: London, UK, 1976. [Google Scholar]
- Raghavan, R.; Joseph, J. Chromatographic Methodsof Analysis-High Performance Lquid Chromatography, Enclopedia or Pharmaceutical Technology; Informa Healthcare USA: New York, NY, USA, 2002. [Google Scholar]
- Young, C.S.; Weigand, R.J. An efficient approach to column selection in HPLC method development. LC-GC N. Am. 2020, 20, 464–473. [Google Scholar]
- Ewing, G.W.; Jordan, J. Instrumental Methods of Chemical Analysis, 5th ed.; Himalaya Publishing House: New Delhi, India, 1955; Volume 27. [Google Scholar]
- Beckett, A.H.; Stenlake, J.B. Practical Pharmaceutical Chemistry; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 1963; Volume 52. [Google Scholar]
- Siddiqui, M.R.; AlOthman, Z.A.; Rahman, N. Analytical techniques in pharmaceutical analysis: A review. Arab. J. Chem. 2017, 10, S1409–S1421. [Google Scholar] [CrossRef] [Green Version]
- Butnariu, M.; Coradini, C.Z. Evaluation of Biologically Active Compounds from Calendula officinalis Flowers using Spectrophotometry. Chem. Cent. J. 2012, 6, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antosiewicz, J.M.; Shugar, D. UV–Vis spectroscopy of tyrosine side-groups in studies of protein structure. Part 1: Basic principles and properties of tyrosine chromophore. Biophys. Rev. 2016, 8, 151–161. [Google Scholar] [CrossRef] [Green Version]
- Braun, C.S.; Kueltzo, L.A.; Russell Middaugh, C. Ultraviolet absorption and circular dichroism spectroscopy of nonviral gene delivery complexes. Methods Mol. Med. 2001, 65, 253–284. [Google Scholar] [CrossRef]
- Giovannetti, R. The Use of Spectrophotometry UV-Vis for the Study of Porphyrins. In Macro to Nano Spectroscopy; IntechOpen: Rijeka, Croatia, 2012. [Google Scholar]
- Owen, T. Fundamentals of UV-Visible Spectroscopy: A Primer; Agilent Technologies: Waldbronn, Germany, 2000; ISBN 9788578110796. [Google Scholar]
- Parnis, J.M.; Oldham, K.B. Beyond the beer-lambert law: The dependence of absorbance on time in photochemistry. J. Photochem. Photobiol. A Chem. 2013, 267, 6–10. [Google Scholar] [CrossRef]
- Allen, H.C.; Brauers, T.; Finlayson-Pitts, B.J. Illustration of Deviations in the Beer-Lambert Law in an Instrumental Analysis Laboratory: Measuring Atmospheric Pollutants by Differential Optical Absorption Spectrometry. J. Chem. Educ. 1997, 74, 1459. [Google Scholar] [CrossRef]
- Ernst, O.; Zor, T. Linearization of the Bradford protein assay. J. Vis. Exp. 2010, 1–6. [Google Scholar] [CrossRef]
- Baker, W.B.; Parthasarathy, A.B.; Busch, D.R.; Mesquita, R.C.; Greenberg, J.H.; Yodh, A.G. Modified Beer-Lambert law for blood flow. Biomed. Opt. Express 2014, 5, 4053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peltonen, L.; Strachan, C. Understanding critical quality attributes for nanocrystals from preparation to delivery. Molecules 2015, 20, 22286–22300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Li, T. Cellular Uptake Mechanism of Paclitaxel Nanocrystals Determined by Confocal Imaging and Kinetic Measurement. AAPS J. 2015, 17, 1126–1134. [Google Scholar] [CrossRef] [Green Version]
- Witika, B.A.; Walker, R.B. Development, manufacture and characterization of niosomes for the delivery for nevirapine. Pharmazie 2019, 74, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Makoni, P.A.; Kasongo, K.W.; Walker, R.B. Short Term Stability Testing of Efavirenz-Loaded Solid Lipid Nanoparticle (SLN) and Nanostructured Lipid Carrier (NLC) Dispersions. Pharmaceutics 2019, 11, 397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerdeira, A.M.; Mazzotti, M.; Gander, B. Pharmaceutical Nanotechnology Miconazole nanosuspensions: Influence of formulation variables on particle size reduction and physical stability. Int. J. Pharm. 2010, 396, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Kumar, S.; Gokhale, R.; Burgess, D.J. Physical stability of nanosuspensions: Investigation of the role of stabilizers on Ostwald ripening. Int. J. Pharm. 2011, 406, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Teeranachaideekul, V.; Junyaprasert, V.B.; Souto, E.B.; Müller, R.H. Development of ascorbyl palmitate nanocrystals applying the nanosuspension technology. Int. J. Pharm. 2008, 354, 227–234. [Google Scholar] [CrossRef]
- Center of Drug Evaluation and Research. Regulatory Classification of Pharmaceutical Co-Crystals, Guidance for Industry; US Food&Drug Administration: Silver Spring, MD, USA, 2018.
- Peltonen, L. Practical guidelines for the characterization and quality control of pure drug nanoparticles and nano-cocrystals in the pharmaceutical industry. Adv. Drug Deliv. Rev. 2018, 131, 101–115. [Google Scholar] [CrossRef] [PubMed]
- Accardo, A.; Tesauro, D.; Roscigno, P.; Gianolio, E.; Paduano, L.; D’Errico, G.; Pedone, C.; Morelli, G. Physicochemical properties of mixed micellar aggregates containing CCK peptides and Gd complexes designed as tumor specific contrast agents in MRI. J. Am. Chem. Soc. 2004, 126, 3097–3107. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Wei, H.; Zhang, X.Z.; Cheng, S.X.; Zhuo, R.X. Thermo-triggered and biotinylated biotin-P(NIPAAm-co-HMAAm)-b-PMMA micelles for controlled drug release. J. Biomed. Mater. Res. Part A 2009, 88, 814–822. [Google Scholar] [CrossRef] [PubMed]
- Toncheva, V.; Schacht, E.; Ng, S.Y.; Barr, J.; Heller, J. Use of block copolymers of poly(ortho esters) and poly (ethylene glycol) micellar carriers as potential tumour targeting systems. J. Drug Target. 2003, 11, 345–353. [Google Scholar] [CrossRef]
- Yang, X.; Li, L.; Wang, Y.; Tan, Y. Preparation, pharmacokinetics and tissue distribution of micelles made of reverse thermo-responsive polymers. Int. J. Pharm. 2009, 370, 210–215. [Google Scholar] [CrossRef]
- Tuomela, A.; Hirvonen, J.; Peltonen, L. Stabilizing agents for drug nanocrystals: Effect on bioavailability. Pharmaceutics 2016, 8, 16. [Google Scholar] [CrossRef] [Green Version]
- Rad, R.T.; Mortazavi, S.A.; Vatanara, A.; Dadashzadeh, S. Enhanced dissolution rate of tadalafil nanoparticles prepared by sonoprecipitation technique: Optimization and physicochemical investigation. Iran. J. Pharm. Res. 2017, 16, 1335–1348. [Google Scholar] [CrossRef]
- Pireddu, R.; Caddeo, C.; Valenti, D.; Marongiu, F.; Scano, A.; Ennas, G.; Lai, F.; Fadda, A.M.; Sinico, C. Diclofenac acid nanocrystals as an effective strategy to reduce in vivo skin inflammation by improving dermal drug bioavailability. Colloids Surfaces B Biointerfaces 2016, 143, 64–70. [Google Scholar] [CrossRef]
- Hoffman, A.S. The origins and evolution of “controlled” drug delivery systems. J. Control Release 2008, 132, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Anhalt, K.; Geissler, S.; Harms, M.; Weigandt, M.; Fricker, G. Development of a new method to assess nanocrystal dissolution based on light scattering. Pharm. Res. 2012, 29, 2887–2901. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, M.; Baptista, B.; Lopes, J.A.; Sarraguça, M.C. Pharmaceutical cocrystallization techniques. Advances and challenges. Int. J. Pharm. 2018, 547, 404–420. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, Y.; Patel, V. Nanosuspension: An approach to enhance solubility of drugs. J. Adv. Pharm. Technol. Res. 2011, 2, 81. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. ICH Topic Q6A Specifications: Test Procedures and Acceptance Criteria for New Drug Substances and New Drug Products: Chemical Substances Step; ECA Academy: London, UK, 2000. [Google Scholar]



{kind=link}
{kind=link}
| Entity to Be Characterized | Techniques * |
|---|---|
| Size and morphology | DLS, SEM, TEM |
| Surface charge | ZP |
| Surface chemical analysis | XPS, EDX |
| Crystal structure | SAXS, PXRD, DSC |
| Growth kinetics | SAXS, NMR, TEM, cryo-TEM, liquid-TEM |
| Agglomeration state | ZP, DLS, DCS, UV-Vis, SEM, Cryo-TEM, TEM |
| Ligand binding/density/mass/surface composition /bulk composition | XPS, FTIR, NMR, |
| Dispersion of NCM in matrices/supports | TEM, SEM |
| Dissolution Testing | (U)HPLC, UV |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Witika, B.A.; Aucamp, M.; Mweetwa, L.L.; Makoni, P.A. Application of Fundamental Techniques for Physicochemical Characterizations to Understand Post-Formulation Performance of Pharmaceutical Nanocrystalline Materials. Crystals 2021, 11, 310. https://doi.org/10.3390/cryst11030310
Witika BA, Aucamp M, Mweetwa LL, Makoni PA. Application of Fundamental Techniques for Physicochemical Characterizations to Understand Post-Formulation Performance of Pharmaceutical Nanocrystalline Materials. Crystals. 2021; 11(3):310. https://doi.org/10.3390/cryst11030310
Chicago/Turabian StyleWitika, Bwalya A., Marique Aucamp, Larry L. Mweetwa, and Pedzisai A. Makoni. 2021. "Application of Fundamental Techniques for Physicochemical Characterizations to Understand Post-Formulation Performance of Pharmaceutical Nanocrystalline Materials" Crystals 11, no. 3: 310. https://doi.org/10.3390/cryst11030310