Halogen Bonding in the Complexes of Brominated Electrophiles with Chloride Anions: From a Weak Supramolecular Interaction to a Covalent Br–Cl Bond

Abstract
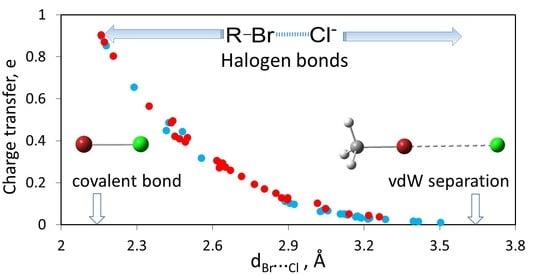
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Crystallization and X-ray Structural Analysis
2.3. Computations
3. Results and Discussion
3.1. Crystallization and X-ray Structural Characterization of the Solid-State Associates of R-Br with Cl−
3.2. Quantum-Mechanical Computations of Halogen-Bonded R–Br…Cl− Complexes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The halogen bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef] [Green Version]
- Gilday, L.C.; Robinson, S.W.; Barendt, T.A.; Langton, M.J.; Mullaney, B.R.; Beer, P.D. Halogen Bonding in Supramolecular Chemistry. Chem. Rev. 2015, 115, 7118–7195. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.-W.; Jiang, Y.-J.; Guo, M.; Hu, G.-X.; Zhang, B.; Liu, H.-C.; Yu, Q.-S. Ab Initio Study of the Complexes of Halogen-Containing Molecules RX (X=Cl, Br, and I) and NH3: Towards Understanding the Nature of Halogen Bonding and the Electron-Accepting Propensities of Covalently Bonded Halogen Atoms. Chem. Eur. J. 2005, 11, 740–751. [Google Scholar] [CrossRef] [PubMed]
- Erdélyi, M. Halogen bonding in solution. Chem. Soc. Rev. 2012, 41, 3547–3557. [Google Scholar] [CrossRef]
- Sarwar, M.G.; Dragisic, B.; Salsberg, L.J.; Gouliaras, C.; Taylor, M.S. Thermodynamics of Halogen Bonding in Solution: Substituent, Structural, and Solvent Effects. J. Am. Chem. Soc. 2010, 132, 1646–1653. [Google Scholar] [CrossRef] [PubMed]
- Weinberger, C.; Hines, R.; Zeller, M.; Rosokha, S.V. Continuum of covalent to intermolecular bonding in the halogen-bonded complexes of 1,4-diazabicyclo[2.2.2]octane with bromine-containing electrophiles. Chem. Commun. 2018, 54, 8060–8063. [Google Scholar] [CrossRef] [PubMed]
- Borley, W.; Watson, B.; Nizhnik, Y.P.; Zeller, M.; Rosokha, S.V. Complexes of Diiodine with Heteroaromatic N-Oxides: Effects of Halogen-Bond Acceptors in Halogen Bonding. J. Phys. Chem. A 2019, 123, 7113–7123. [Google Scholar] [CrossRef] [PubMed]
- Pennington, W.T.; Resnati, G.; Taylor, M.S. Halogen bonding: From self-assembly to materials and biomolecules. CrystEngComm 2013, 15, 3057. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen bonding: An electrostatically-driven highly directional noncovalent interaction. Phys. Chem. Chem. Phys. 2010, 12, 7748–7757. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen bonding and other σ-hole interactions: A perspective. Phys. Chem. Chem. Phys. 2013, 15, 11178–11189. [Google Scholar] [CrossRef]
- Wang, C.; Danovich, D.; Mo, Y.; Shaik, S. On The Nature of the Halogen Bond. J. Chem. Theory Comput. 2014, 10, 3726–3737. [Google Scholar] [CrossRef] [PubMed]
- Thirman, J.; Engelage, E.; Huber, S.M.; Head-Gordon, M. Characterizing the interplay of Pauli repulsion, electrostatics, dispersion and charge transfer in halogen bonding with energy decomposition analysis. Phys. Chem. Chem. Phys. 2018, 20, 905–915. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, V.; Kraka, E.; Cremer, D. The intrinsic strength of the halogen bond: Electrostatic and covalent contributions described by coupled cluster theory. Phys. Chem. Chem. Phys. 2016, 18, 33031–33046. [Google Scholar] [CrossRef] [PubMed]
- Huber, S.M.; Jimenez-Izal, E.; Ugalde, J.M.; Infante, I. Unexpected trends in halogen-bond based noncovalent adducts. Chem. Commun. 2012, 48, 7708. [Google Scholar] [CrossRef] [PubMed]
- Rosokha, S.V.; Stern, C.L.; Ritzert, J.T. Experimental and Computational Probes of the Nature of Halogen Bonding: Complexes of Bromine-Containing Molecules with Bromide Anions. Chem. Eur. J. 2013, 19, 8774–8788. [Google Scholar] [CrossRef]
- Robinson, S.W.; Mustoe, C.L.; White, N.G.; Brown, A.; Thompson, A.L.; Kennepohl, P.; Beer, P.D. Evidence for Halogen Bond Covalency in Acyclic and Interlocked Halogen-Bonding Receptor Anion Recognition. J. Am. Chem. Soc. 2015, 137, 499–507. [Google Scholar] [CrossRef]
- Kellett, C.W.; Kennepohl, P.; Berlinguette, C.P. π covalency in the halogen bond. Nat. Commun. 2020, 11, 3310. [Google Scholar] [CrossRef]
- Grounds, O.; Zeller, M.; Rosokha, S.V. Structural preferences in strong anion-π and halogen-bonded complexes: π- and σ-holes vs frontier orbitals interaction. New J. Chem. 2018, 42, 10572–10583. [Google Scholar] [CrossRef]
- Eraković, M.; Cinčić, D.; Molčanov, K.; Stilinović, V. A Crystallographic charge density study of the partial covalent nature of strong N⋅⋅⋅Br halogen bonds. Angew. Chem. Int. Ed. 2019, 58, 15702–15706. [Google Scholar] [CrossRef]
- Rosokha, S.V. Electron-transfer reactions of halogenated electrophiles: A different look into the nature of halogen bonding. Faraday Discuss. 2017, 203, 315–332. [Google Scholar] [CrossRef]
- Rosokha, S.V.; Traversa, A. From charge transfer to electron transfer in halogen-bonded complexes of electrophilic bromocarbons with halide anions. Phys. Chem. Chem. Phys. 2015, 17, 4989–4999. [Google Scholar] [CrossRef] [PubMed]
- Rosokha, S.V.; Vinakos, M.K. Halogen bond-assisted electron transfer reactions of aliphatic bromosubstituted electrophiles. Phys. Chem. Chem. Phys. 2014, 16, 1809–1813. [Google Scholar] [CrossRef] [PubMed]
- Ruttreddy, R.; Jurcek, O.; Bhowmik, S.; Makela, T.; Rissanen, K. Very strong –N-X+∙∙∙-O-N+ halogen bonds. Chem. Commun. 2016, 52, 2338–2341. [Google Scholar] [CrossRef] [PubMed]
- Suero, M.I.; McNeil, L.E.; Marquez, F. 15N isotopic effects on the Raman spectra of tribromoacetonitrile. J. Raman Spectrosc. 1987, 18, 273–275. [Google Scholar] [CrossRef]
- Heasley, V.L.; Titterington, D.R.; Rold, T.L.; Heasley, G.E. Bromination of nitroalkanes with alkyl hypobromites. J. Org. Chem. 1976, 41, 1285–1287. [Google Scholar] [CrossRef]
- Pfrunder, M.C.; Micallef, A.S.; Rintoul, L.; Arnold, D.P.; Davy, K.J.P.; McMurtrie, J.C. Exploitation of the Menshutkin Reaction for the Controlled Assembly of Halogen Bonded Architectures Incorporating 1,2-Diiodotetrafluorobenzene and 1,3,5-Triiodotrifluorobenzene. Cryst. Growth Des. 2012, 12, 714–724. [Google Scholar] [CrossRef]
- Bruker. Apex3 v2019.1-0, SAINT V8.40A; Bruker AXS Inc.: Madison, WI, USA, 2019. [Google Scholar]
- SHELXTL Suite of Programs; Version 6.14; Bruker Advanced X-ray Solutions; Bruker AXS Inc.: Madison, WI, USA, 2000.
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. A. 2008, 64, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Hübschle, C.; Sheldrick, G.; Dittrich, B. ShelXle: A Qt Graphical user interface for SHELXL. J. Appl. Crystallogr. 2011, 44, 1281–1284. [Google Scholar] [CrossRef] [Green Version]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and. J. Appl. Cryst. 2020, 53, 226–235. [Google Scholar] [CrossRef] [Green Version]
- Gaussian 09, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2009.
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef] [Green Version]
- Kozuch, S.; Martin, J.M.L. Halogen Bonds: Benchmarks and Theoretical Analysis. J. Chem. Theor. Comput. 2013, 9, 1918–1931. [Google Scholar] [CrossRef] [PubMed]
- Bauzá, A.; Alkorta, I.; Frontera, A.; Elguero, J. On the Reliability of Pure and Hybrid DFT Methods for the Evaluation of Halogen, Chalcogen, and Pnicogen Bonds Involving Anionic and Neutral Electron Donors. J. Chem. Theory Comput. 2013, 9, 5201–5210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Z.; Xu, Z.; Zhu, W. Interaction Nature and Computational Methods for Halogen Bonding: A Perspective. J. Chem. Inf. Model. 2020, 60, 2683–2696. [Google Scholar] [CrossRef]
- Rosokha, S.V.; Stern, C.L.; Swartz, A.; Stewart, R. Halogen bonding of electrophilic bromocarbons with pseudohalide anions. Phys. Chem. Chem. Phys. 2014, 16, 12968–12979. [Google Scholar] [CrossRef]
- Watson, B.; Grounds, O.; Borley, W.; Rosokha, S.V. Resolving the halogen vs. hydrogen bonding dichotomy in solutions: Intermolecular complexes of trihalomethanes with halide and pseudohalide anions. Phys. Chem. Chem. Phys. 2018, 20, 21999–22007. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef]
- Boys, S.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Wolters, L.P.; Bickelhaupt, F.M. Halogen Bonding versus Hydrogen Bonding: A Molecular Orbital Perspective. ChemistryOpen 2012, 1, 96–105. [Google Scholar] [CrossRef]
- Shannon, R.D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. Sect. A 1976, 32, 751–767. [Google Scholar] [CrossRef]
- Bruno, I.J.; Cole, J.C.; Edgington, P.R.; Kessler, M.; Macrae, C.F.; McCabe, P.; Pearson, J.; Taylor, R. New software for searching the Cambridge Structural Database and visualising crystal structures. Acta Cryst. B 2002, 58, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Legon, A.; Thorn, J. Equilibrium nuclear quadrupole coupling constants from the rotational spectrum of BrCl: A source of the electric quadrupole moment ratios Q (79 Br)/ Q (81 Br) and Q (35 Cl)/ Q (37 Cl). Chem. Phys. Lett. 1993, 215, 554–560. [Google Scholar] [CrossRef]
- Nizhnik, Y.P.; Sons, A.; Zeller, M.; Rosokha, S.V. Effect of supramolecular architecture on halogen bonding between diiodine and heteroaromatic N-oxides. Cryst. Growth Des. 2018, 18, 1198–1207. [Google Scholar] [CrossRef]
- Weinhold, F.; Landis, C.R. Discovering Chemistry with Natural Bond Orbitals; Wiley: Hoboken, NJ, USA, 2012. [Google Scholar] [CrossRef]
- Yannacone, S.; Oliveira, V.P.; Verma, N.; Kraka, E. A Continuum from Halogen Bonds to Covalent Bonds: Where Do λ3 Iodanes Fit? Inorganics 2019, 7, 47. [Google Scholar] [CrossRef] [Green Version]
- Torubaev, Y.V.; Dolgushin, F.M.; Skabitsky, I.V.; Popova, A.E.; Skabitskiy, I.V. Isomorphic substitution in molecular crystals and geometry of hypervalent tellurium: Comments inspired by a case study of RMeTeI2 and [RMe2Te]+I− (R = Ph, Fc). New J. Chem. 2019, 43, 12225–12232. [Google Scholar] [CrossRef]
- Grabowski, S.J. What Is the Covalency of Hydrogen Bonding? Chem. Rev. 2011, 111, 2597–2625. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, S.J.; Sokalski, W.A.; Dyguda, E.; Leszczyński, J. Quantitative classification of covalent and noncovalent H-bonds. J. Phys. Chem. B 2006, 110, 6444–6446. [Google Scholar] [CrossRef]
- Kertesz, M. Pancake Bonding: An Uunusual pi-stacking interaction. Chem. Eur. J. 2019, 25, 400–416. [Google Scholar] [CrossRef]
- Molčanov, K.; Jelsch, C.; Landeros, B.; Hernández-Trujillo, J.; Wenger, E.; Stilinović, V.; Kojić-Prodić, B.; Escudero-Adán, E.C. Partially Covalent Two-Electron/Multicentric Bonding between Semiquinone Radicals. Cryst. Growth Des. 2018, 19, 391–402. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| R-Br | Counter-Ion | Bonding 1 | dBr…Cl, Å | Refcode 2 |
|---|---|---|---|---|
| CBr3F | Pr4N+ | 3:3 | 3.0820 (10), 3.1266 (9), 3.1728 (10) | This work |
| CBr3COCBr3 | Pr4N+ | 3:3 | 3.0306 (8), 3.0618 (8), 3.2056 (8) | This work |
| CBr3CN | Pr4N+ | 6:3 | 3.0980 (4), 3.1320 (4), 3.1398 (4) | This work |
| CBr4 | Pr4N+ | 3:3 | 3.0650 (7), 3.1769 (7), 3.2431 (7) | This work |
| Et4N+ | 4:4 | 3.090 | VAPVOY | |
| CBr3NO2 | DABCO-CH2Cl+ | 3:3 | 3.1420 (17), 3.1728 (16), 3.1715 (16) | This work |
| Pr4N+ | 3:3 | 2.987, 3.044, 3.269, 3.124, 3.084, 2.986 | WOTTAC | |
| CBr3CONH2 | DABCO-CH2Cl+ | 2:2 | 3.0457 (12), 3.2770 (12) | This work |
| C6F4Br2 3 | C19H20NO+ 3 | 1:2 | 3.169 | PUDBUO |
| 2-BrPYR+ 4 | - | 1:1 | 3.310 | CIGZIC |
| 3-BrPYR+ 4 | 1:1 | 3.359 | CIHBAX | |
| 4-BrPYR+ 4 | 1:1 | 3.312 | CIHBOL | |
| C4H8Br2N+ 5 | 1:1 | 3.347 | BRPYRL | |
| C12H8BrF2+ 6 | 2:2 | 3.049 | YATMEO | |
| BrPIM 7 | Ph4P+ | 2:1 | 2.700, 2.734 | WEYZAB |
| BrSIM 8 | Ph4P+ | 2:1 | 2.822, 2.852 | WEYYOO |
| ClBr | Ph4As+ | 3:1 | 2.603, 2.757, 2.801, 2.632, 2.724, 2.730 | DIDHUW |
| ClBr | Et4N+ | 1:1 | 2.362, 2.396, 2.410, 2.402 | TEACBR |
| ClBr | Ph4P+ 9 | 1:1 | 2.426 | GOLKAV |
| Br2 | NMHDA+ 10 | 1:1 | 2.567 | URUBUF |
| Br+ | 2.139 11 | - |
| R-Br 1 | dBr···Cl, Å | ΔE, kJ mol−1 | R3 | dX–Brcom/dX–Brsep 4 | q(Cl), 5 |
|---|---|---|---|---|---|
| CH3Br | 3.503 | 15.1 | 0.96 | 1.00 | −0.989 |
| CH2BrF | 3.415 | 11.9 | 0.93 | 1.00 | −0.985 |
| CH2Br2 | 3.395 | 9.9 | 0.93 | 1.00 | −0.982 |
| C4H8Br2N+ | 3.284 | −13.1 | 0.90 | 1.01 | −0.974 |
| CHBr3 | 3.223 | 4.5 | 0.88 | 1.00 | −0.967 |
| C6Br2F4 | 3.219 | 2.5 | 0.88 | 1.00 | −0.969 |
| BrCCH | 3.215 | 1.4 | 0.88 | 1.01 | −0.972 |
| 4-BrPYR+ | 3.189 | −17.1 | 0.87 | 1.00 | −0.967 |
| 3-BrPYR+ | 3.185 | −17.2 | 0.87 | 1.00 | −0.966 |
| CBr3CONH2 | 3.174 | −0.1 | 0.87 | 1.00 | −0.959 |
| C2Br2F4 | 3.167 | −4.8 | 0.87 | 1.00 | −0.962 |
| CBr3F | 3.134 | 0.1 | 0.86 | 1.00 | −0.954 |
| CBrCl3 | 3.127 | −0.6 | 0.85 | 1.00 | −0.950 |
| CBr4 | 3.122 | −0.5 | 0.85 | 1.00 | −0.948 |
| CBr3COCBr3 | 3.106 | −1.5 | 0.85 | 1.00 | −0.948 |
| CBr3CN | 3.057 | −5.0 | 0.84 | 1.00 | −0.934 |
| CBr3NO2 | 3.038 | −5.7 | 0.83 | 1.01 | −0.93 |
| C12H8BrF2+ | 3.026 | −42.7 | 0.83 | 1.00 | −0.937 |
| BrSIM | 2.923 | −8.0 | 0.80 | 1.02 | −0.902 |
| BrPIM | 2.905 | −9.5 | 0.79 | 1.02 | −0.897 |
| CBr(NO2)3 | 2.887 | −18.7 | 0.79 | 1.02 | −0.888 |
| BrSAC 2 | 2.556 | −21.4 | 0.70 | 1.08 | −0.682 |
| Br2 | 2.480 | −29.7 | 0.68 | 1.11 | −0.557 |
| BrCl | 2.427 | −42.0 | 0.66 | 1.12 | −0.513 |
| BrF | 2.416 | −73.7 | 0.66 | 1.10 | −0.551 |
| pyrazine-Br+ | 2.289 | −116.3 | 0.63 | 1.22 | −0.345 |
| F5Pyr-Br+ | 2.179 | −209.8 | 0.60 | 1.49 | −0.147 |
| Br+ | 2.160 | −607.9 | 0.59 | N/A | −0.103 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Loy, C.; Zeller, M.; Rosokha, S.V. Halogen Bonding in the Complexes of Brominated Electrophiles with Chloride Anions: From a Weak Supramolecular Interaction to a Covalent Br–Cl Bond. Crystals 2020, 10, 1075. https://doi.org/10.3390/cryst10121075
Loy C, Zeller M, Rosokha SV. Halogen Bonding in the Complexes of Brominated Electrophiles with Chloride Anions: From a Weak Supramolecular Interaction to a Covalent Br–Cl Bond. Crystals. 2020; 10(12):1075. https://doi.org/10.3390/cryst10121075
Chicago/Turabian StyleLoy, Cody, Matthias Zeller, and Sergiy V. Rosokha. 2020. "Halogen Bonding in the Complexes of Brominated Electrophiles with Chloride Anions: From a Weak Supramolecular Interaction to a Covalent Br–Cl Bond" Crystals 10, no. 12: 1075. https://doi.org/10.3390/cryst10121075