
Rare Non-Neuroendocrine Pancreatic Tumours
 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
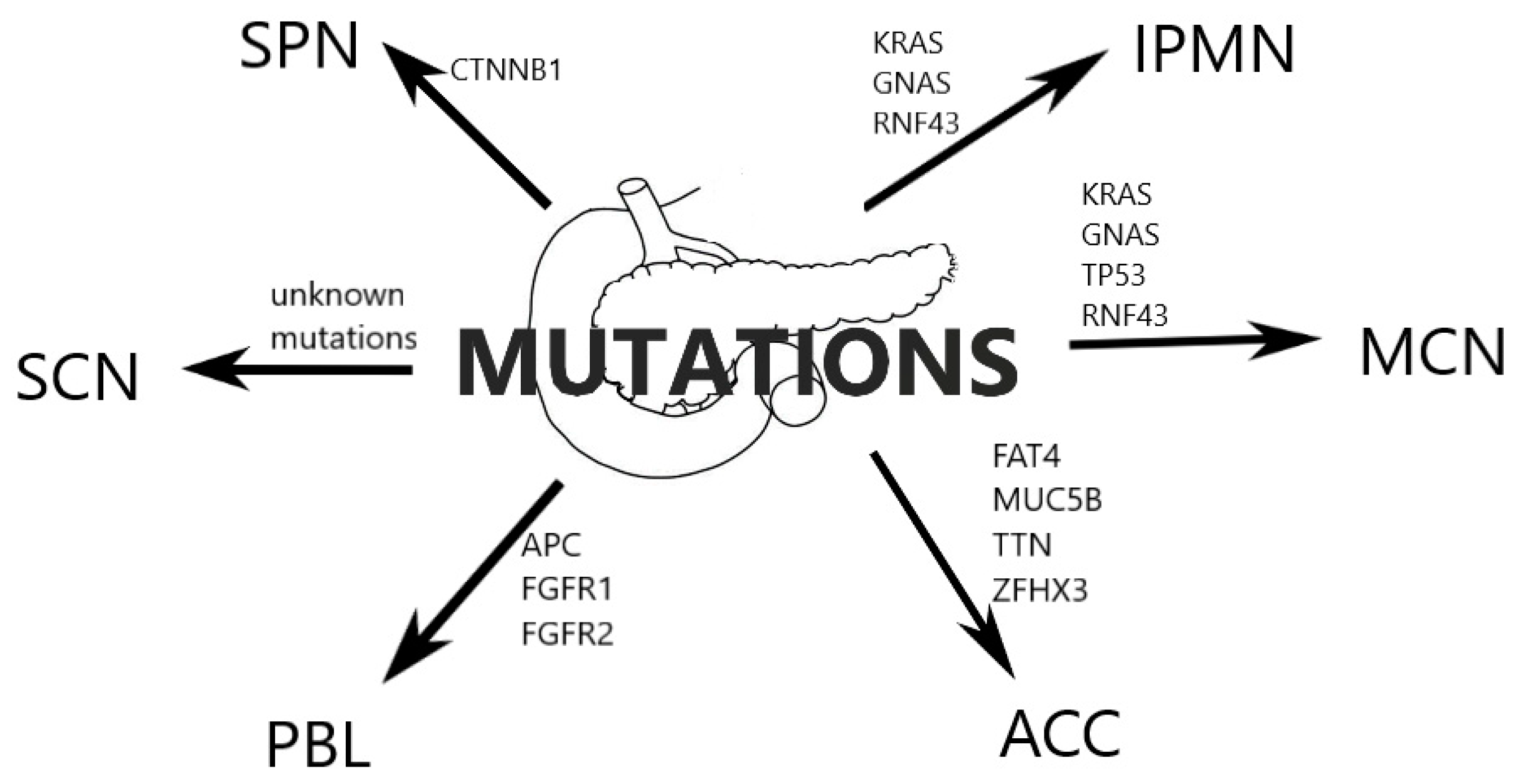
2. Classification
3. IPMN
3.1. Epidemiology, Clinical and Gross Features
3.2. Diagnosis
3.3. Management
4. MCN
4.1. Epidemiology, Clinical and Gross Features
4.2. Malignancy
4.3. Diagnosis
4.4. Differential Diagnosis
4.5. Biomarkers
4.6. Metabolomics
4.7. Genetics
4.8. Management
5. SCN
5.1. Epidemiology, Clinical and Gross Features
5.2. Diagnosis
5.3. Management
6. SPN
6.1. Epidemiology, Clinical and Gross Features
6.2. Diagnosis
6.3. Management
7. ACC
7.1. Epidemiology, Clinical and Gross Features
7.2. Diagnosis
7.3. Genetics
7.4. Management
8. Pancreatoblastoma
8.1. Epidemiology, Clinical and Gross Features
8.2. Diagnosis
8.3. Management
9. Other Rare Non-Neuroendocrine Pancreatic Tumours
9.1. Pancreatic Sarcomas
9.2. Pancreatic Squamous Cell Carcinoma
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Steinman, J.; Zaheer, A.; Kluger, M.D.; Remotti, H.; Hecht, E.M. Rare pancreatic tumors. Abdom. Radiol. 2018, 43, 285–300. [Google Scholar] [CrossRef] [PubMed]
- Walsh, R.M.; Perlmutter, B.C.; Adsay, V.; Reid, M.D.; Baker, M.E.; Stevens, T.; Hue, J.J.; Hardacre, J.M.; Shen, G.Q.; Simon, R.; et al. Advances in the management of pancreatic cystic neoplasms. Curr. Probl. Surg. 2021, 58, 100879. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.W.; Lee, S.K.; Jun, J.H.; Song, T.J.; Park, D.H.; Lee, S.S.; Seo, D.W.; Kim, M.H. Timing and Clinical Features of Spontaneous Decrease in Size of Small Pancreatic Cystic Lesions without High-Risk Stigmata. Gut Liver 2020, 14, 248–256. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, R.S. WHO Classification, Pathol. Website. (n.d.). Available online: https://www.pathologyoutlines.com/topic/pancreaswho.html (accessed on 5 January 2023).
- Castellano-Megías, V.M.; Andrés, C.I.; López-Alonso, G.; Colina-Ruizdelgado, F. Pathological features and diagnosis of intraductal papillary mucinous neoplasm of the pancreas. World J. Gastrointest. Oncol. 2014, 6, 311–324. [Google Scholar] [CrossRef] [PubMed]
- Geramizadeh, B.; Marzban, M.; Shojazadeh, A.; Kadivar, A.; Maleki, Z. Intraductal papillary mucinous neoplasm of the pancreas: Cytomorphology, imaging, molecular profile, and prognosis. Cytopathology 2021, 32, 397–406. [Google Scholar] [CrossRef]
- Fujita, Y.; Hirono, S.; Kawai, M.; Okada, K.-I.; Miyazawa, M.; Kitahata, Y.; Ueno, M.; Hayami, S.; Kobayashi, R.; Yanagisawa, A.; et al. Malignant potential and specific characteristics of pure main duct type intraductal papillary mucinous neoplasm. Eur. J. Surg. Oncol. J. Eur. Soc. Surg. Oncol. Br. Assoc. Surg. Oncol. 2022, 48, 1054–1061. [Google Scholar] [CrossRef]
- Yang, Z.; Shi, G. Comparison of clinicopathologic characteristics and survival outcomes between invasive IPMN and invasive MCN: A population-based analysis. Front. Oncol. 2022, 12, 899761. [Google Scholar] [CrossRef]
- Huang, X.; You, S.; Ding, G.; Liu, X.; Wang, J.; Gao, Y.; Zheng, J. Sites of Distant Metastases and Cancer-Specific Survival in Intraductal Papillary Mucinous Neoplasm with Associated Invasive Carcinoma: A Study of 1178 Patients. Front. Oncol. 2021, 11, 681961. [Google Scholar] [CrossRef]
- Gavazzi, F.; Capretti, G.; Giordano, L.; Ridolfi, C.; Spaggiari, P.; Sollai, M.; Carrara, S.; Nappo, G.; Bozzarelli, S.; Zerbi, A. Pancreatic ductal adenocarcinoma and invasive intraductal papillary mucinous tumor: Different prognostic factors for different overall survival. Dig. Liver Dis. Off. J. Ital. Soc. Gastroenterol. Ital. Assoc. Study Liver 2022, 54, 826–833. [Google Scholar] [CrossRef]
- Holmberg, M.; Ghorbani, P.; Gilg, S.; Del Chiaro, M.; Arnelo, U.; Löhr, J.-M.; Sparrelid, E. Outcome after resection for invasive intraductal papillary mucinous neoplasia is similar to conventional pancreatic ductal adenocarcinoma. Pancreatology 2021, 21, 1371–1377. [Google Scholar] [CrossRef]
- Valsangkar, N.P.; Morales-Oyarvide, V.; Thayer, S.P.; Ferrone, C.R.; Wargo, J.A.; Warshaw, A.L.; Castillo, C.F.-d. 851 resected cystic tumors of the pancreas: A 33-year experience at the Massachusetts General Hospital. Surgery 2012, 152, S4–S12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- European Study Group on Cystic Tumours of the Pancreas. European evidence-based guidelines on pancreatic cystic neoplasms. Gut 2018, 67, 789–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmicheal, J.; Patel, A.; Dalal, V.; Atri, P.; Dhaliwal, A.S.; Wittel, U.; Malafa, M.P.; Talmon, G.; Swanson, B.J.; Singh, S.; et al. Elevating pancreatic cystic lesion stratification: Current and future pancreatic cancer biomarker(s). Biochim. Biophys. Acta. Rev. Cancer 2020, 1873, 188318. [Google Scholar] [CrossRef]
- Shipley, L.C.; Ahmed, A.M. New and emerging technology in the diagnosis and treatment of pancreatic cysts. Transl. Gastroenterol. Hepatol. 2022, 7, 15. [Google Scholar] [CrossRef] [PubMed]
- Das, K.K.; Geng, X.; Brown, J.W.; Morales-Oyarvide, V.; Huynh, T.; Pergolini, I.; Pitman, M.B.; Ferrone, C.; Al Efishat, M.; Haviland, D.; et al. Cross Validation of the Monoclonal Antibody Das-1 in Identification of High-Risk Mucinous Pancreatic Cystic Lesions. Gastroenterology 2019, 157, 720–730.e2. [Google Scholar] [CrossRef]
- Luchini, C.; Grillo, F.; Fassan, M.; Vanoli, A.; Capelli, P.; Paolino, G.; Ingravallo, G.; Renzulli, G.; Doglioni, C.; D’Amuri, A.; et al. Malignant epithelial/exocrine tumors of the pancreas. Pathologica 2020, 112, 210–226. [Google Scholar] [CrossRef]
- Carr, R.A.; Yip-Schneider, M.T.; Dolejs, S.; Hancock, B.A.; Wu, H.; Radovich, M.; Schmidt, C. Pancreatic Cyst Fluid Vascular Endothelial Growth Factor A and Carcinoembryonic Antigen: A Highly Accurate Test for the Diagnosis of Serous Cystic Neoplasm. J. Am. Coll. Surg. 2017, 225, 93–100. [Google Scholar] [CrossRef]
- Wong, N.A.C.S.; Beavers, S.; Gill, P.; Heryet, A.; Linares, J. Calponin and MUC6 complement inhibin as diagnostic immunomarkers of serous cystadenoma in endoscopic ultrasound guided aspiration/biopsy specimens. Histopathology. 2021, 79, 252–259. [Google Scholar] [CrossRef]
- Isobe, T.; Seki, M.; Yoshida, K.; Sekiguchi, M.; Shiozawa, Y.; Shiraishi, Y.; Kimura, S.; Yoshida, M.; Inoue, Y.; Yokoyama, A.; et al. Integrated Molecular Characterization of the Lethal Pediatric Cancer Pancreatoblastoma. Cancer Res. 2018, 78, 865–876. [Google Scholar] [CrossRef] [Green Version]
- Das, K.K.; Xiao, H.; Geng, X.; Fernandez-Del-Castillo, C.; Morales-Oyarvide, V.; Daglilar, E.; Forcione, D.G.; Bounds, B.C.; Brugge, W.R.; Pitman, M.; et al. mAb Das-1 is specific for high-risk and malignant intraductal papillary mucinous neoplasm (IPMN). Gut 2014, 63, 1626–1634. [Google Scholar] [CrossRef]
- Zhou, X.; Sun, Z.; Zhang, M.; Qu, X.; Yang, S.; Wang, L.; Jing, Y.; Li, L.; Deng, W.; Liu, F.; et al. Deficient Rnf43 potentiates hyperactive Kras-mediated pancreatic preneoplasia initiation and malignant transformation. Anim. Model. Exp. Med. 2022, 5, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Vila-Navarro, E.; Vila-Casadesús, M.; Moreira, L.; Duran-Sanchon, S.; Sinha, R.; Ginés, À.; Fernández-Esparrach, G.; Miquel, R.; Cuatrecasas, M.; Castells, A.; et al. MicroRNAs for Detection of Pancreatic Neoplasia: Biomarker Discovery by Next-generation Sequencing and Validation in 2 Independent Cohorts. Ann. Surg. 2017, 265, 1226–1234. [Google Scholar] [CrossRef] [Green Version]
- Yoon, J.G.; Smith, D.; Ojili, V.; Paspulati, R.M.; Ramaiya, N.H.; Tirumani, S.H. Pancreatic cystic neoplasms: A review of current recommendations for surveillance and management. Abdom. Radiol. 2021, 46, 3946–3962. [Google Scholar] [CrossRef] [PubMed]
- Conroy, T.; Hammel, P.; Hebbar, M.; Abdelghani, M.B.; Wei, A.C.; Raoul, J.-L.; Choné, L.; Francois, E.; Artru, P.; Biagi, J.; et al. FOLFIRINOX or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. N. Engl. J. Med. 2018, 379, 2395–2406. [Google Scholar] [CrossRef]
- Kaiser, J.; Scheifele, C.; Hinz, U.; Leonhardt, C.-S.; Hank, T.; Koenig, A.-K.; Tjaden, C.; Hackert, T.; Bergmann, F.; Büchler, M.W.; et al. IPMN-associated pancreatic cancer: Survival, prognostic staging and impact of adjuvant chemotherapy. Eur. J. Surg. Oncol. J. Eur. Soc. Surg. Oncol. Br. Assoc. Surg. Oncol. 2022, 48, 1309–1320. [Google Scholar] [CrossRef] [PubMed]
- Aronsson, L.; Marinko, S.; Ansari, D.; Andersson, R. Adjuvant therapy in invasive intraductal papillary mucinous neoplasm (IPMN) of the pancreas: A systematic review. Ann. Transl. Med. 2019, 7, 689. [Google Scholar] [CrossRef] [PubMed]
- Aizpuru, M.; Starlinger, P.; Nagorney, D.M.; Smoot, R.L.; Truty, M.J.; Kendrick, M.; Cleary, S.P. Contemporary outcomes of pancreaticoduodenectomy for benign and precancerous cystic lesions. HPB Off. J. Int. Hepato Pancreato Biliary Assoc. 2022, 24, 1416–1424. [Google Scholar] [CrossRef]
- Munigala, S.; Javia, S.; Agarwal, B. Etiologic Distribution of Pancreatic Cystic Lesions Identified on Computed Tomography/Magnetic Resonance Imaging. Pancreas 2019, 48, 1092–1097. [Google Scholar] [CrossRef]
- Dinarvand, P.; Lai, J. Solid Pseudopapillary Neoplasm of the Pancreas: A Rare Entity with Unique Features. Arch. Pathol. Lab. Med. 2017, 141, 990–995. [Google Scholar] [CrossRef] [Green Version]
- Hirono, S.; Yamaue, H. Surgical strategy for intraductal papillary mucinous neoplasms of the pancreas. Surg. Today 2020, 50, 50–55. [Google Scholar] [CrossRef] [Green Version]
- Mylonas, K.S.; Doulamis, I.P.; Tsilimigras, D.; Nasioudis, D.; Schizas, D.; Masiakos, P.T.; Kelleher, C.M. Solid pseudopapillary and malignant pancreatic tumors in childhood: A systematic review and evidence quality assessment. Pediatr. Blood Cancer 2018, 65, e27114. [Google Scholar] [CrossRef]
- Farhat, W.; Ammar, H.; Said, M.A.A.; Mizouni, A.; Bouazzi, A.; Abdessaied, N.; Mabrouk, M.B.; Ali, A.B. Solid pseudopapillary neoplasm of the pancreas: A report of 10 cases and literature review. ANZ J. Surg. 2020, 90, 1683–1688. [Google Scholar] [CrossRef] [PubMed]
- Erguibi, D.; El Berni, Y.; Moufakkir, A.; Boufettal, R.; Saad, R.; Chehab, F. Solid pseudopapillary neoplasm of the head of the pancreas: A case report. Ann. Med. Surg. 2021, 69, 102708. [Google Scholar] [CrossRef]
- Maimaijiang, A.; Wang, H.; Li, W.; Wang, Y. Diagnosis and treatment of solid pseudopapillary neoplasm of the pancreas in children: A report of 18 cases. Front. Pediatr. 2022, 10, 899965. [Google Scholar] [CrossRef] [PubMed]
- Bien, E.; Roganovic, J.; Krawczyk, M.A.; Godzinski, J.; Orbach, D.; Cecchetto, G.; Barthlen, W.; Defachelles, A.-S.; Ferrari, A.; Weldon, C.B.; et al. Pancreatoblastoma in children: EXPeRT/PARTNER diagnostic and therapeutic recommendations. Pediatr. Blood Cancer. 2021, 68 (Suppl. S4), e29112. [Google Scholar] [CrossRef] [PubMed]
- Bojanapu, S.; Kasi, A. Pancreatic Mucinous Cystadenoma; NIH: Treasure Island, FL, USA, 2022. [Google Scholar]
- Lee, L. Updates in diagnosis and management of pancreatic cysts. World J. Gastroenterol. 2021, 27, 5700–5714. [Google Scholar] [CrossRef] [PubMed]
- Marchegiani, G.; Andrianello, S.; Crippa, S.; Pollini, T.; Belfiori, G.; Gozzini, L.; Cassalia, F.; Caravati, A.; Luchini, C.; Doglioni, C.; et al. Actual malignancy risk of either operated or non-operated presumed mucinous cystic neoplasms of the pancreas under surveillance. Br. J. Surg. 2021, 108, 1097–1104. [Google Scholar] [CrossRef] [PubMed]
- Yeh, M.M.; Tang, L.H.; Wang, S.; Robert, M.E.; Zheng, W.; Jain, D. Inhibin expression in ovarian-type stroma in mucinous cystic neoplasms of the pancreas. Appl. Immunohistochem. Mol. Morphol. AIMM 2004, 12, 148–152. [Google Scholar] [CrossRef]
- Lam, M.M.; Swanson, P.E.; Upton, M.; Yeh, M. Ovarian-type stroma in hepatobiliary cystadenomas and pancreatic mucinous cystic neoplasms: An immunohistochemical study. Am. J. Clin. Pathol. 2008, 129, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Vullierme, M.-P.; Gregory, J.; Rebours, V.; Cros, J.; Abelhady-Attia, Y.; Vilgrain, V.; Aguilera-Munoz, L.; Laurent, L.; Levy, P.; Sauvanet, A.; et al. MRI is useful to suggest and exclude malignancy in mucinous cystic neoplasms of the pancreas. Eur. Radiol. 2022, 32, 1297–1307. [Google Scholar] [CrossRef]
- Tanaka, M.; Castillo, C.F.-d.; Adsay, V.; Chari, S.; Falconi, M.; Jang, J.-Y.; Kimura, W.; Levy, P.; Pitman, M.B.; Schmidt, C.; et al. International consensus guidelines 2012 for the management of IPMN and MCN of the pancreas. Pancreatology 2012, 12, 183–197. [Google Scholar] [CrossRef] [PubMed]
- Perri, G.; Marchegiani, G.; Frigerio, I.; Dervenis, C.G.; Conlon, K.; Bassi, C.; Salvia, R. Management of Pancreatic Cystic Lesions. Dig. Surg. 2020, 37, 1–9. [Google Scholar] [CrossRef]
- Naganuma, S.; Honda, K.; Noriki, S.; Kimura, S.; Murakami, M.; Koneri, K.; Katayama, K.; Yamaguchi, A.; Itoh, H. Ruptured mucinous cystic neoplasm with an associated invasive carcinoma of pancreatic head in a pregnant woman: Report of a case and review of literature. Pathol. Int. 2011, 61, 28–33. [Google Scholar] [CrossRef]
- Ahmad, M.; Maegawa, F.B.; De La Rosa, E.; Davis, B.; Tyroch, A.; Konstantinidis, I.T. Mucinous cystic neoplasms of the pancreas in the modern era. Experience with 707 patients. Am. J. Surg. 2020, 220, 1433–1437. [Google Scholar] [CrossRef] [PubMed]
- Elta, G.H.; Enestvedt, B.K.; Sauer, B.; Lennon, A.M. ACG Clinical Guideline: Diagnosis and Management of Pancreatic Cysts. Am. J. Gastroenterol. 2018, 113, 464–479. [Google Scholar] [CrossRef]
- Fukasawa, M.; Maguchi, H.; Takahashi, K.; Katanuma, A.; Osanai, M.; Kurita, A.; Ichiya, T.; Tsuchiya, T.; Kin, T. Clinical features and natural history of serous cystic neoplasm of the pancreas. Pancreatology 2010, 10, 695–701. [Google Scholar] [CrossRef] [PubMed]
- Jais, B.; Rebours, V.; Malleo, G.; Salvia, R.; Fontana, M.; Maggino, L.; Bassi, C.; Manfredi, R.; Moran, R.; Lennon, A.; et al. Serous cystic neoplasm of the pancreas: A multinational study of 2622 patients under the auspices of the International Association of Pancreatology and European Pancreatic Club (European Study Group on Cystic Tumors of the Pancreas). Gut 2016, 65, 305–312. [Google Scholar] [CrossRef] [Green Version]
- Amico, E.C.; Salgado, C.T.S.; Emerenciano, L.M.; Filho, G.A.S.F.; Alves, J.R.; de Souza, L.E.O.F.F.; da Silva, J.S.P. Serous cystadenoma of pancreas: Why there is low accuracy in imaging exams? Arq. Bras. Cir. Dig. ABCD = Braz. Arch. Dig. Surg. 2022, 34, e1640. [Google Scholar] [CrossRef]
- Mustafa, S.; Hruban, R.H.; Ali, S.Z. Acinar cell carcinoma of the pancreas: A clinicopathologic and cytomorphologic review. J. Am. Soc. Cytopathol. 2020, 9, 586–595. [Google Scholar] [CrossRef]
- La Rosa, S.; Sessa, F.; Capella, C. Acinar Cell Carcinoma of the Pancreas: Overview of Clinicopathologic Features and Insights into the Molecular Pathology. Front. Med. 2015, 2, 41. [Google Scholar] [CrossRef] [Green Version]
- Wisnoski, N.C.; Townsend, C.M.J.; Nealon, W.H.; Freeman, J.L.; Riall, T.S. 672 patients with acinar cell carcinoma of the pancreas: A population-based comparison to pancreatic adenocarcinoma. Surgery 2008, 144, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, M.; Miura, S.; Hamada, S.; Sano, T.; Matsumoto, R.; Tanaka, Y.; Hongo, S.; Yoshida, N.; Takikawa, T.; Kikuta, K.; et al. Acinar Cell Carcinoma with Morphological Change in One Month. Intern. Med. 2021, 60, 2799–2806. [Google Scholar] [CrossRef] [PubMed]
- La Rosa, S.; Adsay, V.; Albarello, L.; Asioli, S.; Casnedi, S.; Franzi, F.; Marando, A.; Notohara, K.; Sessa, F.; Vanoli, A.; et al. Clinicopathologic study of 62 acinar cell carcinomas of the pancreas: Insights into the morphology and immunophenotype and search for prognostic markers. Am. J. Surg. Pathol. 2012, 36, 1782–1795. [Google Scholar] [CrossRef] [PubMed]
- Qu, Q.; Xin, Y.; Xu, Y.; Yuan, Y.; Deng, K. Imaging and Clinicopathological Features of Acinar Cell Carcinoma. Front. Oncol. 2022, 12, 888679. [Google Scholar] [CrossRef]
- Uhlig, R.; Contreras, H.; Weidemann, S.; Gorbokon, N.; Menz, A.; Büscheck, F.; Luebke, A.M.; Kluth, M.; Hube-Magg, C.; Hinsch, A.; et al. Carboxypeptidase A1 (CPA1) Immunohistochemistry Is Highly Sensitive and Specific for Acinar Cell Carcinoma (ACC) of the Pancreas. Am. J. Surg. Pathol. 2022, 46, 97–104. [Google Scholar] [CrossRef]
- Liu, Y.; Raimondo, M.; Wallace, M.; Mody, K.; Stauffer, J.A.; Zhang, L.; Ji, B.; Bi, Y. Exome Sequencing of Pancreatic Acinar Carcinoma Identified Distinctive Mutation Patterns. Pancreas 2021, 50, 1007–1013. [Google Scholar] [CrossRef]
- Zong, Y.; Qi, C.; Peng, Z.; Shen, L.; Zhou, J. Patients With Acinar Cell Carcinoma of the Pancreas After 2005: A Large Population Study. Pancreas 2020, 49, 781–787. [Google Scholar] [CrossRef]
- Patel, D.J.; Lutfi, W.; Sweigert, P.; Eguia, E.; Abood, G.; Knab, L.; Kuo, P.C.; Baker, M.S. Clinically resectable acinar cell carcinoma of the pancreas: Is there a benefit to adjuvant systemic therapy? Am. J. Surg. 2020, 219, 522–526. [Google Scholar] [CrossRef]
- Xing-Mao, Z.; Hong-Juan, Z.; Qing, L.; Qiang, H. Pancreatic acinar cell carcinoma-case report and literature review. BMC Cancer 2018, 18, 1083. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Yang, W.; Hu, J.; Zhu, Z.; Qin, H.; Han, W.; Wang, H. Diagnosis and treatment of pancreatoblastoma in children: A retrospective study in a single pediatric center. Pediatr. Surg. Int. 2019, 35, 1231–1238. [Google Scholar] [CrossRef]
- Reid, M.D.; Bhattarai, S.; Graham, R.P.; Pehlivanoglu, B.; Sigel, C.S.; Shi, J.; Saqi, A.; Shirazi, M.; Xue, Y.; Basturk, O.; et al. Pancreatoblastoma: Cytologic and histologic analysis of 12 adult cases reveals helpful criteria in their diagnosis and distinction from common mimics. Cancer Cytopathol. 2019, 127, 708–719. [Google Scholar] [CrossRef]
- Omiyale, A.O. Adult pancreatoblastoma: Current concepts in pathology. World J. Gastroenterol. 2021, 27, 4172–4181. [Google Scholar] [CrossRef] [PubMed]
- Shetty, A.S.; Menias, C.O. Rare Pancreatic Tumors. Magn. Reson. Imaging Clin. N. Am. 2018, 26, 421–437. [Google Scholar] [CrossRef]
- Berger, A.K.; Mughal, S.S.; Allgäuer, M.; Springfeld, C.; Hackert, T.; Weber, T.; Naumann, P.; Hutter, B.; Horak, P.; Jahn, A.; et al. Metastatic adult pancreatoblastoma: Multimodal treatment; molecular characterization of a very rare disease. Pancreatology 2020, 20, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Gong, Y.; Ji, M.; Yang, B.; Qiao, Z. Differential diagnosis of pancreatoblastoma (PB) and solid pseudopapillary neoplasms (SPNs) in children by CT and MR imaging. Eur. Radiol. 2021, 31, 2209–2217. [Google Scholar] [CrossRef] [PubMed]
- Simone, C.G.; Toro, T.Z.; Chan, E.; Feely, M.M.; Trevino, J.G.; George, T.J. Characteristics and outcomes of adenosquamous carcinoma of the pancreas. Gastrointest. Cancer Res. 2013, 6, 75–79. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Benign | Serous cystadenoma, NOS (8441/0) |
Intraductal papillary mucinous neoplasm
| |
Mucinous cystic neoplasm
| |
| Malignant | Acinar cell carcinoma (8550/3) |
| Acinar cell cystadenocarcinoma (8551/3) | |
| Mixed acinar-neuroendocrine carcinoma (8154/3) | |
| Mixed acinar-endocrine-ductal carcinoma (8154/3) | |
| Mixed acinar-ductal carcinoma (8552/3) | |
| Pancreatoblastoma (8971/3) | |
Solid pseudopapillary neoplasm of the pancreas (8452/3)
|
| Type | Mucins |
|---|---|
| Gastric | MUC5AC+ MUC6+ |
| Intestinal | MUC2+ CDX2+ |
| Pancreato-biliary | MUC1 strong |
| IPMN | MCN | SCN | ACC | SPN | Pancreatoblastoma | |
|---|---|---|---|---|---|---|
| Incidence | 3–5% | 1–2% | 1–2% | 1% | 1–2% | <1% |
| M/F ratio | 1:1 | 1:20 | 1:2 | 2:1 | 1:10 | 2:1 |
| Malignancy potential | MD 50% BD 15% | 4.4–16.6% | 0% | 100% | 15% | High |
| Median age | 69 | 48 | 61 | 59 | 28 | 5 |
| Location | Head | Body/tail | Body/tail | Head/body/tail | Tail | Head |
| Mean size [mm] | MD 28.8 BD 29.1 | 20–250 | 46.1 | 80–100 | 20–150 | 20–200 |
| Cystic/Solid | Cystic | Cystic | Cystic | Solid (rarely cystic) | Solid | Solid |
| 5-year survival | MD 83% BD 88% | 26–75% (invasive MCN) | 97% | 25–50% | 97% | 95% |
| Biomarkers | CEA > 192 ng/mL High amylase | CEA > 192 ng/mL Low amylase | CEA < 192 ng/mL VEGF-A > 5000 pg/mL | Trypsin Chymotrypsin BCL-10 | CTNNB1 mutations, β-catenin, E-cadherin | High AFP CK AAT |
| Histological features | Tall papillary structures lined by epithelium Mucin production | Ovarian cortical cells Mucin production | Low cuboidal cells with clear cytoplasm Sponge-like structure in microcystic type | Acinar cell differentiation | Sheets of epithelioid cells with scant cytoplasm Pseudopapillary structures | Mixed epithelial and mesenchymal components Vascular and perineural elements |
| IPMN | MCN | SCN | |
|---|---|---|---|
| Biomarkers | |||
| CEA | >192 ng/mL | >192 ng/mL | <192 ng/mL |
| CA19-9 | >50,000 U/mL | >50,000 U/mL | - |
| VEGF-A | - | - | >5000 pg/mL |
| Amylase | High | Low | Low |
| Glucose | <50 mg/dL | <50 mg/dL | >50 mg/dL |
| Mutations | KRAS, GNAS | KRAS, GNAS, TP53, RNF43 | - |
| Histological Features | Papillary structures, atypia | Ovarian-like columnar cells, mucus production | Simple cuboidal epithelial cells |
| Cyst Fluid Viscosity | High | High | Low |
| Marker | Diagnosis | Cut-Off Value | Material | Sn/Sp (%) | Tumours | Notes |
|---|---|---|---|---|---|---|
| CEA | Malignant | >5 µg/L | Serum | 40/92 | IPMN, MCN | 80% of patients with invasive IPMN had increased levels of CEA |
| Cystic | >192 ng/mL | Cystic fluid | 73/84 | IPMN, MCN | ||
| ≤10 ng/ml | Cystic fluid | 96/82 | SCN | |||
| CA19-9 | Malignant | >37 U/mL | Serum | 74/86 | ||
| Mucinous | >50,000 U/mL | Cystic fluid | 75/90 | IPMN, MCN | ||
| CEA/ CA19-9 | Malignant | >5 µg/L/>37 U/mL | Serum | 80/82 | IPMN, MCN | Accuracy of 81% |
| VEGF-A | Serous | >5000 pg/mL | Cystic fluid | 100/84 | SCN | Elevated levels of VEGF-A may also occur in PNETs |
| VEGF-A/CEA | Serous | >5000 pg/mL/≤10 ng/mL | Cystic fluid | 96/100 | SCN | |
| VEGF-C | Serous | >200 pg/mL | Cystic fluid | 100/90 | SCN | |
| MUC6 | Serous | NA | Cystic fluid | 100/100 | SCN | PNET differentiation |
| Calponin | Serous | NA | Cystic fluid | 71/100 | SCN | PNET differentiation |
| Glucose | Mucinous | <66 mg/dL | Cystic fluid | 94/64 | MCN | |
| Amylase | Pseudocyst | >250 U/mL | Cystic fluid | 44/98 | - | High amylase levels may be associated with IPMN |
| IMP3 | Malignant | NA | Cystic fluid | 78/96 | IPMN, MCN | |
| mAb Das-1 | Malignant | NA | Cystic fluid | 88/99 | IPMN | |
| KRAS | Malignant | NA | Cystic fluid | 45/96 | IPMN, MCN | |
| KRAS/GNAS | Malignant | NA | Cystic fluid | 65/100 | IPMN, MCN | |
| Trypsin | Acinar cell | NA | Tissue | NA | ACC | Negative in SPN |
| BCL-10 | Acinar cell | NA | Tissue | NA | ACC | Negative in SPN |
| β-catenin/ E-cadherin | SPN | NA | Tissue | NA | - | β-catenin is expressed in over 90% of SPN |
| CD10 | SPN | NA | Tissue | NA | - | |
| Cytokeratin | Pancreatoblastoma | NA | Tissue | NA | - | |
| AAT | Pancreatoblastoma | NA | Tissue | NA | - |
| IPMN | MCN | SCN | ACC | SPN | Pancreatoblastoma | |
|---|---|---|---|---|---|---|
| Primary/preferred method | Resection (with adjuvant therapy if invasive) | Resection (>40 mm/ symptomatic/ have risk factors) or surveillance [9] | Surveillance | Resection (regardless of tumour size) [29] | Resection [30] | Resection (with resection of hepatic metastases considered) [1] |
| Surgical resection | Pancreatoctomy (with lymph node dissection for invasive IPMN) [31] | Distal pancreatico-splenectomy with or without lymph node dissection [23] | Not recommended | Radical resection [29] | Classic Whipple surgery [32] | Classic Whipple surgery [32] |
| Adjuvant therapy | FOLFIRINOX or gemcitabine [25] | gemcitabine and 5-fluorouracil [9] | NA | FOLFIRINOX [33] | 5FU-based chemotherapy alone or in combination with cisplatin [33,34,35] | PLADO (cisplatin and doxorubicin) [36] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mormul, A.; Włoszek, E.; Nowoszewska, J.; Fudalej, M.; Budzik, M.; Badowska-Kozakiewicz, A.; Deptała, A. Rare Non-Neuroendocrine Pancreatic Tumours. Cancers 2023, 15, 2216. https://doi.org/10.3390/cancers15082216
Mormul A, Włoszek E, Nowoszewska J, Fudalej M, Budzik M, Badowska-Kozakiewicz A, Deptała A. Rare Non-Neuroendocrine Pancreatic Tumours. Cancers. 2023; 15(8):2216. https://doi.org/10.3390/cancers15082216
Chicago/Turabian StyleMormul, Agata, Emilia Włoszek, Julia Nowoszewska, Marta Fudalej, Michał Budzik, Anna Badowska-Kozakiewicz, and Andrzej Deptała. 2023. "Rare Non-Neuroendocrine Pancreatic Tumours" Cancers 15, no. 8: 2216. https://doi.org/10.3390/cancers15082216