
Surface Structure and Electronic Properties of Lu3Al5O12

Abstract
:1. Introduction
2. Computational Methods
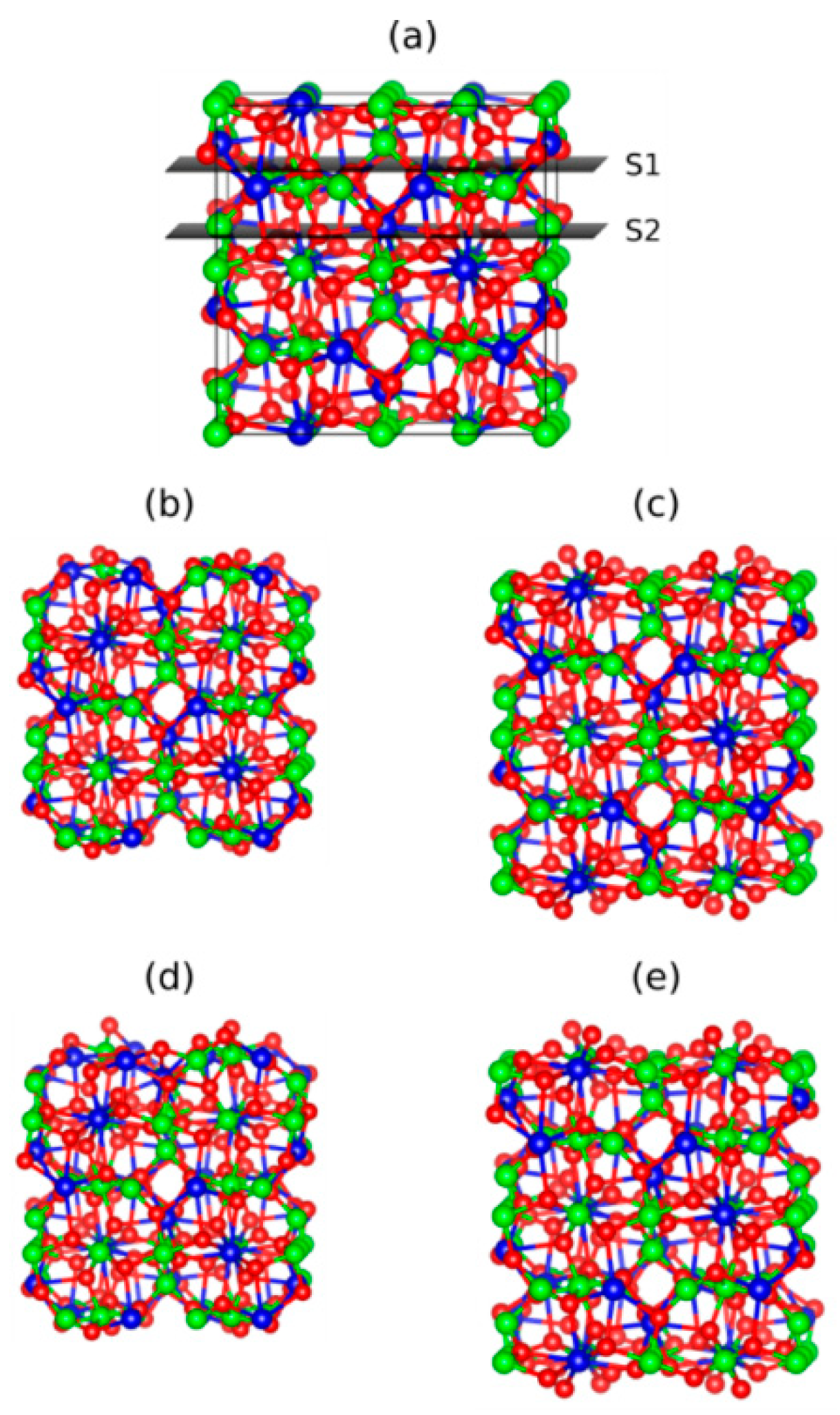
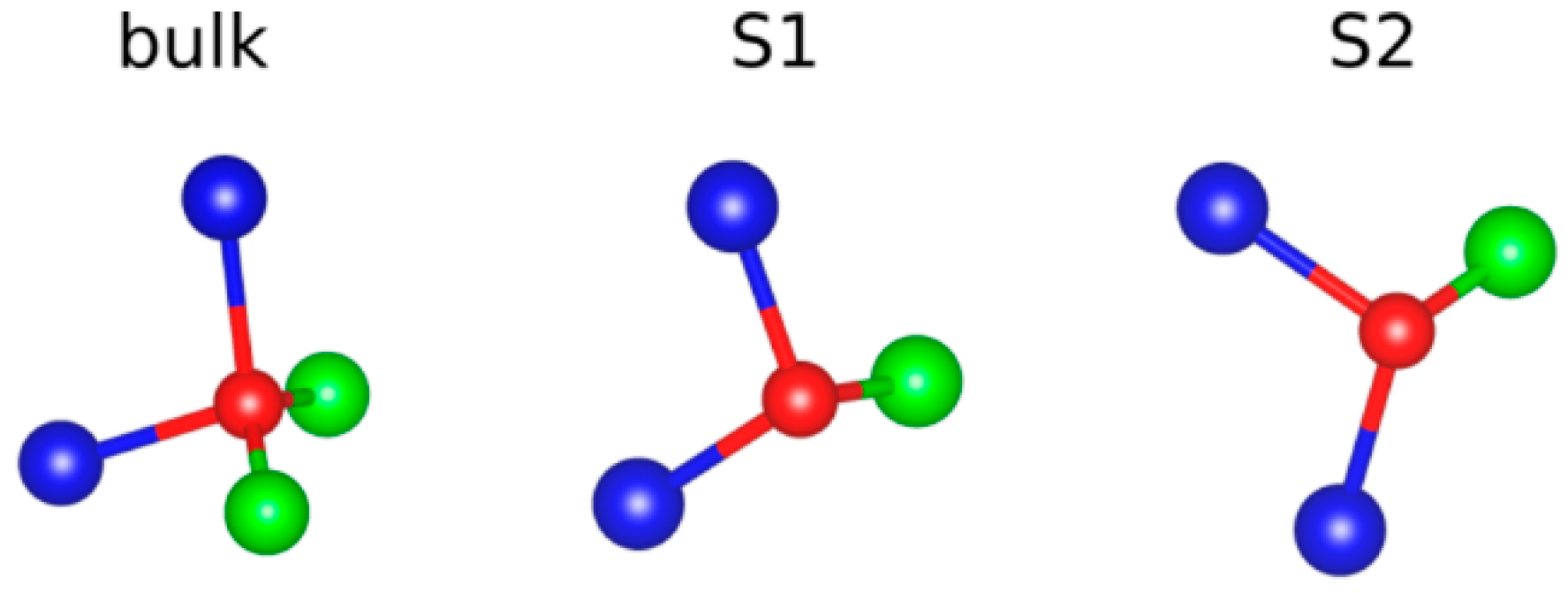
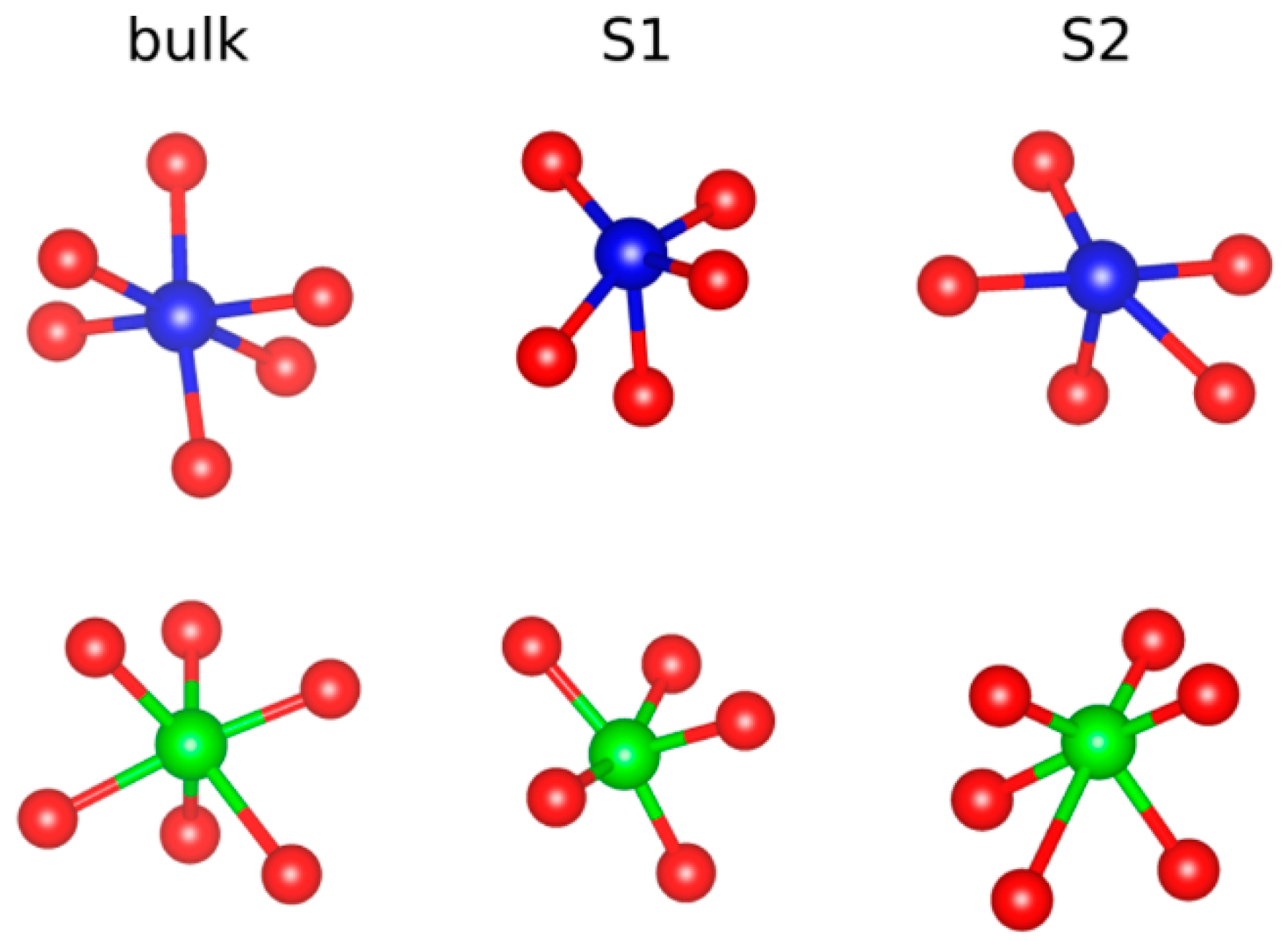
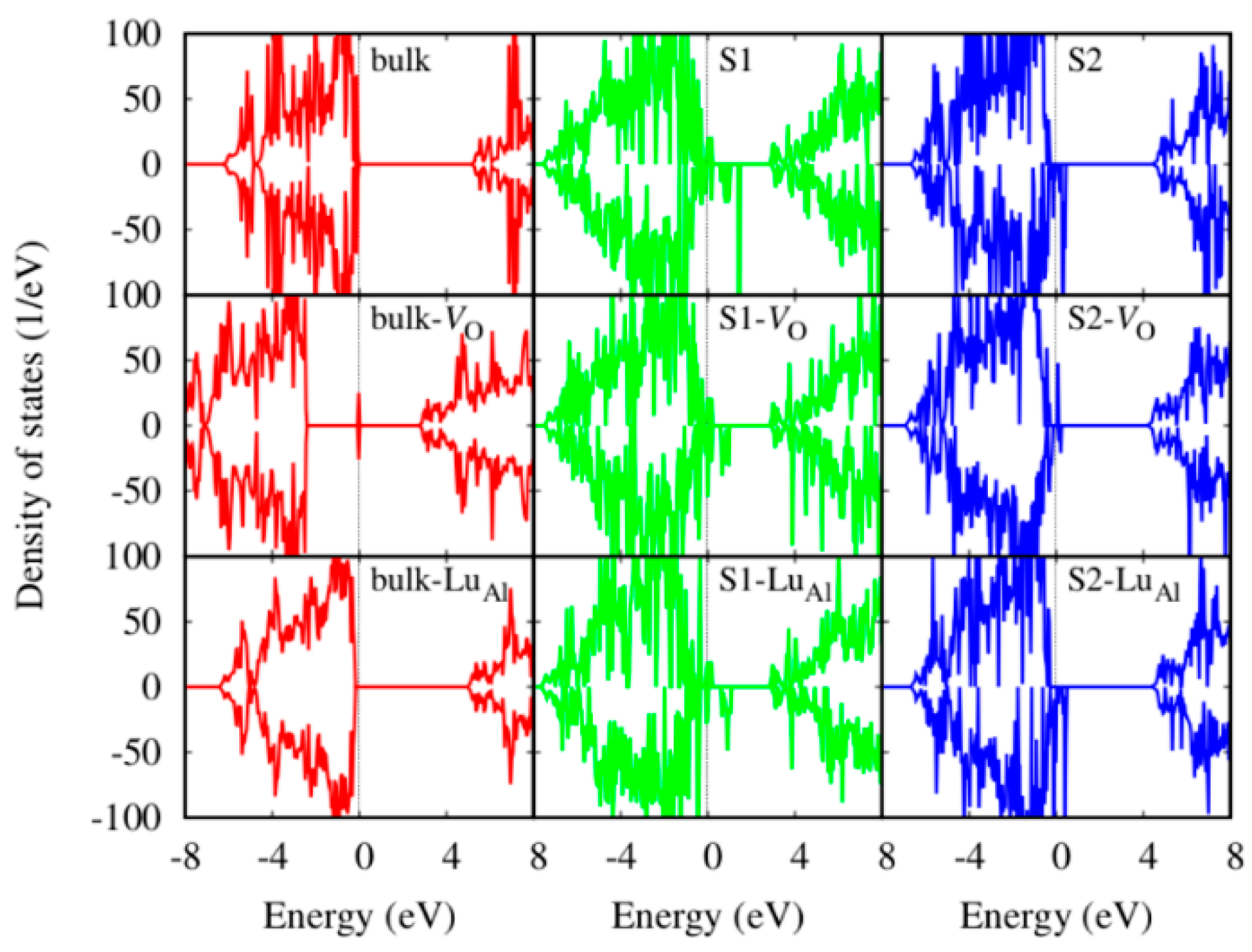
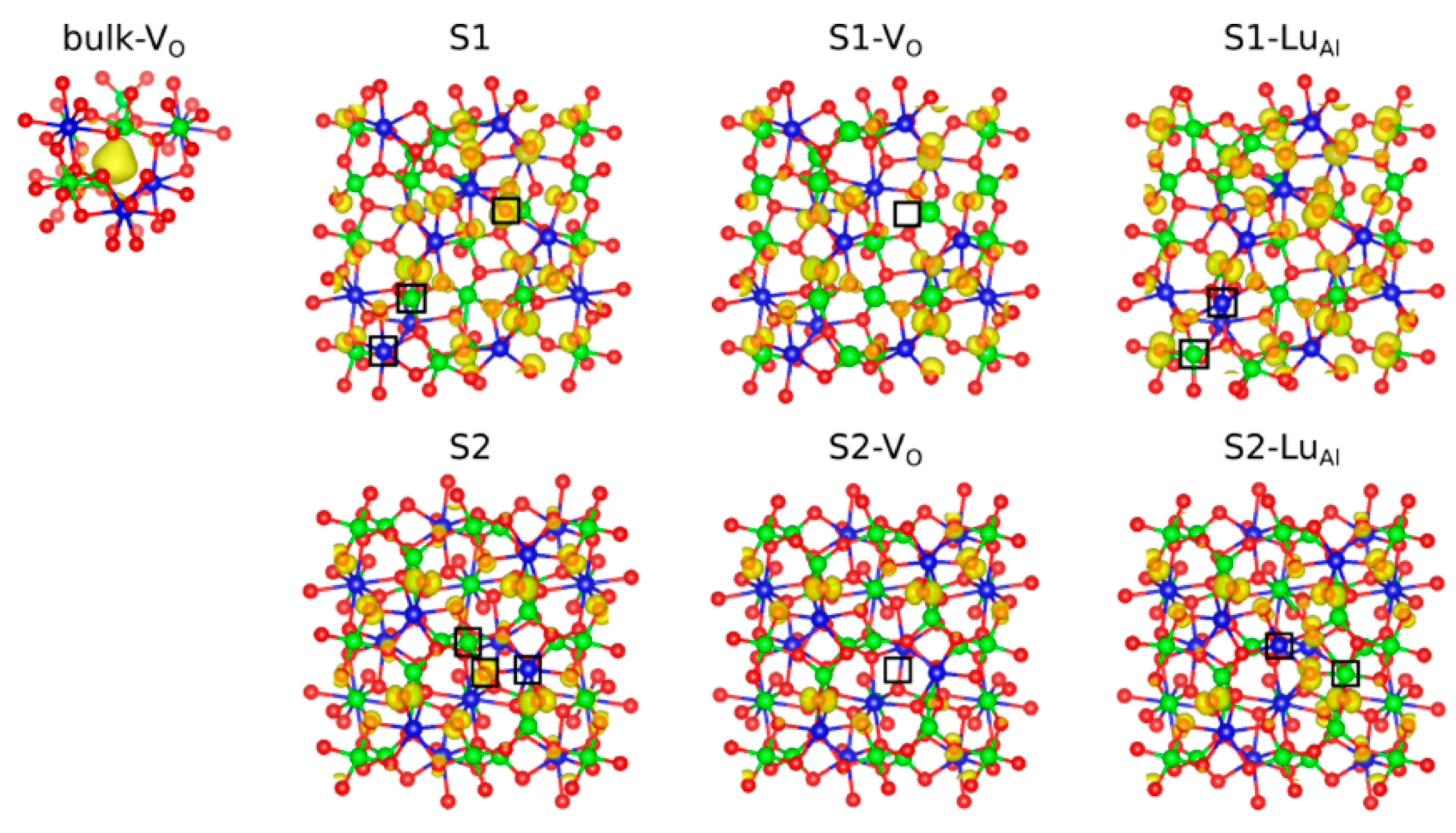
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Mao, J.; Wang, C.; Hong, T.; Yu, Y. Three-nanosecond-equal interval sub-pulse Nd: YAG laser with multi-step active Q-switching. Chin. Opt. Lett. 2021, 19, 071404. [Google Scholar] [CrossRef]
- Tang, K.; Liao, W.; Lin, D.; Li, B.; Chen, W. Self-polarization emission based on coherent combination of intracavity eigenmodes in Nd:YAG/Cr 4 +:YAG lasers. Chin. Opt. Lett. 2021, 19, 3–7. [Google Scholar] [CrossRef]
- Wei, M.; Cheng, T.; Dou, R.; Zhang, Q.; Jiang, H. Superior performance of a 2 kHz pulse Nd: YAG laser based on a gradient-doped crystal. Photonics Res. 2021, 9, 1191–1196. [Google Scholar] [CrossRef]
- Sipes, D.L. Highly efficient neodymium: Yttrium aluminum garnet laser end pumped by a semiconductor laser array. Appl. Phys. Lett. 1985, 47, 74–75. [Google Scholar] [CrossRef]
- Takayuki YANAGIDA Inorganic scintillating materials and scintillation detectors. Proc. Jpn. Acad. Ser. B 2018, 94, 75–97. [CrossRef] [Green Version]
- Dujardin, C.; Auffray, E.; Bourret-Courchesne, E.; Dorenbos, P.; Lecoq, P.; Nikl, M.; Vasil’Ev, A.N.; Yoshikawa, A.; Zhu, R.Y. Needs, trends, and advances in inorganic scintillators. IEEE Trans. Nucl. Sci. 2018, 65, 1977–1997. [Google Scholar] [CrossRef] [Green Version]
- Wolski, D. Properties of the YAG: Ce scintillator. Nucl. Instrum. Methods Phys. Res. Sect. A Accel. Spectrometers Detect. Assoc. Equip. 1994, 345, 461–467. [Google Scholar] [CrossRef]
- Nikl, M. Energy transfer phenomena in the luminescence of wide band-gap scintillators. Phys. Stat. Sol. 2005, 202, 201–206. [Google Scholar] [CrossRef]
- Nikl, M. Scintillation detectors for X-rays. Meas. Sci. Technol. 2006, 17, 37–54. [Google Scholar] [CrossRef]
- Gironnet, J.; Mikhailik, V.B.; Kraus, H.; Marcillac, P.; De Coron, N. Scintillation studies of Bi4Ge3O12 (BGO) down to a temperature of 6 K. Nucl. Instrum. Methods Phys. Res. Sect. A Accel. Spectrometers Detect. Assoc. Equip. 2008, 594, 358–361. [Google Scholar] [CrossRef]
- Nikl, M.; Yoshikawa, A.; Kamada, K.; Nejezchleb, K. Development of luag-based scintillator crystals—A review. Prog. Cryst. Growth Charact. Mater. 2013, 59, 47–72. [Google Scholar] [CrossRef]
- Nikl, M.; Vedda, A.; Fasoli, M.; Fontana, I.; Laguta, V.V.; Mihokova, E.; Pejchal, J.; Rosa, J.; Nejezchleb, K. shallow traps and radiative recombination processes in lu3al5o12: Ce single crystal scintillator. Phys. Rev. B 2007, 76, 195121. [Google Scholar] [CrossRef]
- Nikl, M.; Laguta, V.V.; Vedda, A. Complex oxide scintillators: Material defects and scintillation performance. Phys. Status Solidi B 2008, 245, 1701–1722. [Google Scholar] [CrossRef]
- Kuklja, M.M. Defects in yttrium aluminium perovskite and garnet crystals: Atomistic study. J. Phys. Condens. Matter 2000, 12, 2953–2967. [Google Scholar] [CrossRef]
- Chen, J.; Lu, T.C.; Xu, Y.; Xu, A.G.; Chen, D.Q. Ab initio study of a charged vacancy in yttrium aluminum garnet (Y3Al5O12). J. Phys. Condens. Matter 2008, 20, 325212. [Google Scholar] [CrossRef]
- Popov, A.I.; Kotomin, E.A.; Maier, J. Basic properties of the F -type centers in halides, oxides and perovskites. Nucl. Inst. Methods Phys. Res. B 2010, 268, 3084–3089. [Google Scholar] [CrossRef]
- Kotomin, E.A.; Popov, A.I. Radiation-induced point defects in simple oxides. Nucl. Inst. Methods Phys. Res. B 1998, 141, 1–15. [Google Scholar] [CrossRef]
- Springis, M.; Pujats, A.; Valbis, J. Polarization of luminescence of colour centres in YAG crystals. J. Phys. Condens. Matter 1991, 3, 5457–5461. [Google Scholar] [CrossRef]
- Sun, Y.; Egawa, T.; Shao, C.; Zhang, L.; Yao, X. Quantitative study of F center in high-surface-area anatase titania nanoparticles prepared by MOCVD. J. Phys. Chem. Solids 2004, 65, 1793–1797. [Google Scholar] [CrossRef]
- Lushchik, A.; Lushchik, C.; Popov, A.I.; Schwartz, K.; Shablonin, E.; Vasil’chenko, E. Influence of complex impurity centres on radiation damage in wide-gap metal oxides. Nucl. Inst. Methods Phys. Res. B 2016, 374, 90–96. [Google Scholar] [CrossRef] [Green Version]
- Babin, V.; Blazek, K.; Krasnikov, A.; Nejezchleb, K.; Nikl, M.; Savikhina, T.; Zazubovich, S. Luminescence of undoped LuAG and YAG crystals. Phys. Status Solidi C. 2005, 2, 97–100. [Google Scholar] [CrossRef] [Green Version]
- Zorenko, Y.; Voloshinovskii, A.; Savchyn, V.; Voznyak, T.; Nikl, M.; Nejezchleb, K.; Mikhailin, V.; Kolobanov, V.; Spassky, D. Exciton and antisite defect-related luminescence in Lu3Al5O12 and Y3Al5O12 garnets. Phys. Status Solidi B. 2007, 244, 2180–2189. [Google Scholar] [CrossRef]
- Voznyak, T.; Gorbenko, V.; Zorenko, T.; Voloshinovski, A.; Vistovsky, V.; Nikl, M.; Nejezchleb, K.; Kolobanov, V.; Spasski, D. Luminescence Spectroscopy of Excitons and Antisite Defects in Lu3Al5O12 Single Crystals and Single-Crystal Films. Opt. Spectrosc. 2008, 104, 75–87. [Google Scholar] [CrossRef]
- Marinopoulos, A.G. First-principles study of the formation energies and positron lifetimes of vacancies in the Yttrium-Aluminum Garnet Y3Al5O12. Eur. Phys. J. B 2019, 92, 3–9. [Google Scholar] [CrossRef]
- Jia, Y.; Miglio, A.; Gonze, X.; Mikami, M. Ab-initio study of oxygen vacancy stability in bulk and Cerium-doped lutetium oxyorthosilicate. J. Lumin. 2018, 204, 499–505. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [Green Version]
- Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.; Sidletskiy, O.; Boyaryntseva, Y.; Grynyov, B. Structure and role of carbon-related defects in yttrium aluminum garnet. Opt. Mater. 2021, 111, 110561. [Google Scholar] [CrossRef]
- Hu, C.; Liu, S.; Shi, Y.; Kou, H.; Li, J.; Pan, Y.; Feng, X.; Liu, Q. Antisite defects in nonstoichiometric Lu3Al5O12: Ce ceramic scintillators. Phys. Status Solidi B 2015, 252, 1993–1999. [Google Scholar] [CrossRef]
- Zorenko, Y.; Zorenko, T.; Gorbenko, V.; Pavlyk, B.; Laguta, V.; Nikl, M.; Kolobanov, V.; Spassky, D. Luminescence and ESR characteristics of γ-irradiated Lu 3Al5O12:Ce single crystalline film scintillators. Radiat. Meas. 2010, 45, 419–421. [Google Scholar] [CrossRef]
- Jia, Y.; Miglio, A.; Mikami, M.; Gonze, X. Ab initio study of luminescence in Ce-doped Lu2SiO5: The role of oxygen vacancies on emission color and thermal quenching behavior. Phys. Rev. Mater. 2018, 2, 125202. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Coordination | Length | ||
|---|---|---|---|
| Lu-O | S1 | 6 | |
| S2 | 7 | ||
| bulk | 8 | ||
| Al-O | S1 | 5 | |
| S2 | 4/5 | ||
| bulk | 4/6 |
| Bulk O | S1 O | S2 O | |
|---|---|---|---|
| Coordination | 2Lu + 2Al | 2Lu + 1Al | 2Lu + 1Al |
| Lu-O | 2.33 | 2.16 | 1.90 |
| Al-O | 1.84 | 2.26 | 1.87 |
| Bader charge | −1.5 | −1.2 | |
| Coordination | Length | ||
|---|---|---|---|
| LuAl-O | Bulk | 6 | 2.14 |
| S1 | 5 | 2.17 | |
| S2 | 5 | 2.15 | |
| Al-O | Bulk | 6 | 2.05 |
| S1 | 5 | 1.90 | |
| S2 | 6 | 2.01 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, W.; Jiang, B.; Zhu, J.; Zhang, L. Surface Structure and Electronic Properties of Lu3Al5O12. Crystals 2021, 11, 1433. https://doi.org/10.3390/cryst11111433
Guo W, Jiang B, Zhu J, Zhang L. Surface Structure and Electronic Properties of Lu3Al5O12. Crystals. 2021; 11(11):1433. https://doi.org/10.3390/cryst11111433
Chicago/Turabian StyleGuo, Weian, Benxue Jiang, Jiajie Zhu, and Long Zhang. 2021. "Surface Structure and Electronic Properties of Lu3Al5O12" Crystals 11, no. 11: 1433. https://doi.org/10.3390/cryst11111433