Numerical Simulation of Adsorption of Organic Inhibitors on C-S-H Gel


Abstract
:1. Introduction
2. Model Constructions and Computational Methods
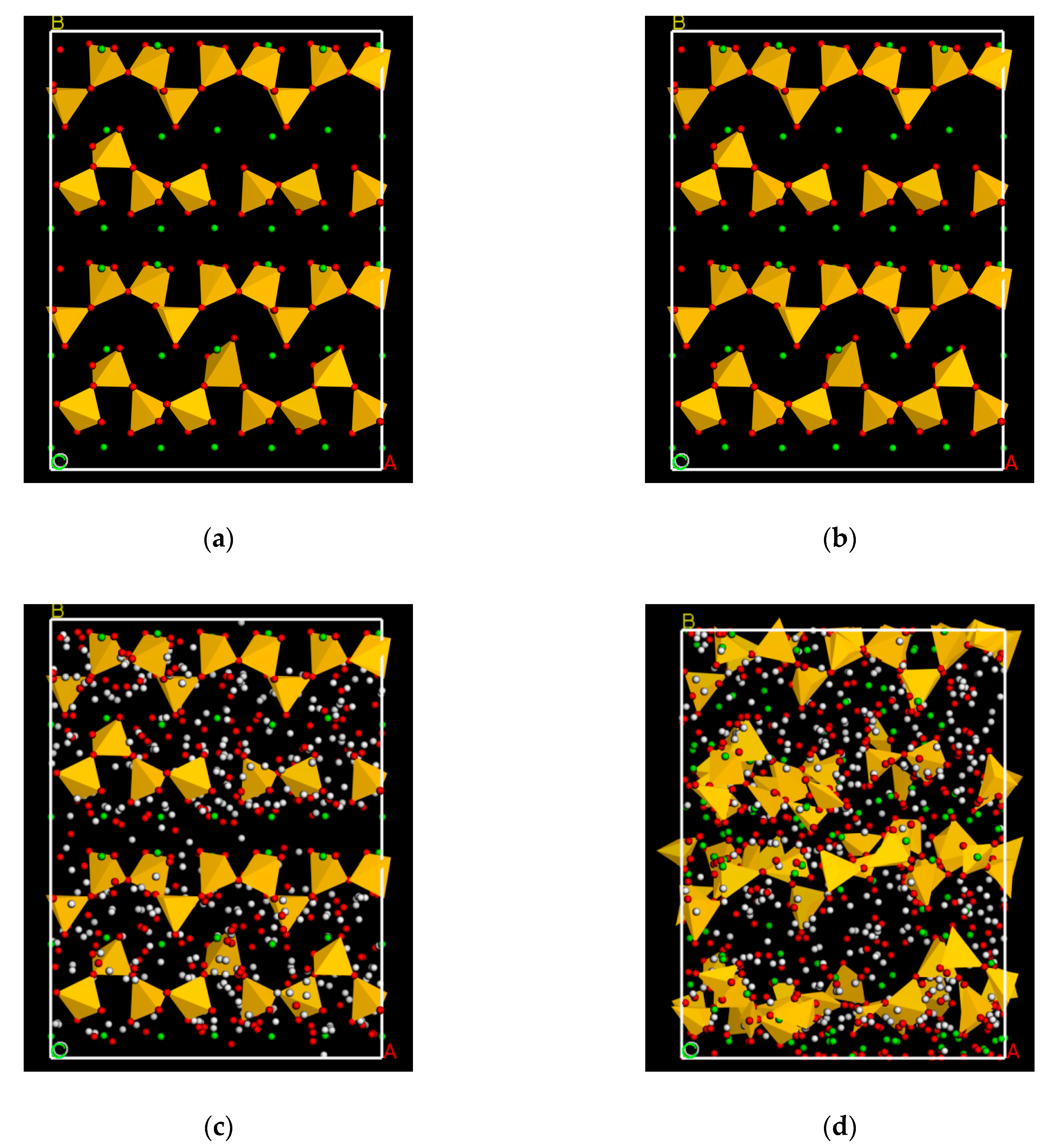
2.1. Model Constructions
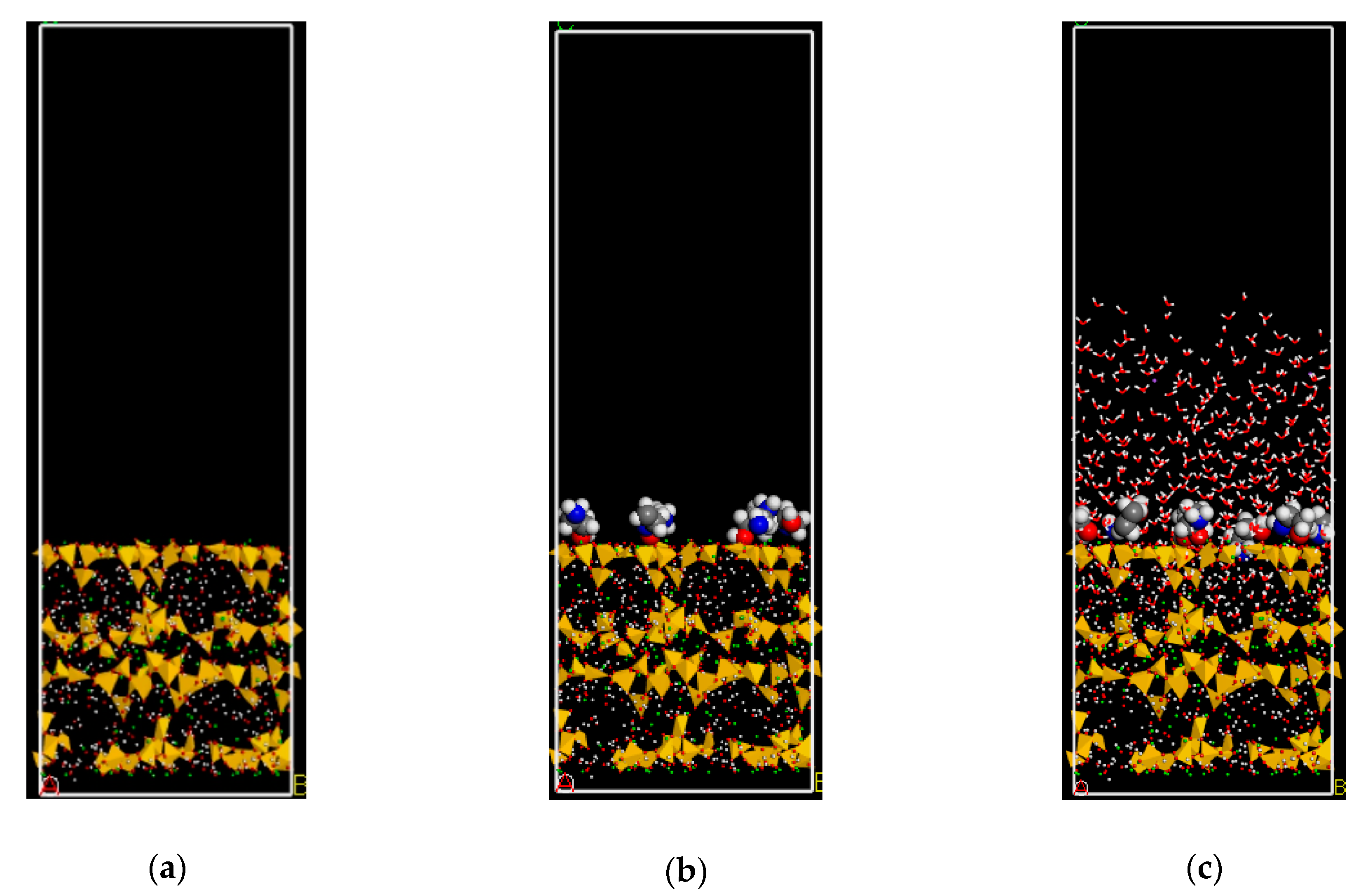
2.2. Surface Adsorption Test
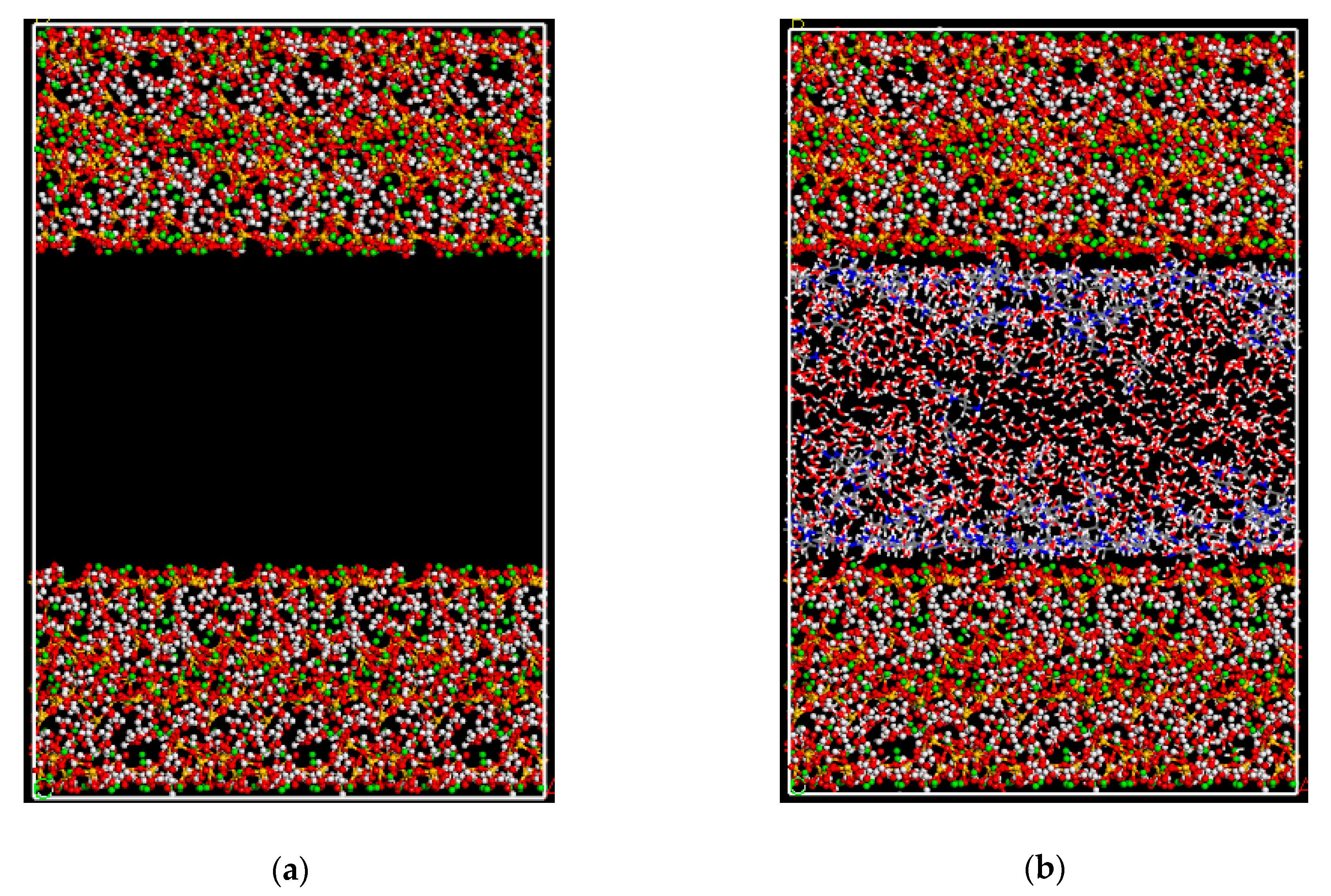
2.3. Pore Adsorption Test
3. Simulation Results of Surface Adsorption System
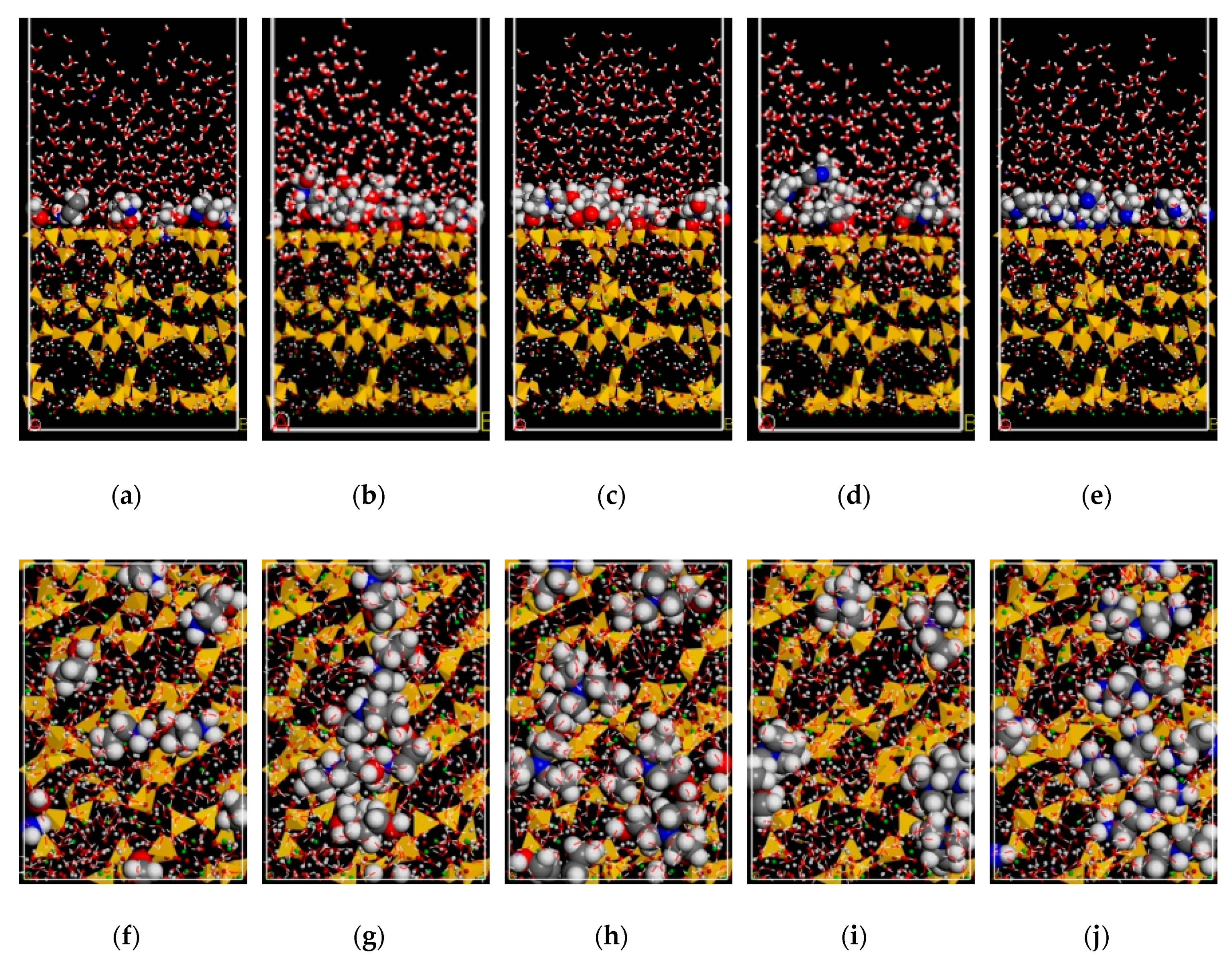
3.1. Adsorption Morphology
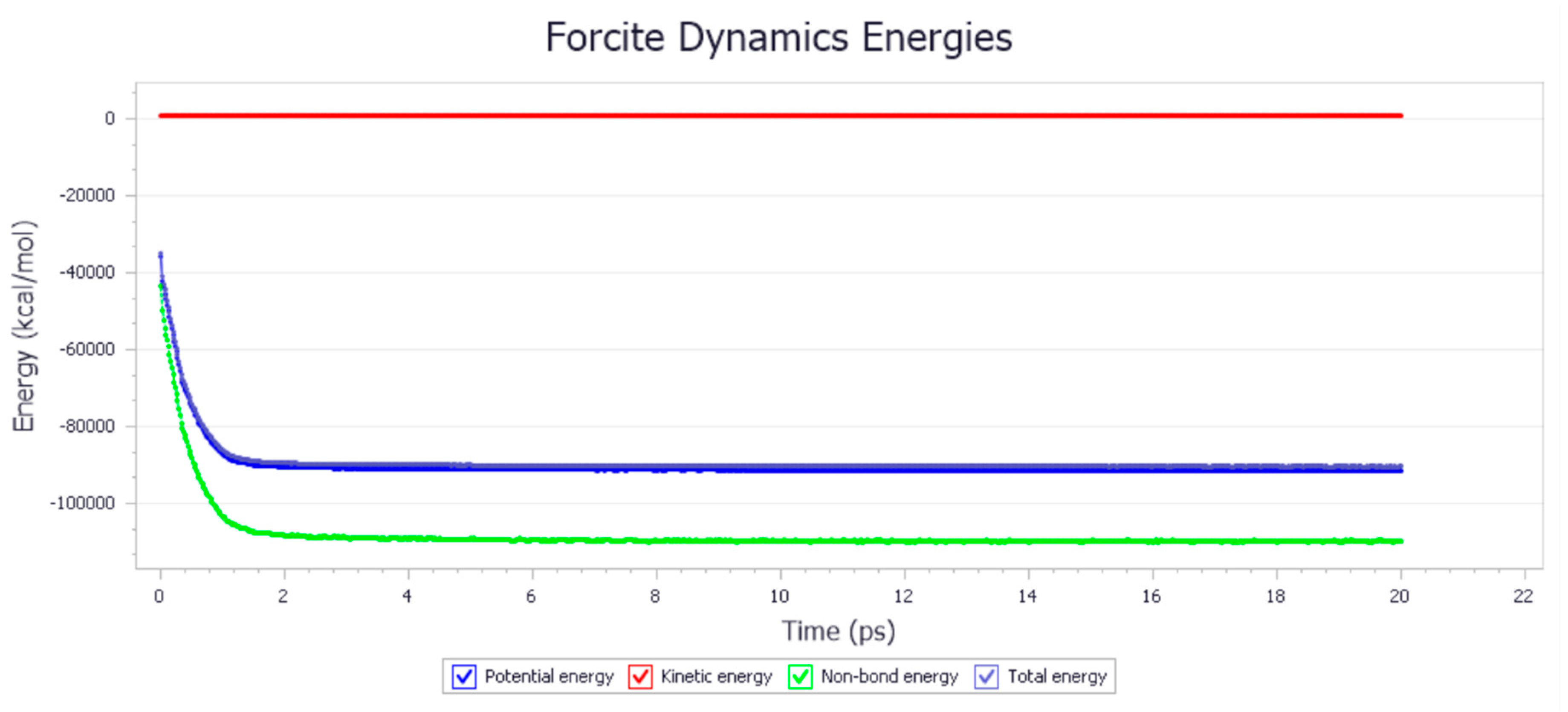
3.2. Energy Analysis
4. The Grant Canonical Monte Carlo System
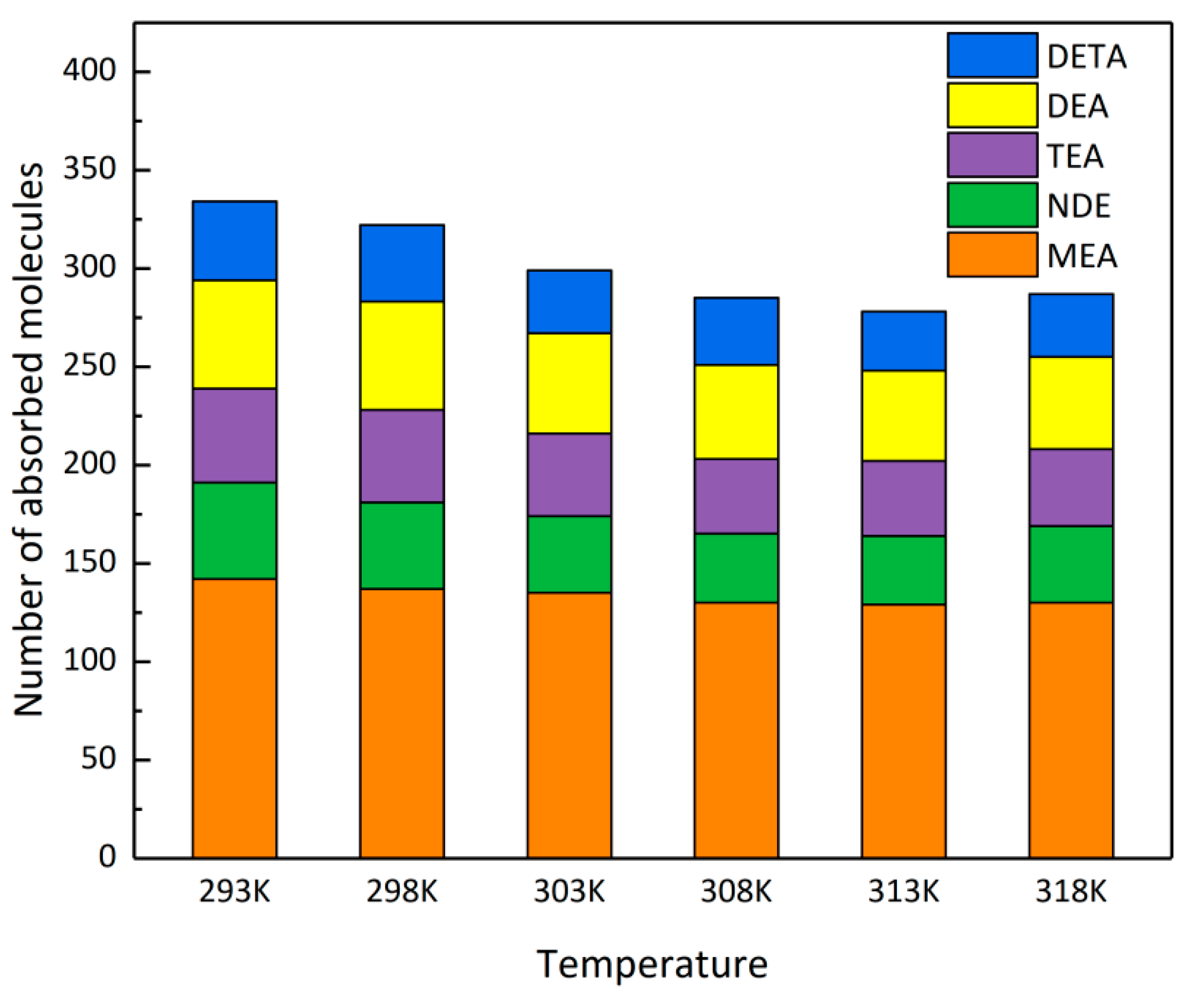
4.1. Adsorption Quantity Analysis
4.2. Energy Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bertolini, L.; Elsener, B.; Pedeferri, P.; Redaelli, E.; Polder, R.B. Corrosion of Steel in Concrete: Prevention, Diagnosis, Repair; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2013; Volume 49, pp. 4113–4133. [Google Scholar]
- Liu, Q.Y.; Song, Z.J.; Cai, H.C.; Zhou, A.P.; Wang, W.Y.; Jiang, L.H.; Liu, Y.Q.; Zhang, Y.J.; Xu, N. Effect of Ultrasonic Parameters on Electrochemical Chloride Removal and Rebar Repassivation of Reinforced Concrete. Materials 2019, 12, 2774. [Google Scholar] [CrossRef] [Green Version]
- Ormellese, M.; Lazzari, L.; Goidanich, S.; Fumagalli, G.; Brenna, A. A study of organic substances as inhibitors for chloride-induced corrosion in concrete. Corros. Sci. 2009, 51, 2959–2968. [Google Scholar] [CrossRef]
- Rakanta, E.; Zafeiropoulou, T.; Batis, G. Corrosion protection of steel with DMEA-based organic inhibitor. Constr. Build. Mater. 2013, 44, 507–513. [Google Scholar] [CrossRef]
- Morris, W.; Vico, A.; Vazquze, M. Corrosion of reinforcing steel by means of concrete resistively measurements. Corros. Sci. 2002, 44, 81–99. [Google Scholar] [CrossRef]
- Liu, Q.Y.; Song, Z.J.; Han, H. A novel green reinforcement corrosion inhibitor extracted from waste Platanus acerifolia leaves. Constr. Build. Mater. 2020, 260, 119695. [Google Scholar] [CrossRef]
- Khaled, K.F.; El-Maghraby, A. Experimental, Monte Carlo and molecular dynamics simulations to investigate corrosion inhibition of mild steel in hydrochloric acid solutions. Arab. J. Chem. 2014, 7, 319–326. [Google Scholar] [CrossRef]
- Dehghani, A.; Bahlakeh, G.; Ramezanzadeh, B.; Karati, M.R. Potential role of a novel green eco-friendly inhibitor in corrosion inhibition of mild steel in HCl solution: Detailed macro/micro-scale experimental and computational explorations. Constr. Build. Mater. 2020, 245, 118464. [Google Scholar] [CrossRef]
- Sulaiman, K.O.; Onawole, A.T.; Faye, O.; Shuaib, D.T. Understanding the corrosion inhibition of mild steel by selected green compounds using chemical quantum based assessments and molecular dynamics simulations. J. Mol. Liq. 2019, 279, 342–350. [Google Scholar] [CrossRef]
- Zhu, Y.Y.; Ma, Y.W.; Hu, J.; Zhang, Z.M.; Huang, J.Z.; Wang, Y.Y.; Wang, H.; Cai, W.X.; Huang, H.L.; Yu, Q.J.; et al. Adsorption of organic core-shell corrosion inhibitors on cement particles and their influence on early age properties of fresh cement paste. Cem. Concr. Res. 2020, 130, 106000. [Google Scholar] [CrossRef]
- Alibakhshi, E.; Ramezanzadeh, M.; Bahlakeh, G.; Ramezanzadeh, B.; Mahdavian, M.; Motamedi, M. Glycyrrhiza glabra leaves extract as a green corrosion inhibitor for mild steel in 1 M hydrochloric acid solution: Experimental, molecular dynamics, Monte Carlo and quantum mechanics study. J. Mol. Liq. 2018, 255, 185–198. [Google Scholar] [CrossRef]
- Ormellese, M.; Perez, E.A.; Raffaini, G.; Ganazzoli, F.; Lazzari, L. Inhibition mechanism in concrete by organic substances: An experimental and theoretical study. J. Mater. Sci. Eng. 2010, 13, 66–77. [Google Scholar]
- Zheng, H.; Li, W.; Ma, F.; Kong, Q. The performance of a surface-applied corrosion inhibitor for the carbon steel in saturated Ca(OH)2 solutions. Cem. Concr. Res. 2014, 55, 102–108. [Google Scholar] [CrossRef]
- Li, Z. Structure of concrete. In Proceedings of the Advanced Concrete Technology; John Wiley & Sons Inc.: Hoboken, NJ, USA, 2011; pp. 140–163. [Google Scholar]
- Puibasset, J.; Pellenq, J.M. Water adsorption on hydrophilic mesoporous and plane silica substrates: A grand canonical Monte Carlo simulation study. J. Chem. Phys. 2003, 118, 5613–5622. [Google Scholar] [CrossRef]
- Ma, H.; Li, Z. Realistic pore structure of Portland cementpaste: Experimental study and numerical simulation. Comput. Concr. 2013, 11, 317–336. [Google Scholar] [CrossRef]
- Mehta, P.K.; Paulo, J.M. Concrete: Microstructure, Properties, and Materials; Prentice-Hall: Upper Saddle River, NJ, USA, 2013. [Google Scholar]
- Allen, A.J.; Thomas, J.J. Analysis of C–S–H gel and cement paste by small-angle neutron scattering. Cem. Concr. Res. 2007, 37, 319–324. [Google Scholar] [CrossRef] [Green Version]
- Ji, Q.; Pellenq, J.M.; Vliet, K.J.V. Comparison of computational water models for simulation of calcium-silicate-hydrate. Comput. Mater. Sci. 2012, 53, 234–240. [Google Scholar] [CrossRef]
- Bonhoure, I.; Wieland, E.; Scheidegger, A.M.; Ochs, M.; Kunz, D. EXAFS study of Sn(IV) immobilization by hardened cement paste and calcium silicate hydrates. Environ. Sci. Technol. 2003, 37, 2184–2191. [Google Scholar] [CrossRef]
- Gauffinet, S.; Finot, E.; Lesniewska, E.; Nonat, A. Studies of CSH growth on C3S surface during its early hydration. In Proceedings of the 20th International Conference on Cement Microscopy, Guadalajara, Mexico, 19–23 April 1998. [Google Scholar]
- Grangeon, S.; Claret, F.; Linard, Y.; Chiaberge, C. X-ray diffraction: A powerful tool to probe and understand the structure of nanocrystalline calcium silicate hydrates. Acta Crystallogr. B Struct. Sci. Cryst. Eng. Mater. 2013, 69, 465–473. [Google Scholar] [CrossRef] [Green Version]
- Cong, X.; Kirkpatrick, R. 29Si MAS NMR study of the structure of calcium silicate hydrate. Adv. Cem. Based Mater. 1996, 3, 144–156. [Google Scholar] [CrossRef]
- Pellenq, R.J.; Kushima, A.; Shahsavari, R.; Van Vliet, K.; Buehler, M.J.; Yip, S.; Ulm, F.-J. A realistic molecular model of cement hydrates. Proc. Natl. Acad. Sci. USA 2009, 106, 16102–16107. [Google Scholar] [CrossRef] [Green Version]
- Hamid, S. The crystal structure of the 11 Å natural tobermorite Ca2.25 Si3 O7.5 (OH)1.5 1H2 O. Z. Fur Krist. 1981, 154, 189–198. [Google Scholar]
- Murray, S.J.; Subramani, V.J.; Selvam, R.P.; Hall, K.D. Molecular dynamics to understand the mechanical behavior of cement Paste. J. Transp. Res. Board 2010, 2142, 75–82. [Google Scholar] [CrossRef]
- Selvam, R.P.; Subramani, V.J.; Murray, S.; Hall, K.D. Potential Application of Nanotechnology on Cement Based Materials; The National Academies of Sciences, Engineering, and Medicine: Keck Center, DC, USA, 2009. [Google Scholar]
- Manzano, H.; Moeini, S.; Marinelli, F.; van Duin, A.C.T.; Ulm, F.-J.; Pellenq, R.J.-M. Confined water dissociation in microporous defective silicates: Mechanism, dipole distribution, and impact on substrate properties. J. Am. Chem. Soc. 2011, 134, 2208–2215. [Google Scholar] [CrossRef] [PubMed]
- Puibasset, J.; Pellenq, R. Grand canonical monte carlo simulation study of water adsorption in silicalite at 300 K. J. Phys. Chem. B 2008, 112, 6390–6397. [Google Scholar] [CrossRef]
- Valleau, J.P.; Cohen, L.K. Primitive model electrolytes. I. Grand canonical Monte Carlo computations. J. Chem. Phys. 1980, 72, 5935–5941. [Google Scholar] [CrossRef]
- Oelkers, E.H.; Golubev, S.V.; Chairat, C.; Pokrovsky, O.S.; Schott, J. The surface chemistry of multi-oxide silicates. Geochim. Et Cosmochim. Acta 2009, 73, 4617–4634. [Google Scholar] [CrossRef]
- Longo, R.; Cho, K.; Brüner, P.; Welle, A.; Gerdes, A.; Thissen, P. Carbonation of wollastonite(001) competing hydration: Microscopic insights from ion spectroscopy and density functional theory. ACS Appl. Mater. Interfaces 2015, 7, 4706–4712. [Google Scholar] [CrossRef]
- Giraudo, N.; Thissen, P. Carbonation Competing Functionalization on Calcium-Silicate-Hydrates: Investigation of Four Promising Surface-Activation Techniques. ACS Sustain. Chem. Eng. 2016, 4, 3985–3994. [Google Scholar] [CrossRef]
- Sun, H. Simulation of organic adsorption on metallic materials. J. Phys. Chem. B 1998, 102, 7338. [Google Scholar] [CrossRef]
- Musa, A.Y.; Jalgham, R.T.T.; Mohamad, A.B. Molecular dynamic and quantum chemical calculations for phthalazine derivatives as corrosion inhibitors of mild steel in 1M HCl. Corros. Sci. 2012, 56, 176–183. [Google Scholar] [CrossRef]
- Hou, D.; Jia, Y.; Yu, J.; Wang, P.; Liu, Q.F. Transport Properties of Sulfate and Chloride Ions Confined between Calcium Silicate Hydrate Surfaces: A Molecular Dynamics Study. J. Phys. Chem. C 2018, 122, 28021–28032. [Google Scholar] [CrossRef]
- Diamanti, M.V.; Pérez Rosales, E.A.; Raffaini, G.; Ganazzoli, F.; Brenna, A.; Pedeferri, M.; Ormellese, M. Molecular modelling and electrochemical evaluation of organic inhibitors in concrete. Corros. Sci. 2015, 100, 231–241. [Google Scholar] [CrossRef] [Green Version]
- Mendonca, G.L.F.; Costa, S.N.; Freire, V.N.; Casciano, P.N.S.; Correia, A.N.; de Lima-Neto, P. Understanding the corrosion inhibition of carbon steel and copper in sulphuric acid medium by amino acids using electrochemical techniques allied to molecular modelling methods. Corros. Sci. 2017, 115, 41–55. [Google Scholar] [CrossRef]
- Gustincic, D.; Kokalj, A. DFT Study of Azole Corrosion Inhibitors on Cu2O Model of Oxidized Copper Surfaces: I. Molecule-Surface and Cl-Surface Bonding. Metals 2018, 8, 311. [Google Scholar] [CrossRef] [Green Version]
- Guo, L.; Tan, J.; Kaya, S.; Leng, S.; Li, Q.; Zhang, F. Multidimensional insights into the corrosion inhibition of 3,3-dithiodipropionic acid on Q235 steel in H2SO4 medium: A combined experimental and in silico investigation. J. Colloid Interface Sci. 2020, 570, 116–124. [Google Scholar] [CrossRef]
- Zhu, Z.-S.; Li, L.-Q.; Gao, Y.-C.; Zhang, J.-F.; Song, J.-F.; Qin, Y.-X.; Liu, X.-Y. Analysis of Multi-component Organic Gas Adsorption Behaviors and Selectivity of Adsorbent. Nat. Gas Chem. Ind. 2008, 33, 47–51. [Google Scholar]
- Abdollahi, F.; Razmkhah, M.; Moosavi, F. The role of hydrogen bond interaction on molecular orientation of alkanolamines through temperature and pressure variation: A mixed molecular dynamics and quantum mechanics study. Comput. Mater. Sci. 2017, 131, 239–249. [Google Scholar] [CrossRef]
- Button, J.K.; Gubbins, K.E.; Tanaka, H.; Nakanishi, K. Molecular dynamics simulation of hydrogen bonding in monoethanolamine. Fluid Phase Equilibria 1996, 116, 320–325. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Abbreviation | Molecular Formula | Molecular Mass | Molecular Structure |
|---|---|---|---|---|
| Ethanolamine | MEA | C2HNO | 61.08 |  |
| Diethanolamine | DEA | C4H11NO2 | 105.14 |  |
| Triethylolamine | TEA | C6H15NO3 | 149.19 |  |
| N,N-dimethylethanolamine | NDE | C4H11NO | 89.14 |  |
| Diethyleneteiamine | DETA | C4H13N3 | 103.17 |  |
| Molecule | EAds | Esol | EAds − Esol |
|---|---|---|---|
| MEA | −697.496064 | −92.595926 | −604.900138 |
| NDE | −629.069637 | −143.23066 | −485.838977 |
| TEA | −756.114049 | −199.036085 | −557.077964 |
| DEA | −671.377916 | −105.307953 | −566.069963 |
| DETA | −658.629254 | −188.678217 | −469.951037 |
| Molecule | 293 K | 298 K | 303 K | 308 K | 313 K | 318 K |
|---|---|---|---|---|---|---|
| MEA to H2O | 12.93 | 12.54 | 12.19 | 11.68 | 11.46 | 11.12 |
| NDE to H2O | 4.46 | 4.03 | 3.52 | 3.15 | 3.11 | 3.16 |
| TEA to H2O | 4.37 | 4.30 | 3.79 | 3.42 | 3.38 | 3.34 |
| DEA to H2O | 5.01 | 5.03 | 4.61 | 4.31 | 4.09 | 4.02 |
| DETA to H2O | 3.64 | 3.57 | 2.89 | 3.06 | 2.67 | 2.65 |
| Molecule | Temperature | Total Energy | Van Der Waals Energy | Electrostatic Energy | Intramolecular Energy |
|---|---|---|---|---|---|
| MEA | 298 K | −64690.74324 | 2951.46269 | −78902.78028 | 11260.57435 |
| 303 K | −64530.30697 | 2898.92290 | −78303.21347 | 11073.98360 | |
| 308 K | −64075.01570 | 2467.56340 | −77464.32230 | 10921.74313 | |
| 313 K | −63639.92284 | 2366.96724 | −76879.99999 | 10873.10984 | |
| NDE | 298 K | −51813.29218 | 2257.08450 | −65106.40411 | 11036.02744 |
| 303 K | −51011.18774 | 2247.36929 | −63938.87839 | 11080.32136 | |
| 308 K | −49461.25152 | 2175.76475 | −62563.41327 | 10926.39701 | |
| 313 K | −49008.36233 | 2068.31950 | −61393.39248 | 10316.71066 | |
| TEA | 298 K | −53083.22706 | 2715.29208 | −67080.09967 | 11281.58053 |
| 303 K | −52678.75286 | 2680.23054 | −66935.38242 | 11206.39902 | |
| 308 K | −52069.37783 | 2615.87605 | −65897.47767 | 11012.22379 | |
| 313 K | −52092.91453 | 2677.50214 | −65938.40445 | 11067.98778 | |
| DEA | 298 K | −53338.16669 | 2593.65200 | −66600.79470 | 11208.97600 |
| 303 K | −53020.39933 | 2603.99637 | −65289.85228 | 11065.45658 | |
| 308 K | −52974.72470 | 2544.07300 | −64940.22606 | 10721.42836 | |
| 313 K | −51087.92460 | 2507.73629 | −64016.20586 | 10830.54496 | |
| DETA | 298 K | −53106.88604 | 2703.01003 | −64217.20122 | 11448.15528 |
| 303 K | −51976.23648 | 2604.64465 | −63585.48501 | 11004.60387 | |
| 308 K | −508903.83110 | 2592.05362 | −62751.71009 | 11525.82537 | |
| 313 K | −50057.24012 | 2562.15990 | −61100.04881 | 11021.49893 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, Z.; Cai, H.; Liu, Q.; Liu, X.; Pu, Q.; Zang, Y.; Xu, N. Numerical Simulation of Adsorption of Organic Inhibitors on C-S-H Gel. Crystals 2020, 10, 742. https://doi.org/10.3390/cryst10090742
Song Z, Cai H, Liu Q, Liu X, Pu Q, Zang Y, Xu N. Numerical Simulation of Adsorption of Organic Inhibitors on C-S-H Gel. Crystals. 2020; 10(9):742. https://doi.org/10.3390/cryst10090742
Chicago/Turabian StyleSong, Zijian, Huanchun Cai, Qingyang Liu, Xing Liu, Qi Pu, Yingjie Zang, and Na Xu. 2020. "Numerical Simulation of Adsorption of Organic Inhibitors on C-S-H Gel" Crystals 10, no. 9: 742. https://doi.org/10.3390/cryst10090742