
Accelerated Weathering and Carbonation (Mild to Intensified) of Natural Canadian Silicates (Kimberlite and Wollastonite) for CO2 Sequestration †


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
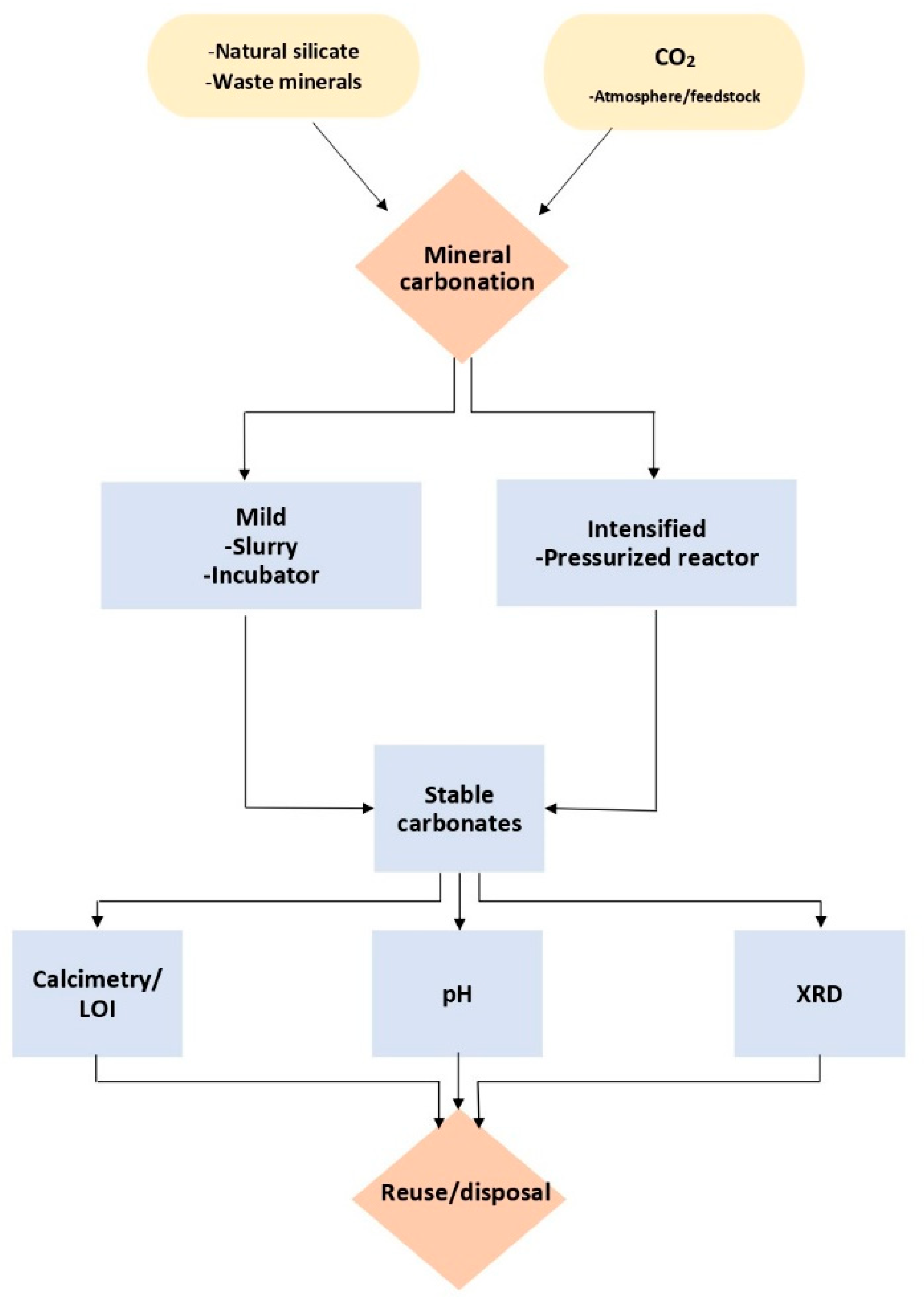
2. Materials and Methods
2.1. Minerals
2.1.1. Kimberlite Tailings
2.1.2. Wollastonite Ore
2.2. Accelerated Weathering and Carbonation Experiments
2.2.1. Incubator Process
2.2.2. Slurry Process
2.2.3. Pressurized Slurry Process
2.3. Analytical Tests
2.3.1. pH Test
2.3.2. Furnace Test
2.3.3. Calcimeter Test
2.3.4. XRD Analysis
3. Results and Discussion
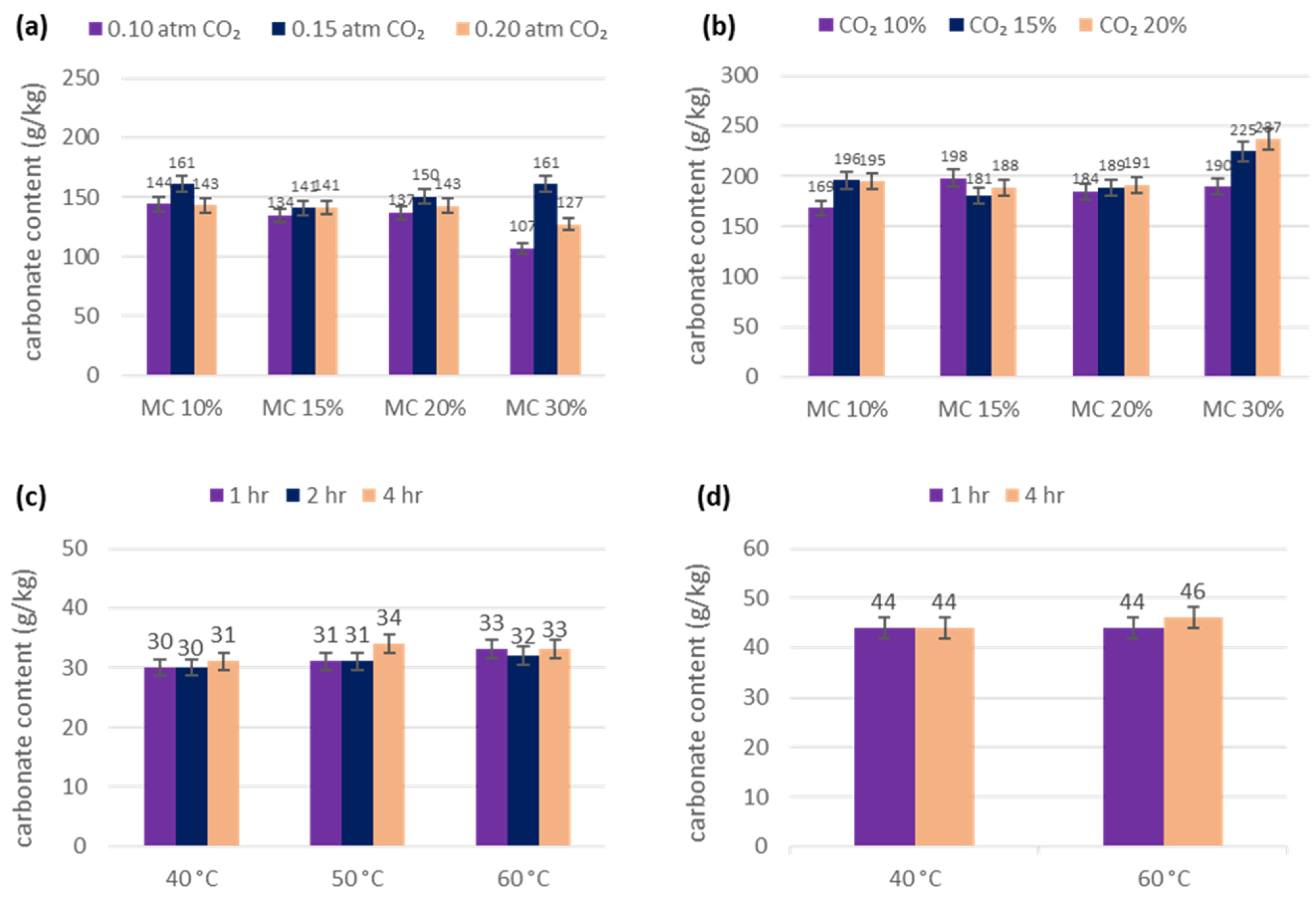
3.1. pH Test
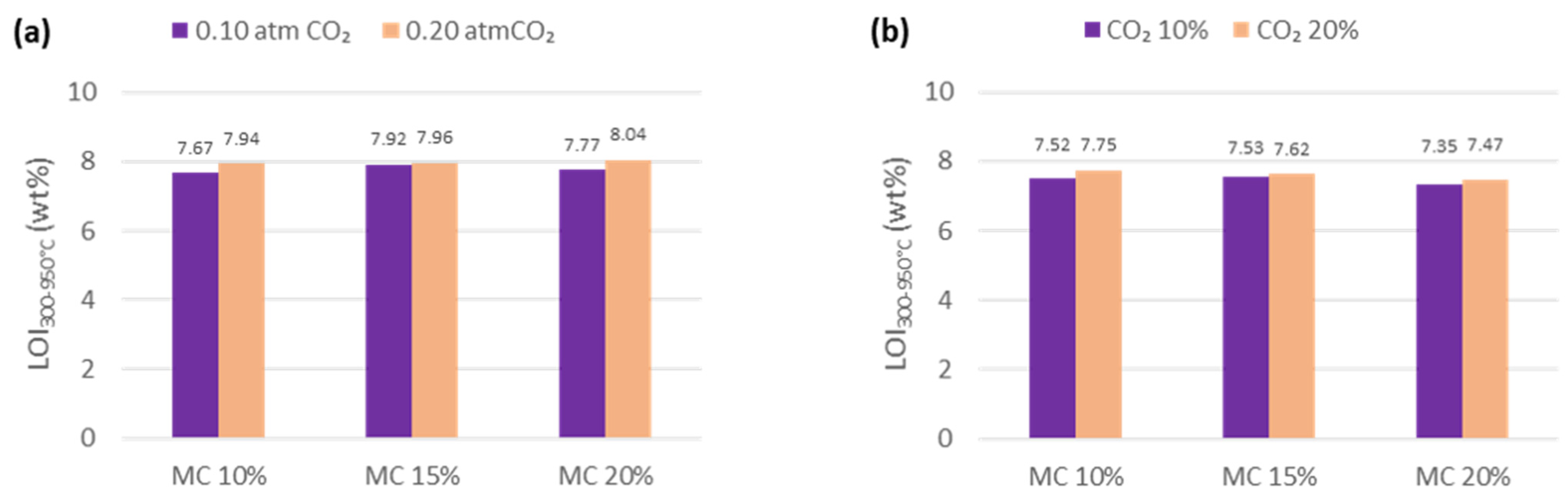
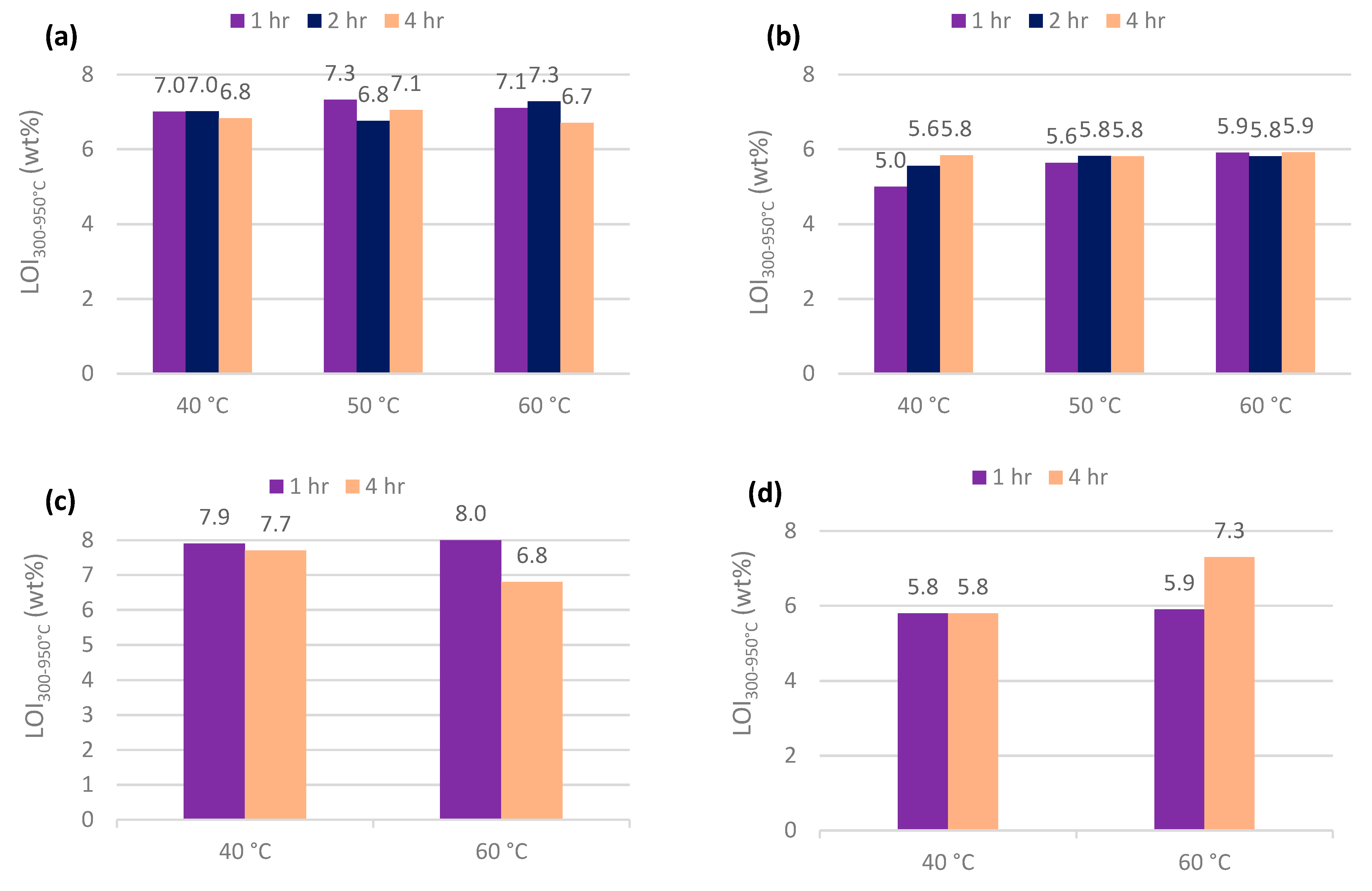
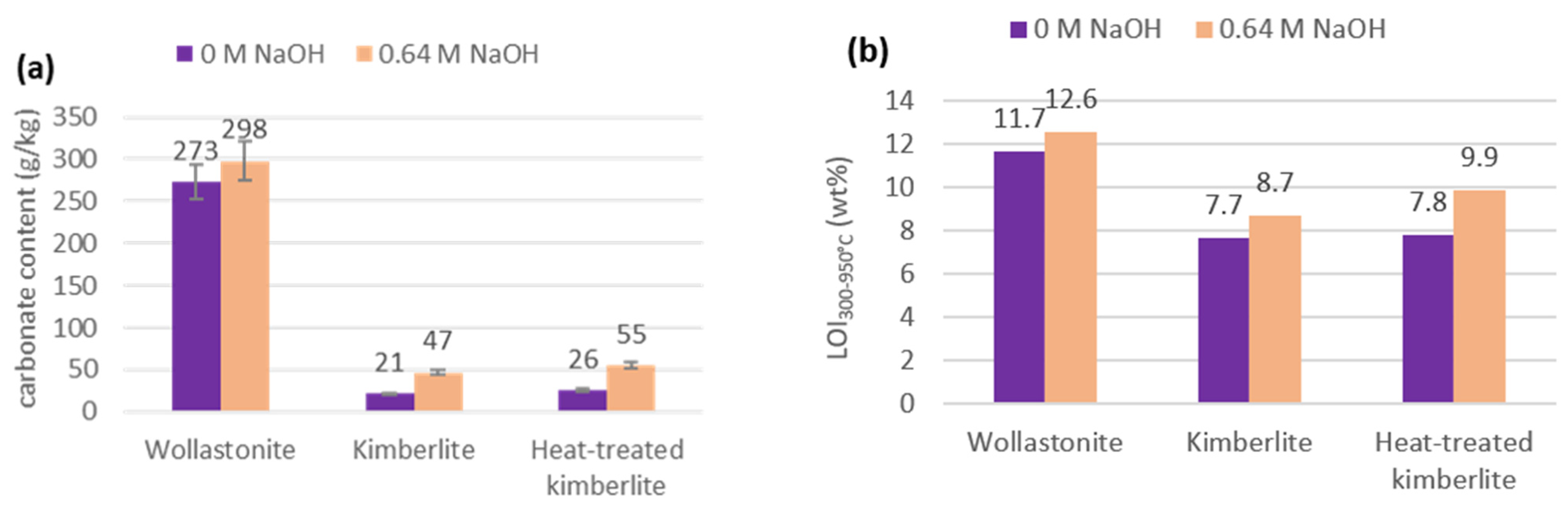
3.2. Furnace and Calcimeter Tests
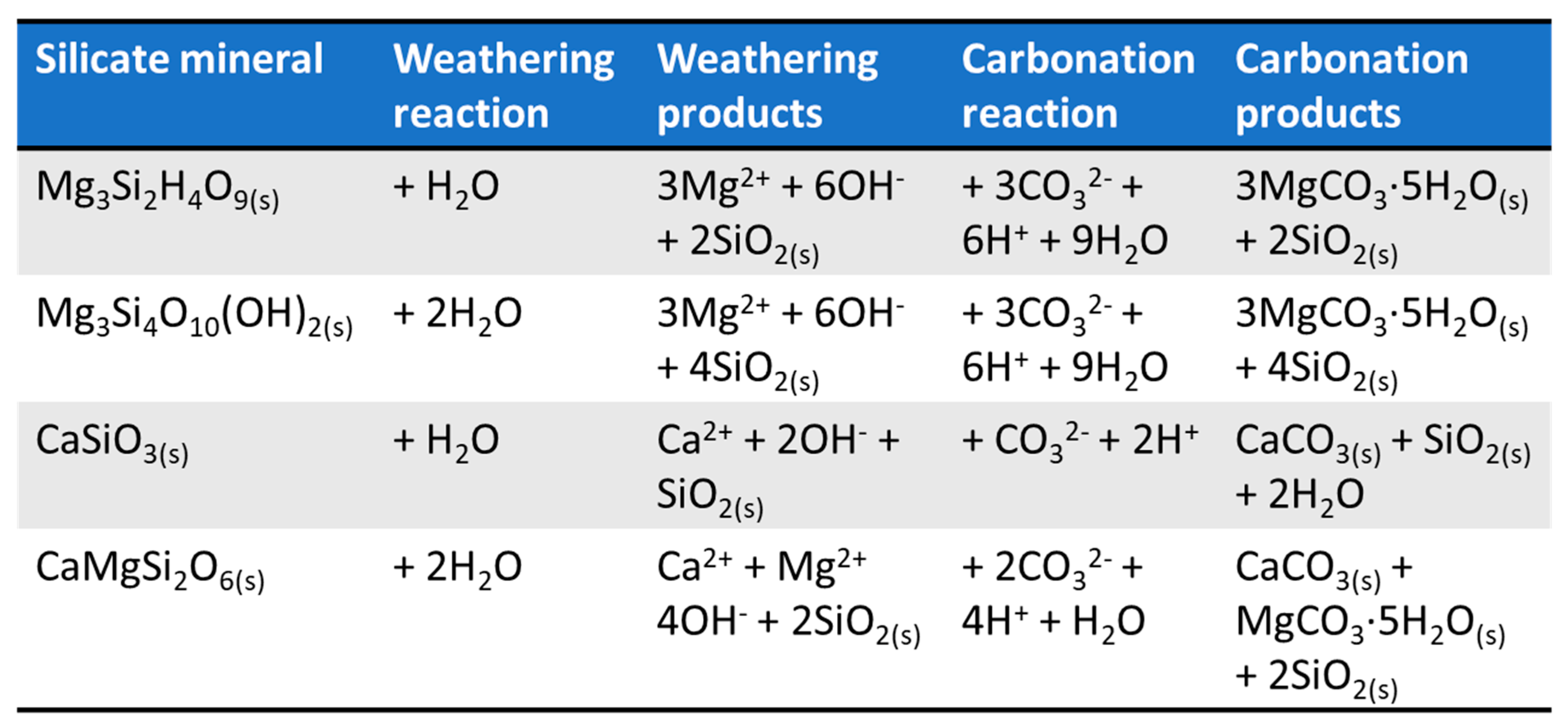
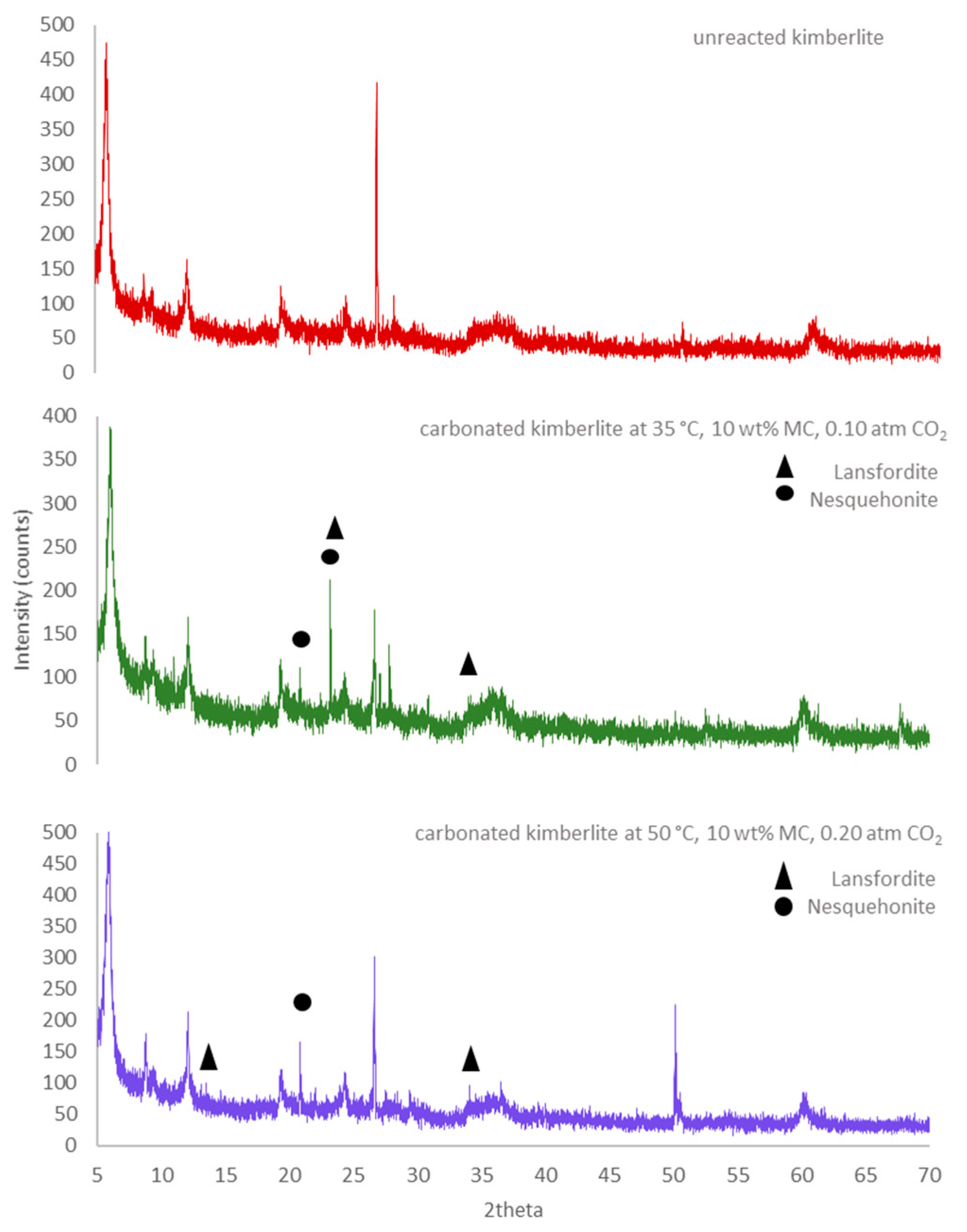
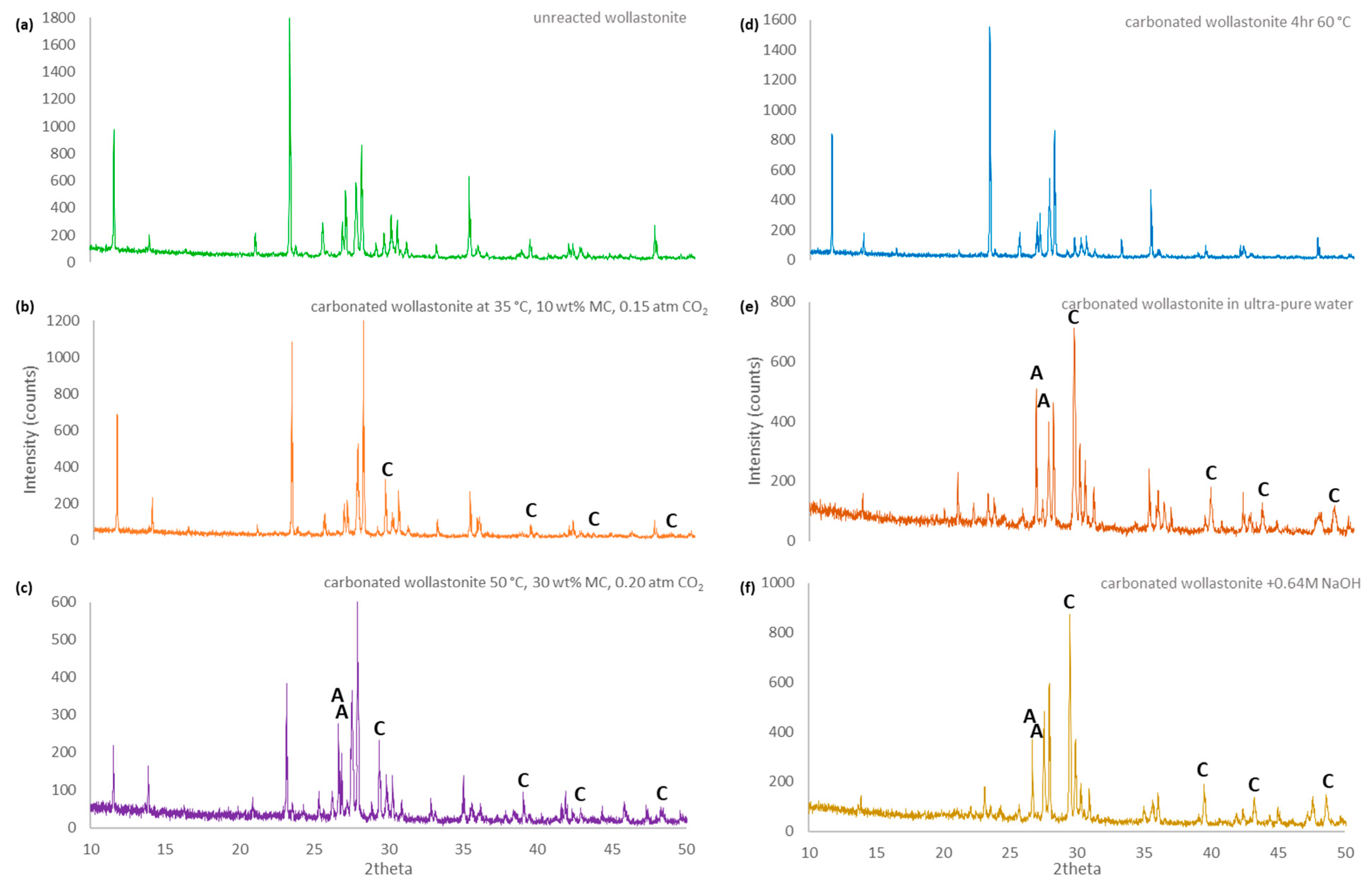
3.3. XRD Analysis
3.4. Perspectives on Tailings and Ore Carbonation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- UNFCCC Secretariat Welcomes IPCC’s Global Warming of 1.5 °C Report. Available online: https://unfccc.int/news/unfccc-secretariat-welcomes-ipcc-s-global-warming-of-15degc-report (accessed on 11 December 2021).
- Pan, S.Y.; Chen, Y.H.; Fan, L.-S.; Kim, H.; Gao, X.; Ling, T.-C.; Chiang, P.-C.; Pei, S.-L.; Gu, G. CO2 mineralization and utilization by alkaline solid wastes for potential carbon reduction. Nat. Sustain. 2020, 3, 399–405. [Google Scholar] [CrossRef]
- Snæbjörnsdóttir, S.Ó.; Sigfússon, B.; Marieni, C.; Goldberg, D.; Gislason, S.R.; Oelkers, E.H. Carbon dioxide storage through mineral carbonation. Nat. Rev. Earth Environ. 2020, 1, 90–102. [Google Scholar] [CrossRef] [Green Version]
- Zhang, N.; Santos, R.M.; Smith, S.M.; Šiller, L. Acceleration of CO2 mineralisation of alkaline brines with nickel nanoparticles catalysts in continuous tubular reactor. Chem. Eng. J. 2019, 377, 120479. [Google Scholar] [CrossRef]
- Boyjoo, Y.; Pareek, V.; Liu, J. Synthesis of micro and nano-sized calcium carbonate particles and their applications. J. Mater. Chem. A 2014, 2, 14270–14288. [Google Scholar] [CrossRef]
- Ncongwane, M.; Broadhurst, J.; Petersen, J. Assessment of the potential carbon footprint of engineered processes for the mineral carbonation of PGM tailings. Int. J. Greenh. Gas Control 2018, 77, 70–81. [Google Scholar] [CrossRef] [Green Version]
- Huijgen, W.; Comans, R. Carbon Dioxide Sequestration by Mineral Carbonation; Energy Research Centre of the Netherlands (ECN): Petten, The Netherlands, 2003. [Google Scholar]
- Fricker, K.J.; Park, A.H. Effect of H2O on Mg(OH)2 carbonation pathways for combined CO2 capture and storage. Chem. Eng. Sci. 2013, 100, 332–341. [Google Scholar] [CrossRef]
- Nduagu, E.; Bergerson, J.; Zevenhoven, R. Life cycle assessment of CO2 sequestration in magnesium silicate rock—A comparative study. Energy Convers. Manag. 2012, 55, 116–126. [Google Scholar] [CrossRef]
- Monasterio-Guillot, L.; Fernandez-Martinez, A.; Ruiz-Agudo, E.; Rodriguez-Navarro, C. Carbonation of calcium-magnesium pyroxenes: Physical-chemical controls and effects of reaction-driven fracturing. Geochim. Cosmochim. Acta 2021, 304, 258–280. [Google Scholar] [CrossRef]
- Wang, F.; Dreisinger, D.; Jarvis, M.; Hitchins, T. Kinetic evaluation of mineral carbonation of natural silicate samples. Chem. Eng. J. 2021, 404, 126522. [Google Scholar] [CrossRef]
- Ashraf, W.; Olek, J. Elucidating the accelerated carbonation products of calcium silicates using multi-technique approach. J. CO2 Util. 2018, 23, 61–74. [Google Scholar] [CrossRef]
- Ding, W.; Fu, L.; Ouyang, J.; Yang, H. CO2 mineral sequestration by wollastonite carbonation. Phys. Chem. Miner. 2014, 41, 489–496. [Google Scholar] [CrossRef]
- Ding, W.; Yang, H.; Ouyang, J.; Long, H. Modified wollastonite sequestrating CO2 and exploratory application of the carbonation products. RSC Adv. 2016, 6, 78090–78099. [Google Scholar] [CrossRef]
- Yadav, S.; Mehra, A. Mathematical modelling and experimental study of carbonation of wollastonite in the aqueous media. J. CO2 Util. 2019, 31, 181–191. [Google Scholar] [CrossRef]
- Di Lorenzo, F.; Ruiz-Agudo, C.; Ibañez-Velasco, A.; Gil-San Millán, R.; Navarro, J.; Ruiz-Agudo, E.; Rodriguez-Navarro, C. The Carbonation of Wollastonite: A Model Reaction to Test Natural and Biomimetic Catalysts for Enhanced CO2 Sequestration. Minerals 2018, 8, 209. [Google Scholar] [CrossRef] [Green Version]
- Min, Y.; Li, Q.; Voltolini, M.; Kneafsey, T.; Jun, Y.-S. Wollastonite Carbonation in Water-Bearing Supercritical CO2: Effects of Particle Size. Environ. Sci. Technol. 2017, 51, 13044–13053. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, J.L.; Wilson, S.A.; Morgan, B.; Harrison, A.; Turvey, C.C.; Paterson, D.; Dipple, G.M.; Southam, G. Accelerating Mineral Carbonation in Ultramafic Mine Tailings via Direct CO2 Reaction and Heap Leaching with Potential for Base Metal Enrichment and Recovery. Econ. Geol. 2020, 115, 303–323. [Google Scholar] [CrossRef]
- Ramli, N.A.A.; Kusin, F.M.; Molahid, V.L.M. Influencing Factors of the Mineral Carbonation Process of Iron Ore Mining Waste in Sequestering Atmospheric Carbon Dioxide. Sustainability 2021, 13, 1866. [Google Scholar] [CrossRef]
- Dlugogorski, B.Z.; Balucan, R.D. Dehydroxylation of serpentine minerals: Implications for mineral carbonation. Renew. Sustain. Energy Rev. 2013, 32, 353–367. [Google Scholar] [CrossRef] [Green Version]
- Haque, F.; Santos, R.M.; Chiang, Y.W. Optimizing inorganic carbon sequestration and crop yield with wollastonite soil amendment in a microplot study. Front. Plant Sci. 2020, 11, 1012. [Google Scholar] [CrossRef]
- Bodor, M.; Santos, R.; Kriskova, L.; Elsen, J.; Vlad, M.; Van Gerven, T. Susceptibility of mineral phases of steel slags towards carbonation: Mineralogical, morphological and chemical assessment. Eur. J. Mineral. 2013, 25, 533–549. [Google Scholar] [CrossRef]
- Santos, R.M.; Van Bouwel, J.; Vandevelde, E.; Mertens, G.; Elsen, J.; Van Gerven, T. Accelerated mineral carbonation of stainless steel slags for CO2 storage and waste valorization: Effect of process parameters on geochemical properties. Int. J. Greenh. Gas Control 2013, 17, 32–45. [Google Scholar] [CrossRef] [Green Version]
- Santos, R.M.; Bodor, M.; Dragomir, P.N.; Vraciu, A.G.; Vlad, M.; Van Gerven, T. Magnesium chloride as a leaching and aragonite-promoting self-regenerative additive for the mineral carbonation of calcium-rich materials. Miner. Eng. 2014, 59, 71–81. [Google Scholar] [CrossRef] [Green Version]
- Santos, R.M.; van Audenaerde, A.; Chiang, Y.W.; Iacobescu, R.I.; Knops, P.; van Gerven, T. Nickel extraction from olivine: Effect of carbonation pre-treatment. Metals 2015, 5, 1620–1644. [Google Scholar] [CrossRef] [Green Version]
- Dudhaiya, A.; Haque, F.; Fantucci, H.; Santos, R.M. Characterization of physically fractionated wollastonite-amended agricultural soils. Minerals 2019, 9, 635. [Google Scholar] [CrossRef] [Green Version]
- Soong, Y.; Goodman, A.L.; Mccarthy-Jones, J.R.; Baltrus, J.P. Experimental and simulation studies on mineral trapping of CO2 with brine. Energy Convers. Manag. 2004, 45, 1845–1859. [Google Scholar] [CrossRef]
- Li, Y.; Chen, X.; Huang, W.; Yang, J. Below the Room Temperature Measurements of CO2 Solubilities in Six Physical Absorbents. J. Chem. Thermodyn. 2018, 122, 113–141. [Google Scholar] [CrossRef]
- Morgan, B.; Wilson, S.A.; Madsen, I.C.; Gozukara, Y.M.; Habsuda, J. Increased thermal stability of nesquehonite (MgCO3·3H2O) in the presence of humidity and CO2: Implications for low-temperature CO2 storage. Int. J. Greenh. Gas Control 2015, 39, 366–376. [Google Scholar] [CrossRef]
- Zhang, Z.; Zheng, Y.; Ni, Y.; Liu, Z.; Chen, J.; Liang, X. Temperature-and pH-dependent morphology and FT-IR analysis of magnesium carbonate hydrates. J. Phys. Chem. B 2006, 110, 12969–12973. [Google Scholar] [CrossRef] [PubMed]
- Mervine, E.M.; Wilson, S.A.; Power, I.M.; Dipple, G.M.; Turvey, C.C.; Hamilton, J.L.; Vanderzee, S.; Raudsepp, M.; Southam, C.; Matter, J.M.; et al. Potential for offsetting diamond mine carbon emissions through mineral carbonation of process kimberlite: An assessment of De Beers mine sites in South Africa and Canada. Miner. Petrol. 2018, 112, 755–765. [Google Scholar] [CrossRef]
- Ryu, K.W.; Jo, H.; Choi, S.H.; Chae, S.C.; Jang, Y.N. Changes in mineral assemblages during serpentine carbonation. Appl. Clay Sci. 2016, 134, 62–67. [Google Scholar] [CrossRef]
- Chang, R.; Kim, S.; Lee, S.; Choi, S.; Kim, M.; Park, Y. Calcium Carbonate Precipitation for CO2 Storage and Utilization: A Review of the Carbonate Crystallization and Polymorphism. Front. Energy Res. 2017, 5, 17. [Google Scholar] [CrossRef] [Green Version]
- Georgakopoulos, E.; Santos, R.M.; Chiang, Y.W.; Manovic, V. Two-way valorization of blast furnace slag: Synthesis of precipitated calcium carbonate and zeolitic heavy metal adsorbent. J. Vis. Exp. 2017, 120, e55062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakravarthy, A.C.; Chalouati, S.; Chai, Y.E.; Fantucci, H.; Santos, R.M. Valorization of Kimberlite Tailings by Carbon Capture and Utilization (CCU) Method. Minerals 2020, 10, 611. [Google Scholar] [CrossRef]
- Haque, F.; Santos, R.M.; Chiang, Y.W. Urban farming with enhanced rock weathering as a prospective climate stabilization wedge. Environ. Sci. Technol. 2021, 55, 13575–13578. [Google Scholar] [CrossRef] [PubMed]
- Hopkinson, L.; Rutt, K.; Cressey, G. The Transformation of Nesquehonite to Hydromagnesite in the System CaO-MgO-H2O-CO2: An Experimental Spectroscopic Study. J. Geol. 2018, 116, 387–400. [Google Scholar] [CrossRef] [Green Version]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chai, Y.E.; Chalouati, S.; Fantucci, H.; Santos, R.M. Accelerated Weathering and Carbonation (Mild to Intensified) of Natural Canadian Silicates (Kimberlite and Wollastonite) for CO2 Sequestration. Crystals 2021, 11, 1584. https://doi.org/10.3390/cryst11121584
Chai YE, Chalouati S, Fantucci H, Santos RM. Accelerated Weathering and Carbonation (Mild to Intensified) of Natural Canadian Silicates (Kimberlite and Wollastonite) for CO2 Sequestration. Crystals. 2021; 11(12):1584. https://doi.org/10.3390/cryst11121584
Chicago/Turabian StyleChai, Ye Eun, Salma Chalouati, Hugo Fantucci, and Rafael M. Santos. 2021. "Accelerated Weathering and Carbonation (Mild to Intensified) of Natural Canadian Silicates (Kimberlite and Wollastonite) for CO2 Sequestration" Crystals 11, no. 12: 1584. https://doi.org/10.3390/cryst11121584