
Deficiency of iPLA2β Primes Immune Cells for Proinflammation: Potential Involvement in Age-Related Mesenteric Lymph Node Lymphoma

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
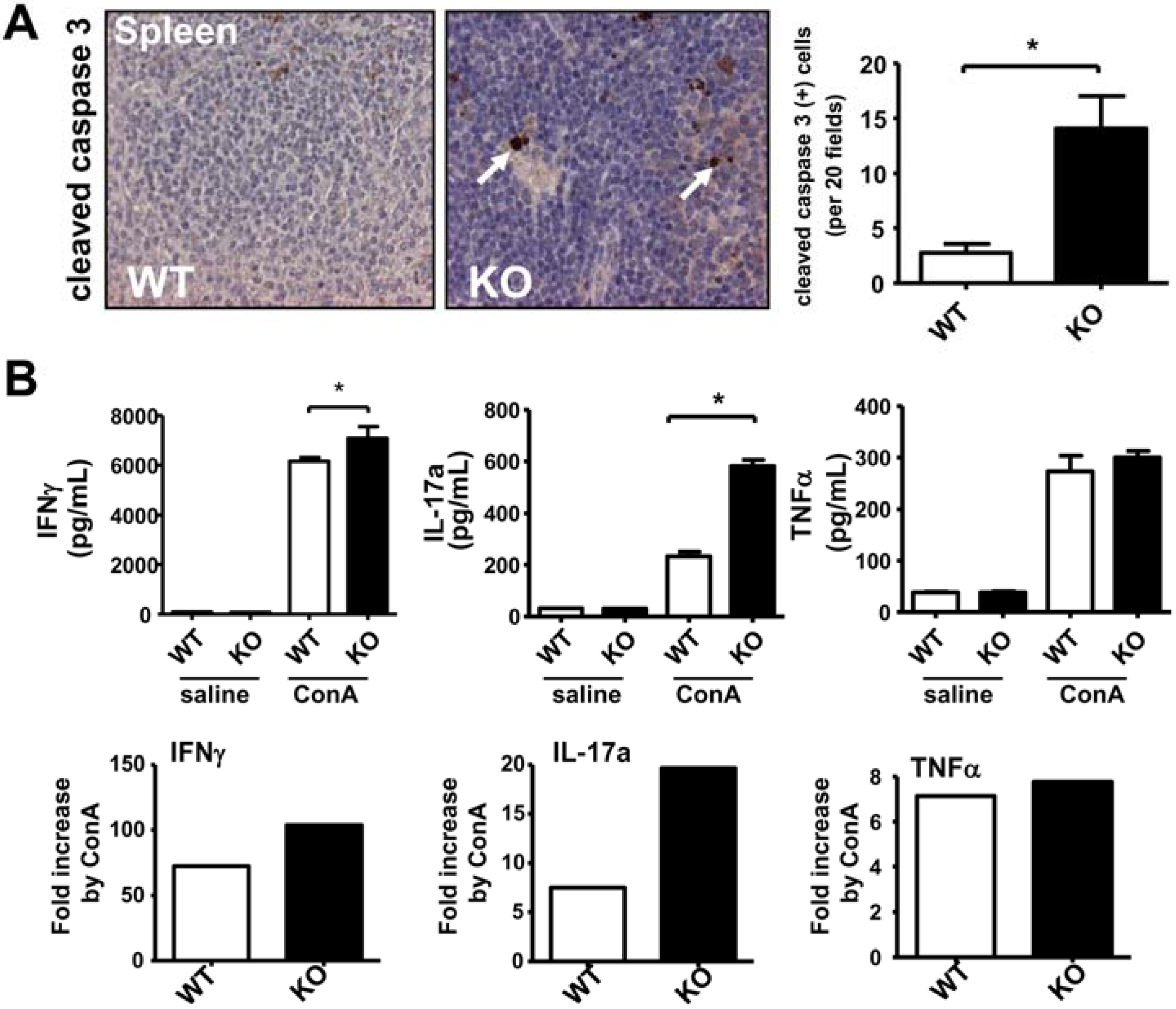
2.1. Deficiency of iPLA2β Increases Apoptosis in Spleen and Primes Splenocytes for Th1/Th17 Response

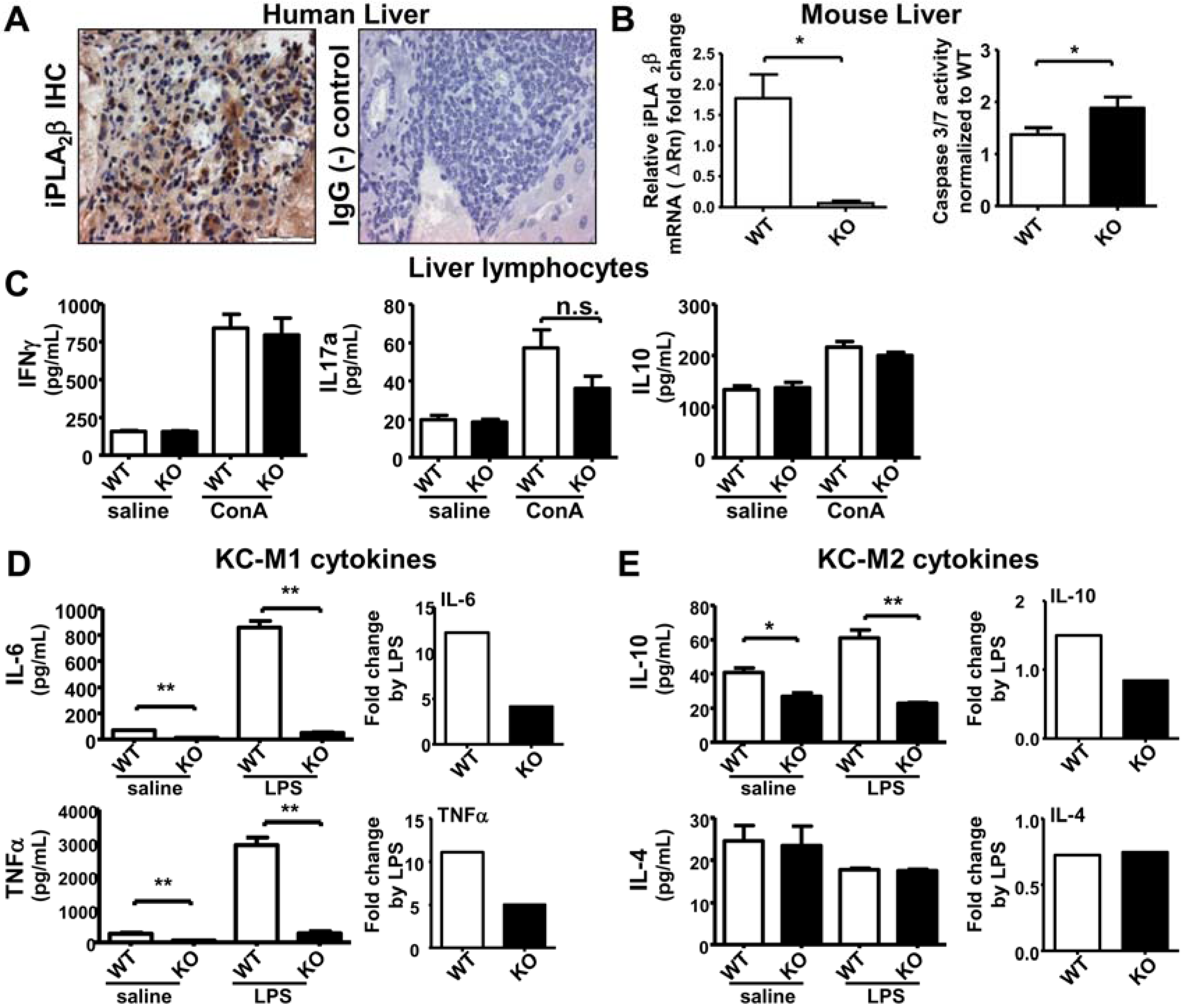
2.2. iPLA2β Deficiency Increases Apoptosis in Liver Associated with Suppressed Cytokine Release by KC

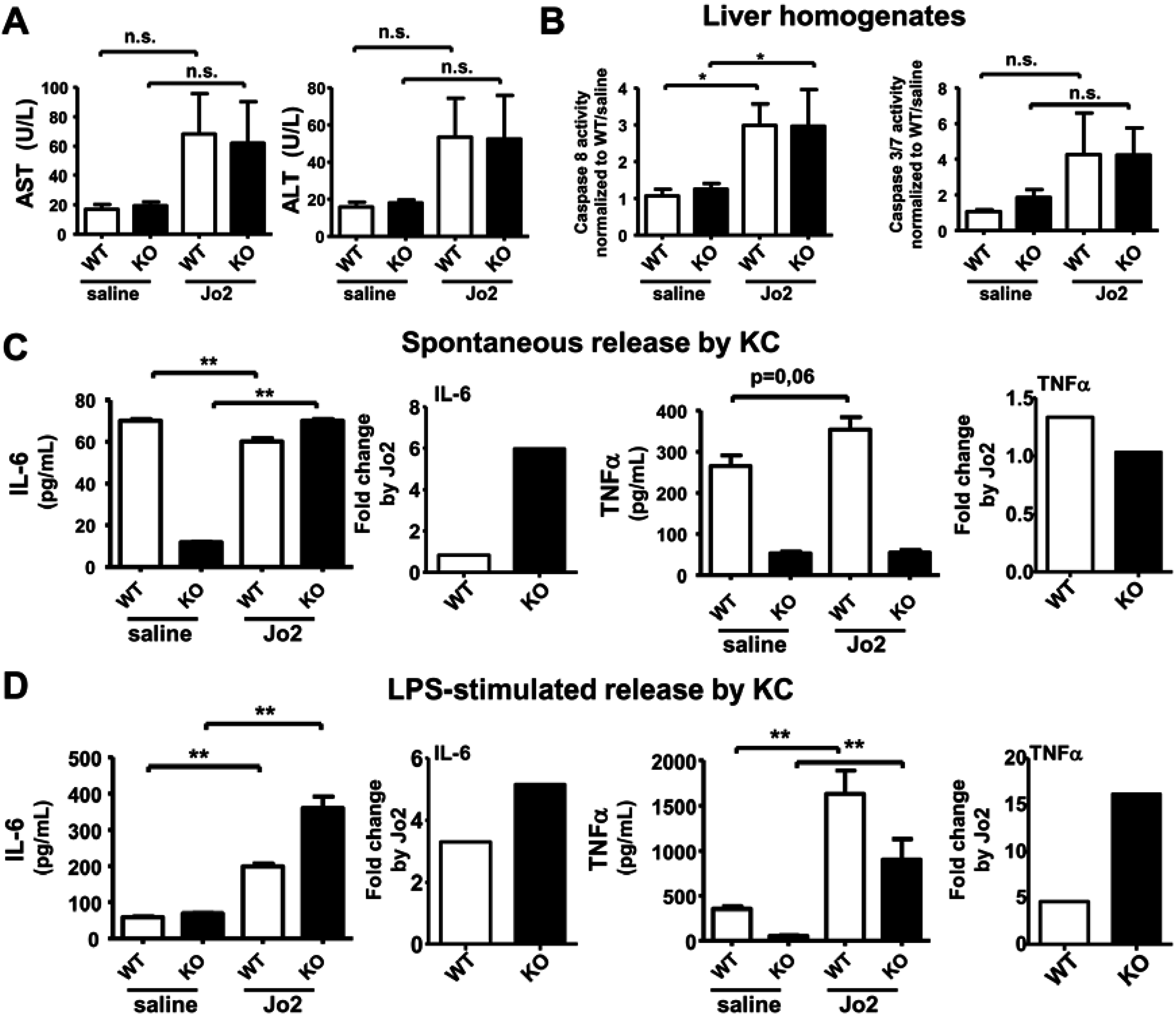
2.3. Sublethal Dose CD95/FasL Treatment Primes Mutant KC for Enhanced M1 Cytokine Release

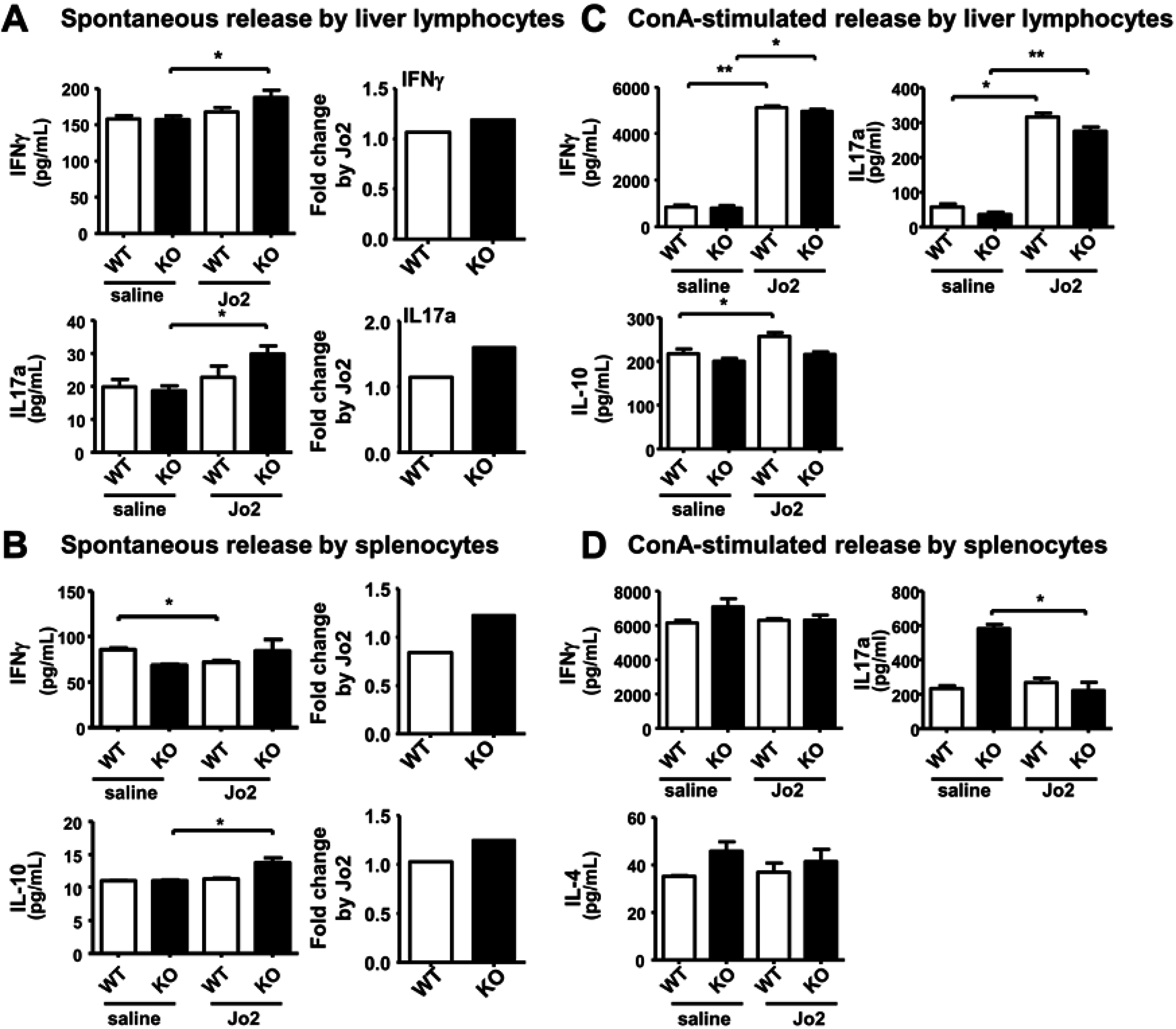
2.4. Sublethal-Dose CD95/FasL Treatment Primes Mutant Liver Lymphocytes for a Weak Increase in Th1 Cytokine Release
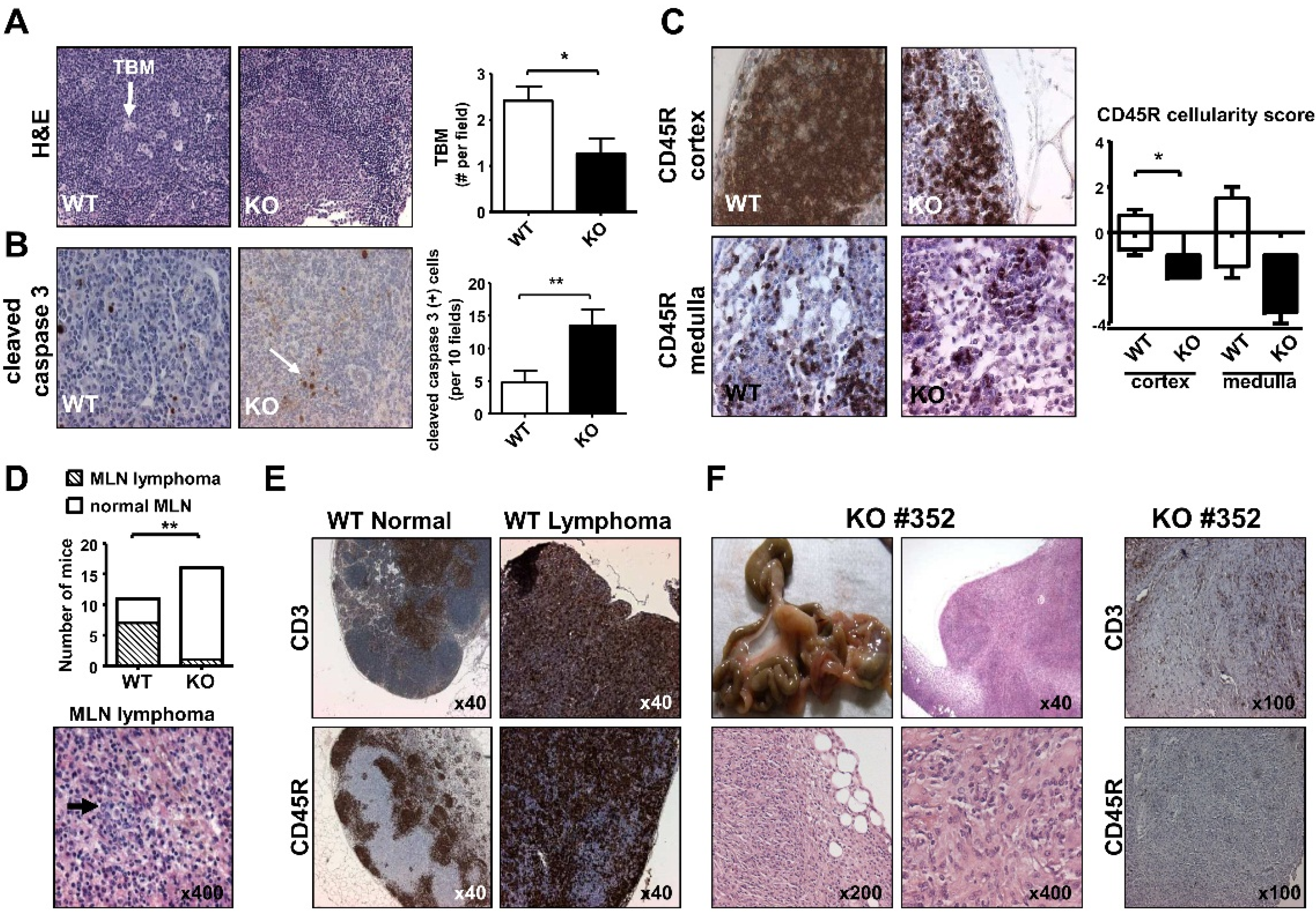
2.5. Mesenteric Lymph Node Abnormalities of Aged iPLA2β-Deficient Mice


3. Experimental Section
3.1. Animals and Treatment
3.2. Biochemical Assays
3.3. Histology and Immunohistochemistry (IHC)
3.4. Cell Isolation and Cell Culture
3.5. Determination of Cytokine Release
3.6. Quantitative qRT-PCR
3.7. Statistics
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Aggarwal, B.B.; Vijayalekshmi, R.V.; Sung, B. Targeting inflammatory pathways for prevention and therapy of cancer: Short-term friend, long-term foe. Clin. Cancer Res. 2009, 15, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Kotas, M.E.; Medzhitov, R. Homeostasis, inflammation, and disease susceptibility. Cell 2015, 160, 816–827. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed]
- Ramanadham, S.; Ali, T.; Ashley, J.W.; Bone, R.N.; Hancock, W.D.; Lei, X. Calcium-independent phospholipases A2 and their roles in biological processes and diseases. J. Lipid Res. 2015, 56, 1643–1668. [Google Scholar] [CrossRef] [PubMed]
- Balsinde, J.; Balboa, M.A. Cellular regulation and proposed biological functions of group VIA calcium-independent phospholipase A2 in activated cells. Cell. Signal. 2005, 17, 1052–1062. [Google Scholar] [CrossRef] [PubMed]
- Balsinde, J.; Bianco, I.D.; Ackermann, E.J.; Conde-Frieboes, K.; Dennis, E.A. Inhibition of calcium-independent phospholipase A2 prevents arachidonic acid incorporation and phospholipid remodeling in P388D1 macrophages. Proc. Natl. Acad. Sci. USA 1995, 92, 8527–8531. [Google Scholar] [CrossRef] [PubMed]
- Perez, R.; Matabosch, X.; Llebaria, A.; Balboa, M.A.; Balsinde, J. Blockade of arachidonic acid incorporation into phospholipids induces apoptosis in U937 promonocytic cells. J. Lipid Res. 2006, 47, 484–491. [Google Scholar] [CrossRef] [PubMed]
- Bao, S.; Miller, D.J.; Ma, Z.; Wohltmann, M.; Eng, G.; Ramanadham, S.; Moley, K.; Turk, J. Male mice that do not express group VI phospholipase A2 produce spermatozoa with impaired motility and have greatly reduced fertility. J. Biol. Chem. 2004, 279, 38194–38200. [Google Scholar] [CrossRef] [PubMed]
- Bao, S.; Song, H.; Wohltmann, M.; Ramanadham, S.; Jin, W.; Bohrer, A.; Turk, J. Insulin secretory responses and phospholipid composition of pancreatic islets from mice that do not express Group VIA phospholipase A2 and effects of metabolic stress on glucose homeostasis. J. Biol. Chem. 2006, 281, 20958–20973. [Google Scholar] [CrossRef] [PubMed]
- Ramanadham, S.; Yarasheski, K.E.; Silva, M.J.; Wohltmann, M.; Novack, D.V.; Christiansen, B.; Tu, X.; Zhang, S.; Lei, X.; Turk, J. Age-related changes in bone morphology are accelerated in group VIA phospholipase A2 (iPLA2β)-null mice. Am. J. Pathol. 2008, 172, 868–881. [Google Scholar] [CrossRef] [PubMed]
- Shinzawa, K.; Sumi, H.; Ikawa, M.; Matsuoka, Y.; Okabe, M.; Sakoda, S.; Tsujimoto, Y. Neuroaxonal dystrophy caused by group VIA phospholipase A2 deficiency in mice: A model of human neurodegenerative disease. J. Neurosci. 2008, 28, 2212–2220. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Tuma, S.; Katava, N.; Pathil-Warth, A.; Stremmel, W.; Chamulitrat, W. Deficiencies of Calcium-independent Phospholipase A2 B in vivo Causes Reduced Systemic Lipids and Lipoproteins Concomitant with Increased Hepatic Apoptosis and Inflammation. In Proceeding of The International Liver CongressTM (EASL), Barcelona, Spain, 18–22 April 2012.
- Jiao, L.; Gan-Schreier, H.; Tuma-Kellner, S.; Stremmel, W.; Chamulitrat, W. Sensitization to autoimmune hepatitis in group VIA calcium-independent phospholipase A2-null mice led to duodenal villous atrophy with apoptosis, goblet cell hyperplasia and leaked bile acids. Biochim. Biophys. Acta 2015, 1852, 1646–1657. [Google Scholar] [CrossRef] [PubMed]
- Jiao, L.; Inhoffen, J.; Gan-Schreier, H.; Tuma-Kellner, S.; Stremmel, W.; Sun, Z.; Chamulitrat, W. Deficiency of Group VIA Phospholipase A2 (iPLA2β) Renders Susceptibility for Chemical-Induced Colitis. Dig. Dis. Sci. 2015, in press. [Google Scholar] [CrossRef] [PubMed]
- Lauber, K.; Bohn, E.; Kröber, S.M.; Xiao, Y.J.; Blumenthal, S.G.; Lindemann, R.K.; Marini, P.; Wiedig, C.; Zobywalski, A.; Baksh, S.; et al. Apoptotic cells induce migration of phagocytes via caspase-3-mediated release of a lipid attraction signal. Cell 2003, 113, 717–730. [Google Scholar] [CrossRef]
- Mishra, R.S.; Carnevale, K.A.; Cathcart, M.K. iPLA2β: Front and center in human monocyte chemotaxis to MCP-1. J. Exp. Med. 2008, 205, 347–359. [Google Scholar] [CrossRef] [PubMed]
- Poon, I.K.; Lucas, C.D.; Rossi, A.G.; Ravichandran, K.S. Apoptotic cell clearance: Basic biology and therapeutic potential. Nat. Rev. Immunol. 2014, 14, 166–180. [Google Scholar] [CrossRef] [PubMed]
- Truman, L.A.; Ford, C.A.; Pasikowska, M.; Pound, J.D.; Wilkinson, S.J.; Dumitriu, I.E.; Melville, L.; Melrose, L.A.; Ogden, C.A.; Nibbs, R.; et al. CX3CL1/fractalkine is released from apoptotic lymphocytes to stimulate macrophage chemotaxis. Blood 2008, 112, 5026–5036. [Google Scholar] [CrossRef] [PubMed]
- Elliott, M.R.; Chekeni, F.B.; Trampont, P.C.; Lazarowski, E.R.; Kadl, A.; Walk, S.F.; Park, D.; Woodson, R.I.; Ostankovich, M.; Sharma, P.; et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature 2009, 461, 282–286. [Google Scholar] [CrossRef] [PubMed]
- Scaffidi, P.; Misteli, T.; Bianchi, M.E. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 2002, 418, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Albert, M.L. Death-defying immunity: Do apoptotic cells influence antigen processing and presentation? Nat. Rev. Immunol. 2004, 4, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Le, L.Q.; Kabarowski, J.H.; Weng, Z.; Satterthwaite, A.B.; Harvill, E.T.; Jensen, E.R.; Miller, J.F.; Witte, O.N. Mice lacking the orphan G protein-coupled receptor G2A develop a late-onset autoimmune syndrome. Immunity 2001, 14, 561–571. [Google Scholar] [CrossRef]
- O’Shea, J.J.; Ma, A.; Lipsky, P. Cytokines and autoimmunity. Nat. Rev. Immunol. 2002, 2, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Dranoff, G. Cytokines in cancer pathogenesis and cancer therapy. Nat. Rev. Cancer 2004, 4, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.W.; Karin, M. A cytokine-mediated link between innate immunity, inflammation, and cancer. J. Clin. Investig. 2007, 117, 1175–1183. [Google Scholar] [CrossRef] [PubMed]
- Park, D.R.; Thomsen, A.R.; Frevert, C.W.; Pham, U.; Skerrett, S.J.; Kiener, P.A.; Liles, W.C. Fas (CD95) induces proinflammatory cytokine responses by human monocytes and monocyte-derived macrophages. J. Immunol. 2003, 170, 6209–6216. [Google Scholar] [CrossRef] [PubMed]
- Cesta, M.F. Normal structure, function, and histology of the spleen. Toxicol. Pathol. 2006, 34, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Czaja, A.J. Review article: Chemokines as orchestrators of autoimmune hepatitis and potential therapeutic targets. Aliment. Pharmacol. Ther. 2014, 40, 261–279. [Google Scholar] [CrossRef] [PubMed]
- Harrington, L.E.; Hatton, R.D.; Mangan, P.R.; Turner, H.; Murphy, T.L.; Murphy, K.M.; Weaver, C.T. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat. Immunol. 2005, 6, 1123–1132. [Google Scholar] [CrossRef] [PubMed]
- Dardalhon, V.; Korn, T.; Kuchroo, V.K.; Anderson, A.C. Role of Th1 and Th17 cells in organ-specific autoimmunity. J. Autoimmun. 2008, 31, 252–256. [Google Scholar] [CrossRef] [PubMed]
- Brand, S. Crohn's disease: Th1, Th17 or both? The change of a paradigm: New immunological and genetic insights implicate Th17 cells in the pathogenesis of Crohn’s disease. Gut 2009, 58, 1152–1167. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Rhee, K.J.; Albesiano, E.; Rabizadeh, S.; Wu, X.; Yen, H.R.; Huso, D.L.; Brancati, F.L.; Wick, E.; McAllister, F.; et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat. Med. 2009, 15, 1016–1022. [Google Scholar] [CrossRef] [PubMed]
- Mocellin, S.; Rossi, C.R.; Pilati, P.; Nitti, D. Tumor necrosis factor, cancer and anticancer therapy. Cytokine Growth Factor Rev. 2005, 16, 35–53. [Google Scholar] [CrossRef] [PubMed]
- Benchetrit, F.; Ciree, A.; Vives, V.; Warnier, G.; Gey, A.; Sautès-Fridman, C.; Fossiez, F.; Haicheur, N.; Fridman, W.H.; Tartour, E. Interleukin-17 inhibits tumor cell growth by means of a T-cell-dependent mechanism. Blood 2002, 99, 2114–2121. [Google Scholar] [CrossRef] [PubMed]
- Zamarron, B.F.; Chen, W. Dual roles of immune cells and their factors in cancer development and progression. Int. J. Biol. Sci. 2011, 7, 651–658. [Google Scholar] [CrossRef] [PubMed]
- Enzler, T.; Gillessen, S.; Manis, J.P.; Ferguson, D.; Fleming, J.; Alt, F.W.; Mihm, M.; Dranoff, G. Deficiencies of GM-CSF and interferon gamma link inflammation and cancer. J. Exp. Med. 2003, 197, 1213–1219. [Google Scholar] [CrossRef] [PubMed]
- Racanelli, V.; Rehermann, B. The liver as an immunological organ. Hepatology 2006, 43, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Mackay, I.R. Hepatoimmunology: A perspective. Immunol. Cell Biol. 2002, 80, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Laskin, D.L. Macrophages and inflammatory mediators in chemical toxicity: A battle of forces. Chem. Res. Toxicol. 2009, 22, 1376–1385. [Google Scholar] [CrossRef] [PubMed]
- Lucas, M.; Stuart, L.M.; Savill, J.; Lacy-Hulbert, A. Apoptotic cells and innate immune stimuli combine to regulate macrophage cytokine secretion. J. Immunol. 2003, 171, 2610–2615. [Google Scholar] [CrossRef] [PubMed]
- Huynh, M.L.; Fadok, V.A.; Henson, P.M. Phosphatidylserine-dependent ingestion of apoptotic cells promotes TGF-beta1 secretion and the resolution of inflammation. J. Clin. Investig. 2002, 109, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Fadok, V.A.; Bratton, D.L.; Konowal, A.; Freed, P.W.; Westcott, J.Y.; Henson, P.M. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J. Clin. Investig. 1998, 101, 890–898. [Google Scholar] [CrossRef] [PubMed]
- Moran, J.M.; Buller, R.M.; McHowat, J.; Turk, J.; Wohltmann, M.; Gross, R.W.; Corbett, J.A. Genetic and pharmacologic evidence that calcium-independent phospholipase A2β regulates virus-induced inducible nitric-oxide synthase expression by macrophages. J. Biol. Chem. 2005, 280, 28162–28168. [Google Scholar] [CrossRef] [PubMed]
- Daigle, I.; Rückert, B.; Schnetzler, G.; Simon, H.U. Induction of the IL-10 gene via the fas receptor in monocytes—An anti-inflammatory mechanism in the absence of apoptosis. Eur. J. Immunol. 2000, 30, 2991–2997. [Google Scholar] [CrossRef]
- Fickert, P.; Trauner, M.; Fuchsbichler, A.; Zollner, G.; Wagner, M.; Marschall, H.U.; Zatloukal, K.; Denk, H. Oncosis represents the main type of cell death in mouse models of cholestasis. J. Hepatol. 2005, 42, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Bahjat, F.R.; Dharnidharka, V.R.; Fukuzuka, K.; Morel, L.; Crawford, J.M.; Clare-Salzler, M.J.; Moldawer, L.L. Reduced susceptibility of nonobese diabetic mice to TNF-α and d-galactosamine-mediated hepatocellular apoptosis and lethality. J. Immunol. 2000, 165, 6559–6567. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, F.; Pontisso, P.; Pettenazzo, E.; Benevegnù, L.; Vario, A.; Chemello, L.; Alberti, A.; Valente, M. Liver cell apoptosis in chronic hepatitis C correlates with histological but not biochemical activity or serum HCV-RNA levels. Hepatology 2000, 31, 1153–1159. [Google Scholar] [CrossRef] [PubMed]
- Tinel, M.; Berson, A.; Vadrot, N.; Descatoire, V.; Grodet, A.; Feldmann, G.; Thénot, J.P.; Pessayre, D. Subliminal Fas stimulation increases the hepatotoxicity of acetaminophen and bromobenzene in mice. Hepatology 2004, 39, 655–666. [Google Scholar] [CrossRef] [PubMed]
- Cullen, S.P.; Henry, C.M.; Kearney, C.J.; Logue, S.E.; Feoktistova, M.; Tynan, G.A.; Lavelle, E.C.; Leverkus, M.; Martin, S.J. Fas/CD95-induced chemokines can serve as "find-me" signals for apoptotic cells. Mol. Cell 2013, 49, 1034–1048. [Google Scholar] [CrossRef] [PubMed]
- Vergani, D.; Mieli-Vergani, G. Aetiopathogenesis of autoimmune hepatitis. World J. Gastroenterol. 2008, 14, 3306–3312. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Huang, J.; Liu, Y.; Ai, G.; Yan, W.; Wang, X.; Ning, Q. IL-17 contributes to autoimmune hepatitis. J. Huazhong Univ. Sci. Technol. Med. Sci. 2010, 30, 443–446. [Google Scholar] [CrossRef] [PubMed]
- Ward, J.M. Lymphomas and leukemias in mice. Exp. Toxicol. Pathol. 2006, 57, 377–381. [Google Scholar] [CrossRef] [PubMed]
- Ward, J.M.; Anver, M.R.; Haines, D.C.; Benveniste, R.E. Chronic active hepatitis in mice caused by Helicobacter hepaticus. Am. J. Pathol. 1994, 145, 959–968. [Google Scholar] [PubMed]
- Fox, J.G.; Ge, Z.; Whary, M.T.; Erdman, S.E.; Horwitz, B.H. Helicobacter hepaticus infection in mice: Models for understanding lower bowel inflammation and cancer. Mucosal Immunol. 2011, 4, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Roshak, A.K.; Capper, E.A.; Stevenson, C.; Eichman, C.; Marshall, L.A. Human calcium-independent phospholipase A2 mediates lymphocyte proliferation. J. Biol. Chem. 2000, 275, 35692–35698. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Wilkins, P.; Hu, W.; Murthy, K.S.; Chen, J.; Lee, Z.; Oyesanya, R.; Wu, J.; Barbour, S.E.; Fang, X. Inhibition of calcium-independent phospholipase A2 suppresses proliferation and tumorigenicity of ovarian carcinoma cells. Biochem. J. 2007, 406, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Street, S.E.; Trapani, J.A.; MacGregor, D.; Smyth, M.J. Suppression of lymphoma and epithelial malignancies effected by interferon gamma. J. Exp. Med. 2002, 196, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Hilbert, D.M.; Kopf, M.; Mock, B.A.; Köhler, G.; Rudikoff, S. Interleukin 6 is essential for in vivo development of B lineage neoplasms. J. Exp. Med. 1995, 182, 243–248. [Google Scholar] [CrossRef] [PubMed]
- Kogan, S.C.; Ward, J.M.; Anver, M.R.; Berman, J.J.; Brayton, C.; Cardiff, R.D.; Carter, J.S.; de Coronado, S.; Downing, J.R.; Fredrickson, T.N.; et al. Hematopathology subcommittee of the Mouse Models of Human Cancers Consortium. Bethesda proposals for classification of nonlymphoid hematopoietic neoplasms in mice. Blood 2002, 100, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Lahaie, R.G.; Chiba, N.; Fallone, C. Meeting review—Helicobacter pylori: Basic mechanisms to clinical cure 2000. Can. J. Gastroenterol. 2000, 14, 856–861. [Google Scholar] [PubMed]
- Ward, J.M.; Nikolov, N.P.; Tschetter, J.R.; Kopp, J.B.; Gonzales, F.J.; Kimura, S.; Siegel, R.M. Progressive glomerulonephritis and histiocytic sarcoma associated with macrophage functional defects in CYP1B1-deficient mice. Toxicol. Pathol. 2004, 32, 710–718. [Google Scholar] [CrossRef] [PubMed]
- Henry, J.B. Clinical Diagnosis and Management by Laboratory Methods; Henry, J.B., Ed.; Saunders Company: Philadelphia, PA, USA, 1979; Volume 1, p. 60. [Google Scholar]
- Thavasu, P.W.; Longhurst, S.; Joel, S.P.; Slevin, M.L.; Balkwill, F.R. Measuring cytokine levels in blood. Importance of anticoagulants, processing, and storage conditions. J. Immunol. Methods 1992, 153, 115–124. [Google Scholar] [CrossRef]
- Schreiber, R.; Taschler, U.; Wolinski, H.; Seper, A.; Tamegger, S.N.; Graf, M.; Kohlwein, S.D.; Haemmerle, G.; Zimmermann, R.; Zechner, R.; et al. Esterase 22 and β-glucuronidase hydrolyze retinoids in mouse liver. J. Lipid Res. 2009, 50, 2514–2523. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Zhang, S.; Xu, L.; Song, B.; Huang, G.; Lu, J.; Guan, S. Suppression of T-cell activation in vitro and in vivo by cordycepin from Cordycepsmilitaris. J. Surg. Res. 2013, 185, 912–922. [Google Scholar] [CrossRef] [PubMed]
- Tian, Z.; Sun, R.; Wie, H.; Gao, B. Impaired natural killer (NK) cell activity in leptin receptor deficient mice: Leptin as a critical regulator in NK cell development and activation. Biochem. Biophys. Res. Commun. 2002, 298, 297–302. [Google Scholar] [CrossRef]
- Elliott, M.R.; Ravichandran, K.S. Clearance of apoptotic cells: Implications in health and disease. J. Cell Biol. 2010, 189, 1059–1070. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Inhoffen, J.; Tuma-Kellner, S.; Straub, B.; Stremmel, W.; Chamulitrat, W. Deficiency of iPLA2β Primes Immune Cells for Proinflammation: Potential Involvement in Age-Related Mesenteric Lymph Node Lymphoma. Cancers 2015, 7, 2427-2442. https://doi.org/10.3390/cancers7040901
Inhoffen J, Tuma-Kellner S, Straub B, Stremmel W, Chamulitrat W. Deficiency of iPLA2β Primes Immune Cells for Proinflammation: Potential Involvement in Age-Related Mesenteric Lymph Node Lymphoma. Cancers. 2015; 7(4):2427-2442. https://doi.org/10.3390/cancers7040901
Chicago/Turabian StyleInhoffen, Johannes, Sabine Tuma-Kellner, Beate Straub, Wolfgang Stremmel, and Walee Chamulitrat. 2015. "Deficiency of iPLA2β Primes Immune Cells for Proinflammation: Potential Involvement in Age-Related Mesenteric Lymph Node Lymphoma" Cancers 7, no. 4: 2427-2442. https://doi.org/10.3390/cancers7040901