
Complement Activation and Up-Regulated Expression of Anaphylatoxin C3a/C3aR in Glioblastoma: Deciphering the Links with TGF-β and VEGF
 , , , and
, , , and
Abstract
:Simple Summary
Abstract
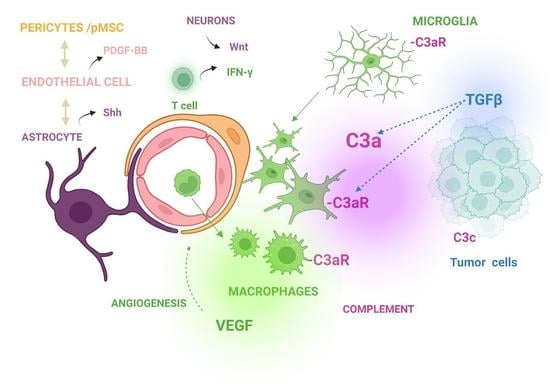
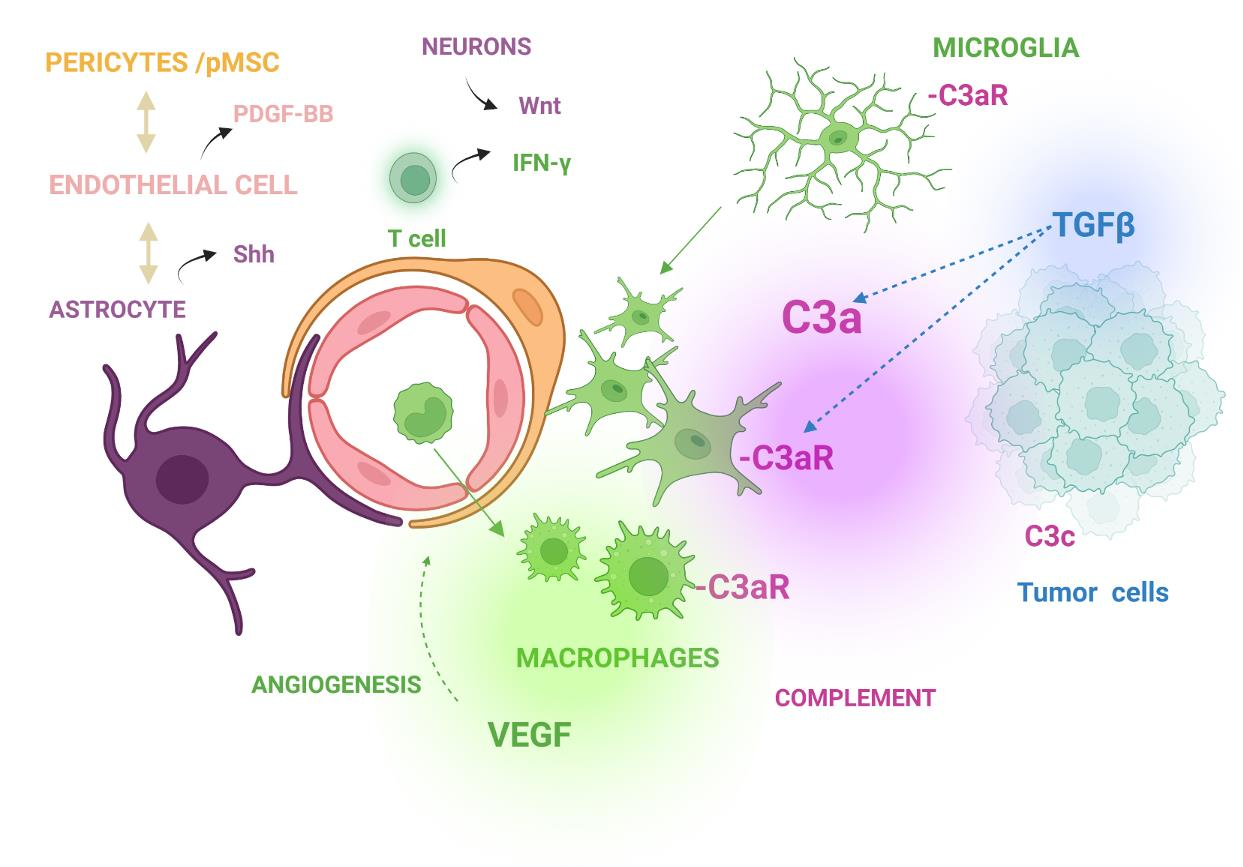
1. Introduction
2. Materials and Methods
2.1. Tissue Samples
2.2. Immunohistochemistry Experiments
2.3. Immunofluorescence Experiments
2.4. Cells and Stimulatory Conditions
2.5. Quantitative RT-PCR Analysis
2.6. Statistical Analysis
3. Results
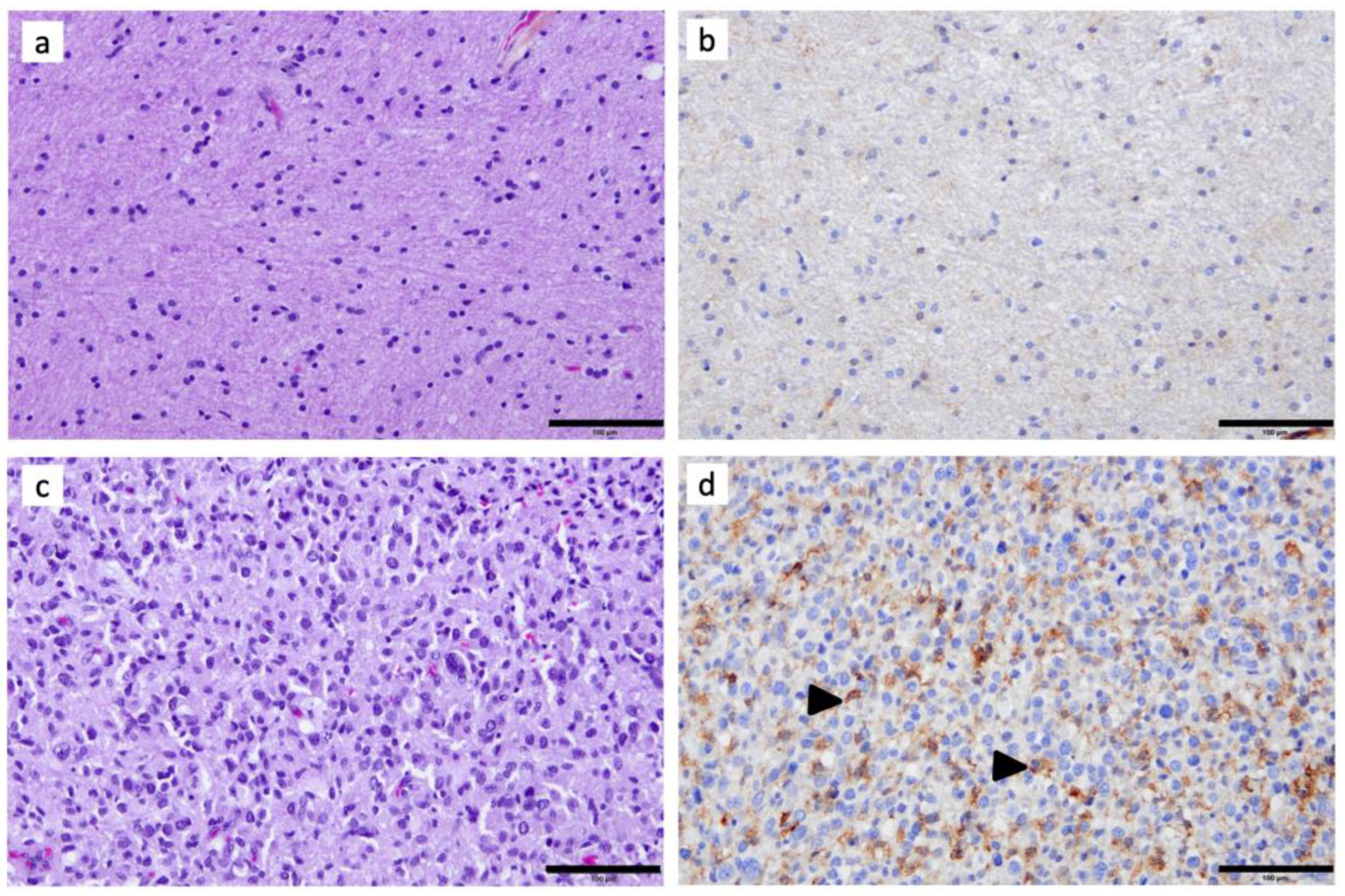
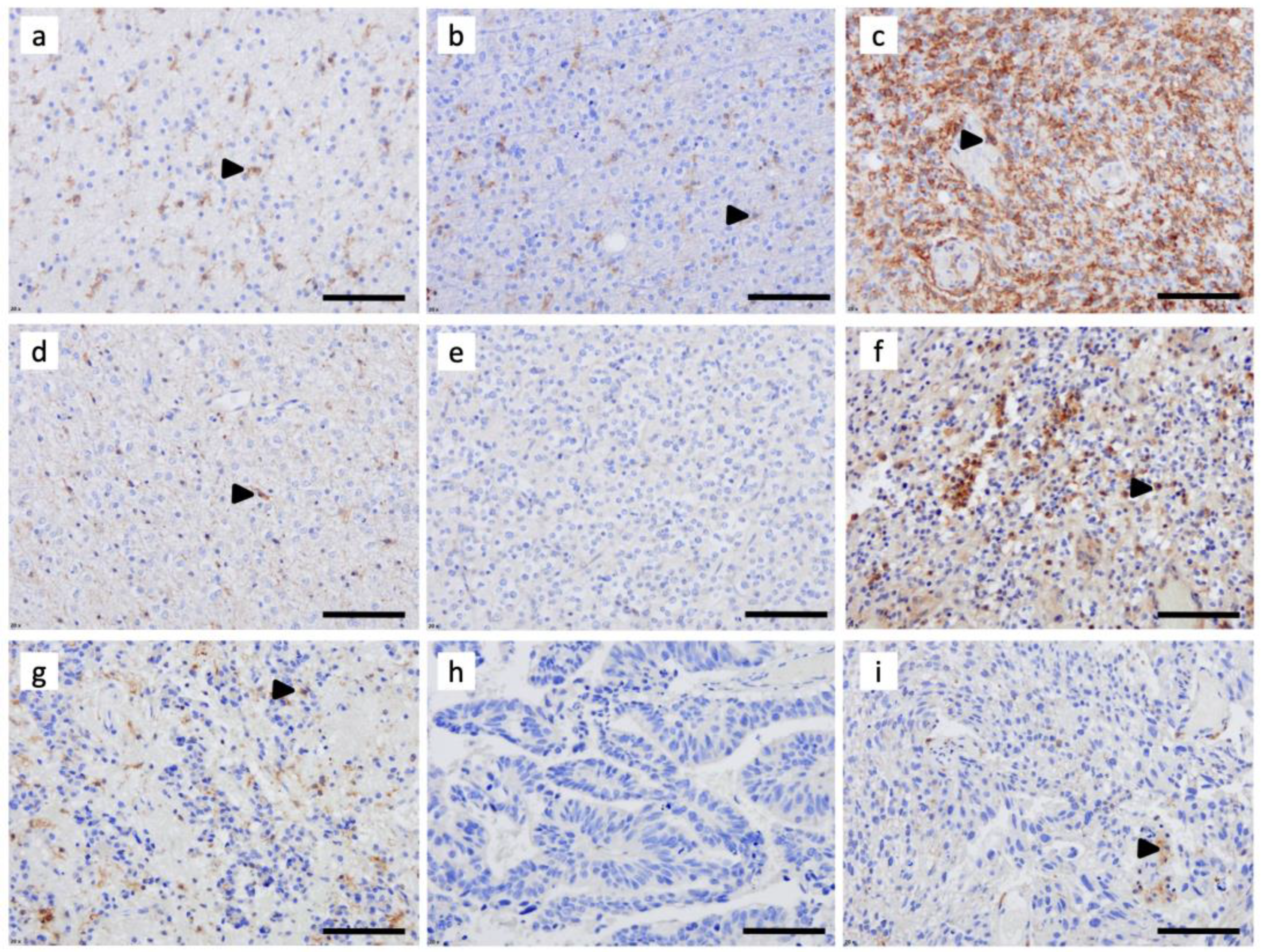
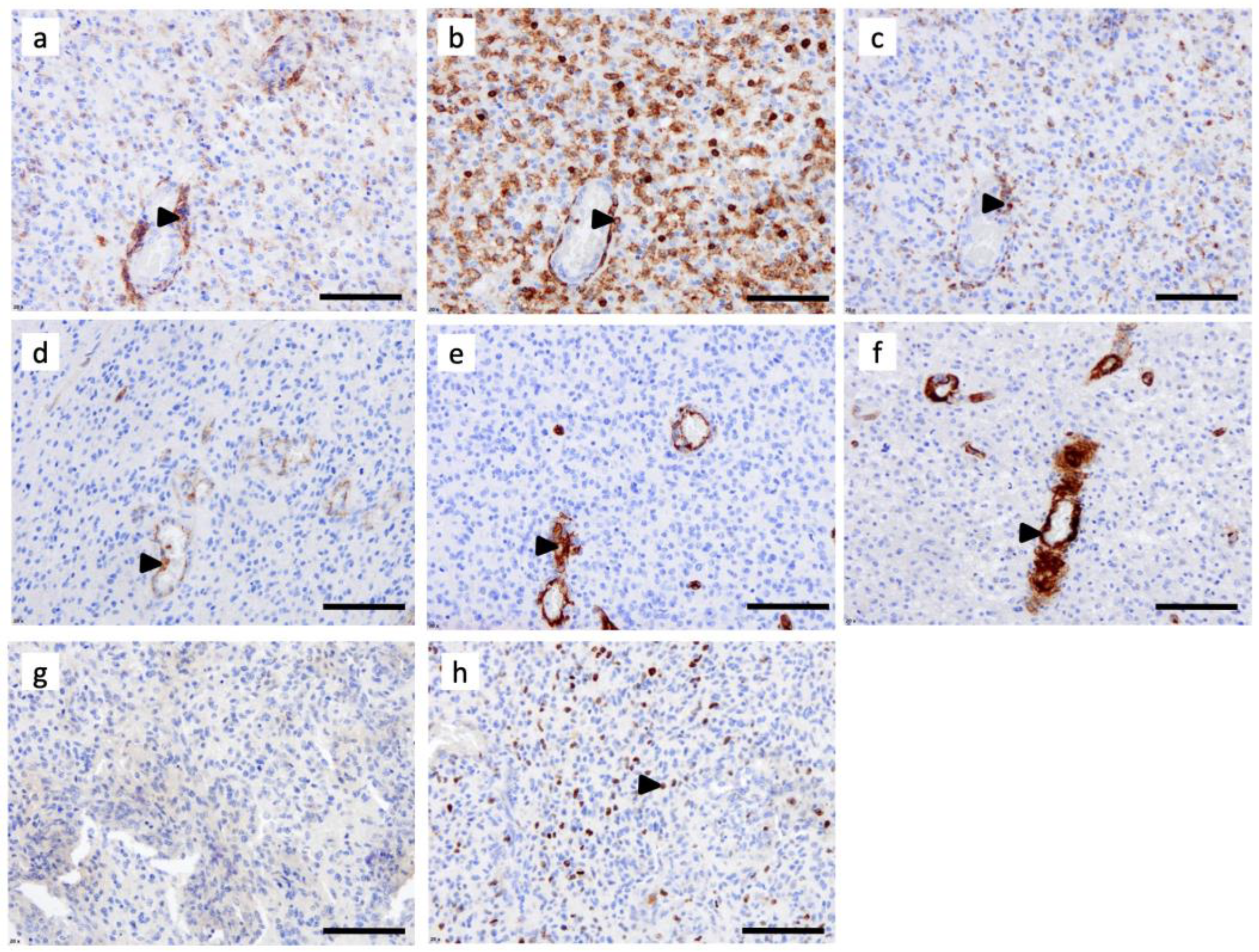
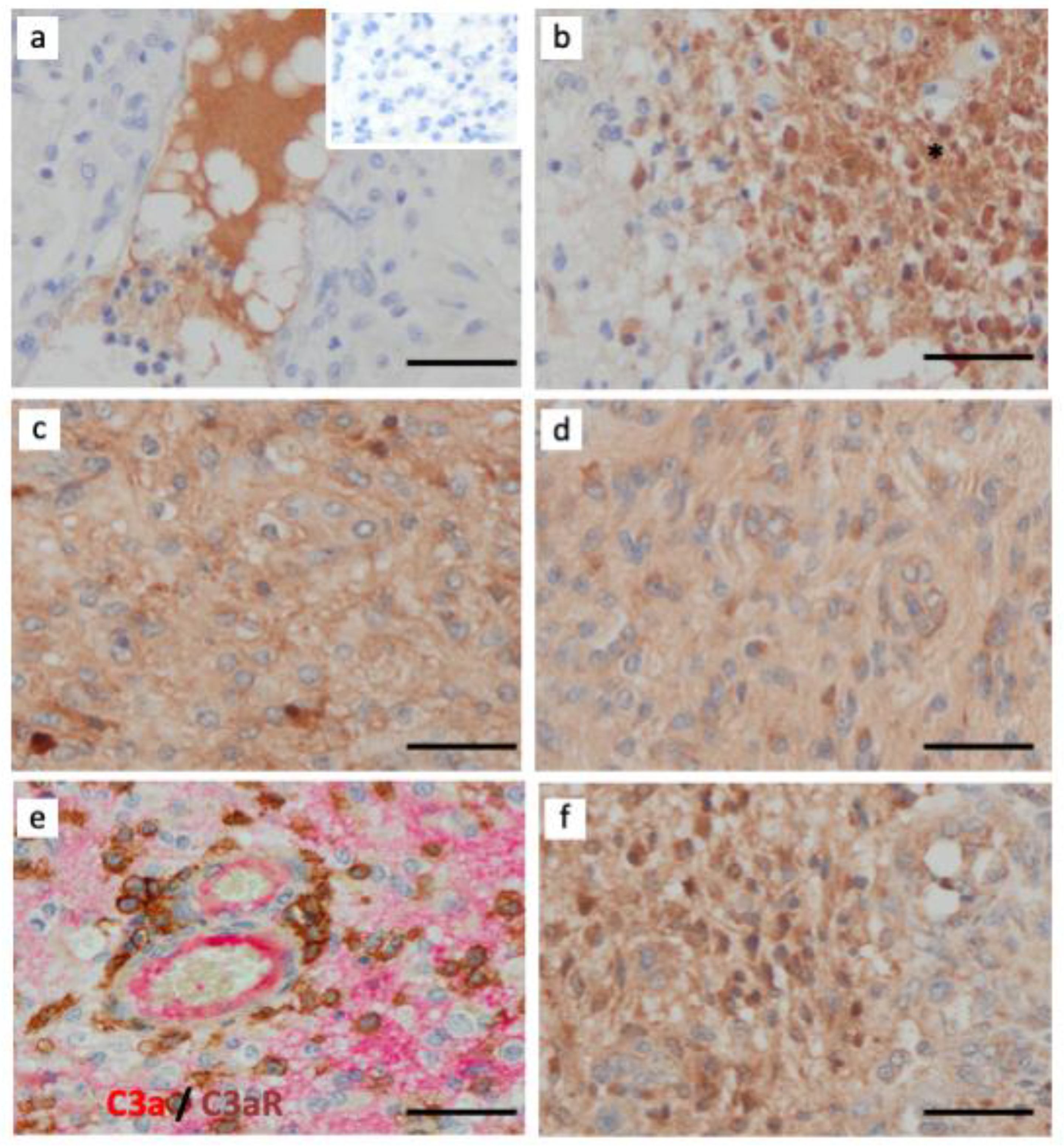
3.1. C3aR Is Highly Expressed in Glioblastoma, IDHwt and in Astrocytoma, IDH-Mutant, Grade 4
3.2. C3aR Is Less Expressed in Other Brain Tumors
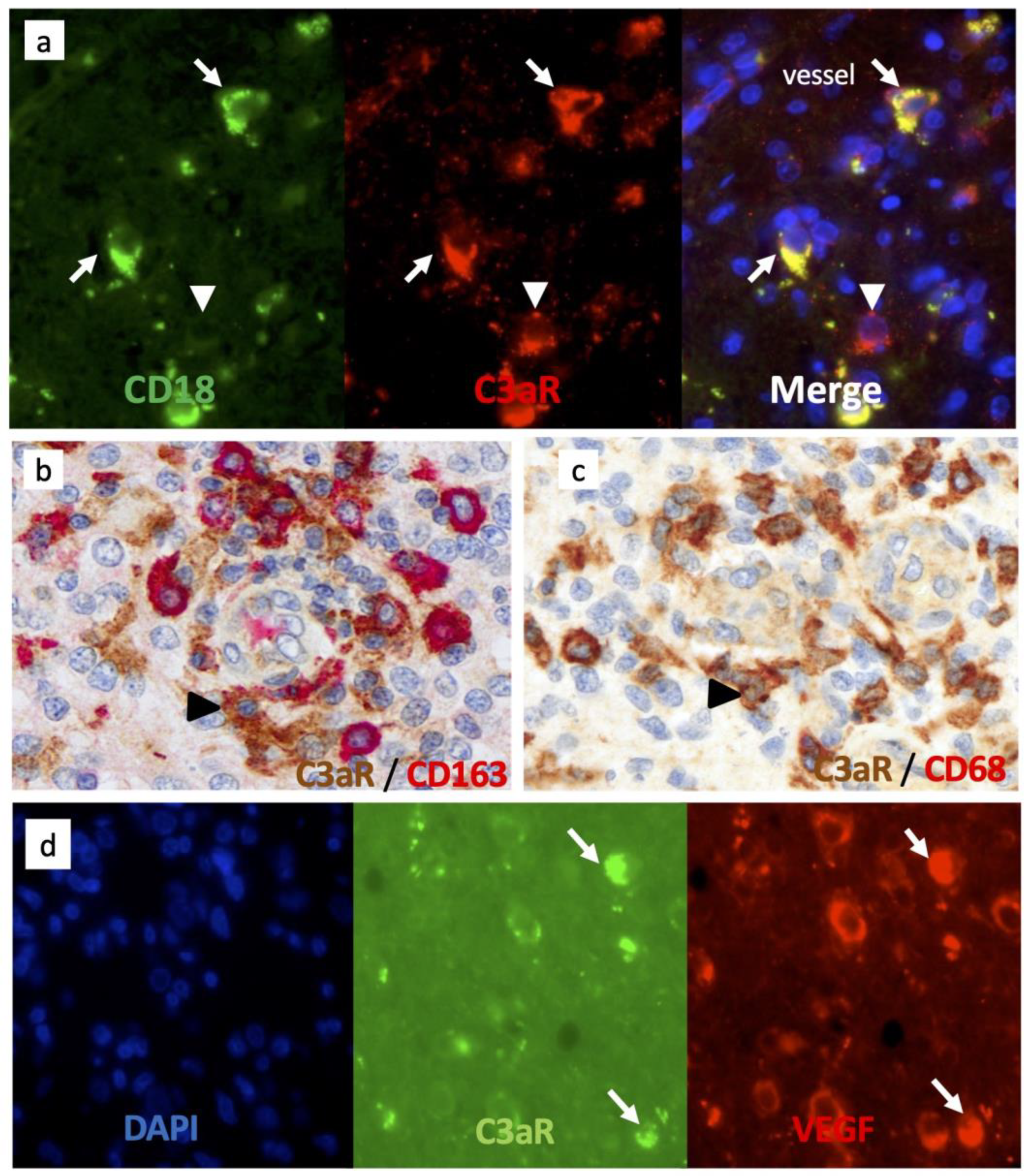
3.3. Tumor-Associated Macrophages Express High Levels of C3aR in GBM
3.4. C3aR Tumor-Associated Macrophages Also Express VEGF in GBM
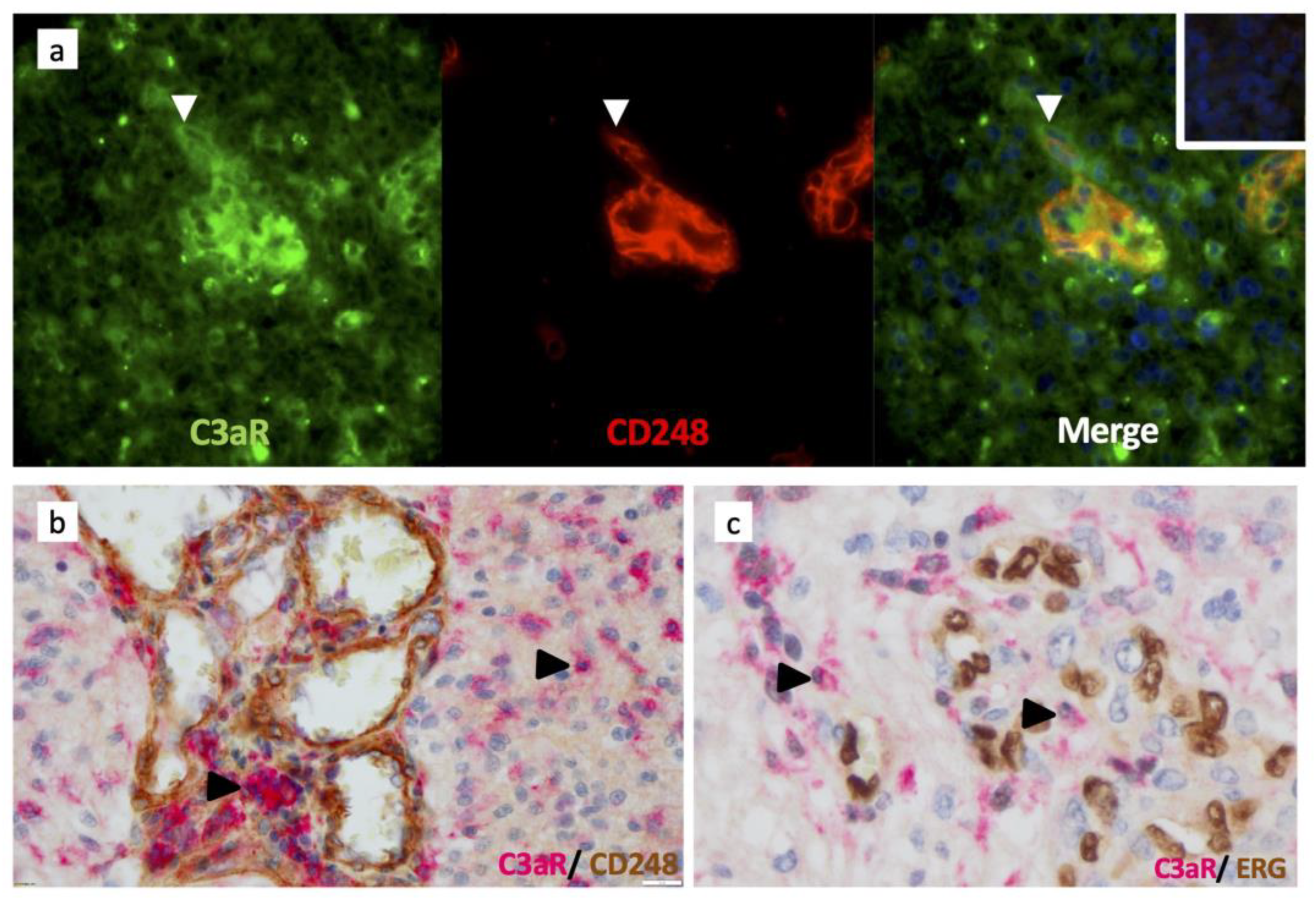
3.5. C3aR Is Not Expressed in Perivascular Mesenchymal Cells/Pericytes or Endothelial Cells
3.6. The Alternative Complement Pathway Is Activated to Generate C3a
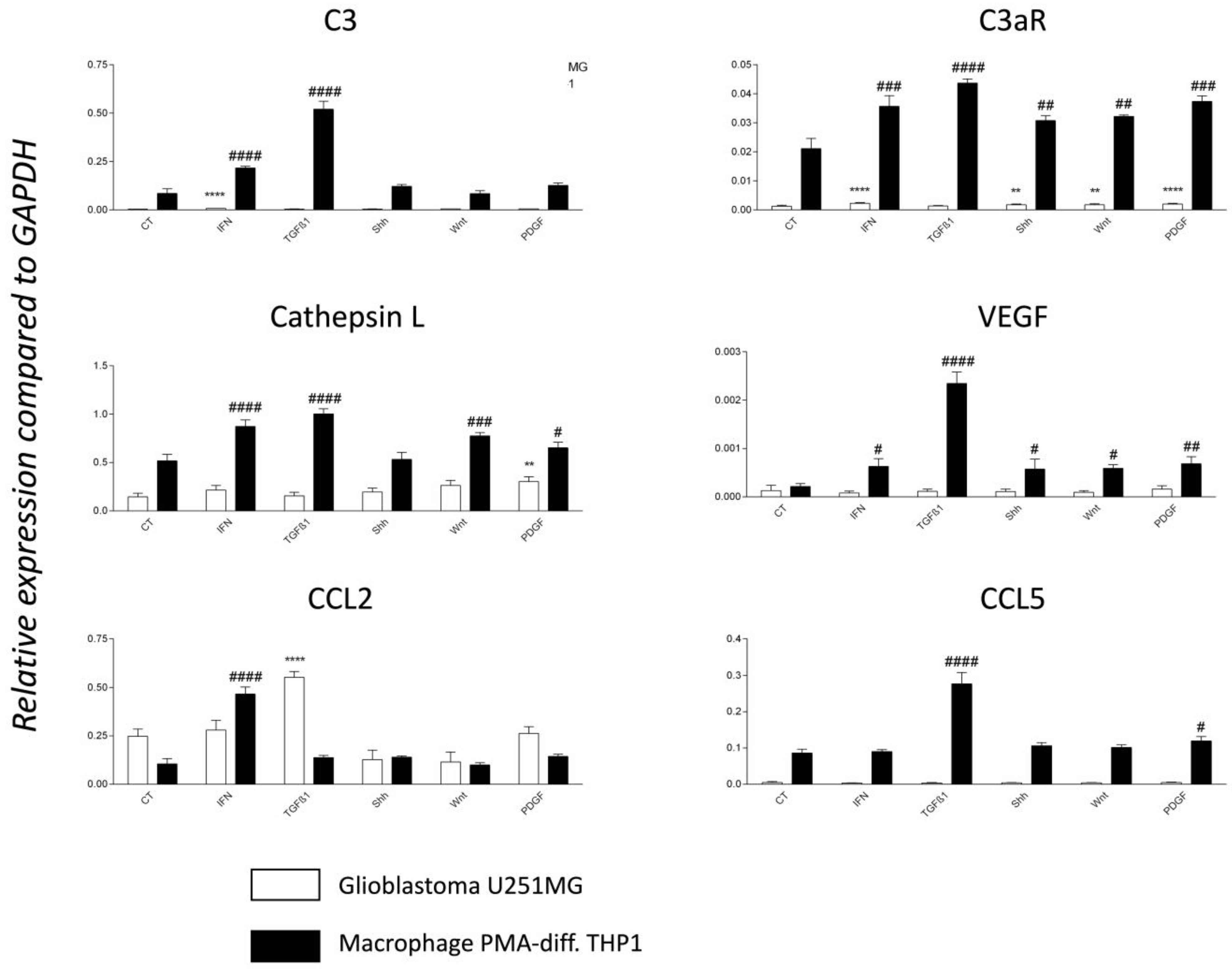
3.7. TGF-1β Is a Major Regulator of C3 and C3aR Expression in PMA-Differentiated THP1 Cell Line In Vitro
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Walport, M.J. Advances in Immunology: Complement (First of Two Parts). N. Engl. J. Med. 2001, 344, 1058–1066. [Google Scholar] [CrossRef] [PubMed]
- Gasque, P. Complement: A Unique Innate Immune Sensor for Danger Signals. Mol. Immunol. 2004, 41, 1089–1098. [Google Scholar] [CrossRef] [PubMed]
- Kolev, M.; Le Friec, G.; Kemper, C. Complement—Tapping into New Sites and Effector Systems. Nat. Rev. Immunol. 2014, 14, 811–820. [Google Scholar] [CrossRef]
- Ricklin, D.; Hajishengallis, G.; Yang, K.; Lambris, J.D. Complement: A Key System for Immune Surveillance and Homeostasis. Nat. Immunol. 2010, 11, 785–797. [Google Scholar] [CrossRef]
- Aderem, A.; Underhill, D.M. Mechanisms of Phagocytosis in Macrophages. Annu. Rev. Immunol. 1999, 17, 593–623. [Google Scholar] [CrossRef]
- Xia, Y.; Vetvicka, V.; Yan, J.; Hanikyrova, M.; Mayadas, T.; Ross, G.D. The Beta-Glucan-Binding Lectin Site of Mouse CR3 (CD11b/CD18) and Its Function in Generating a Primed State of the Receptor That Mediates Cytotoxic Activation in Response to IC3b-Opsonized Target Cells. J. Immunol. 1999, 162, 2281–2290. [Google Scholar] [CrossRef] [PubMed]
- Meri, S.; Morgan, B.; Davies, A.; Daniels, R.; Olavesen, M.; Waldmann, H.; Lachmann, P. Human Protectin (Cd59), an 18,000-20,000 Mw Complement Lysis Restricting Factor, Inhibits C5b-8 Catalyzed Insertion of C9 into Lipid Bilayers. Immunology 1990, 71, 1–9. [Google Scholar]
- Zipfel, P.F.; Skerka, C. Complement Regulators and Inhibitory Proteins. Nat. Rev. Immunol. 2009, 9, 729–740. [Google Scholar] [CrossRef]
- Reis, E.S.; Mastellos, D.C.; Ricklin, D.; Mantovani, A.; Lambris, J.D. Complement in Cancer: Untangling an Intricate Relationship. Nat. Rev. Immunol. 2018, 18, 5–18. [Google Scholar] [CrossRef]
- Klos, A.; Tenner, A.J.; Johswich, K.-O.; Ager, R.R.; Reis, E.S.; Köhl, J. The Role of the Anaphylatoxins in Health and Disease. Mol. Immunol. 2009, 46, 2753–2766. [Google Scholar] [CrossRef]
- Coulthard, L.G.; Woodruff, T.M. Is the Complement Activation Product C3a a Proinflammatory Molecule? Re-Evaluating the Evidence and the Myth. J. Immunol. 2015, 194, 3542–3548. [Google Scholar] [CrossRef] [PubMed]
- Gerard, N.P.; Gerard, C. Complement in Allergy and Asthma. Curr. Opin. Immunol. 2002, 14, 705–708. [Google Scholar] [CrossRef] [PubMed]
- Hugli, T.; Gerard, C.; Kawahara, M.; Scheetz, M.; Barton, R.; Briggs, S.; Koppel, G.; Russell, S. Isolation of 3 Separate Anaphylatoxins from Complement-Activated Human-Serum. Mol. Cell. Biochem. 1981, 41, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Elward, K.; Gasque, P. “Eat Me” and “Don’t Eat Me” Signals Govern the Innate Immune Response and Tissue Repair in the CNS: Emphasis on the Critical Role of the Complement System. Mol. Immunol. 2003, 40, 85–94. [Google Scholar] [CrossRef]
- Morgan, B.P.; Gasque, P. Expression of Complement in the Brain: Role in Health and Disease. Immunol. Today 1996, 17, 461–466. [Google Scholar] [CrossRef]
- Stephan, A.H.; Barres, B.A.; Stevens, B. The Complement System: An Unexpected Role in Synaptic Pruning During Development and Disease. In Annual Review of Neuroscience; Hyman, S.E., Ed.; Annual Reviews: Palo Alto, CA, USA, 2012; Volume 35, pp. 369–389. ISBN 978-0-8243-2435-3. [Google Scholar]
- Farina, C.; Aloisi, F.; Meinl, E. Astrocytes Are Active Players in Cerebral Innate Immunity. Trends Immunol. 2007, 28, 138–145. [Google Scholar] [CrossRef]
- Akiyama, H.; Barger, S.; Barnum, S.; Bradt, B.; Bauer, J.; Cole, G.M.; Cooper, N.R.; Eikelenboom, P.; Emmerling, M.; Fiebich, B.L.; et al. Inflammation and Alzheimer’s Disease. Neurobiol. Aging 2000, 21, 383–421. [Google Scholar] [CrossRef]
- Nataf, S.; Stahel, P.F.; Davoust, N.; Barnum, S.R. Complement Anaphylatoxin Receptors on Neurons: New Tricks for Old Receptors? Trends Neurosci. 1999, 22, 397–402. [Google Scholar] [CrossRef]
- Shellard, A.; Mayor, R. Chemotaxis during Neural Crest Migration. Semin. Cell Dev. Biol. 2016, 55, 111–118. [Google Scholar] [CrossRef]
- Gasque, P.; Dean, Y.D.; McGreal, E.P.; VanBeek, J.; Morgan, B.P. Complement Components of the Innate Immune System in Health and Disease in the CNS. Immunopharmacology 2000, 49, 171–186. [Google Scholar] [CrossRef]
- Schraufstatter, I.; Khaldoyanidi, S.; DiScipio, R. Complement Activation in the Context of Stem Cells and Tissue Repair. World J. Stem Cells 2015, 7, 1090–1108. [Google Scholar] [CrossRef] [PubMed]
- van der Vlis, T.A.M.B.; Kros, J.M.; Mustafa, D.A.M.; van Wijck, R.T.A.; Ackermans, L.; van Hagen, P.M.; van der Spek, P.J. The Complement System in Glioblastoma Multiforme. Acta Neuropathol. Commun. 2018, 6, 91. [Google Scholar] [CrossRef]
- Afshar-Kharghan, V. The Role of the Complement System in Cancer. J. Clin. Investig. 2017, 127, 780–789. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.S.; Vasquez, H.G.; Rupaimoole, R.; Pradeep, S.; Wu, S.; Zand, B.; Han, H.-D.; Rodriguez-Aguayo, C.; Bottsford-Miller, J.; Huang, J.; et al. Autocrine Effects of Tumor-Derived Complement. Cell Rep 2014, 6, 1085–1095. [Google Scholar] [CrossRef] [PubMed]
- Wen, P.Y.; Kesari, S. Malignant Gliomas in Adults. N. Engl. J. Med. 2008, 359, 492–507. [Google Scholar] [CrossRef] [PubMed]
- Pyonteck, S.M.; Akkari, L.; Schuhmacher, A.J.; Bowman, R.L.; Sevenich, L.; Quail, D.F.; Olson, O.C.; Quick, M.L.; Huse, J.T.; Teijeiro, V.; et al. CSF-1R Inhibition Alters Macrophage Polarization and Blocks Glioma Progression. Nat. Med. 2013, 19, 1264–1272. [Google Scholar] [CrossRef] [PubMed]
- Lewis, C.E.; Pollard, J.W. Distinct Role of Macrophages in Different Tumor Microenvironments. Cancer Res. 2006, 66, 605–612. [Google Scholar] [CrossRef] [PubMed]
- Solinas, G.; Germano, G.; Mantovani, A.; Allavena, P. Tumor-Associated Macrophages (TAM) as Major Players of the Cancer-Related Inflammation. J. Leukoc. Biol. 2009, 86, 1065–1073. [Google Scholar] [CrossRef]
- Davidson, S.; Efremova, M.; Riedel, A.; Mahata, B.; Pramanik, J.; Huuhtanen, J.; Kar, G.; Vento-Tormo, R.; Hagai, T.; Chen, X.; et al. Single-Cell RNA Sequencing Reveals a Dynamic Stromal Niche That Supports Tumor Growth. Cell Rep. 2020, 31, 107628. [Google Scholar] [CrossRef]
- Consonni, F.M.; Bleve, A.; Totaro, M.G.; Storto, M.; Kunderfranco, P.; Termanini, A.; Pasqualini, F.; Ali, C.; Pandolfo, C.; Sgambelluri, F.; et al. Heme Catabolism by Tumor-Associated Macrophages Controls Metastasis Formation. Nat. Immunol. 2021, 22, 595–606. [Google Scholar] [CrossRef]
- Magrini, E.; Di Marco, S.; Mapelli, S.N.; Perucchini, C.; Pasqualini, F.; Donato, A.; de la Luz Guevara Lopez, M.; Carriero, R.; Ponzetta, A.; Colombo, P.; et al. Complement Activation Promoted by the Lectin Pathway Mediates C3aR-Dependent Sarcoma Progression and Immunosuppression. Nat. Cancer 2021, 2, 218–232. [Google Scholar] [CrossRef] [PubMed]
- Nabizadeh, J.A.; Manthey, H.D.; Steyn, F.J.; Chen, W.; Widiapradja, A.; Md Akhir, F.N.; Boyle, G.M.; Taylor, S.M.; Woodruff, T.M.; Rolfe, B.E. The Complement C3a Receptor Contributes to Melanoma Tumorigenesis by Inhibiting Neutrophil and CD4+ T Cell Responses. J. Immunol. 2016, 196, 4783–4792. [Google Scholar] [CrossRef]
- Qiang, L.; Yang, Y.; Ma, Y.-J.; Chen, F.-H.; Zhang, L.-B.; Liu, W.; Qi, Q.; Lu, N.; Tao, L.; Wang, X.-T.; et al. Isolation and Characterization of Cancer Stem like Cells in Human Glioblastoma Cell Lines. Cancer Lett. 2009, 279, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Gasque, P.; Singhrao, S.K.; Neal, J.W.; Wang, P.; Sayah, S.; Fontaine, M.; Morgan, B.P. The Receptor for Complement Anaphylatoxin C3a Is Expressed by Myeloid Cells and Nonmyeloid Cells in Inflamed Human Central Nervous System: Analysis in Multiple Sclerosis and Bacterial Meningitis. J. Immunol. 1998, 160, 3543–3554. [Google Scholar] [CrossRef] [PubMed]
- Monsinjon, T.; Gasque, P.; Chan, P.; Ischenko, A.; Brady, J.J.; Fontaine, M.C. Regulation by Complement C3a and C5a Anaphylatoxins of Cytokine Production in Human Umbilical Vein Endothelial Cells. FASEB J. 2003, 17, 1003–1014. [Google Scholar] [CrossRef]
- Hambardzumyan, D.; Gutmann, D.H.; Kettenmann, H. The Role of Microglia and Macrophages in Glioma Maintenance and Progression. Nat. Neurosci. 2016, 19, 20–27. [Google Scholar] [CrossRef]
- Legler, D.F.; Loetscher, M.; Jones, S.A.; Dahinden, C.A.; Arock, M.; Moser, B. Expression of High- and Low-Affinity Receptors for C3a on the Human Mast Cell Line, HMC-1. Eur. J. Immunol. 1996, 26, 753–758. [Google Scholar] [CrossRef]
- Schraufstatter, I.U.; Discipio, R.G.; Zhao, M.; Khaldoyanidi, S.K. C3a and C5a Are Chemotactic Factors for Human Mesenchymal Stem Cells, Which Cause Prolonged ERK1/2 Phosphorylation. J. Immunol. 2009, 182, 3827–3836. [Google Scholar] [CrossRef]
- Afzali, B.; Kemper, C. Fibroblast Tissue Priming-Not so Nice to C You! Immunity 2021, 54, 847–850. [Google Scholar] [CrossRef]
- Rao, J.S. Molecular Mechanisms of Glioma Invasiveness: The Role of Proteases. Nat. Rev. Cancer 2003, 3, 489–501. [Google Scholar] [CrossRef]
- Zhao, Z.; Nelson, A.R.; Betsholtz, C.; Zlokovic, B.V. Establishment and Dysfunction of the Blood-Brain Barrier. Cell 2015, 163, 1064–1078. [Google Scholar] [CrossRef]
- Zhu, H.; Yu, X.; Zhang, S.; Shu, K. Targeting the Complement Pathway in Malignant Glioma Microenvironments. Front. Cell Dev. Biol. 2021, 9, 657472. [Google Scholar] [CrossRef] [PubMed]
- Makela, K.; Helen, P.; Haapasalo, H.; Paavonen, T. Complement Activation in Astrocytomas: Deposition of C4d and Patient Outcome. BMC Cancer 2012, 12, 565. [Google Scholar] [CrossRef]
- Bouwens, T.A.M.; Trouw, L.A.; Veerhuis, R.; Dirven, C.M.F.; Lamfers, M.L.M.; Al-Khawaja, H. Complement Activation in Glioblastoma Multiforme Pathophysiology: Evidence from Serum Levels and Presence of Complement Activation Products in Tumor Tissue. J. Neuroimmunol. 2015, 278, 271–276. [Google Scholar] [CrossRef]
- Mangogna, A.; Belmonte, B.; Agostinis, C.; Zacchi, P.; Iacopino, D.G.; Martorana, A.; Rodolico, V.; Bonazza, D.; Zanconati, F.; Kishore, U.; et al. Prognostic Implications of the Complement Protein C1q in Gliomas. Front. Immunol. 2019, 10, 2366. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-Cell RNA-Seq Highlights Intratumoral Heterogeneity in Primary Glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef] [PubMed]
- Lim, E.-J.; Kim, S.; Oh, Y.; Suh, Y.; Kaushik, N.; Lee, J.-H.; Lee, H.-J.; Kim, M.-J.; Park, M.-J.; Kim, R.-K.; et al. Crosstalk between GBM Cells and Mesenchymal Stemlike Cells Promotes the Invasiveness of GBM through the C5a/P38/ZEB1 Axis. Neuro-Oncology 2020, 22, 1452–1462. [Google Scholar] [CrossRef]
- Plate, K.; Breier, G.; Weich, H.; Risau, W. Vascular Endothelial Growth-Factor Is a Potential Tumor Angiogenesis Factor in Human Gliomas Invivo. Nature 1992, 359, 845–848. [Google Scholar] [CrossRef]
- Zhou, W.; Ke, S.Q.; Huang, Z.; Flavahan, W.; Fang, X.; Paul, J.; Wu, L.; Sloan, A.E.; McLendon, R.E.; Li, X.; et al. Periostin Secreted by Glioblastoma Stem Cells Recruits M2 Tumour-Associated Macrophages and Promotes Malignant Growth. Nat. Cell Biol. 2015, 17, 170–182. [Google Scholar] [CrossRef]
- Habermann, J.K.; Roblick, U.J.; Luke, B.T.; Prieto, D.A.; Finlay, W.J.J.; Podust, V.N.; Roman, J.M.; Oevermann, E.; Schiedeck, T.; Homann, N.; et al. Increased Serum Levels of Complement C3a Anaphylatoxin Indicate the Presence of Colorectal Tumors. Gastroenterology 2006, 131, 1020–1029. [Google Scholar] [CrossRef]
- Gasque, P.; Julen, N.; Ischenko, A.M.; Picot, C.; Mauger, C.; Chauzy, C.; Ripoche, J.; Fontaine, M. Expression of Complement Components of the Alternative Pathway by Glioma Cell Lines. J. Immunol. 1992, 149, 1381–1387. [Google Scholar] [CrossRef] [PubMed]
- Barnum, S.R.; Ishii, Y.; Agrawal, A.; Volanakis, J.E. Production and Interferon-Gamma-Mediated Regulation of Complement Component C2 and Factors B and D by the Astroglioma Cell Line U105-MG. Biochem. J. 1992, 287 Pt 2, 595–601. [Google Scholar] [CrossRef] [PubMed]
- Constam, D.; Philipp, J.; Malipiero, U.; Tendijke, P.; Schachner, M.; Fontana, A. Differential Expression of Transforming Growth Factor-Beta-1, Factor-Beta-2, and Factor-Beta-3 by Glioblastoma Cells, Astrocytes, and Microglia. J. Immunol. 1992, 148, 1404–1410. [Google Scholar] [CrossRef] [PubMed]
- Wesolowska, A.; Kwiatkowska, A.; Slomnicki, L.; Dembinski, M.; Master, A.; Sliwa, M.; Franciszkiewicz, K.; Chouaib, S.; Kaminska, B. Microglia-Derived TGF-Beta as an Important Regulator of Glioblastoma Invasion—An Inhibition of TGF-Beta-Dependent Effects by ShRNA against Human TGF-Beta Type II Receptor. Oncogene 2008, 27, 918–930. [Google Scholar] [CrossRef]
- Elward, K.; Griffiths, M.; Mizuno, M.; Harris, C.L.; Neal, J.W.; Morgan, B.P.; Gasque, P. CD46 Plays a Key Role in Tailoring Innate Immune Recognition of Apoptotic and Necrotic Cells. J. Biol. Chem. 2005, 280, 36342–36354. [Google Scholar] [CrossRef]
- Levicar, N.; Dewey, R.A.; Daley, E.; Bates, T.E.; Davies, D.; Kos, J.; Pilkington, G.J.; Lah, T.T. Selective Suppression of Cathepsin L by Antisense CDNA Impairs Human Brain Tumor Cell Invasion in Vitro and Promotes Apoptosis. Cancer Gene Ther. 2003, 10, 141–151. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, C.; Li, Y.; Miwa, T.; Liu, C.; Cui, W.; Song, W.-C.; Du, J. Complement C3a Signaling Facilitates Skeletal Muscle Regeneration by Regulating Monocyte Function and Trafficking. Nat. Commun. 2017, 8, 2078. [Google Scholar] [CrossRef]
- De Palma, M.; Venneri, M.A.; Galli, R.; Sergi, L.S.; Politi, L.S.; Sampaolesi, M.; Naldini, L. Tie2 Identifies a Hematopoietic Monocytes Required for Tumor Lineage of Proangiogenic Vessel Formation and a Mesenchymal Population of Pericyte Progenitors. Cancer Cell 2005, 8, 211–226. [Google Scholar] [CrossRef]
- Long, Q.; Cao, X.; Bian, A.; Li, Y. C3a Increases VEGF and Decreases PEDF MRNA Levels in Human Retinal Pigment Epithelial Cells. BioMed Res. Int. 2016, 2016, 6958752. [Google Scholar] [CrossRef]
- Ricklin, D.; Reis, E.S.; Mastellos, D.C.; Gros, P.; Lambris, J.D. Complement Component C3-The “Swiss Army Knife” of Innate Immunity and Host Defense. Immunol. Rev. 2016, 274, 33–58. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Sequence (5′-3′) | Reverse Sequence (5′-3′) |
|---|---|---|
| GAPDH | TGCGTCGCCAGCCGAG | AGTTAAAAGCAGCCCTGGTGA |
| CCL5/RANTES | TCCTCATTGCTACTGCCCTC | TCGGGTGACAAAGACGACTG |
| CCL2/MCP1 | CTGCTCATAGCAGCCACCTT | CTTGAAGATCACAGCTTCTTTGGG |
| C3 | TCAACCACAAGCTGCTACCC | CTGGCCCATGTTGACGAGTT |
| C3aR | AGACAGGACTCGTGGAGACA | CTCAGCAGAGAAAGACGCCA |
| Cathepsin L | CTGGCAACGAGAGCGTCTAC | CCATCCTTCTTCATTCATGCCG |
| VEGF-A | ACAACAAATGTGAATGCAGACCA | GAGGCTCCAGGGCATTAGAC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ah-Pine, F.; Malaterre-Septembre, A.; Bedoui, Y.; Khettab, M.; Neal, J.W.; Freppel, S.; Gasque, P. Complement Activation and Up-Regulated Expression of Anaphylatoxin C3a/C3aR in Glioblastoma: Deciphering the Links with TGF-β and VEGF. Cancers 2023, 15, 2647. https://doi.org/10.3390/cancers15092647
Ah-Pine F, Malaterre-Septembre A, Bedoui Y, Khettab M, Neal JW, Freppel S, Gasque P. Complement Activation and Up-Regulated Expression of Anaphylatoxin C3a/C3aR in Glioblastoma: Deciphering the Links with TGF-β and VEGF. Cancers. 2023; 15(9):2647. https://doi.org/10.3390/cancers15092647
Chicago/Turabian StyleAh-Pine, Franck, Axelle Malaterre-Septembre, Yosra Bedoui, Mohamed Khettab, James W. Neal, Sébastien Freppel, and Philippe Gasque. 2023. "Complement Activation and Up-Regulated Expression of Anaphylatoxin C3a/C3aR in Glioblastoma: Deciphering the Links with TGF-β and VEGF" Cancers 15, no. 9: 2647. https://doi.org/10.3390/cancers15092647