Quantum Crystallography in the Last Decade: Developments and Outlooks


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
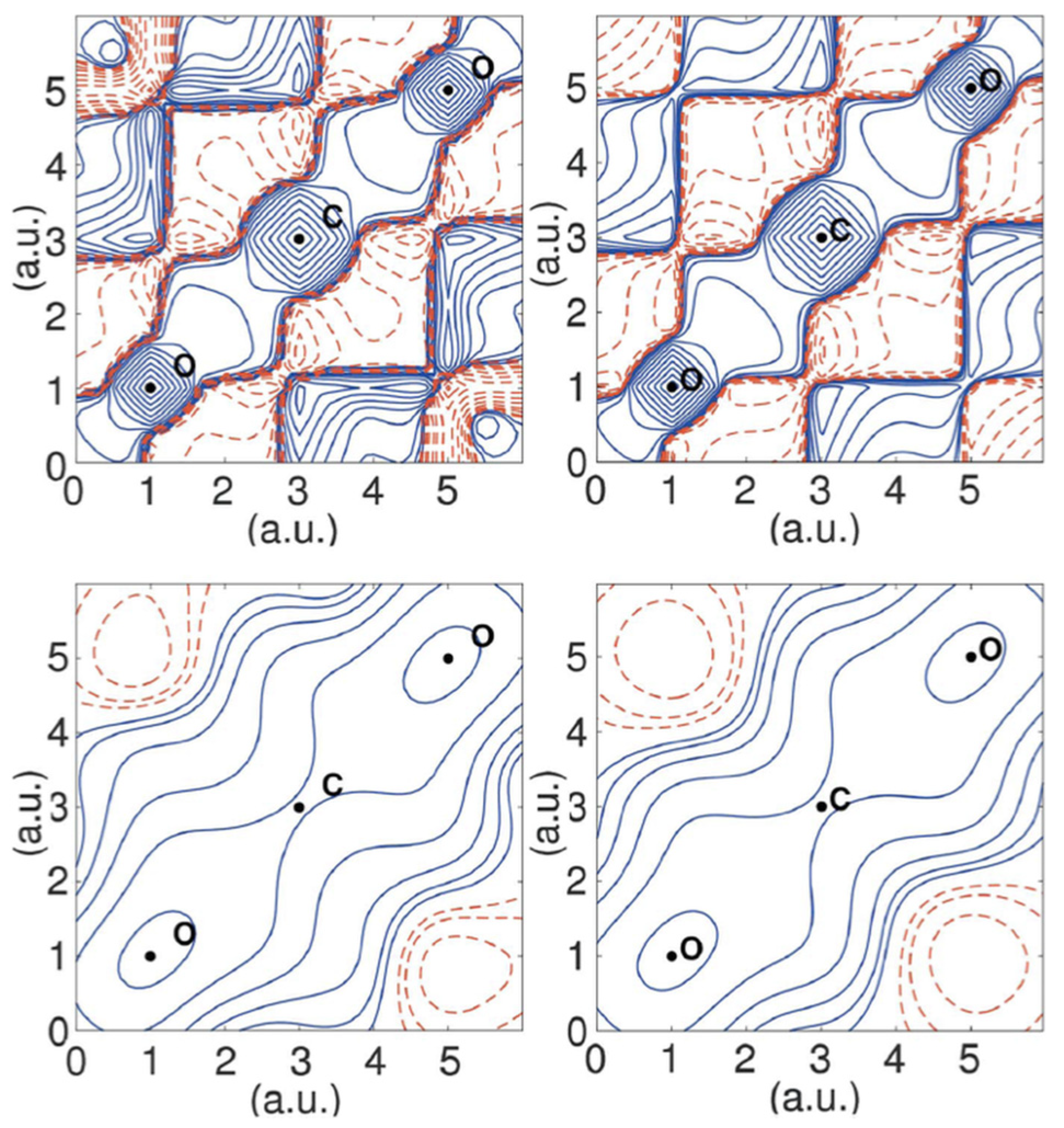
2. Fitting the Wavefunction
3. Fitting the Density
4. Conclusions and Outlook
Author Contributions
Funding
Conflicts of Interest
References
- Debye, P. Zerstreuung von Röntgenstrahlen. Ann. Phys. 1915, 46, 809–823. [Google Scholar] [CrossRef] [Green Version]
- Bohr, N.I. On the constitution of atoms and molecules. Lond. Edinb. Dublin Philos. Mag. J. Sci. 1913, 26, 1–25. [Google Scholar] [CrossRef] [Green Version]
- Hartree, D. The atomic structure factor in the intensity of reflexion of X-rays by crystals. Lond. Edinb. Dublin Philos. Mag. J. Sci. 1925, 50, 289–306. [Google Scholar] [CrossRef]
- Bragg, W.L.; James, R.; Bosanquet, C. The distribution of electrons around the nucleus in the sodium and chlorine atoms. Lond. Edinb. Dublin Philos. Mag. J. Sci. 1922, 44, 433–449. [Google Scholar] [CrossRef]
- Waller, I.; Hartree, D.R. On the Intensity of Total Scattering of X-Rays. Proc. Royal. Soc. Lond. Ser. A 1929, 124, 119–142. [Google Scholar]
- Weiss, R.J.; De Marco, J.J. X-ray determination of the number of 3d electrons in Cu, Ni, Co, Fe, and Cr. Rev. Mod. Phys. 1958, 30, 59–62. [Google Scholar] [CrossRef]
- Weiss, R.J.; Phillips, W.C. X-Ray Determination of the Electron Momentum Density in Diamond, Graphite, and Carbon Black. Phys. Rev. 1968, 176, 900–904. [Google Scholar] [CrossRef]
- Weiss, R.J. Spin Density in Cobalt. Phys. Rev. Lett. 1963, 11, 264–265. [Google Scholar] [CrossRef]
- Weiss, R.J.; AzároffL, V. X-Ray Determination of Electron Distributions. Phys. Today 1967, 20, 103. [Google Scholar] [CrossRef]
- Massa, L.; Huang, L.; Karle, J. Quantum crystallography and the use of kernel projector matrices. Int. J. Quantum Chem. 1995, 56, 371–384. [Google Scholar] [CrossRef]
- Huang, L.; Massa, L.; Karle, J. Quantum crystallography applied to crystalline maleic anhydride. Int. J. Quantum Chem. 1999, 73, 439–450. [Google Scholar] [CrossRef]
- Dawson, B. A general structure factor formalism for interpreting accurate X-ray and neutron diffraction data. Proc. R. Soc. Lond. Ser. A Math. Phys. Sci. 1967, 298, 255–263. [Google Scholar] [CrossRef]
- Stewart, R.F. Generalized X-Ray Scattering Factors. J. Chem. Phys. 1969, 51, 4569–4577. [Google Scholar] [CrossRef]
- Stewart, R.F. Electron population analysis with generalized X-ray-scattering factors—Higher multipoles. J. Chem. Phys. 1973, 58, 1668–1676. [Google Scholar] [CrossRef]
- Stewart, R.F. Electron population analysis with rigid pseudoatoms. Acta Crystallogr. Sect. A Cryst. Phys. Diffr. Theor. Gen. Crystallogr. 1976, 32, 565–574. [Google Scholar] [CrossRef]
- Hansen, N.K.; Coppens, P. Testing aspherical atom refinements on small-molecule data sets. Acta Crystallogr. Sect. A Cryst. Phys. Diffr. Theor. Gen. Crystallogr. 1978, 34, 909–921. [Google Scholar] [CrossRef]
- Hirshfeld, F.L. Space partitioning of the charge density. Isr. J. Chem. 1977, 16, 198–201. [Google Scholar] [CrossRef]
- Bultinck, P.; Van Alseneoy, C.; Ayers, P.W.; Carbó-Dorca, R.J. Critical analysis and extension of the Hirshfeld atoms in molecules. Chem. Phys. 2007, 26, 144111. [Google Scholar] [CrossRef]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld surface analysis. CrystEngComm 2009, 11, 19–32. [Google Scholar] [CrossRef]
- Jayatilaka, D.; Dittrich, B. X-ray structure refinement using aspherical atomic density functions obtained from quantum-mechanical calculations. Acta Cryst. 2008, A64, 383–393. [Google Scholar] [CrossRef]
- Coppens, P.; Willoughby, T.V.; Csonka, L.N. Electron Population Analysis of Accurate Diffraction Data. I. Formalisms and Restrictions. Acta Cryst. 1971, A27, 248–256. [Google Scholar] [CrossRef]
- Clinton, W.L.; Nakhleh, J.; Wunderlich, F. Direct Determination of Pure-State Density Matrices. I. Some Simple Introductory Calculations. Phys. Rev. 1969, 177, 1–6. [Google Scholar] [CrossRef]
- Clinton, W.L.; Galli, A.J.; Massa, L.J. Direct Determination of Pure-State Density Matrices. II. Construction of Constrained Idempotent One-Body Densities. Phys. Rev. 1969, 177, 7–13. [Google Scholar] [CrossRef]
- Clinton, W.L.; Henderson, G.A.; Prestia, J.V. Direct Determination of Pure-State Density Matrices. III. Purely Theoretical Densities via an Electrostatic-Virial Theorem. Phys. Rev. 1969, 177, 13–18. [Google Scholar] [CrossRef]
- Clinton, W.L.; Lamers, G.B. Direct Determination of Pure-State Density Matrices. IV. Investigation of Another Constraint and Another Application of the P Equations. Phys. Rev. 1969, 177, 19–27. [Google Scholar] [CrossRef]
- Clinton, W.L.; Galli, A.J.; Henderson, G.A.; Lamers, G.B.; Massa, L.J.; Zarur, J. Direct Determination of Pure-State Density Matrices. V. Constrained Eigenvalue Problems. Phys. Rev. 1969, 177, 27–33. [Google Scholar] [CrossRef]
- Clinton, W.L.; Massa, L.J. The cusp condition: Constraint on the electron density matrix. Int. J. Quantum Chem. 1972, 6, 519–523. [Google Scholar] [CrossRef]
- Clinton, W.L.; Massa, L.J. Determination of the Electron Density Matrix from X-Ray Diffraction Data. Phys. Rev. Lett. 1972, 29, 1363–1366. [Google Scholar] [CrossRef]
- Clinton, W.L.; Frishberg, C.A.; Massa, L.J.; Oldfield, P.A. Methods for obtaining an electron-density matrix from X-ray diffraction data. Int. J. Quantum Chem. 2009, 7, 505–514. [Google Scholar] [CrossRef]
- Frishberg, C.; Massa, L.J. Idempotent density matrices for correlated systems from x-ray-diffraction structure factors. Phys. Rev. B 1981, 24, 7018–7024. [Google Scholar] [CrossRef]
- Nosanow, L.H. Theory of Quantum Crystals. Phys. Rev. 1966, 146, 120–133. [Google Scholar] [CrossRef]
- Capelli, S.C.; Bürgi, H.-B.; Dittrich, B.; Grabowsky, S.; Jayatilaka, D. Hirshfeld atom refinement. IUCrJ 2014, 1, 361–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woińska, M.; Jayatilaka, D.; Spackman, M.A.; Edwards, A.; Dominiak, P.M.; Woźniak, K.; Nishibori, E.; Sugimoto, K.; Grabowsky, S. Hirshfeld atom refinement for modelling strong hydrogen bonds. Acta Crystallogr. Sect. A 2014, 70, 483–498. [Google Scholar] [CrossRef] [PubMed]
- Woińska, M.; Grabowsky, S.; Dominiak, P.M.; Woźniak, K.; Jayatilaka, D. Hydrogen atoms can be located accurately and precisely by x-ray crystallography. Sci. Adv. 2016, 2, e1600192. [Google Scholar] [CrossRef] [Green Version]
- Fugel, M.; Jayatilaka, D.; Hupf, E.; Overgaard, J.; Hathwar, V.R.; Macchi, P.; Turner, M.J.; Howard, J.A.K.; Dolomanov, O.V.; Puschmann, H.; et al. Probing the accuracy and precision of Hirshfeld atom refinement with HARt interfaced with Olex2. IUCrJ 2018, 5, 32–44. [Google Scholar] [CrossRef] [PubMed]
- Wieduwilt, E.K.; Macetti, G.; Malaspina, L.A.; Jayatilaka, D.; Grabowsky, S.; Genoni, A. Post-Hartree-Fock methods for Hirshfeld atom refinement: Are they necessary? Investigation of a strongly hydrogen-bonded molecular crystal. J. Mol. Struct. 2020, 1209, 127934. [Google Scholar] [CrossRef]
- Hibbs, D.E.; Huke, J.P.; Waller, M.P. A new orbital-based model for the analysis of experimental molecular charge densities: An application to (Z)-N-methyl-C-phenylnitrone. Phys. Chem. Chem. Phys. 2005, 7, 1772–1778. [Google Scholar] [CrossRef]
- Waller, M.P.; Howard, S.T.; Platts, J.A.; Piltz, R.O.; Willock, D.J.; Hibbs, D.E. Novel Properties form Experimental Charge Densities: An Application to the Zwitterionic Neurotransmitter Taurine. Chem. Eur. J. 2006, 12, 7603–7614. [Google Scholar] [CrossRef]
- Tanaka, K. X-ray analysis of wavefunctions by the least-squares method incorporating orthonormality. I. General formalism. Acta Crystallogr. Sect. A 1988, 44, 1002–1008. [Google Scholar] [CrossRef]
- Tanaka, K. X-ray molecular orbital analysis. I. Quantum mechanical and crystallographic framework. Acta Crystallogr. Sect. A 2018, 74, 345–356. [Google Scholar] [CrossRef]
- Jayatilaka, D. Wave Function for Beryllium from X-Ray Diffraction Data. Phys. Rev. Lett. 1998, 80, 798–801. [Google Scholar] [CrossRef]
- Jayatilaka, D.; Grimwood, D.J. Wavefunctions Derived from Experiment. I. Motivation and Theory. Acta Crystallogr. Sect. A 2001, 57, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, T.L. Hohenberg-Kohn theorem for nonlocal external potentials. Phys. Rev. B 1975, 12, 2111–2120. [Google Scholar] [CrossRef]
- Coleman, A.J. Structure of Fermion Density Matrices. Rev. Mod. Phys. 1963, 35, 668–686. [Google Scholar] [CrossRef]
- Henderson, G.A. One-electron properties as variational parameters. J. Chem. Phys. 1976, 65, 619–622. [Google Scholar] [CrossRef]
- Grimwood, D.; Jayatilaka, D. Wavefunctions derived from experiment. II. A wavefunction for oxalic acid dihydrate. Acta Crystallogr. Sect. A 2001, 57, 87–100. [Google Scholar] [CrossRef]
- Bytheway, I.; Grimwood, D.; Jayatilaka, D. Wavefunctions derived from experiment. III. Topological analysis of crystal fragments. Acta Crystallogr. Sect. A 2002, 58, 232–243. [Google Scholar] [CrossRef]
- Bytheway, I.; Grimwood, D.; Figgis, B.N.; Chandler, G.S.; Jayatilaka, D. Wavefunctions derived from experiment. IV. Investigation of the crystal environment of ammonia. Acta Crystallogr. Sect. A 2002, 58, 244–251. [Google Scholar] [CrossRef]
- Grimwood, D.; Bytheway, I.; Jayatilaka, D. Wave functions derived from experiment. V. Investigation of electron densities, electrostatic potentials, and electron localization functions for noncentrosymmetric crystals. J. Comput. Chem. 2003, 24, 470–483. [Google Scholar] [CrossRef]
- Jayatilaka, D. Using Wave functions to Get More Information out of Diffraction Experiments. In Modern Charge-Density Analysis; Gatti, C., Macchi, P., Eds.; Springer: Dordrecht, The Netherlands, 2012; pp. 213–257. [Google Scholar]
- Whitten, A.E.; Jayatilaka, D.; Spackman, M.A. Effective molecular polarizabilities and crystal refractive indices estimated from x-ray diffraction data. J. Chem. Phys. 2006, 125, 174505. [Google Scholar] [CrossRef] [Green Version]
- Jayatilaka, D.; Munshi, P.; Turner, M.J.; Howard, J.A.K.; Spackman, M.A. Refractive indices for molecular crystals from the response of X-ray constrained Hartree–Fock wavefunctions. Phys. Chem. Chem. Phys. 2009, 11, 7209–7218. [Google Scholar] [CrossRef] [PubMed]
- Hickstein, D.D.; Cole, J.M.; Turner, M.J.; Jayatilaka, D. Modeling electron density distributions from X-ray diffraction to derive optical properties: Constrained wavefunction versus multipole refinement. J. Chem. Phys. 2013, 139, 064108. [Google Scholar] [CrossRef] [PubMed]
- Cole, J.M.; Hickstein, D.D. Molecular origins of nonlinear optical activity in zinc tris(thiourea)sulfate revealed by high-resolution x-ray diffraction data and ab initio calculations. Phys. Rev. B 2013, 88, 184105. [Google Scholar] [CrossRef]
- Ernst, M.; Genoni, A.; Macchi, P. Analysis of crystal field effects and interactions using X-ray restrained ELMOs. J. Mol. Struct. 2020, 1209, 127975. [Google Scholar] [CrossRef]
- Genoni, A.; Dos Santos, L.H.R.; Meyer, B.; Macchi, P. Can X-ray constrained Hartree-Fock wavefunctions retrieve electron correlation? IUCrJ 2017, 4, 136–146. [Google Scholar] [CrossRef] [Green Version]
- Jayatilaka, D.; Grimwood, D. Electron localization functions obtained from X-ray constrained Hartree-Fock wavefunctions for molecular crystals of ammonia, urea and alloxan. Acta Crystallogr. Sect. A 2004, 60, 111–119. [Google Scholar] [CrossRef]
- Grabowsky, S.; Jayatilaka, D.; Mebs, S.; Luger, P. The Electron Localizability Indicator from X-Ray Diffraction Data – A First Application to a Series of Epoxide Derivatives. Chem. Eur. J. 2010, 16, 12818–12821. [Google Scholar] [CrossRef]
- Grabowsky, S.; Weber, M.; Jayatilaka, D.; Chen, Y.-S.; Grabowski, M.T.; Brehme, R.; Hesse, M.; Schirmeister, T.; Luger, P. Reactivity Differences between α,β-Unsaturated Carbonyls and Hydrazones Investigated by Experimental and Theoretical Electron Density and Electron Localizability Analyses. J. Phys. Chem. A 2011, 115, 12715–12732. [Google Scholar] [CrossRef] [PubMed]
- Grabowsky, S.; Luger, P.; Buschmann, J.; Schneider, T.; Schirmeister, T.; Sobolev, A.N.; Jayatilaka, D. The Significance of Ionic Bonding in Sulfur Dioxide: Bond Orders from X-ray Diffraction Data. Angew. Chem. Int. Ed. 2012, 51, 6776–6779. [Google Scholar] [CrossRef]
- Fugel, M.; Malaspina, L.A.; Pal, R.; Thomas, S.P.; Shi, M.W.; Spackman, M.A.; Sugimoto, K.; Grabowsky, S. Revisiting a historical concept by using quantum crystallography: Are phosphate, sulfate and perchlorate anions hypervalent? Chem. Eur. J. 2019, 25, 6523–6532. [Google Scholar] [CrossRef]
- Thomas, S.P.; Jayatilaka, D.; Row, T.N.G. S···O chalcogen bonding in sulfa drugs: Insights from multipole charge density and X-ray wavefunction of acetazolamide. Phys. Chem. Chem. Phys. 2015, 17, 25411–25420. [Google Scholar] [CrossRef] [PubMed]
- Hudák, M.; Jayatilaka, D.; Perašínová, L.; Biskupič, S.; Kožíšek, J.; Bucinsky, L. X-ray constrained unrestricted Hartree–Fock and Douglas–Kroll–Hess wavefunctions. Acta Crystallogr. Sect. A 2009, 66, 78–92. [Google Scholar] [CrossRef] [PubMed]
- Bucinsky, L.; Biskupič, S.; Jayatilaka, D. Study of the picture change error at the 2nd order Douglas Kroll Hess level of theory. Electron and spin density and structure factors of the Bis[bis(methoxycarbimido) aminato] copper (II) complex. Chem. Phys. 2012, 395, 44–53. [Google Scholar] [CrossRef]
- Bucinsky, L.; Jayatilaka, D.; Grabowsky, S. Importance of Relativistic Effects and Electron Correlation in Structure Factors and Electron Density of Diphenyl Mercury and Triphenyl Bismuth. J. Phys. Chem. A 2016, 120, 6650–6669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bučinský, L.; Jayatilaka, D.; Grabowsky, S. Relativistic Quantum Crystallography of Diphenyl and Dicyano Mercury. Theoretical Structure Factors and Hirshfeld Atom Refinement. Acta Crystallogr. Sect. A 2019, 75, 705–717. [Google Scholar] [CrossRef] [PubMed]
- Stoll, H.; Wagenblast, G.; Preu, H. On the use of local basis sets for localized molecular orbitals. Theor. Chem. Accounts 1980, 57, 169–178. [Google Scholar] [CrossRef]
- Fornili, A.; Sironi, M.; Raimondi, M. Determination of extremely localized molecular orbitals and their application to quantum mechanics/molecular mechanics methods and to the study of intramolecular hydrogen bonding. J. Mol. Struct. THEOCHEM 2003, 632, 157–172. [Google Scholar] [CrossRef]
- Genoni, A.; Sironi, M. A novel approach to relax extremely localized molecular orbitals: The extremely localized molecular orbital valence bond method. Theor. Chem. Acc. 2004, 112, 254–262. [Google Scholar] [CrossRef]
- Genoni, A.; Fornili, A.; Sironi, M. Optimal Virtual Orbitals to Relax Wavefunctions Built Up with Transferred Extremely Localized Molecular Orbitals. J. Comput. Chem. 2005, 26, 827–835. [Google Scholar] [CrossRef] [PubMed]
- Genoni, A.; Ghitti, M.; Pieraccini, S.; Sironi, M. A novel extremely localized molecular orbitals based technique for the one-electron density matrix computation. Chem. Phys. Lett. 2005, 415, 256–260. [Google Scholar] [CrossRef]
- Sironi, M.; Genoni, A.; Civera, M.; Pieraccini, S.; Ghitti, M. Extremely localized molecular orbitals: Theory and applications. Theor. Chem. Acc. 2007, 117, 685–698. [Google Scholar] [CrossRef]
- Sironi, M.; Ghitti, M.; Genoni, A.; Saladino, G.; Pieraccini, S. DENPOL: A new program to determine electron densities of polypeptides using extremely localized molecular orbitals. J. Mol. Struct. THEOCHEM 2009, 898, 8–16. [Google Scholar] [CrossRef]
- Genoni, A. Molecular Orbitals Strictly Localized on Small Molecular Fragments from X-ray Diffraction Data. J. Phys. Chem. Lett. 2013, 4, 1093–1099. [Google Scholar] [CrossRef] [PubMed]
- Genoni, A. X-ray Constrained Extremely Localized Molecular Orbitals: Theory and Critical Assessment of the New Technique. J. Chem. Theory Comput. 2013, 9, 3004–3019. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, L.H.R.; Genoni, A.; Macchi, P. Unconstrained and X-ray constrained extremely localized molecular orbitals: Analysis of the reconstructed electron density. Acta Crystallogr. Sect. A 2014, 70, 532–551. [Google Scholar] [CrossRef] [Green Version]
- Genoni, A.; Meyer, B. X-Ray Constrained Wave Functions: Fundamentals and Effects of the Molecular Orbitals Localization. Adv. Quantum Chem. 2016, 73, 333–362. [Google Scholar] [CrossRef]
- Meyer, B.; Guillot, B.; Ruiz-Lopez, M.F.; Genoni, A. Libraries of Extremely Localized Molecular Orbitals. 1. Model Molecules Approximation and Molecular Orbitals Transferability. J. Chem. Theory Comput. 2016, 12, 1052–1067. [Google Scholar] [CrossRef]
- Meyer, B.; Guillot, B.; Ruiz-Lopez, M.F.; Jelsch, C.; Genoni, A. Libraries of Extremely Localized Molecular Orbitals. 2. Comparison with the Pseudoatoms Transferability. J. Chem. Theory Comput. 2016, 12, 1068–1081. [Google Scholar] [CrossRef]
- Meyer, B.; Genoni, A. Libraries of Extremely Localized Molecular Orbitals. 3. Construction and Preliminary Assessment of the New Databanks. J. Phys. Chem. A 2018, 122, 8965–8981. [Google Scholar] [CrossRef]
- Macetti, G.; Genoni, A. Quantum Mechanics/Extremely Localized Molecular Orbital Method: A Fully Quantum Mechanical Embedding Approach for Macromolecules. J. Phys. Chem. A 2019, 123, 9420–9428. [Google Scholar] [CrossRef]
- Macetti, G.; Wieduwilt, E.K.; Assfeld, X.; Genoni, A. Localized Molecular Orbital-Based Embedding Scheme for Correlated Methods. J. Chem. Theory Comput. 2020. [Google Scholar] [CrossRef] [PubMed]
- Arias-Olivares, D.; Wieduwilt, E.K.; Contreras-Garcia, J.; Genoni, A. NCI-ELMO: A New Method to Quickly and Accurately Detect Noncovalent Interactions in Biosystems. J. Chem. Theory Comput. 2019, 15, 6456–6470. [Google Scholar] [CrossRef] [PubMed]
- Malaspina, L.A.; Wieduwilt, E.K.; Bergmann, J.; Kleemiss, F.; Meyer, B.; Ruiz-López, M.F.; Pal, R.; Hupf, E.; Beckmann, J.; Piltz, R.O.; et al. Fast and Accurate Quantum Crystallography: From Small to Large, from Light to Heavy. J. Phys. Chem. Lett. 2019, 10, 6973–6982. [Google Scholar] [CrossRef] [PubMed]
- Genoni, A. A first-prototype multi-determinant X-ray constrained wavefunction approach: The X-ray constrained extremely localized molecular orbital–valence bond method. Acta Crystallogr. Sect. A 2017, 73, 312–316. [Google Scholar] [CrossRef] [PubMed]
- Casati, N.; Genoni, A.; Meyer, B.; Krawczuk, A.; Macchi, P. Exploring charge density analysis in crystals at high pressure: Data collection, data analysis and advanced modelling. Acta Crystallogr. Sect. B 2017, 73, 584–597. [Google Scholar] [CrossRef] [Green Version]
- Casati, N.; Kleppe, A.; Jephcoat, A.P.; Macchi, P. Putting pressure on aromaticity along with in situ experimental electron density of a molecular crystal. Nat. Commun. 2016, 7, 10901. [Google Scholar] [CrossRef] [Green Version]
- Genoni, A.; Franchini, D.; Pieraccini, S.; Sironi, M. X-ray Constrained Spin-Coupled Wavefunction: A New Tool to Extract Chemical Information from X-ray Diffraction Data. Chem. A Eur. J. 2018, 24, 15507–15511. [Google Scholar] [CrossRef]
- Genoni, A.; Macetti, G.; Franchini, D.; Pieraccini, S.; Sironi, M. X-ray constrained spin-coupled technique: Theoretical details and further assessment of the method. Acta Crystallogr. Sect. A 2019, 75, 778–797. [Google Scholar] [CrossRef] [Green Version]
- Cooper, D.L.; Gerratt, J.; Raimondi, M. Applications of spin-coupled valence bond theory. Chem. Rev. 1991, 91, 929–964. [Google Scholar] [CrossRef]
- Cooper, D.L.; Gerratt, J.; Raimondi, M. The electronic structure of the benzene molecule. Nature 1986, 323, 699–701. [Google Scholar] [CrossRef]
- Cooper, D.L.; Gerrat, J.; Raimondi, M.; Sironi, M.; Thorsteinsson, T. Expansion of the spin-coupled wavefunction in Slater determinants. Theor. Chim. Acta 1993, 85, 261–270. [Google Scholar] [CrossRef]
- Woińska, M.; Jayatilaka, D.; Dittrich, B.; Flaig, R.; Luger, P.; Woźniak, K.; Dominiak, P.M.; Grabowsky, S. Validation of X-ray Wavefunction Refinement. ChemPhysChem 2017, 18, 3334–3351. [Google Scholar] [CrossRef] [PubMed]
- Schmider, H.; Smith, V.H., Jr.; Weyrich, W. Determination of electron densities and one-matrices from experimental information. Trans. Am. Crystallogr. Assoc. 1990, 26, 125–140. [Google Scholar]
- Schmider, H.; Smith, V.H.; Weyrich, W. Reconstruction of the one-particle density matrix from expectation values in position and momentum space. J. Chem. Phys. 1992, 96, 8986–8994. [Google Scholar] [CrossRef]
- Weyrich, W. An electronic position and momentum density study of chemical bonding in TiO2 (Rutile). Lect. Ser. Comput. Comput. Sci. 2006, 5, 1–3. [Google Scholar]
- Gillet, J.-M.; Cortona, P.; Becker, P.J. Joint refinement of a local wave-function model from Compton and Bragg scattering data. Phys. Rev. B 2001, 63, 235115. [Google Scholar] [CrossRef]
- Gillet, J.-M.; Becker, P.J. Position and momentum densities. Complementarity at work: Refining a quantum model from different data sets. J. Phys. Chem. Solids 2004, 65, 2017–2023. [Google Scholar] [CrossRef]
- Gillet, J.-M. Determination of a one-electron reduced density matrix using a coupled pseudo-atom model and a set of complementary scattering data. Acta Crystallogr. Sect. A 2007, 63, 234–238. [Google Scholar] [CrossRef]
- De Bruyne, B.; Gillet, J.-M. Inferring the one-electron reduced density matrix of molecular crystals from experimental data sets through semidefinite programming. Acta Crystallogr. Sect. A 2020, 76, 1–6. [Google Scholar] [CrossRef]
- Kibalin, I.A.; Yan, Z.; Voufack, A.B.; Gueddida, S.; Gillon, B.; Gukasov, A.; Porcher, F.; Bataille, A.M.; Morini, F.; Claiser, N.; et al. Spin density in YTiO3: I. Joint refinement of polarized neutron diffraction and magnetic x-ray diffraction data leading to insights into orbital ordering. Phys. Rev. B 2017, 96, 054426. [Google Scholar] [CrossRef] [Green Version]
- Yan, Z.; Kibalin, I.A.; Claiser, N.; Gueddida, S.; Gillon, B.; Gukasov, A.; Voufack, A.B.; Morini, F.; Sakurai, Y.; Brancewicz, M.; et al. Spin density in YTiO3: II. Momentum-space representation of electron spin density supported by position-space results. Phys. Rev. B 2017, 96, 054427. [Google Scholar] [CrossRef]
- Gueddida, S.; Yan, Z.; Gillet, J.-M. Development of a joint refinement model for the spin-resolved one-electron reduced density matrix using different data sets. Acta Crystallogr. Sect. A 2018, 74, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Gueddida, S.; Yan, Z.; Kibalin, I.; Voufack, A.B.; Claiser, N.; Souhassou, M.; LeComte, C.; Gillon, B.; Gillet, J.-M. Joint refinement model for the spin resolved one-electron reduced density matrix of YTiO3 using magnetic structure factors and magnetic Compton profiles data. J. Chem. Phys. 2018, 148, 164106. [Google Scholar] [CrossRef] [PubMed]
- Volkov, A.; Abramov, Y.A.; Coppens, P. Critical examination of the radial functions in the Hansen-Coppens multipole model through topological analysis of primary and refined theoretical densities. Acta Crystallogr. Sect. A 2001, 57, 395–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volkov, A.; Abramov, Y.A.; Coppens, P. Density-optimized radial exponents for X-ray charge-density refinement from ab initio crystal calculations. Acta Crystallogr. Sect. A 2001, 57, 272–282. [Google Scholar] [CrossRef] [Green Version]
- Pichon-Pesme, V.; Lecomte, C.; Lachekar, H. On Building a Data Bank of Transferable Experimental Electron Density Parameters Applicable to Polypeptides. J. Phys. Chem. 1995, 99, 6242–6250. [Google Scholar] [CrossRef]
- Domagała, S.; Fournier, B.; Liebschner, D.; Guillot, B.; Jelsch, C. An Improved Experimental Databank of Transferable Multipolar Atom Models–ELMAM2. Construction Details and Applications. Acta Crystallogr. Sect. A 2012, 68, 337–351. [Google Scholar] [CrossRef] [Green Version]
- Volkov, A.; Li, X.; Koritsanszky, T.; Coppens, P. Ab InitioQuality Electrostatic Atomic and Molecular Properties Including Intermolecular Energies from a Transferable Theoretical Pseudoatom Databank. J. Phys. Chem. A 2004, 108, 4283–4300. [Google Scholar] [CrossRef]
- Dittrich, B.; Hubschle, C.B.; Luger, P.; Spackman, M.A. Introduction and validation of an invariom database for amino-acid, peptide and protein molecules. Acta Crystallogr. Sect. A 2006, 62, 1325–1335. [Google Scholar] [CrossRef]
- Jarzembska, K.N.; Dominiak, P.M. New version of the theoretical databank of transferable aspherical pseudoatoms, UBDB2011 – towards nucleic acid modelling. Acta Crystallogr. Sect. A 2011, 68, 139–147. [Google Scholar] [CrossRef]
- Gruza, B.; Chodkiewicz, M.L.; Krzeszczakowska, J.; Dominiak, P.M. Refinement of organic crystal structures with multipolar electron scattering factors. Acta Crystallogr. Sect. A 2020, 76, 92–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, A.; Tiana, D.; Scherer, W.; Batke, K.; Eickerling, G.; Svendsen, H.; Bindzus, N.; Iversen, B.B. Experimental and Theoretical Charge Density Studies at Subatomic Resolution. J. Phys. Chem. A 2011, 115, 13061–13071. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, M.; Claiser, N.; Pillet, S.; Chumakov, Y.; Becker, P.; Gillet, J.-M.; Gillon, B.; Lecomte, C.; Souhassou, M. Experimental determination of spin-dependent electron density by joint refinement of X-ray and polarized neutron diffraction data. Acta Crystallogr. Sect. A 2012, 68, 675–686. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, M.; Gillon, B.; Claiser, N.; Gillet, J.-M.; Lecomte, C.; Souhassou, M. First spin-resolved electron distributions in crystals from combined polarized neutron and X-ray diffraction experiments. IUCrJ 2014, 1, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Voufack, A.B.; Claiser, N.; Lecomte, C.; Pillet, S.; Pontillon, Y.; Gillon, B.; Yan, Z.; Gillet, J.-M.; Marazzi, M.; Genoni, A.; et al. When combined X-ray and polarized neutron diffraction data challenge high-level calculations: Spin-resolved electron density of an organic radical. Acta Crystallogr. Sect. B 2017, 73, 544–549. [Google Scholar] [CrossRef] [PubMed]
- Wahlberg, N.; Bindzus, N.; Bjerg, L.; Becker, J.; Christensen, S.; Dippel, A.-C.; Jørgensen, M.R.V.; Iversen, B.B. Powder X-ray Diffraction Electron Density of Cubic Boron Nitride. J. Phys. Chem. C 2015, 119, 6164–6173. [Google Scholar] [CrossRef]
- Gajda, R.; Stachowicz, M.; Makal, A.; Sutuła, S.; Parafiniuk, J.; Fertey, P.; Woźniak, K. Experimental charge density of grossular under pressure—A feasibility study. IUCrJ 2020, 7, 383–392. [Google Scholar] [CrossRef] [Green Version]
- Eikeland, E.Z.; Borup, M.; Thomsen, M.K.; Roelsgaard, M.; Overgaard, J.; Spackman, M.A.; Iversen, B.B. Single-Crystal High-Pressure X-ray Diffraction Study of Host Structure Compression in Clathrates of Dianin’s Compound. Cryst. Growth Des. 2020. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Genoni, A.; Macchi, P. Quantum Crystallography in the Last Decade: Developments and Outlooks. Crystals 2020, 10, 473. https://doi.org/10.3390/cryst10060473
Genoni A, Macchi P. Quantum Crystallography in the Last Decade: Developments and Outlooks. Crystals. 2020; 10(6):473. https://doi.org/10.3390/cryst10060473
Chicago/Turabian StyleGenoni, Alessandro, and Piero Macchi. 2020. "Quantum Crystallography in the Last Decade: Developments and Outlooks" Crystals 10, no. 6: 473. https://doi.org/10.3390/cryst10060473