In Situ TEM Crystallization of Amorphous Iron Particles


Abstract
:
1. Introduction
2. Materials and Methods
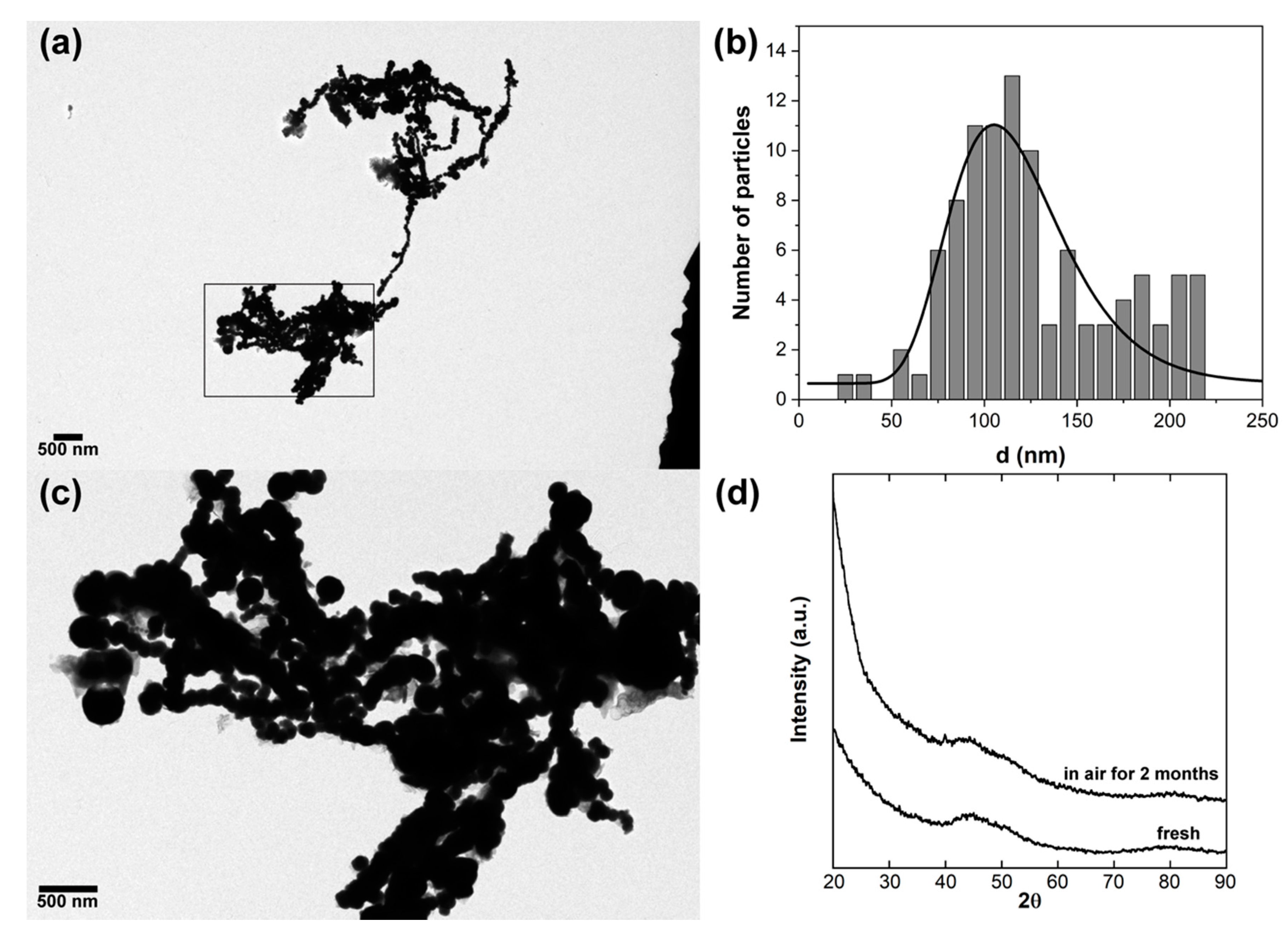
2.1. Synthesis of the Iron Amorphous Particles
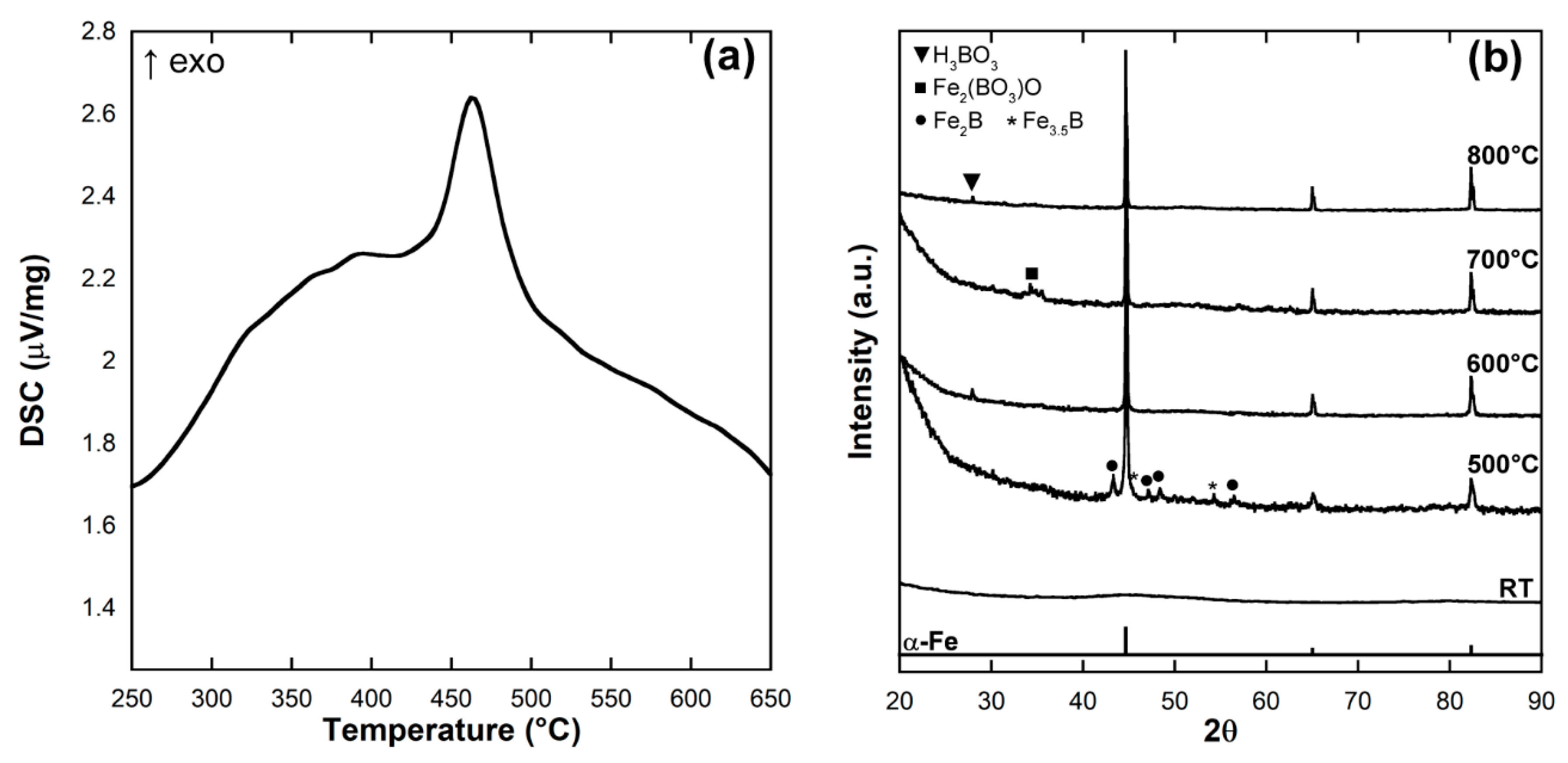
2.2. Differential Scanning Calorimetry and X-ray Diffraction
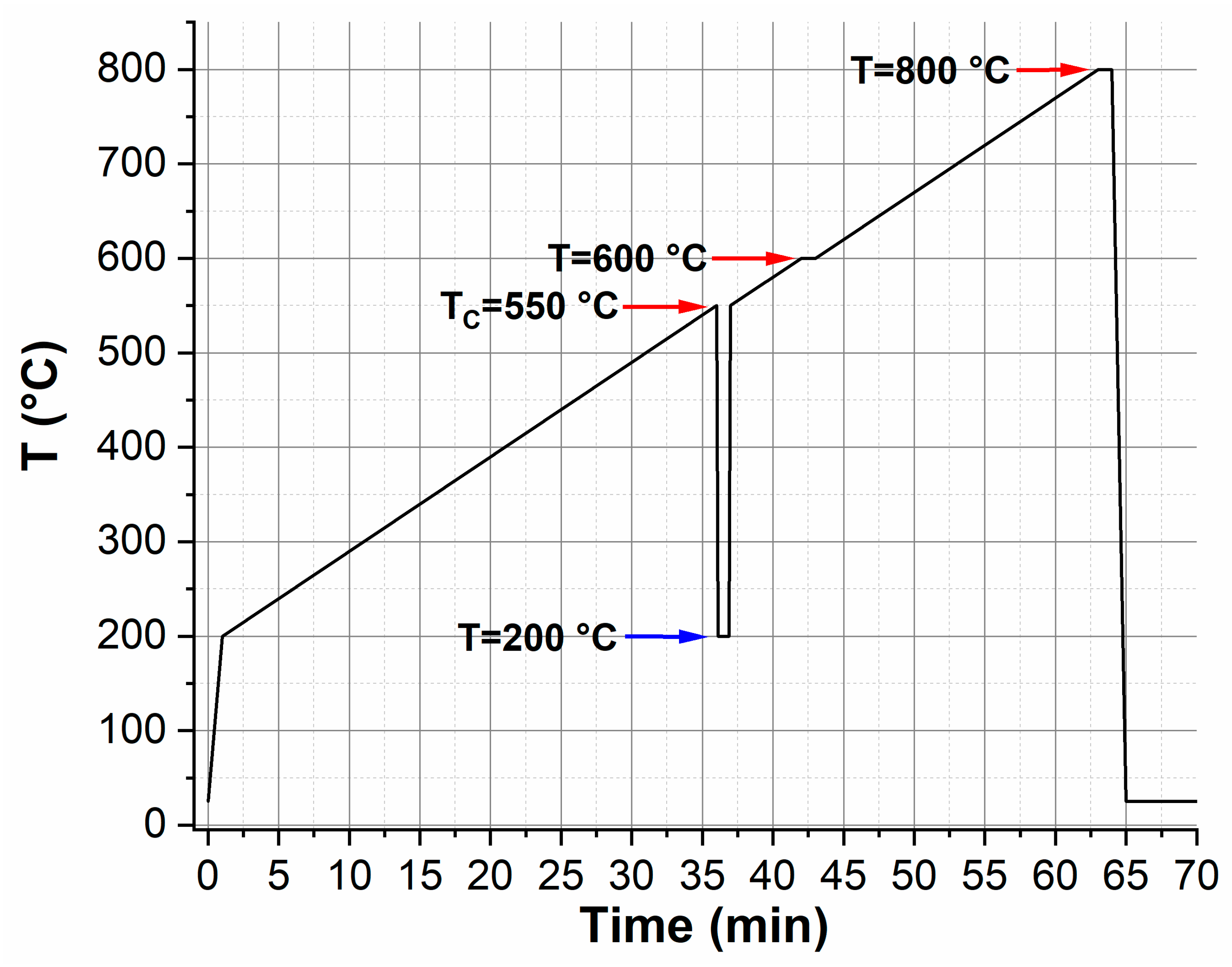
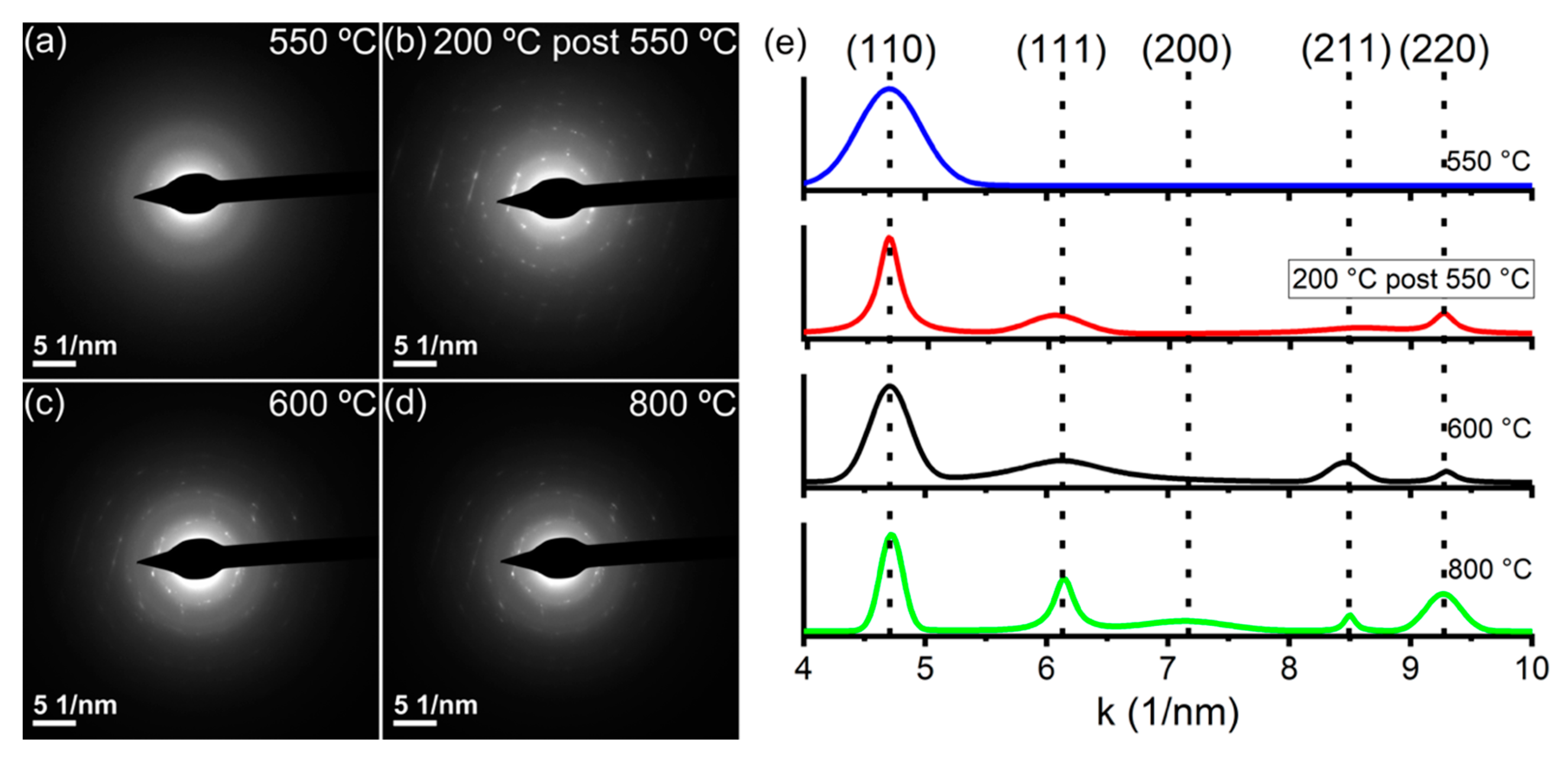
2.3. Conventional and In Situ Heating Transmission Electron Microscopy
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Klement, W.; Willens, R.H.; Duwez, P.O.L. Non-crystalline structure in solidified gold-silicon alloys. Nature 1960, 187, 869–870. [Google Scholar] [CrossRef]
- Inoue, A.; Zhang, T.; Zhang, W.; Takeuchi, A. Bulk Nd-Fe-Al amorphous alloys with hard magnetic properties. Mater. Trans. JIM 1996, 37, 99–108. [Google Scholar] [CrossRef] [Green Version]
- Tiberto, P.; Baricco, M.; Olivetti, E.; Piccin, R. Magnetic properties of bulk metallic glasses. Adv. Eng. Mater. 2007, 7, 468–474. [Google Scholar] [CrossRef]
- Buschow, K.H.J.; Beekmans, N.M. Thermal stability and electronic properties of amorphous Zr-Co and Zr-Ni alloys. Phys. Rev. B 1979, 19, 3843. [Google Scholar] [CrossRef]
- Qin, C.L.; Zhang, W.; Asami, K.; Kimura, H.; Wang, X.M.; Inoue, A. A novel Cu-based BMG composite with high corrosion resistance and excellent mechanical properties. Acta Mater. 2006, 54, 3713–3719. [Google Scholar] [CrossRef]
- Peker, A.; Johnson, W.L. A highly processable metallic glass-Zr41.2Ti13.8Cu12.5Ni10.0Be22.5. Appl. Phys. Lett. 1993, 63, 2342–2344. [Google Scholar] [CrossRef] [Green Version]
- Shin, J.; Waheed, A.; Agapiou, K.; Winkenwerder, W.A.; Kim, H.-W.; Jones, R.A.; Hwang, G.S.; Ekerdt, J.G. Growth of ultrathin films of amorphous ruthenium−phosphorus alloys using a single source CVD precursor. J. Am. Chem. Soc. 2006, 128, 16510–16511. [Google Scholar] [CrossRef]
- Rehn, L.E.; Okamoto, P.R.; Pearson, J.; Bhadra, R.; Grimsditch, M. Solid-state amorphization of Zr3Al: Evidence of an elastic instability and first-order phase transformation. Phys. Rev. Lett. 1987, 59, 2987–2990. [Google Scholar] [CrossRef] [Green Version]
- Limthongkul, P.; Jang, Y.-I.; Dudney, N.J.; Chiang, Y.-M. Electrochemically-driven solid-state amorphization in lithium-silicon alloys and implications for lithium storage. Acta Mater. 2003, 51, 1103–1113. [Google Scholar] [CrossRef]
- Grant, W.A. Amorphous metals and ion implantation. J. Vac. Sci. Technol. 1978, 15, 1644–1649. [Google Scholar] [CrossRef]
- Suryanarayana, C. Mechanical alloying and milling. Progr. Mater. Sci. 2001, 46, 1–184. [Google Scholar] [CrossRef]
- Jiles, D.C. Recent advances and future directions in magnetic materials. Acta Mater. 2003, 51, 5907–5939. [Google Scholar] [CrossRef]
- Huber, D.L. Synthesis, properties, and application of iron nanoparticles. Small 2005, 1, 482–501. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.H.; Chang, C.T.; Wang, A.; Shen, B.L. Development of quaternary Fe-based bulk metallic glasses with high saturation magnetization above 1.6 T. J. Non Cryst. Solids 2012, 358, 1443–1446. [Google Scholar] [CrossRef]
- Kong, F.L.; Chang, C.T.; Inoue, A.; Shalaan, E.; Al-Marzouki, F. Fe-based amorphous soft magnetic alloys with high saturation magnetization and good bending ductility. J. Alloy. Compd. 2014, 615, 163–166. [Google Scholar] [CrossRef]
- Mchenry, M.E.; Laughlin, D.E. Nano-scale materials development for future magnetic applications. Acta Mater. 2000, 48, 223–238. [Google Scholar] [CrossRef]
- Fang, J.X.; Vainio, U.; Puff, W.; Würschum, R.; Wang, X.L.; Wang, D.; Ghafari, M.; Jiang, F.; Sun, J.; Hahn, H.; et al. Atomic structure and structural stability of Sc75Fe25 nanoglasses. Nano Lett. 2012, 12, 458–463. [Google Scholar] [CrossRef]
- Grinstaff, M.W.; Cichowlas, A.A.; Choe, S.-B.; Suslick, K.S. Effect of cavitation on amorphous metal synthesis. Ultrasonics 1992, 30, 168–172. [Google Scholar] [CrossRef]
- Grinstaff, M.W.; Salamon, M.B.; Suslick, K.S. Magnetic properties of amorphous iron. Phys. Rev. B 1993, 48, 269–273. [Google Scholar] [CrossRef]
- Yang, B.; Yang, X.Y.; Li, X.P.; Cao, Y.; Yu, R.H. Surface modification and enhanced performances of chemically synthesized nanosized amorphous Fe particles. J. Supercond. Nov. Magn. 2015, 28, 2177–2182. [Google Scholar] [CrossRef]
- Yang, X.Y.; Yang, B.; Li, X.P.; Cao, Y.; Yu, R.H. Structural-controlled chemical synthesis of nanosized amorphous Fe particles and their improved performances. J. Alloy. Compd. 2015, 651, 551–556. [Google Scholar] [CrossRef]
- Casu, A.; Lamberti, A.; Stassi, S.; Falqui, A. Crystallization of TiO2 nanotubes by in situ heating TEM. Nanomaterials 2018, 8, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stassi, S.; Lamberti, A.; Roppolo, I.; Casu, A.; Bianco, S.; Scaiola, D.; Falqui, A.; Pirri, C.F.; Ricciardi, C. Evolution of nanomechanical properties and crystallinity of individual titanium dioxide nanotube resonators. Nanotechnology 2017, 29, 085702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.; Yin, P.; Zhu, X.; OuYang, C.O.; Xie, Y. Synthesis of hematite (α-Fe2O3) nanorods: Diameter-size and shape effects on their applications in magnetism, lithium ion battery, and gas sensors. J. Phys. Chem. B 2006, 110, 17806–17812. [Google Scholar] [CrossRef]
- Chen, M.; Tang, B.; Nikles, D.E. Preparation of iron nanoparticles by reduction of acicular β-FeOOH particles. IEEE Trans. Magn. 1998, 34, 1141–1143. [Google Scholar] [CrossRef]
- Jagodzinski, H. HP Klug und LE Alexander: X-ray Diffraction Procedures for Polycrystalline and Amorphous Materials, 2. Auflage. John Wiley & Sons, New York-Sydney-Toronto 1974, 966 Seiten, Preis:£ 18.55. Ber. Bunsenges. Phys. Chem. 1975, 79, 553. [Google Scholar]
- Granqvist, C.G.; Buhrman, R.A. Ultrafine metal particles. J. Appl. Phys. 1976, 47, 2200–2219. [Google Scholar] [CrossRef]
- Blanco-Mantecón, M.; O’Grady, K. Grain size and blocking distributions in fine particle iron oxide nanoparticles. J. Magn. Magn. Mater. 1999, 203, 50–53. [Google Scholar] [CrossRef]
- Zhang, X.F.; Dong, X.L.; Huang, H.; Lv, B.; Lei, J.P.; Ma, S.; Liu, W.; Zhang, Z.D. Synthesis, structure and magnetic properties of B2O3/H3BO3-coated Fe nanocapsules. Mat. Sci. Eng. B 2007, 143, 76–80. [Google Scholar] [CrossRef]
- Wang, W.K.; Iwasaki, H.; Fukamichi, K. Effect of high pressure on the crystallization of an amorphous Fe83B17 alloy. J. Mater. Sci. 1980, 15, 2701–2708. [Google Scholar] [CrossRef]
- Imura, T.; Suwa, M.; Fujii, K. Effects of ultrahigh pressure on the crystallization temperature of Ni80P20 amorphous alloys. Mater. Sci. Eng. 1988, 97, 247–251. [Google Scholar] [CrossRef]
- Cedergren, M.; Bäckström, G. Crystallization temperature of amorphous Fe80B20 under pressure. J. Non-Cryst. Solids. 1978, 30, 69–76. [Google Scholar] [CrossRef]
- Wang, W.K.; Iwasaki, H.; Suryanarayana, C.; Masumoto, T.; Toyota, N.; Fukase, T.; Kogiku, F. Crystallization characteristics of an amorphous Nb81Si19 alloy under high pressure and formation of the A15 phase. J. Mater. Sci. 1982, 17, 1523–1532. [Google Scholar] [CrossRef]
- Ye, F.; Lu, K. Pressure effect on polymorphous crystallization kinetics in amorphous selenium. Acta Mater. 1998, 46, 5965–5971. [Google Scholar] [CrossRef] [Green Version]
- Ye, F.; Lu, K. Pressure effect on crystallization kinetics of an Al-La-Ni amorphous alloy. Acta Mater. 1999, 47, 2449–2454. [Google Scholar] [CrossRef]
- Zhang, T.; Song, Z.; Sun, M.; Liu, B.; Feng, S.; Chen, B. Investigation of electron beam induced phase change in Si2Sb2Te5 material. Appl. Phys. A 2008, 90, 451–455. [Google Scholar] [CrossRef]
- Kooi, B.J.; Groot, W.M.G.; De Hosson, J.T.M. In situ transmission electron microscopy study of the crystallization of Ge2Sb2Te5. J. Appl. Phys. 2004, 95, 451–455. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Temperature | <d>XRD (nm) |
|---|---|
| 500 °C | 55 ± 1 |
| 600 °C | 110 ± 4 |
| 700 °C | 180 ± 14 |
| 800 °C | 320 ± 22 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Falqui, A.; Loche, D.; Casu, A. In Situ TEM Crystallization of Amorphous Iron Particles. Crystals 2020, 10, 41. https://doi.org/10.3390/cryst10010041
Falqui A, Loche D, Casu A. In Situ TEM Crystallization of Amorphous Iron Particles. Crystals. 2020; 10(1):41. https://doi.org/10.3390/cryst10010041
Chicago/Turabian StyleFalqui, Andrea, Danilo Loche, and Alberto Casu. 2020. "In Situ TEM Crystallization of Amorphous Iron Particles" Crystals 10, no. 1: 41. https://doi.org/10.3390/cryst10010041