
Lipid Metabolic Reprogramming in Embryonal Neoplasms with MYCN Amplification

Abstract
:Simple Summary
Abstract
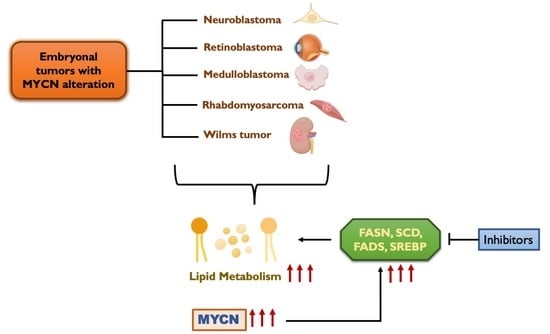
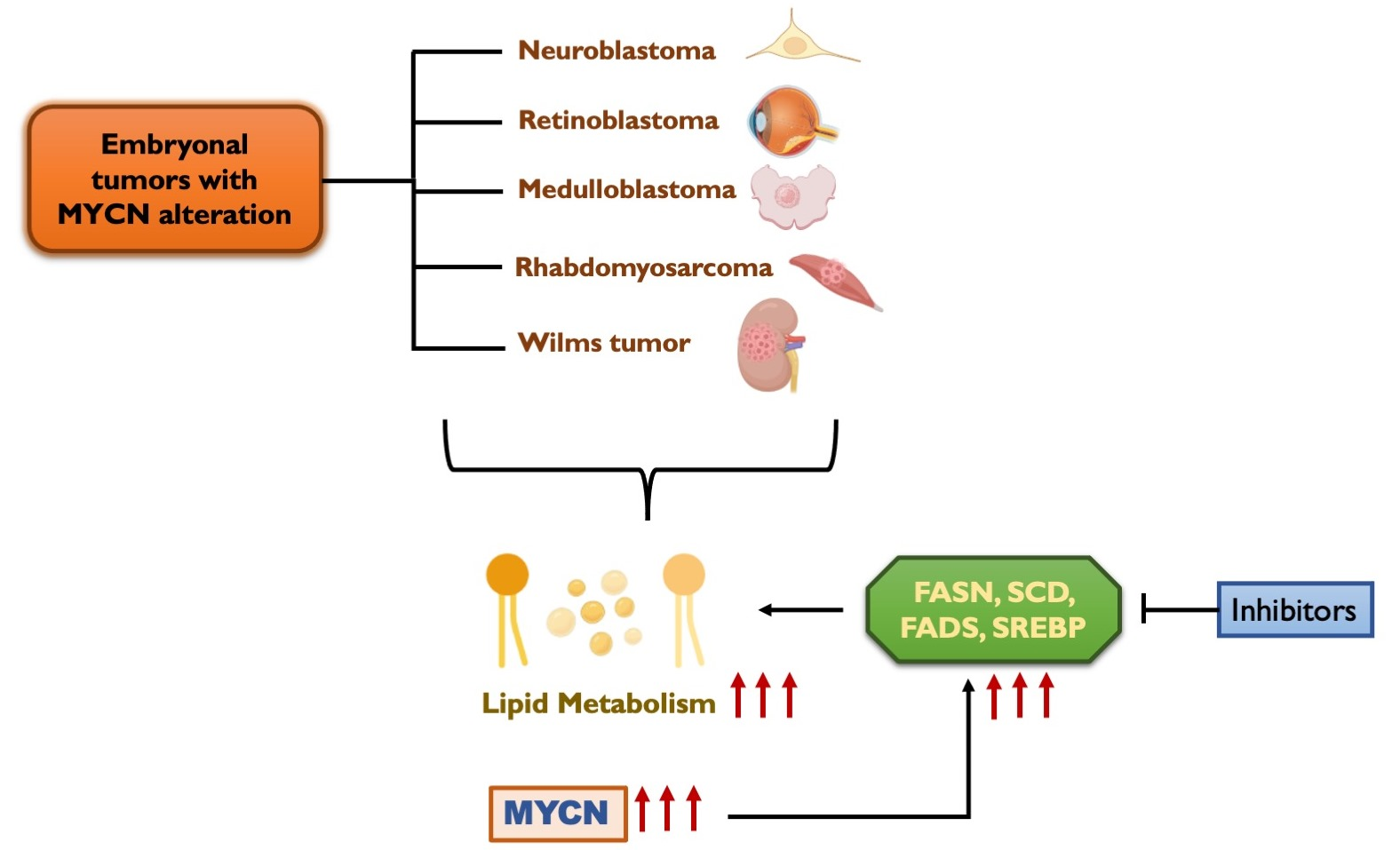{kind=link}
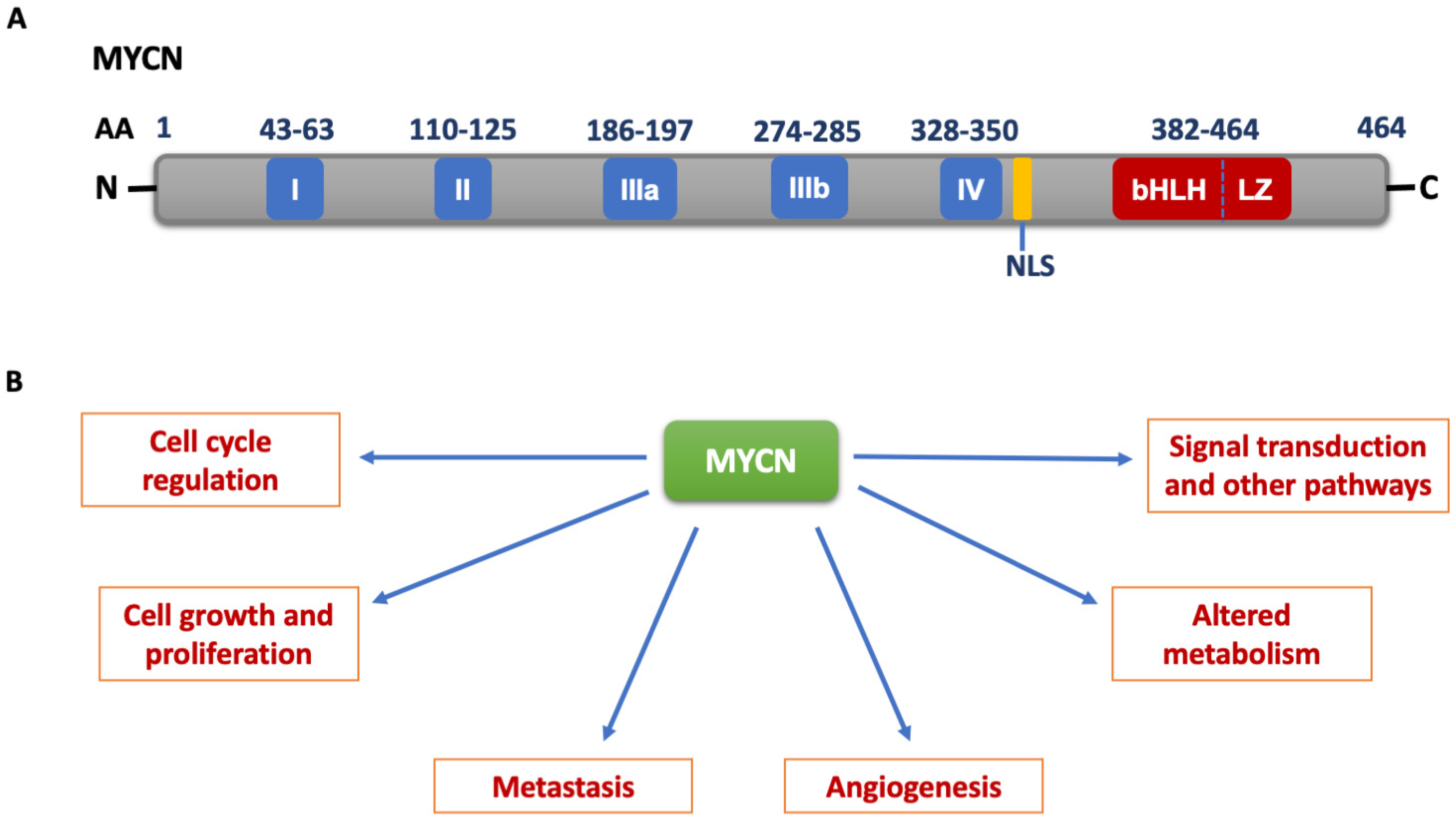{kind=link}
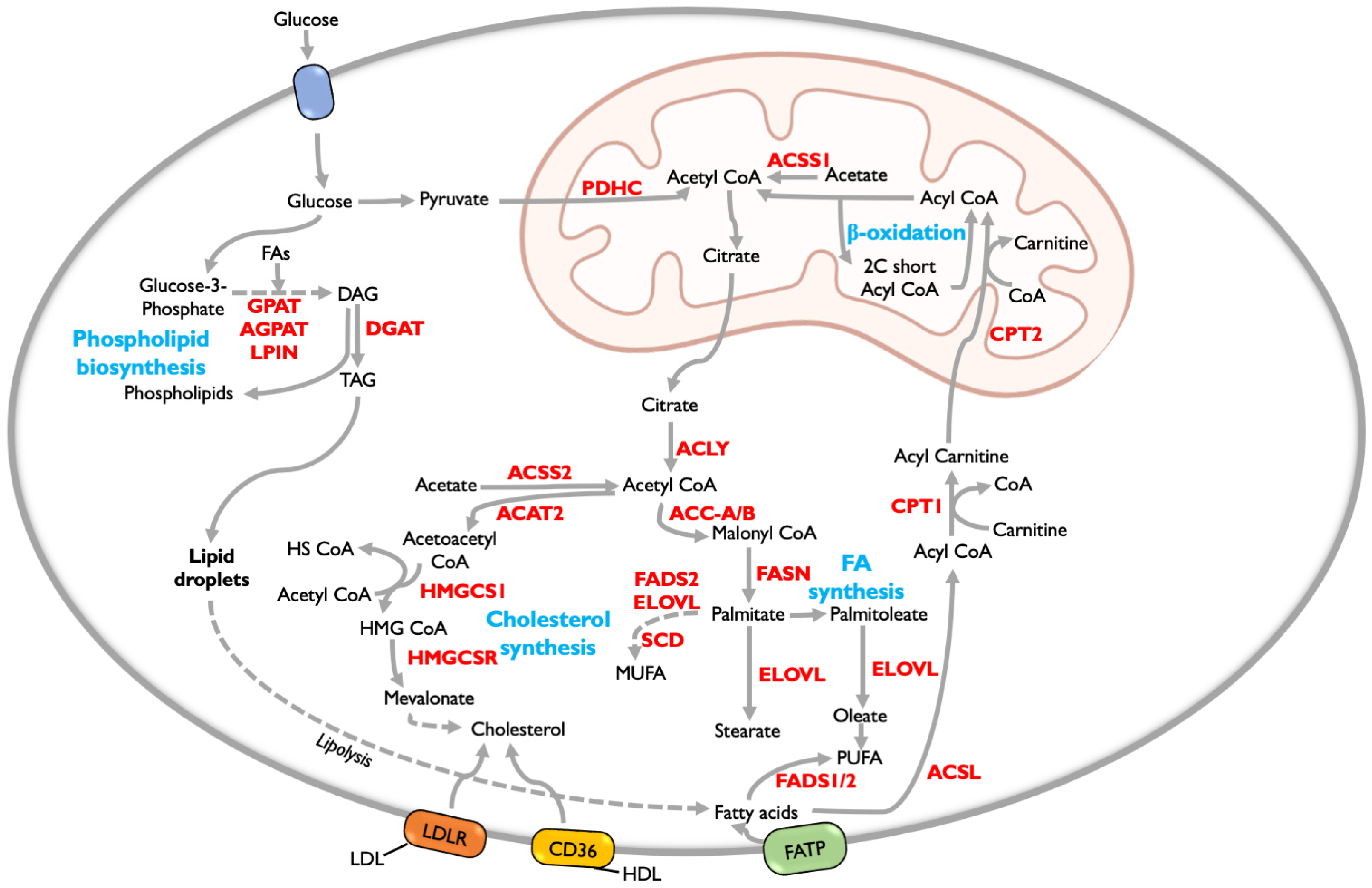{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. MYCN Structure and Function
3. Deregulation of MYCN in Embryonal Neoplasms
4. Metabolic Reprogramming in Cancer and MYCN
5. Lipid Metabolism

5.1. Fatty Acid Metabolism
5.2. Fatty Acid Oxidation (FAO)
5.3. Phospholipid Synthesis
5.4. Cholesterol Synthesis
6. Lipid Metabolism in Embryonal Tumors
6.1. Neuroblastoma
6.2. Retinoblastoma
6.3. Medulloblastoma
6.4. Rhabdomyosarcoma
6.5. Wilms Tumor
7. Targeting Lipid Metabolism as a Therapeutic Strategy
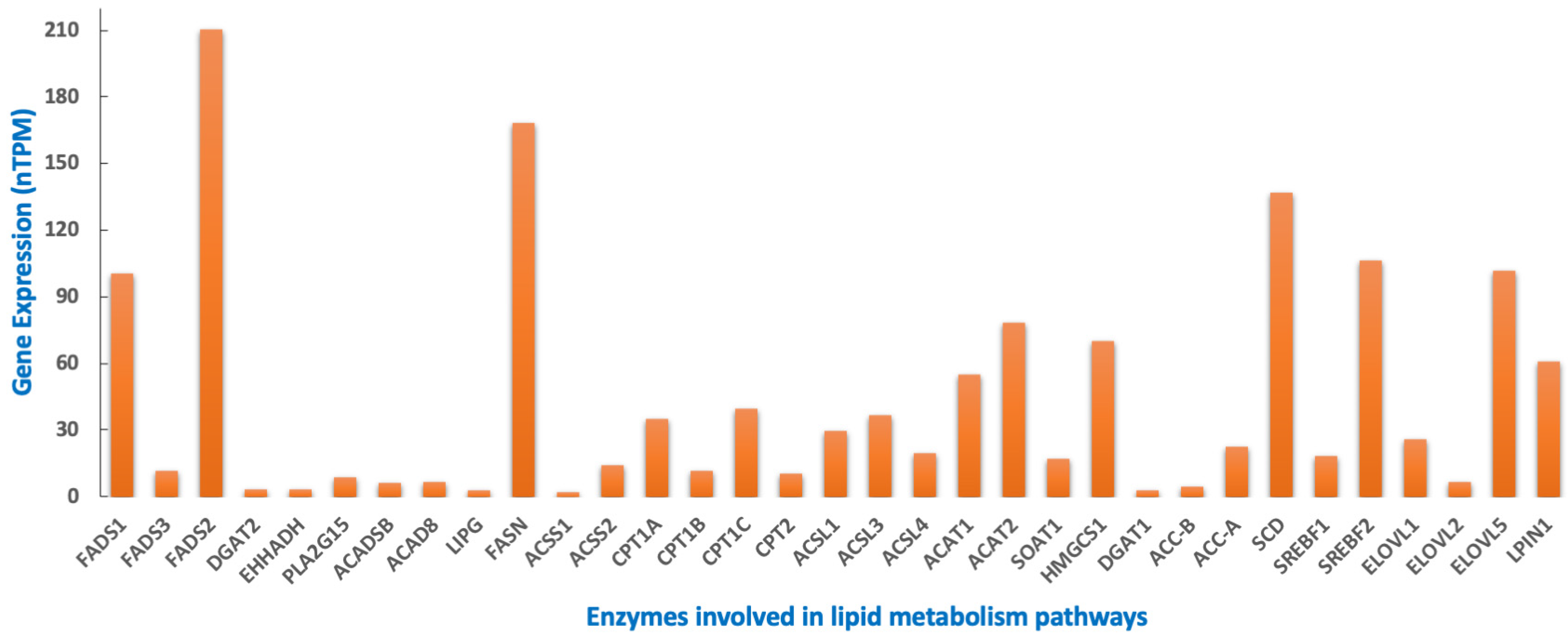
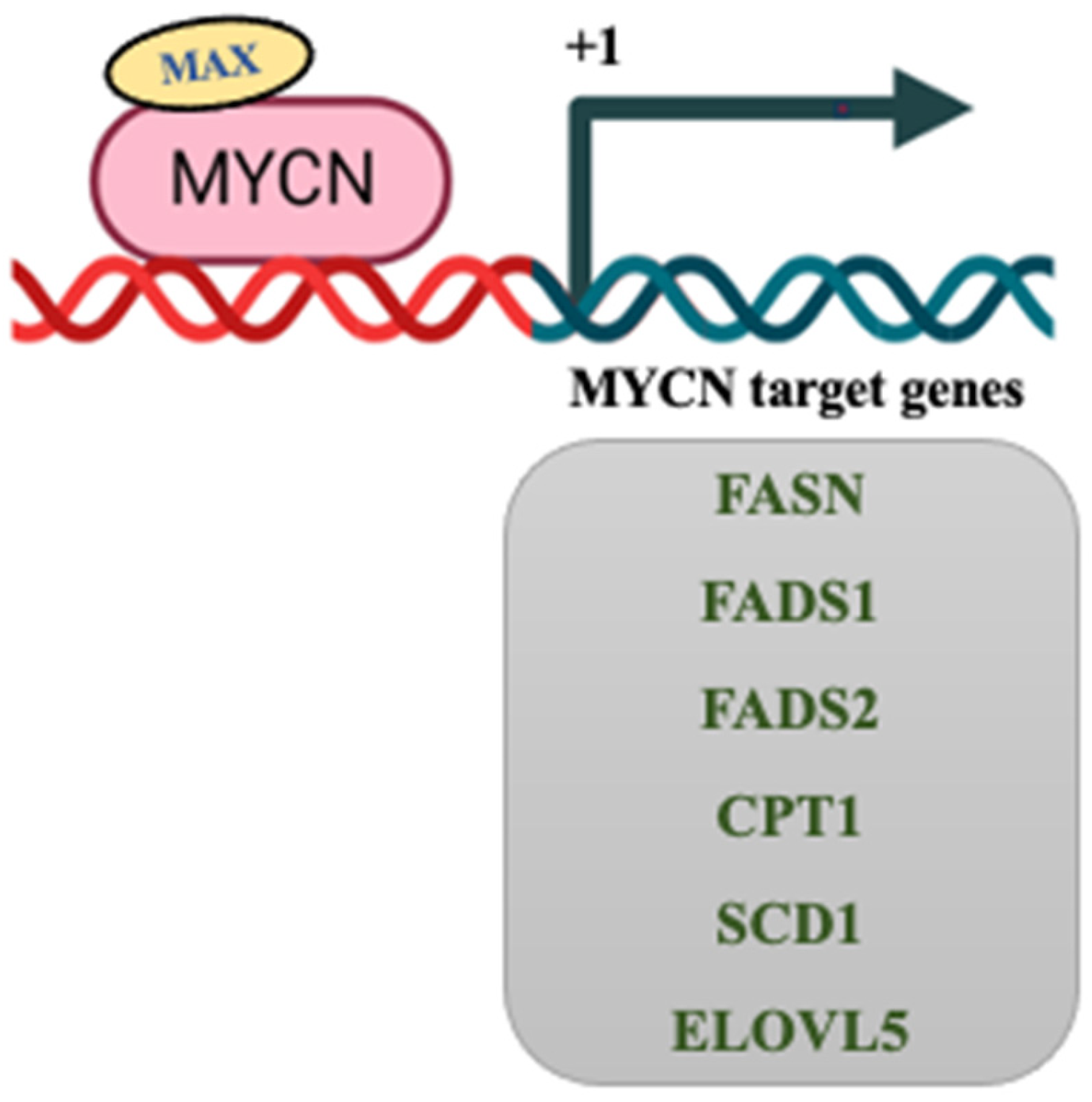
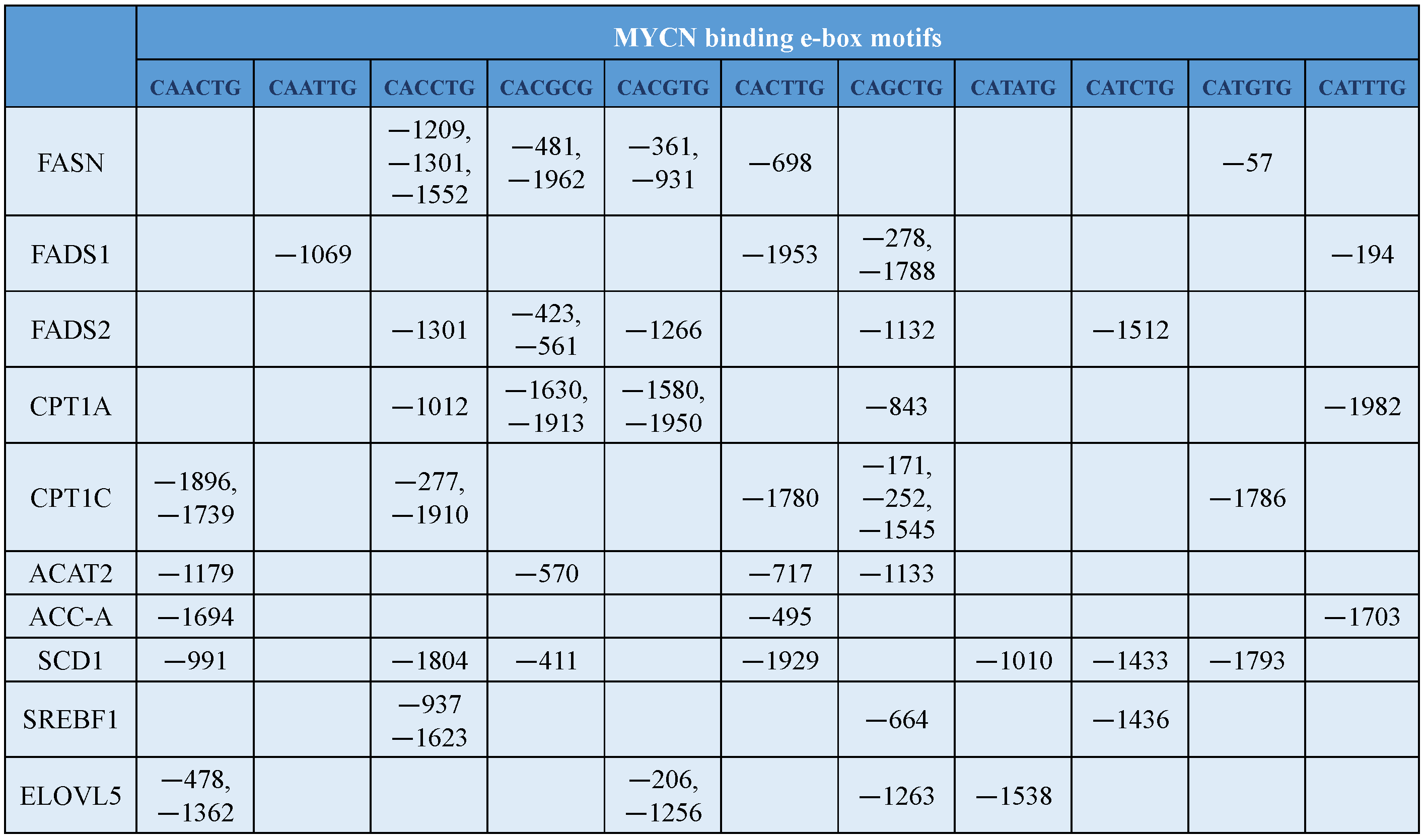
8. MYCN Regulation of Lipid Metabolism
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Duesberg, P.H.; Bister, K.; Vogt, P.K. The RNA of Avian Acute Leukemia Virus MC29. Proc. Natl. Acad. Sci. USA 1977, 74, 4320–4324. [Google Scholar] [CrossRef] [Green Version]
- Sheiness, D.; Fanshier, L.; Bishop, J.M. Identification of Nucleotide Sequences Which May Encode the Oncogenic Capacity of Avian Retrovirus MC29. J. Virol. 1978, 28, 600–610. [Google Scholar] [CrossRef] [Green Version]
- Bister, K.; Jansen, H.W. Oncogenes in Retroviruses and Cells: Biochemistry and Molecular Genetics. Adv. Cancer Res. 1986, 47, 99–188. [Google Scholar] [CrossRef]
- Schwab, M.; Alitalo, K.; Klempnauer, K.H.; Varmus, H.E.; Bishop, J.M.; Gilbert, F.; Brodeur, G.; Goldstein, M.; Trent, J. Amplified DNA with Limited Homology to Myc Cellular Oncogene Is Shared by Human Neuroblastoma Cell Lines and a Neuroblastoma Tumour. Nature 1983, 305, 245–248. [Google Scholar] [CrossRef]
- Nau, M.M.; Brooks, B.J.; Battey, J.; Sausville, E.; Gazdar, A.F.; Kirsch, I.R.; McBride, O.W.; Bertness, V.; Hollis, G.F.; Minna, J.D. L-Myc, a New Myc-Related Gene Amplified and Expressed in Human Small Cell Lung Cancer. Nature 1985, 318, 69–73. [Google Scholar] [CrossRef]
- Meyer, N.; Penn, L.Z. Reflecting on 25 Years with MYC. Nat. Rev. Cancer 2008, 8, 976–990. [Google Scholar] [CrossRef]
- Dang, C.V. MYC on the Path to Cancer. Cell 2012, 149, 22–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, M.; Weiss, W.A. Neuroblastoma and MYCN. Cold Spring Harb. Perspect. Med. 2013, 3, a014415. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, M.S.; Hart, J.R.; Vogt, P.K. A Brave New MYC-Amplified World. Aging 2015, 7, 459–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burrell, R.A.; McGranahan, N.; Bartek, J.; Swanton, C. The Causes and Consequences of Genetic Heterogeneity in Cancer Evolution. Nature 2013, 501, 338–345. [Google Scholar] [CrossRef]
- Kohl, N.E.; Kanda, N.; Schreck, R.R.; Bruns, G.; Latt, S.A.; Gilbert, F.; Alt, F.W. Transposition and Amplification of Oncogene-Related Sequences in Human Neuroblastomas. Cell 1983, 35, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Brodeur, G.M.; Seeger, R.C.; Schwab, M.; Varmus, H.E.; Bishop, J.M. Amplification of N-Myc in Untreated Human Neuroblastomas Correlates with Advanced Disease Stage. Science 1984, 224, 1121–1124. [Google Scholar] [CrossRef]
- Seeger, R.C.; Brodeur, G.M.; Sather, H.; Dalton, A.; Siegel, S.E.; Wong, K.Y.; Hammond, D. Association of Multiple Copies of the N- Myc Oncogene with Rapid Progression of Neuroblastomas. N. Engl. J. Med. 1985, 313, 1111–1116. [Google Scholar] [CrossRef] [PubMed]
- Little, C.D.; Nau, M.M.; Carney, D.N.; Gazdar, A.F.; Minna, J.D. Amplification and Expression of the C-Myc Oncogene in Human Lung Cancer Cell Lines. Nature 1983, 306, 194–196. [Google Scholar] [CrossRef] [PubMed]
- Nau, M.M.; Brooks, B.J.; Carney, D.N.; Gazdar, A.F.; Battey, J.F.; Sausville, E.A.; Minna, J.D. Human Small-Cell Lung Cancers Show Amplification and Expression of the N-Myc Gene. Proc. Natl. Acad. Sci. USA 1986, 83, 1092–1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz-Pérez, M.V.; Henley, A.B.; Arsenian-Henriksson, M. The MYCN Protein in Health and Disease. Genes 2017, 8, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beltran, H. The N-Myc Oncogene: Maximizing Its Targets, Regulation, and Therapeutic Potential. Mol. Cancer Res. 2014, 12, 815–822. [Google Scholar] [CrossRef] [Green Version]
- Follis, A.V.; Hammoudeh, D.I.; Daab, A.T.; Metallo, S.J. Small-Molecule Perturbation of Competing Interactions between c-Myc and Max. Bioorg. Med. Chem. Lett. 2009, 19, 807–810. [Google Scholar] [CrossRef]
- Hu, J.; Banerjee, A.; Goss, D.J. Assembly of b/HLH/z Proteins c-Myc, Max, and Mad1 with Cognate DNA: Importance of Protein-Protein and Protein-DNA Interactions. Biochemistry 2005, 44, 11855–11863. [Google Scholar] [CrossRef] [Green Version]
- Mustata, G.; Follis, A.V.; Hammoudeh, D.I.; Metallo, S.J.; Wang, H.; Prochownik, E.V.; Lazo, J.S.; Bahar, I. Discovery of Novel Myc-Max Heterodimer Disruptors with a Three-Dimensional Pharmacophore Model. J. Med. Chem. 2009, 52, 1247–1250. [Google Scholar] [CrossRef] [Green Version]
- Sauvé, S.; Naud, J.-F.; Lavigne, P. The Mechanism of Discrimination between Cognate and Non-Specific DNA by Dimeric b/HLH/LZ Transcription Factors. J. Mol. Biol. 2007, 365, 1163–1175. [Google Scholar] [CrossRef] [PubMed]
- Blackwell, T.K.; Huang, J.; Ma, A.; Kretzner, L.; Alt, F.W.; Eisenman, R.N.; Weintraub, H. Binding of Myc Proteins to Canonical and Noncanonical DNA Sequences. Mol. Cell. Biol. 1993, 13, 5216–5224. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V.; Lee, W.M. Identification of the Human C-Myc Protein Nuclear Translocation Signal. Mol. Cell. Biol. 1988, 8, 4048–4054. [Google Scholar] [CrossRef] [PubMed]
- Kato, G.J.; Barrett, J.; Villa-Garcia, M.; Dang, C. V An Amino-Terminal c-Myc Domain Required for Neoplastic Transformation Activates Transcription. Mol. Cell. Biol. 1990, 10, 5914–5920. [Google Scholar] [CrossRef]
- Madden, S.K.; de Araujo, A.D.; Gerhardt, M.; Fairlie, D.P.; Mason, J.M. Taking the Myc out of Cancer: Toward Therapeutic Strategies to Directly Inhibit c-Myc. Mol. Cancer 2021, 20, 3. [Google Scholar] [CrossRef]
- Cowling, V.H.; Chandriani, S.; Whitfield, M.L.; Cole, M.D. A Conserved Myc Protein Domain, MBIV, Regulates DNA Binding, Apoptosis, Transformation, and G2 Arrest. Mol. Cell. Biol. 2006, 26, 4226–4239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herbst, A.; Hemann, M.T.; Tworkowski, K.A.; Salghetti, S.E.; Lowe, S.W.; Tansey, W.P. A Conserved Element in Myc That Negatively Regulates Its Proapoptotic Activity. EMBO Rep. 2005, 6, 177–183. [Google Scholar] [CrossRef] [Green Version]
- Kohl, N.E.; Legouy, E.; DePinho, R.A.; Nisen, P.D.; Smith, R.K.; Gee, C.E.; Alt, F.W. Human N-Myc Is Closely Related in Organization and Nucleotide Sequence to c-Myc. Nature 1986, 319, 73–77. [Google Scholar] [CrossRef]
- Sarid, J.; Halazonetis, T.D.; Murphy, W.; Leder, P. Evolutionarily Conserved Regions of the Human C-Myc Protein Can Be Uncoupled from Transforming Activity. Proc. Natl. Acad. Sci. USA 1987, 84, 170–173. [Google Scholar] [CrossRef] [Green Version]
- Stanton, L.W.; Schwab, M.; Bishop, J.M. Nucleotide Sequence of the Human N-Myc Gene. Proc. Natl. Acad. Sci. USA 1986, 83, 1772–1776. [Google Scholar] [CrossRef] [Green Version]
- Reisman, D.; Elkind, N.B.; Roy, B.; Beamon, J.; Rotter, V. C-Myc Trans-Activates the P53 Promoter through a Required Downstream CACGTG Motif. Cell Growth Differ. 1993, 4, 57–65. [Google Scholar] [PubMed]
- Lüscher, B. Function and Regulation of the Transcription Factors of the Myc/Max/Mad Network. Gene 2001, 277, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, P.C.; Frank, S.R.; Wang, L.; Schroeder, M.; Liu, S.; Greene, J.; Cocito, A.; Amati, B. Genomic Targets of the Human C-Myc Protein. Genes Dev. 2003, 17, 1115–1129. [Google Scholar] [CrossRef] [Green Version]
- Shohet, J.M. Redefining Functional MYCN Gene Signatures in Neuroblastoma. Proc. Natl. Acad. Sci. USA 2012, 109, 19041–19042. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Chen, S.S.; Clarke, S.; Veschi, V.; Thiele, C.J. Targeting MYCN in Pediatric and Adult Cancers. Front. Oncol. 2021, 10, 623679. [Google Scholar] [CrossRef]
- Liu, R.; Shi, P.; Wang, Z.; Yuan, C.; Cui, H. Molecular Mechanisms of MYCN Dysregulation in Cancers. Front. Oncol. 2020, 10, 625332. [Google Scholar] [CrossRef]
- Shields, C.L.; Shields, J.A. Diagnosis and Management of Retinoblastoma. Cancer Control 2004, 11, 317–327. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.H.; Murphree, A.L.; Benedict, W.F. Expression and Amplification of the N-Myc Gene in Primary Retinoblastoma. Nature 1984, 309, 458–460. [Google Scholar] [CrossRef]
- Rushlow, D.E.; Mol, B.M.; Kennett, J.Y.; Yee, S.; Pajovic, S.; Thériault, B.L.; Prigoda-Lee, N.L.; Spencer, C.; Dimaras, H.; Corson, T.W.; et al. Characterisation of Retinoblastomas without RB1 Mutations: Genomic, Gene Expression, and Clinical Studies. Lancet Oncol. 2013, 14, 327–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sradhanjali, S.; Rout, P.; Tripathy, D.; Kaliki, S.; Rath, S.; Modak, R.; Mittal, R.; Chowdary, T.K.; Reddy, M.M. The Oncogene MYCN Modulates Glycolytic and Invasive Genes to Enhance Cell Viability and Migration in Human Retinoblastoma. Cancers 2021, 13, 5248. [Google Scholar] [CrossRef]
- Treger, T.D.; Brok, J.; Pritchard-Jones, K. Biology and Treatment of Wilms’ Tumours in Childhood. Rev. Oncol. Hématol. Pédiatr. 2016, 4, 170–181. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Ye, B.; Ding, J.; Yu, Y.; Alptekin, A.; Thangaraju, M.; Prasad, P.D.; Ding, Z.-C.; Park, E.J.; Choi, J.-H.; et al. Metabolic Reprogramming by MYCN Confers Dependence on the Serine-Glycine-One-Carbon Biosynthetic Pathway. Cancer Res. 2019, 79, 3837–3850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, L.; Mohammad, M.A.; Milazzo, G.; Moreno-Smith, M.; Patel, T.D.; Zorman, B.; Badachhape, A.; Hernandez, B.E.; Wolf, A.B.; Zeng, Z.; et al. MYCN-Driven Fatty Acid Uptake Is a Metabolic Vulnerability in Neuroblastoma. Nat. Commun. 2022, 13, 3728. [Google Scholar] [CrossRef] [PubMed]
- Bansal, M.; Gupta, A.; Ding, H.-F. MYCN and Metabolic Reprogramming in Neuroblastoma. Cancers 2022, 14, 4113. [Google Scholar] [CrossRef]
- Dang, C.V.; Reddy, E.P.; Shokat, K.M.; Soucek, L. Drugging the “undruggable” Cancer Targets. Nat. Rev. Cancer 2017, 17, 502–508. [Google Scholar] [CrossRef]
- Borgenvik, A.; Čančer, M.; Hutter, S.; Swartling, F.J. Targeting MYCN in Molecularly Defined Malignant Brain Tumors. Front. Oncol. 2020, 10, 626751. [Google Scholar] [CrossRef]
- Aldape, K.; Brindle, K.M.; Chesler, L.; Chopra, R.; Gajjar, A.; Gilbert, M.R.; Gottardo, N.; Gutmann, D.H.; Hargrave, D.; Holland, E.C.; et al. Challenges to Curing Primary Brain Tumours. Nat. Rev. Clin. Oncol. 2019, 16, 509–520. [Google Scholar] [CrossRef] [Green Version]
- Wolpaw, A.J.; Bayliss, R.; Büchel, G.; Dang, C.V.; Eilers, M.; Gustafson, W.C.; Hansen, G.H.; Jura, N.; Knapp, S.; Lemmon, M.A.; et al. Drugging the “Undruggable” MYCN Oncogenic Transcription Factor: Overcoming Previous Obstacles to Impact Childhood Cancers. Cancer Res. 2021, 81, 1627–1632. [Google Scholar] [CrossRef]
- Puissant, A.; Frumm, S.M.; Alexe, G.; Bassil, C.F.; Qi, J.; Chanthery, Y.H.; Nekritz, E.A.; Zeid, R.; Gustafson, W.C.; Greninger, P.; et al. Targeting MYCN in Neuroblastoma by BET Bromodomain Inhibition. Cancer Discov. 2013, 3, 308–323. [Google Scholar] [CrossRef] [Green Version]
- Delmore, J.E.; Issa, G.C.; Lemieux, M.E.; Rahl, P.B.; Shi, J.; Jacobs, H.M.; Kastritis, E.; Gilpatrick, T.; Paranal, R.M.; Qi, J.; et al. BET Bromodomain Inhibition as a Therapeutic Strategy to Target C-Myc. Cell 2011, 146, 904–917. [Google Scholar] [CrossRef] [Green Version]
- Kato, F.; Fiorentino, F.P.; Alibés, A.; Perucho, M.; Sánchez-Céspedes, M.; Kohno, T.; Yokota, J. MYCL Is a Target of a BET Bromodomain Inhibitor, JQ1, on Growth Suppression Efficacy in Small Cell Lung Cancer Cells. Oncotarget 2016, 7, 77378–77388. [Google Scholar] [CrossRef] [Green Version]
- Gustafson, W.C.; Meyerowitz, J.G.; Nekritz, E.A.; Chen, J.; Benes, C.; Charron, E.; Simonds, E.F.; Seeger, R.; Matthay, K.K.; Hertz, N.T.; et al. Drugging MYCN through an Allosteric Transition in Aurora Kinase A. Cancer Cell 2014, 26, 414–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Healy, J.R.; Hart, L.S.; Shazad, A.L.; Gagliardi, M.E.; Tsang, M.; Elias, J.; Ruden, J.; Farrel, A.; Rokita, J.L.; Li, Y.; et al. Limited Antitumor Activity of Combined BET and MEK Inhibition in Neuroblastoma. Pediatr. Blood Cancer 2020, 67, e28267. [Google Scholar] [CrossRef] [PubMed]
- Mossé, Y.P.; Fox, E.; Teachey, D.T.; Reid, J.M.; Safgren, S.L.; Carol, H.; Lock, R.B.; Houghton, P.J.; Smith, M.A.; Hall, D.; et al. A Phase II Study of Alisertib in Children with Recurrent/Refractory Solid Tumors or Leukemia: Children’s Oncology Group Phase I and Pilot Consortium (ADVL0921). Clin. Cancer Res. 2019, 25, 3229–3238. [Google Scholar] [CrossRef] [Green Version]
- Iniguez, A.B.; Alexe, G.; Wang, E.J.; Roti, G.; Patel, S.; Chen, L.; Kitara, S.; Conway, A.; Robichaud, A.L.; Stolte, B.; et al. Resistance to Epigenetic-Targeted Therapy Engenders Tumor Cell Vulnerabilities Associated with Enhancer Remodeling. Cancer Cell 2018, 34, 922–938.e7. [Google Scholar] [CrossRef] [Green Version]
- Fong, C.Y.; Gilan, O.; Lam, E.Y.N.; Rubin, A.F.; Ftouni, S.; Tyler, D.; Stanley, K.; Sinha, D.; Yeh, P.; Morison, J.; et al. BET Inhibitor Resistance Emerges from Leukaemia Stem Cells. Nature 2015, 525, 538–542. [Google Scholar] [CrossRef] [PubMed]
- Kurimchak, A.M.; Shelton, C.; Duncan, K.E.; Johnson, K.J.; Brown, J.; O’Brien, S.; Gabbasov, R.; Fink, L.S.; Li, Y.; Lounsbury, N.; et al. Resistance to BET Bromodomain Inhibitors Is Mediated by Kinome Reprogramming in Ovarian Cancer. Cell Rep. 2016, 16, 1273–1286. [Google Scholar] [CrossRef] [Green Version]
- Rathert, P.; Roth, M.; Neumann, T.; Muerdter, F.; Roe, J.-S.; Muhar, M.; Deswal, S.; Cerny-Reiterer, S.; Peter, B.; Jude, J.; et al. Transcriptional Plasticity Promotes Primary and Acquired Resistance to BET Inhibition. Nature 2015, 525, 543–547. [Google Scholar] [CrossRef] [Green Version]
- DeBose-Boyd, R.A. Significance and Regulation of Lipid Metabolism. Semin. Cell Dev. Biol. 2018, 81, 97. [Google Scholar] [CrossRef] [PubMed]
- Molendijk, J.; Robinson, H.; Djuric, Z.; Hill, M.M. Lipid Mechanisms in Hallmarks of Cancer. Mol. Omics 2020, 16, 6–18. [Google Scholar] [CrossRef] [PubMed]
- Ohotski, J.; Edwards, J.; Elsberger, B.; Watson, C.; Orange, C.; Mallon, E.; Pyne, S.; Pyne, N.J. Identification of Novel Functional and Spatial Associations between Sphingosine Kinase 1, Sphingosine 1-Phosphate Receptors and Other Signaling Proteins That Affect Prognostic Outcome in Estrogen Receptor-Positive Breast Cancer. Int. J. Cancer 2013, 132, 605–616. [Google Scholar] [CrossRef]
- Beloribi-Djefaflia, S.; Vasseur, S.; Guillaumond, F. Lipid Metabolic Reprogramming in Cancer Cells. Oncogenesis 2016, 5, e189. [Google Scholar] [CrossRef] [Green Version]
- Kohe, S.E.; Bennett, C.D.; Gill, S.K.; Wilson, M.; McConville, C.; Peet, A.C. Metabolic Profiling of the Three Neural Derived Embryonal Pediatric Tumors Retinoblastoma, Neuroblastoma and Medulloblastoma, Identifies Distinct Metabolic Profiles. Oncotarget 2018, 9, 11336–11351. [Google Scholar] [CrossRef] [Green Version]
- Bacci, M.; Lorito, N.; Smiriglia, A.; Morandi, A. Fat and Furious: Lipid Metabolism in Antitumoral Therapy Response and Resistance. Trends Cancer 2021, 7, 198–213. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Outschoorn, U.E.; Peiris-Pagés, M.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Cancer Metabolism: A Therapeutic Perspective. Nat. Rev. Clin. Oncol. 2017, 14, 11–31. [Google Scholar] [CrossRef]
- Chen, C.-L.; Uthaya Kumar, D.B.; Punj, V.; Xu, J.; Sher, L.; Tahara, S.M.; Hess, S.; Machida, K. NANOG Metabolically Reprograms Tumor-Initiating Stem-like Cells through Tumorigenic Changes in Oxidative Phosphorylation and Fatty Acid Metabolism. Cell Metab. 2016, 23, 206–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Condello, S.; Thomes-Pepin, J.; Ma, X.; Xia, Y.; Hurley, T.D.; Matei, D.; Cheng, J.-X. Lipid Desaturation Is a Metabolic Marker and Therapeutic Target of Ovarian Cancer Stem Cells. Cell Stem Cell 2017, 20, 303–314. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Zhang, Z.; Song, L.; Gao, J.; Liu, Y. Lipid Metabolism of Cancer Stem Cells (Review). Oncol. Lett. 2022, 23, 119. [Google Scholar] [CrossRef] [PubMed]
- Pascual, G.; Avgustinova, A.; Mejetta, S.; Martín, M.; Castellanos, A.; Attolini, C.S.-O.; Berenguer, A.; Prats, N.; Toll, A.; Hueto, J.A.; et al. Targeting Metastasis-Initiating Cells through the Fatty Acid Receptor CD36. Nature 2017, 541, 41–45. [Google Scholar] [CrossRef]
- Tirinato, L.; Liberale, C.; di Franco, S.; Candeloro, P.; Benfante, A.; la Rocca, R.; Potze, L.; Marotta, R.; Ruffilli, R.; Rajamanickam, V.P.; et al. Lipid Droplets: A New Player in Colorectal Cancer Stem Cells Unveiled by Spectroscopic Imaging. Stem Cells 2015, 33, 35–44. [Google Scholar] [CrossRef] [Green Version]
- Noto, A.; de Vitis, C.; Pisanu, M.E.; Roscilli, G.; Ricci, G.; Catizone, A.; Sorrentino, G.; Chianese, G.; Taglialatela-Scafati, O.; Trisciuoglio, D.; et al. Stearoyl-CoA-Desaturase 1 Regulates Lung Cancer Stemness via Stabilization and Nuclear Localization of YAP/TAZ. Oncogene 2017, 36, 4573–4584. [Google Scholar] [CrossRef]
- Sorrentino, G.; Ruggeri, N.; Specchia, V.; Cordenonsi, M.; Mano, M.; Dupont, S.; Manfrin, A.; Ingallina, E.; Sommaggio, R.; Piazza, S.; et al. Metabolic Control of YAP and TAZ by the Mevalonate Pathway. Nat. Cell Biol. 2014, 16, 357–366.e5. [Google Scholar] [CrossRef]
- Pisanu, M.E.; Noto, A.; de Vitis, C.; Morrone, S.; Scognamiglio, G.; Botti, G.; Venuta, F.; Diso, D.; Jakopin, Z.; Padula, F.; et al. Blockade of Stearoyl-CoA-Desaturase 1 Activity Reverts Resistance to Cisplatin in Lung Cancer Stem Cells. Cancer Lett. 2017, 406, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Broadfield, L.A.; Pane, A.A.; Talebi, A.; Swinnen, J.V.; Fendt, S.-M. Lipid Metabolism in Cancer: New Perspectives and Emerging Mechanisms. Dev. Cell 2021, 56, 1363–1393. [Google Scholar] [CrossRef]
- Currie, E.; Schulze, A.; Zechner, R.; Walther, T.C.; Farese, R.V. Cellular Fatty Acid Metabolism and Cancer. Cell Metab. 2013, 18, 153–161. [Google Scholar] [CrossRef] [Green Version]
- Papandreou, I.; Cairns, R.A.; Fontana, L.; Lim, A.L.; Denko, N.C. HIF-1 Mediates Adaptation to Hypoxia by Actively Downregulating Mitochondrial Oxygen Consumption. Cell Metab. 2006, 3, 187–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaidi, N.; Swinnen, J.V.; Smans, K. ATP-Citrate Lyase: A Key Player in Cancer Metabolism. Cancer Res. 2012, 72, 3709–3714. [Google Scholar] [CrossRef] [Green Version]
- Wei, J.; Leit, S.; Kuai, J.; Therrien, E.; Rafi, S.; Harwood, H.J.; DeLaBarre, B.; Tong, L. An Allosteric Mechanism for Potent Inhibition of Human ATP-Citrate Lyase. Nature 2019, 568, 566–570. [Google Scholar] [CrossRef] [PubMed]
- Xie, S.; Zhou, F.; Wang, J.; Cao, H.; Chen, Y.; Liu, X.; Zhang, Z.; Dai, J.; He, X. Functional Polymorphisms of ATP Citrate Lyase Gene Predicts Clinical Outcome of Patients with Advanced Colorectal Cancer. World J. Surg. Oncol. 2015, 13, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calvisi, D.F.; Wang, C.; Ho, C.; Ladu, S.; Lee, S.A.; Mattu, S.; Destefanis, G.; Delogu, S.; Zimmermann, A.; Ericsson, J.; et al. Increased Lipogenesis, Induced by AKT-MTORC1-RPS6 Signaling, Promotes Development of Human Hepatocellular Carcinoma. Gastroenterology 2011, 140, 1071–1083. [Google Scholar] [CrossRef] [Green Version]
- Pietrocola, F.; Galluzzi, L.; Bravo-San Pedro, J.M.; Madeo, F.; Kroemer, G. Acetyl Coenzyme A: A Central Metabolite and Second Messenger. Cell Metab. 2015, 21, 805–821. [Google Scholar] [CrossRef] [Green Version]
- Nishi, K.; Suzuki, M.; Yamamoto, N.; Matsumoto, A.; Iwase, Y.; Yamasaki, K.; Otagiri, M.; Yumita, N. Glutamine Deprivation Enhances Acetyl-CoA Carboxylase Inhibitor-Induced Death of Human Pancreatic Cancer Cells. Anticancer Res. 2018, 38, 6683–6689. [Google Scholar] [CrossRef] [PubMed]
- Li, E.-Q.; Zhao, W.; Zhang, C.; Qin, L.-Z.; Liu, S.-J.; Feng, Z.-Q.; Wen, X.; Chen, C.-P. Synthesis and Anti-Cancer Activity of ND-646 and Its Derivatives as Acetyl-CoA Carboxylase 1 Inhibitors. Eur. J. Pharm. Sci. 2019, 137, 105010. [Google Scholar] [CrossRef]
- Lally, J.S.V.; Ghoshal, S.; DePeralta, D.K.; Moaven, O.; Wei, L.; Masia, R.; Erstad, D.J.; Fujiwara, N.; Leong, V.; Houde, V.P.; et al. Inhibition of Acetyl-CoA Carboxylase by Phosphorylation or the Inhibitor ND-654 Suppresses Lipogenesis and Hepatocellular Carcinoma. Cell Metab. 2019, 29, 174–182.e5. [Google Scholar] [CrossRef] [Green Version]
- Migita, T.; Ruiz, S.; Fornari, A.; Fiorentino, M.; Priolo, C.; Zadra, G.; Inazuka, F.; Grisanzio, C.; Palescandolo, E.; Shin, E.; et al. Fatty Acid Synthase: A Metabolic Enzyme and Candidate Oncogene in Prostate Cancer. J. Natl. Cancer Inst. 2009, 101, 519–532. [Google Scholar] [CrossRef]
- Tao, B.-B.; He, H.; Shi, X.; Wang, C.; Li, W.; Li, B.; Dong, Y.; Hu, G.-H.; Hou, L.-J.; Luo, C.; et al. Up-Regulation of USP2a and FASN in Gliomas Correlates Strongly with Glioma Grade. J. Clin. Neurosci. 2013, 20, 717–720. [Google Scholar] [CrossRef]
- Tomek, K.; Wagner, R.; Varga, F.; Singer, C.F.; Karlic, H.; Grunt, T.W. Blockade of Fatty Acid Synthase Induces Ubiquitination and Degradation of Phosphoinositide-3-Kinase Signaling Proteins in Ovarian Cancer. Mol. Cancer Res. 2011, 9, 1767–1779. [Google Scholar] [CrossRef] [Green Version]
- De Carvalho, C.C.C.R.; Caramujo, M.J. The Various Roles of Fatty Acids. Molecules 2018, 23, 2583. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.Y.; Kothapalli, K.S.D.; Brenna, J.T. Desaturase and Elongase-Limiting Endogenous Long-Chain Polyunsaturated Fatty Acid Biosynthesis. Curr. Opin. Clin. Nutr. Metab. Care 2016, 19, 103–110. [Google Scholar] [CrossRef]
- Ackerman, D.; Tumanov, S.; Qiu, B.; Michalopoulou, E.; Spata, M.; Azzam, A.; Xie, H.; Simon, M.C.; Kamphorst, J.J. Triglycerides Promote Lipid Homeostasis during Hypoxic Stress by Balancing Fatty Acid Saturation. Cell Rep. 2018, 24, 2596–2605.e5. [Google Scholar] [CrossRef] [Green Version]
- Ackerman, D.; Simon, M.C. Hypoxia, Lipids, and Cancer: Surviving the Harsh Tumor Microenvironment. Trends Cell Biol. 2014, 24, 472–478. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.S.; Stockwell, B.R. Ferroptosis: Death by Lipid Peroxidation. Trends Cell Biol. 2016, 26, 165–176. [Google Scholar] [CrossRef] [Green Version]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Peck, B.; Schulze, A. Lipid Desaturation—The next Step in Targeting Lipogenesis in Cancer? FEBS J. 2016, 283, 2767–2778. [Google Scholar] [CrossRef] [PubMed]
- Young, R.M.; Ackerman, D.; Quinn, Z.L.; Mancuso, A.; Gruber, M.; Liu, L.; Giannoukos, D.N.; Bobrovnikova-Marjon, E.; Diehl, J.A.; Keith, B.; et al. Dysregulated MTORC1 Renders Cells Critically Dependent on Desaturated Lipids for Survival under Tumor-like Stress. Genes Dev. 2013, 27, 1115–1131. [Google Scholar] [CrossRef] [Green Version]
- Griffiths, B.; Lewis, C.A.; Bensaad, K.; Ros, S.; Zhang, Q.; Ferber, E.C.; Konisti, S.; Peck, B.; Miess, H.; East, P.; et al. Sterol Regulatory Element Binding Protein-Dependent Regulation of Lipid Synthesis Supports Cell Survival and Tumor Growth. Cancer Metab. 2013, 1, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vriens, K.; Christen, S.; Parik, S.; Broekaert, D.; Yoshinaga, K.; Talebi, A.; Dehairs, J.; Escalona-Noguero, C.; Schmieder, R.; Cornfield, T.; et al. Evidence for an Alternative Fatty Acid Desaturation Pathway Increasing Cancer Plasticity. Nature 2019, 566, 403–406. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.-F.; Zhang, K.-L.; Zhang, X.-J.; Hu, Y.-J.; Li, P.; Shang, C.-Z.; Wan, J.-B. Abnormalities in Plasma Phospholipid Fatty Acid Profiles of Patients with Hepatocellular Carcinoma. Lipids 2015, 50, 977–985. [Google Scholar] [CrossRef] [PubMed]
- Miryaghoubzadeh, J.; Darabi, M.; Madaen, K.; Shaaker, M.; Mehdizadeh, A.; Hajihosseini, R. Tissue Fatty Acid Composition in Human Urothelial Carcinoma. Br. J. Biomed. Sci. 2013, 70, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Thysell, E.; Surowiec, I.; Hörnberg, E.; Crnalic, S.; Widmark, A.; Johansson, A.I.; Stattin, P.; Bergh, A.; Moritz, T.; Antti, H.; et al. Metabolomic Characterization of Human Prostate Cancer Bone Metastases Reveals Increased Levels of Cholesterol. PLoS ONE 2010, 5, e14175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baró, L.; Hermoso, J.-C.; Núñez, M.-C.; Jiménez-Rios, J.-A.; Gil, A. Abnormalities in Plasma and Red Blood Cell Fatty Acid Profiles of Patients with Colorectal Cancer. Br. J. Cancer 1998, 77, 1978–1983. [Google Scholar] [CrossRef]
- Cvetković, Z.; Vučić, V.; Cvetković, B.; Petrović, M.; Ristić-Medić, D.; Tepšić, J.; Glibetić, M. Abnormal Fatty Acid Distribution of the Serum Phospholipids of Patients with Non-Hodgkin Lymphoma. Ann. Hematol. 2010, 89, 775–782. [Google Scholar] [CrossRef]
- Yang, J.; Zhu, S.; Lin, G.; Song, C.; He, Z. Vitamin D Enhances Omega-3 Polyunsaturated Fatty Acids-Induced Apoptosis in Breast Cancer Cells. Cell Biol. Int. 2017, 41, 890–897. [Google Scholar] [CrossRef]
- Kim, S.; Jing, K.; Shin, S.; Jeong, S.; Han, S.-H.; Oh, H.; Yoo, Y.-S.; Han, J.; Jeon, Y.-J.; Heo, J.-Y.; et al. Ω3-Polyunsaturated Fatty Acids Induce Cell Death through Apoptosis and Autophagy in Glioblastoma Cells: In Vitro and in Vivo. Oncol. Rep. 2017, 39, 239–246. [Google Scholar] [CrossRef] [Green Version]
- Olivecrona, G. Role of Lipoprotein Lipase in Lipid Metabolism. Curr. Opin. Lipidol. 2016, 27, 233–241. [Google Scholar] [CrossRef]
- Bogie, J.F.J.; Haidar, M.; Kooij, G.; Hendriks, J.J.A. Fatty Acid Metabolism in the Progression and Resolution of CNS Disorders. Adv. Drug Deliv. Rev. 2020, 159, 198–213. [Google Scholar] [CrossRef]
- Carracedo, A.; Cantley, L.C.; Pandolfi, P.P. Cancer Metabolism: Fatty Acid Oxidation in the Limelight. Nat. Rev. Cancer 2013, 13, 227–232. [Google Scholar] [CrossRef]
- Padanad, M.S.; Konstantinidou, G.; Venkateswaran, N.; Melegari, M.; Rindhe, S.; Mitsche, M.; Yang, C.; Batten, K.; Huffman, K.E.; Liu, J.; et al. Fatty Acid Oxidation Mediated by Acyl-CoA Synthetase Long Chain 3 Is Required for Mutant KRAS Lung Tumorigenesis. Cell Rep. 2016, 16, 1614–1628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camarda, R.; Zhou, A.Y.; Kohnz, R.A.; Balakrishnan, S.; Mahieu, C.; Anderton, B.; Eyob, H.; Kajimura, S.; Tward, A.; Krings, G.; et al. Inhibition of Fatty Acid Oxidation as a Therapy for MYC-Overexpressing Triple-Negative Breast Cancer. Nat. Med. 2016, 22, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Quan, J.; Bode, A.M.; Luo, X. ACSL Family: The Regulatory Mechanisms and Therapeutic Implications in Cancer. Eur. J. Pharmacol. 2021, 909, 174397. [Google Scholar] [CrossRef] [PubMed]
- Schlaepfer, I.R.; Joshi, M. CPT1A-Mediated Fat Oxidation, Mechanisms, and Therapeutic Potential. Endocrinology 2020, 161, bqz046. [Google Scholar] [CrossRef]
- Liu, J.; Li, Y.; Xiao, Q.; Li, Y.; Peng, Y.; Gan, Y.; Shu, G.; Yi, H.; Yin, G. Identification of CPT2 as a Prognostic Biomarker by Integrating the Metabolism-Associated Gene Signature in Colorectal Cancer. BMC Cancer 2022, 22, 1038. [Google Scholar] [CrossRef]
- Lin, M.; Lv, D.; Zheng, Y.; Wu, M.; Xu, C.; Zhang, Q.; Wu, L. Downregulation of CPT2 Promotes Tumorigenesis and Chemoresistance to Cisplatin in Hepatocellular Carcinoma. Onco Targets Ther. 2018, 11, 3101–3110. [Google Scholar] [CrossRef] [Green Version]
- Pike, L.S.; Smift, A.L.; Croteau, N.J.; Ferrick, D.A.; Wu, M. Inhibition of Fatty Acid Oxidation by Etomoxir Impairs NADPH Production and Increases Reactive Oxygen Species Resulting in ATP Depletion and Cell Death in Human Glioblastoma Cells. Biochim. Biophys. Acta (BBA)-Bioenerg. 2011, 1807, 726–734. [Google Scholar] [CrossRef] [Green Version]
- Carracedo, A.; Weiss, D.; Leliaert, A.K.; Bhasin, M.; de Boer, V.C.J.; Laurent, G.; Adams, A.C.; Sundvall, M.; Song, S.J.; Ito, K.; et al. A Metabolic Prosurvival Role for PML in Breast Cancer. J. Clin. Investig. 2012, 122, 3088–3100. [Google Scholar] [CrossRef] [PubMed]
- Vance, J.E. Phospholipid Synthesis and Transport in Mammalian Cells. Traffic 2015, 16, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Marchan, R.; Büttner, B.; Lambert, J.; Edlund, K.; Glaeser, I.; Blaszkewicz, M.; Leonhardt, G.; Marienhoff, L.; Kaszta, D.; Anft, M.; et al. Glycerol-3-Phosphate Acyltransferase 1 Promotes Tumor Cell Migration and Poor Survival in Ovarian Carcinoma. Cancer Res. 2017, 77, 4589–4601. [Google Scholar] [CrossRef] [Green Version]
- Fernández, L.P.; Ramos-Ruiz, R.; Herranz, J.; Martín-Hernández, R.; Vargas, T.; Mendiola, M.; Guerra, L.; Reglero, G.; Feliu, J.; Ramírez de Molina, A. The Transcriptional and Mutational Landscapes of Lipid Metabolism-Related Genes in Colon Cancer. Oncotarget 2018, 9, 5919–5930. [Google Scholar] [CrossRef] [Green Version]
- Niesporek, S.; Denkert, C.; Weichert, W.; Köbel, M.; Noske, A.; Sehouli, J.; Singer, J.W.; Dietel, M.; Hauptmann, S. Expression of Lysophosphatidic Acid Acyltransferase Beta (LPAAT-β) in Ovarian Carcinoma: Correlation with Tumour Grading and Prognosis. Br. J. Cancer 2005, 92, 1729–1736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, J.; Zhang, F.; Rachel Tay, L.W.; Boroda, S.; Nian, W.; Levental, K.R.; Levental, I.; Harris, T.E.; Chang, J.T.; Du, G. Lipin-1 Regulation of Phospholipid Synthesis Maintains Endoplasmic Reticulum Homeostasis and Is Critical for Triple-negative Breast Cancer Cell Survival. FASEB J. 2017, 31, 2893–2904. [Google Scholar] [CrossRef] [Green Version]
- Fan, X.; Weng, Y.; Bai, Y.; Wang, Z.; Wang, S.; Zhu, J.; Zhang, F. Lipin-1 Determines Lung Cancer Cell Survival and Chemotherapy Sensitivity by Regulation of Endoplasmic Reticulum Homeostasis and Autophagy. Cancer Med. 2018, 7, 2541–2554. [Google Scholar] [CrossRef] [PubMed]
- Blunsom, N.J.; Cockcroft, S. CDP-Diacylglycerol Synthases (CDS): Gateway to Phosphatidylinositol and Cardiolipin Synthesis. Front. Cell Dev. Biol. 2020, 8, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horibata, Y.; Mitsuhashi, S.; Shimizu, H.; Maejima, S.; Sakamoto, H.; Aoyama, C.; Ando, H.; Sugimoto, H. The Phosphatidylcholine Transfer Protein StarD7 Is Important for Myogenic Differentiation in Mouse Myoblast C2C12 Cells and Human Primary Skeletal Myoblasts. Sci. Rep. 2020, 10, 2845. [Google Scholar] [CrossRef] [Green Version]
- Sha, B.; Luo, M. PI Transfer Protein: The Specific Recognition of Phospholipids and Its Functions. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 1999, 1441, 268–277. [Google Scholar] [CrossRef]
- Madni, Z.K.; Tripathi, S.K.; Salunke, D.M. Structural Insights into the Lipid Transfer Mechanism of a Non-specific Lipid Transfer Protein. Plant J. 2020, 102, 340–352. [Google Scholar] [CrossRef]
- Baxter, A.A.; Hulett, M.D.; Poon, I.K. The Phospholipid Code: A Key Component of Dying Cell Recognition, Tumor Progression and Host–Microbe Interactions. Cell Death Differ. 2015, 22, 1893–1905. [Google Scholar] [CrossRef] [Green Version]
- Jansen, M.; Treutner, K.-H.; Lynen Jansen, P.; Otto, J.; Schmitz, B.; Mueller, S.; Weiss, C.; Tietze, L.; Schumpelick, V. Phospholipids Reduce the Intraperitoneal Adhesion of Colonic Tumor Cells in Rats and Adhesion on Extracellular Matrix in Vitro. Int. J. Color. Dis. 2004, 19, 525–532. [Google Scholar] [CrossRef]
- Roe, N.D.; Handzlik, M.K.; Li, T.; Tian, R. The Role of Diacylglycerol Acyltransferase (DGAT) 1 and 2 in Cardiac Metabolism and Function. Sci. Rep. 2018, 8, 4983. [Google Scholar] [CrossRef] [Green Version]
- Sezgin, E.; Levental, I.; Mayor, S.; Eggeling, C. The Mystery of Membrane Organization: Composition, Regulation and Roles of Lipid Rafts. Nat. Rev. Mol. Cell. Biol. 2017, 18, 361–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, J.; Yang, H.; Song, B.-L. Mechanisms and Regulation of Cholesterol Homeostasis. Nat. Rev. Mol. Cell. Biol. 2020, 21, 225–245. [Google Scholar] [CrossRef]
- Huang, C.-S.; Yu, X.; Fordstrom, P.; Choi, K.; Chung, B.C.; Roh, S.-H.; Chiu, W.; Zhou, M.; Min, X.; Wang, Z. Cryo-EM Structures of NPC1L1 Reveal Mechanisms of Cholesterol Transport and Ezetimibe Inhibition. Sci. Adv. 2020, 6, eabb1989. [Google Scholar] [CrossRef]
- De Gonzalo-Calvo, D.; López-Vilaró, L.; Nasarre, L.; Perez-Olabarria, M.; Vázquez, T.; Escuin, D.; Badimon, L.; Barnadas, A.; Lerma, E.; Llorente-Cortés, V. Intratumor Cholesteryl Ester Accumulation Is Associated with Human Breast Cancer Proliferation and Aggressive Potential: A Molecular and Clinicopathological Study. BMC Cancer 2015, 15, 460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saraon, P.; Trudel, D.; Kron, K.; Dmitromanolakis, A.; Trachtenberg, J.; Bapat, B.; van der Kwast, T.; Jarvi, K.A.; Diamandis, E.P. Evaluation and Prognostic Significance of ACAT1 as a Marker of Prostate Cancer Progression. Prostate 2014, 74, 372–380. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-W.; Chou, C.-T.; Chang, C.-C.; Li, Y.-J.; Chen, S.-T.; Lin, I.-C.; Kok, S.-H.; Cheng, S.-J.; Lee, J.-J.; Wu, T.-S.; et al. HMGCS2 Enhances Invasion and Metastasis via Direct Interaction with PPARα to Activate Src Signaling in Colorectal Cancer and Oral Cancer. Oncotarget 2017, 8, 22460–22476. [Google Scholar] [CrossRef] [Green Version]
- Bengtsson, E.; Nerjovaj, P.; Wangefjord, S.; Nodin, B.; Eberhard, J.; Uhlén, M.; Borgquist, S.; Jirström, K. HMG-CoA Reductase Expression in Primary Colorectal Cancer Correlates with Favourable Clinicopathological Characteristics and an Improved Clinical Outcome. Diagn. Pathol. 2014, 9, 78. [Google Scholar] [CrossRef] [Green Version]
- Borgquist, S.; Jögi, A.; Pontén, F.; Rydén, L.; Brennan, D.J.; Jirström, K. Prognostic Impact of Tumour-Specific HMG-CoA Reductase Expression in Primary Breast Cancer. Breast Cancer Res. 2008, 10, R79. [Google Scholar] [CrossRef] [Green Version]
- Rysman, E.; Brusselmans, K.; Scheys, K.; Timmermans, L.; Derua, R.; Munck, S.; van Veldhoven, P.P.; Waltregny, D.; Daniëls, V.W.; Machiels, J.; et al. De Novo Lipogenesis Protects Cancer Cells from Free Radicals and Chemotherapeutics by Promoting Membrane Lipid Saturation. Cancer Res. 2010, 70, 8117–8126. [Google Scholar] [CrossRef] [Green Version]
- Louis, C.U.; Shohet, J.M. Neuroblastoma: Molecular Pathogenesis and Therapy. Annu. Rev. Med. 2015, 66, 49–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fusco, P.; Esposito, M.; Tonini, G. Chromosome Instability in Neuroblastoma (Review). Oncol. Lett. 2018, 16, 6887–6894. [Google Scholar] [CrossRef] [Green Version]
- Brodeur, G.M. Neuroblastoma: Biological Insights into a Clinical Enigma. Nat. Rev. Cancer 2003, 3, 203–216. [Google Scholar] [CrossRef] [PubMed]
- Mossé, Y.P.; Laudenslager, M.; Longo, L.; Cole, K.A.; Wood, A.; Attiyeh, E.F.; Laquaglia, M.J.; Sennett, R.; Lynch, J.E.; Perri, P.; et al. Identification of ALK as a Major Familial Neuroblastoma Predisposition Gene. Nature 2008, 455, 930–935. [Google Scholar] [CrossRef] [Green Version]
- Slade, R.F.; Hunt, D.A.; Pochet, M.M.; Venema, V.J.; Hennigar, R.A. Characterization and Inhibition of Fatty Acid Synthase in Pediatric Tumor Cell Lines. Anticancer. Res. 2003, 23, 1235–1243. [Google Scholar] [PubMed]
- Ruiz-Pérez, M.V.; Sainero-Alcolado, L.; Oliynyk, G.; Matuschek, I.; Balboni, N.; Ubhayasekera, S.J.K.A.; Snaebjornsson, M.T.; Makowski, K.; Aaltonen, K.; Bexell, D.; et al. Inhibition of Fatty Acid Synthesis Induces Differentiation and Reduces Tumor Burden in Childhood Neuroblastoma. iScience 2021, 24, 102128. [Google Scholar] [CrossRef]
- Ding, Y.; Yang, J.; Ma, Y.; Yao, T.; Chen, X.; Ge, S.; Wang, L.; Fan, X. MYCN and PRC1 Cooperatively Repress Docosahexaenoic Acid Synthesis in Neuroblastoma via ELOVL2. J. Exp. Clin. Cancer Res. 2019, 38, 498. [Google Scholar] [CrossRef]
- Zirath, H.; Frenzel, A.; Oliynyk, G.; Segerström, L.; Westermark, U.K.; Larsson, K.; Munksgaard Persson, M.; Hultenby, K.; Lehtiö, J.; Einvik, C.; et al. MYC Inhibition Induces Metabolic Changes Leading to Accumulation of Lipid Droplets in Tumor Cells. Proc. Natl. Acad. Sci. USA 2013, 110, 10258–10263. [Google Scholar] [CrossRef] [Green Version]
- Oliynyk, G.; Ruiz-Pérez, M.V.; Sainero-Alcolado, L.; Dzieran, J.; Zirath, H.; Gallart-Ayala, H.; Wheelock, C.E.; Johansson, H.J.; Nilsson, R.; Lehtiö, J.; et al. MYCN-Enhanced Oxidative and Glycolytic Metabolism Reveals Vulnerabilities for Targeting Neuroblastoma. iScience 2019, 21, 188–204. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Xia, Y.; Ding, J.; Ye, B.; Zhao, E.; Choi, J.-H.; Alptekin, A.; Yan, C.; Dong, Z.; Huang, S.; et al. Transcriptional Profiling Reveals a Common Metabolic Program in High-Risk Human Neuroblastoma and Mouse Neuroblastoma Sphere-Forming Cells. Cell Rep. 2016, 17, 609–623. [Google Scholar] [CrossRef] [Green Version]
- Uhlen, M.; Oksvold, P.; Fagerberg, L.; Lundberg, E.; Jonasson, K.; Forsberg, M.; Zwahlen, M.; Kampf, C.; Wester, K.; Hober, S.; et al. Towards a Knowledge-Based Human Protein Atlas. Nat. Biotechnol. 2010, 28, 1248–1250. [Google Scholar] [CrossRef]
- Kazantzis, M.; Stahl, A. Fatty Acid Transport Proteins, Implications in Physiology and Disease. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2012, 1821, 852–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friend, S.H.; Bernards, R.; Rogelj, S.; Weinberg, R.A.; Rapaport, J.M.; Albert, D.M.; Dryja, T.P. A Human DNA Segment with Properties of the Gene That Predisposes to Retinoblastoma and Osteosarcoma. Nature 1986, 323, 643–646. [Google Scholar] [CrossRef] [Green Version]
- Dryja, T.P.; Friend, S.; Weinberg, R.A. Genetic Sequences That Predispose to Retinoblastoma and Osteosarcoma. Symp. Fundam. Cancer Res. 1986, 39, 115–119. [Google Scholar]
- Xu, X.L.; Fang, Y.; Lee, T.C.; Forrest, D.; Gregory-Evans, C.; Almeida, D.; Liu, A.; Jhanwar, S.C.; Abramson, D.H.; Cobrinik, D. Retinoblastoma Has Properties of a Cone Precursor Tumor and Depends Upon Cone-Specific MDM2 Signaling. Cell 2009, 137, 1018–1031. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.L.; Singh, H.P.; Wang, L.; Qi, D.-L.; Poulos, B.K.; Abramson, D.H.; Jhanwar, S.C.; Cobrinik, D. Rb Suppresses Human Cone-Precursor-Derived Retinoblastoma Tumours. Nature 2014, 514, 385–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohe, S.; Brundler, M.-A.; Jenkinson, H.; Parulekar, M.; Wilson, M.; Peet, A.C.; McConville, C.M. Metabolite Profiling in Retinoblastoma Identifies Novel Clinicopathological Subgroups. Br. J. Cancer 2015, 113, 1216–1224. [Google Scholar] [CrossRef] [Green Version]
- Sahoo, S.; Ravi Kumar, R.K.; Nicolay, B.; Mohite, O.; Sivaraman, K.; Khetan, V.; Rishi, P.; Ganesan, S.; Subramanyan, K.; Raman, K.; et al. Metabolite Systems Profiling Identifies Exploitable Weaknesses in Retinoblastoma. FEBS Lett. 2019, 593, 23–41. [Google Scholar] [CrossRef]
- Camassei, F.D.; Cozza, R.; Acquaviva, A.; Jenkner, A.; Ravà, L.; Gareri, R.; Donfrancesco, A.; Bosman, C.; Vadala`, P.; Hadjistilianou, T.; et al. Expression of the Lipogenic Enzyme Fatty Acid Synthase (FAS) in Retinoblastoma and Its Correlation with Tumor Aggressiveness. Investig. Opthalmol. Vis. Sci. 2003, 44, 2399. [Google Scholar] [CrossRef] [Green Version]
- Yorek, M.A.; Figard, P.H.; Kaduce, T.L.; Spector, A.A. A Comparison of Lipid Metabolism in Two Human Retinoblastoma Cell Lines. Investig. Ophthalmol. Vis. Sci. 1985, 26, 1148–1154. [Google Scholar]
- Sangeetha, M.; Deepa, P.R.; Rishi, P.; Khetan, V.; Krishnakumar, S. Global Gene Deregulations in FASN Silenced Retinoblastoma Cancer Cells: Molecular and Clinico-Pathological Correlations. J. Cell Biochem. 2015, 116, 2676–2694. [Google Scholar] [CrossRef] [PubMed]
- Vandhana, S.; Coral, K.; Jayanthi, U.; Deepa, P.R.; Krishnakumar, S. Biochemical Changes Accompanying Apoptotic Cell Death in Retinoblastoma Cancer Cells Treated with Lipogenic Enzyme Inhibitors. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2013, 1831, 1458–1466. [Google Scholar] [CrossRef]
- Northcott, P.A.; Korshunov, A.; Witt, H.; Hielscher, T.; Eberhart, C.G.; Mack, S.; Bouffet, E.; Clifford, S.C.; Hawkins, C.E.; French, P.; et al. Medulloblastoma Comprises Four Distinct Molecular Variants. J. Clin. Oncol. 2011, 29, 1408–1414. [Google Scholar] [CrossRef]
- Kool, M.; Korshunov, A.; Remke, M.; Jones, D.T.W.; Schlanstein, M.; Northcott, P.A.; Cho, Y.-J.; Koster, J.; Schouten-van Meeteren, A.; van Vuurden, D.; et al. Molecular Subgroups of Medulloblastoma: An International Meta-Analysis of Transcriptome, Genetic Aberrations, and Clinical Data of WNT, SHH, Group 3, and Group 4 Medulloblastomas. Acta Neuropathol. 2012, 123, 473–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Northcott, P.A.; Jones, D.T.W.; Kool, M.; Robinson, G.W.; Gilbertson, R.J.; Cho, Y.-J.; Pomeroy, S.L.; Korshunov, A.; Lichter, P.; Taylor, M.D.; et al. Medulloblastomics: The End of the Beginning. Nat. Rev. Cancer 2012, 12, 818–834. [Google Scholar] [CrossRef] [Green Version]
- Bhatia, B.; Hsieh, M.; Kenney, A.M.; Nahlé, Z. Mitogenic Sonic Hedgehog Signaling Drives E2F1-Dependent Lipogenesis in Progenitor Cells and Medulloblastoma. Oncogene 2011, 30, 410–422. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.; Liu, J.; Eldridge, R.C.; Gaul, D.A.; Paine, M.R.L.; Uppal, K.; MacDonald, T.J.; Fernández, F.M. Lipidome Signatures of Metastasis in a Transgenic Mouse Model of Sonic Hedgehog Medulloblastoma. Anal. Bioanal. Chem. 2020, 412, 7017–7027. [Google Scholar] [CrossRef] [PubMed]
- Park, A.K.; Lee, J.Y.; Cheong, H.; Ramaswamy, V.; Park, S.-H.; Kool, M.; Phi, J.H.; Choi, S.A.; Cavalli, F.; Taylor, M.D.; et al. Subgroup-Specific Prognostic Signaling and Metabolic Pathways in Pediatric Medulloblastoma. BMC Cancer 2019, 19, 571. [Google Scholar] [CrossRef]
- Blüml, S.; Margol, A.S.; Sposto, R.; Kennedy, R.J.; Robison, N.J.; Vali, M.; Hung, L.T.; Muthugounder, S.; Finlay, J.L.; Erdreich-Epstein, A.; et al. Molecular Subgroups of Medulloblastoma Identification Using Noninvasive Magnetic Resonance Spectroscopy. Neuro Oncol. 2016, 18, 126–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, R.E.; Zhang, L.; Peri, S.; Kuo, Y.-M.; Du, F.; Egleston, B.L.; Ng, J.M.Y.; Andrews, A.J.; Astsaturov, I.; Curran, T.; et al. Statins Synergize with Hedgehog Pathway Inhibitors for Treatment of Medulloblastoma. Clin. Cancer Res. 2018, 24, 1375–1388. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.; Mahmud, I.; Pokhrel, R.; Murad, R.; Yuan, M.; Stapleton, S.; Bettegowda, C.; Jallo, G.; Eberhart, C.G.; Garrett, T.; et al. Medulloblastoma Cerebrospinal Fluid Reveals Metabolites and Lipids Indicative of Hypoxia and Cancer-Specific RNAs. Acta Neuropathol. Commun. 2022, 10, 25. [Google Scholar] [CrossRef] [PubMed]
- Bennett, C.D.; Kohe, S.E.; Gill, S.K.; Davies, N.P.; Wilson, M.; Storer, L.C.D.; Ritzmann, T.; Paine, S.M.L.; Scott, I.S.; Nicklaus-Wollenteit, I.; et al. Tissue Metabolite Profiles for the Characterisation of Paediatric Cerebellar Tumours. Sci. Rep. 2018, 8, 11992. [Google Scholar] [CrossRef] [PubMed]
- Marabitti, V.; Giansanti, M.; de Mitri, F.; Gatto, F.; Mastronuzzi, A.; Nazio, F. Pathological Implications of Metabolic Reprogramming and Its Therapeutic Potential in Medulloblastoma. Front. Cell Dev. Biol. 2022, 10, 1007641. [Google Scholar] [CrossRef]
- Tech, K.; Deshmukh, M.; Gershon, T.R. Adaptations of Energy Metabolism during Cerebellar Neurogenesis Are Co-Opted in Medulloblastoma. Cancer Lett. 2015, 356, 268–272. [Google Scholar] [CrossRef] [Green Version]
- Tech, K.; Gershon, T.R. Energy Metabolism in Neurodevelopment and Medulloblastoma. Transl. Pediatr. 2015, 4, 12–19. [Google Scholar] [CrossRef]
- Lupien, L.; Boynton, A.; Chacon, M.; Kumbhani, R.; Gionet, G.; Goodale, A.; Root, D.; Keshishian, H.; Robinson, M.; Carr, S.; et al. MEDB-73. Lipid Metabolism as a Therapeutic Vulnerability in BET Inhibitor-Resistant Medulloblastoma. Neuro Oncol. 2022, 24, i123. [Google Scholar] [CrossRef]
- Martinelli, S.; McDowell, H.P.; Vigne, S.D.; Kokai, G.; Uccini, S.; Tartaglia, M.; Dominici, C. RAS Signaling Dysregulation in Human Embryonal Rhabdomyosarcoma. Genes Chromosomes Cancer 2009, 48, 975–982. [Google Scholar] [CrossRef]
- Zhu, B.; Davie, J.K. New Insights into Signalling-Pathway Alterations in Rhabdomyosarcoma. Br. J. Cancer 2015, 112, 227–231. [Google Scholar] [CrossRef]
- Missiaglia, E.; Williamson, D.; Chisholm, J.; Wirapati, P.; Pierron, G.; Petel, F.; Concordet, J.-P.; Thway, K.; Oberlin, O.; Pritchard-Jones, K.; et al. PAX3/FOXO1 Fusion Gene Status Is the Key Prognostic Molecular Marker in Rhabdomyosarcoma and Significantly Improves Current Risk Stratification. J. Clin. Oncol. 2012, 30, 1670–1677. [Google Scholar] [CrossRef]
- Skapek, S.X.; Anderson, J.; Barr, F.G.; Bridge, J.A.; Gastier-Foster, J.M.; Parham, D.M.; Rudzinski, E.R.; Triche, T.; Hawkins, D.S. PAX-FOXO1 Fusion Status Drives Unfavorable Outcome for Children with Rhabdomyosarcoma: A Children’s Oncology Group Report. Pediatr. Blood Cancer 2013, 60, 1411–1417. [Google Scholar] [CrossRef] [Green Version]
- Mercado, G.E.; Xia, S.J.; Zhang, C.; Ahn, E.H.; Gustafson, D.M.; Laé, M.; Ladanyi, M.; Barr, F.G. Identification of PAX3-FKHR-Regulated Genes Differentially Expressed between Alveolar and Embryonal Rhabdomyosarcoma: Focus on MYCN as a Biologically Relevant Target. Genes Chromosomes Cancer 2008, 47, 510–520. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Wang, Y.-D.; Wu, J.; Cui, J.; Chen, T. Carnitine Palmitoyltransferase 1A (CPT1A): A Transcriptional Target of PAX3-FKHR and Mediates PAX3-FKHR–Dependent Motility in Alveolar Rhabdomyosarcoma Cells. BMC Cancer 2012, 12, 154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyagaki, S.; Kikuchi, K.; Mori, J.; Lopaschuk, G.D.; Iehara, T.; Hosoi, H. Inhibition of Lipid Metabolism Exerts Antitumor Effects on Rhabdomyosarcoma. Cancer Med. 2021, 10, 6442–6455. [Google Scholar] [CrossRef]
- Szychot, E.; Apps, J.; Pritchard-Jones, K. Wilms’ Tumor: Biology, Diagnosis and Treatment. Transl. Pediatr. 2014, 3, 12–24. [Google Scholar] [CrossRef] [PubMed]
- Metzger, M.L.; Dome, J.S. Current Therapy for Wilms’ Tumor. Oncologist 2005, 10, 815–826. [Google Scholar] [CrossRef] [Green Version]
- Williams, R.D.; Al-Saadi, R.; Natrajan, R.; Mackay, A.; Chagtai, T.; Little, S.; Hing, S.N.; Fenwick, K.; Ashworth, A.; Grundy, P.; et al. Molecular Profiling Reveals Frequent Gain of MYCN and Anaplasia-Specific Loss of 4q and 14q in Wilms Tumor. Genes Chromosomes Cancer 2011, 50, 982–995. [Google Scholar] [CrossRef]
- Williams, R.D.; Al-Saadi, R.; Chagtai, T.; Popov, S.; Messahel, B.; Sebire, N.; Gessler, M.; Wegert, J.; Graf, N.; Leuschner, I.; et al. Subtype-Specific FBXW7 Mutation and MYCN Copy Number Gain in Wilms’ Tumor. Clin. Cancer Res. 2010, 16, 2036–2045. [Google Scholar] [CrossRef] [Green Version]
- Williams, R.D.; Chagtai, T.; Alcaide-German, M.; Apps, J.; Wegert, J.; Popov, S.; Vujanic, G.; van Tinteren, H.; van den Heuvel-Eibrink, M.M.; Kool, M.; et al. Multiple Mechanisms of MYCN Dysregulation in Wilms Tumour. Oncotarget 2015, 6, 7232–7243. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Du, G.; Wu, Y.; Zhang, Y.; Guo, F.; Liu, W.; Wu, R. Association between Different Levels of Lipid Metabolism related Enzymes and Fatty Acid Synthase in Wilms’ Tumor. Int. J. Oncol. 2019, 56, 568–580. [Google Scholar] [CrossRef]
- Hammer, E.; Ernst, F.D.; Thiele, A.; Karanam, N.K.; Kujath, C.; Evert, M.; Völker, U.; Barthlen, W. Kidney Protein Profiling of Wilms’ Tumor Patients by Analysis of Formalin-Fixed Paraffin-Embedded Tissue Samples. Clin. Chim. Acta 2014, 433, 235–241. [Google Scholar] [CrossRef]
- Rae, F.K.; Martinez, G.; Gillinder, K.R.; Smith, A.; Shooter, G.; Forrest, A.R.; Grimmond, S.M.; Little, M.H. Anlaysis of Complementary Expression Profiles Following WT1 Induction versus Repression Reveals the Cholesterol/Fatty Acid Synthetic Pathways as a Possible Major Target of WT1. Oncogene 2004, 23, 3067–3079. [Google Scholar] [CrossRef] [Green Version]
- Loomba, R.; Mohseni, R.; Lucas, K.J.; Gutierrez, J.A.; Perry, R.G.; Trotter, J.F.; Rahimi, R.S.; Harrison, S.A.; Ajmera, V.; Wayne, J.D.; et al. TVB-2640 (FASN Inhibitor) for the Treatment of Nonalcoholic Steatohepatitis: FASCINATE-1, a Randomized, Placebo-Controlled Phase 2a Trial. Gastroenterology 2021, 161, 1475–1486. [Google Scholar] [CrossRef]
- Dean, E.J.; Falchook, G.S.; Patel, M.R.; Brenner, A.J.; Infante, J.R.; Arkenau, H.-T.; Borazanci, E.H.; Lopez, J.S.; Pant, S.; Schmid, P.; et al. Preliminary Activity in the First in Human Study of the First-in-Class Fatty Acid Synthase (FASN) Inhibitor, TVB-2640. J. Clin. Oncol. 2016, 34, 2512. [Google Scholar] [CrossRef]
- Ventura, R.; Mordec, K.; Waszczuk, J.; Wang, Z.; Lai, J.; Fridlib, M.; Buckley, D.; Kemble, G.; Heuer, T.S. Inhibition of de Novo Palmitate Synthesis by Fatty Acid Synthase Induces Apoptosis in Tumor Cells by Remodeling Cell Membranes, Inhibiting Signaling Pathways, and Reprogramming Gene Expression. EBioMedicine 2015, 2, 808–824. [Google Scholar] [CrossRef] [Green Version]
- Kridel, S.J.; Axelrod, F.; Rozenkrantz, N.; Smith, J.W. Orlistat Is a Novel Inhibitor of Fatty Acid Synthase with Antitumor Activity. Cancer Res. 2004, 64, 2070–2075. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Xu, C.; Sun, M.; Luo, D.; Liao, D.; Cao, D. Acetyl-CoA Carboxylase-α Inhibitor TOFA Induces Human Cancer Cell Apoptosis. Biochem. Biophys. Res. Commun. 2009, 385, 302–306. [Google Scholar] [CrossRef] [Green Version]
- Beckers, A.; Organe, S.; Timmermans, L.; Scheys, K.; Peeters, A.; Brusselmans, K.; Verhoeven, G.; Swinnen, J.V. Chemical Inhibition of Acetyl-CoA Carboxylase Induces Growth Arrest and Cytotoxicity Selectively in Cancer Cells. Cancer Res. 2007, 67, 8180–8187. [Google Scholar] [CrossRef] [Green Version]
- Von Roemeling, C.A.; Marlow, L.A.; Wei, J.J.; Cooper, S.J.; Caulfield, T.R.; Wu, K.; Tan, W.W.; Tun, H.W.; Copland, J.A. Stearoyl-CoA Desaturase 1 Is a Novel Molecular Therapeutic Target for Clear Cell Renal Cell Carcinoma. Clin. Cancer Res. 2013, 19, 2368–2380. [Google Scholar] [CrossRef] [Green Version]
- Huang, G.-M.; Jiang, Q.-H.; Cai, C.; Qu, M.; Shen, W. SCD1 Negatively Regulates Autophagy-Induced Cell Death in Human Hepatocellular Carcinoma through Inactivation of the AMPK Signaling Pathway. Cancer Lett. 2015, 358, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Ren, J.; Yang, L.; Li, Y.; Fu, J.; Li, Y.; Tian, Y.; Qiu, F.; Liu, Z.; Qiu, Y. Stearoyl-CoA Desaturase-1 Mediated Cell Apoptosis in Colorectal Cancer by Promoting Ceramide Synthesis. Sci. Rep. 2016, 6, 19665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tesfay, L.; Paul, B.T.; Konstorum, A.; Deng, Z.; Cox, A.O.; Lee, J.; Furdui, C.M.; Hegde, P.; Torti, F.M.; Torti, S.V. Stearoyl-CoA Desaturase 1 Protects Ovarian Cancer Cells from Ferroptotic Cell Death. Cancer Res. 2019, 79, 5355–5366. [Google Scholar] [CrossRef]
- She, K.; Fang, S.; Du, W.; Fan, X.; He, J.; Pan, H.; Huang, L.; He, P.; Huang, J. SCD1 Is Required for EGFR-Targeting Cancer Therapy of Lung Cancer via Re-Activation of EGFR/PI3K/AKT Signals. Cancer Cell Int. 2019, 19, 103. [Google Scholar] [CrossRef] [Green Version]
- Pinkham, K.; Park, D.J.; Hashemiaghdam, A.; Kirov, A.B.; Adam, I.; Rosiak, K.; da Hora, C.C.; Teng, J.; Cheah, P.S.; Carvalho, L.; et al. Stearoyl CoA Desaturase Is Essential for Regulation of Endoplasmic Reticulum Homeostasis and Tumor Growth in Glioblastoma Cancer Stem Cells. Stem Cell Rep. 2019, 12, 712–727. [Google Scholar] [CrossRef] [Green Version]
- Fritz, V.; Benfodda, Z.; Rodier, G.; Henriquet, C.; Iborra, F.; Avancès, C.; Allory, Y.; de la Taille, A.; Culine, S.; Blancou, H.; et al. Abrogation of de Novo Lipogenesis by Stearoyl-CoA Desaturase 1 Inhibition Interferes with Oncogenic Signaling and Blocks Prostate Cancer Progression in Mice. Mol. Cancer Ther. 2010, 9, 1740–1754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, Y.; Sun, S.; Wang, J.; Fei, F.; Dong, Z.; Ke, A.-W.; He, R.; Wang, L.; Zhang, L.; Ji, M.-B.; et al. Canonical Wnt Signaling Remodels Lipid Metabolism in Zebrafish Hepatocytes Following Ras Oncogenic Insult. Cancer Res. 2018, 78, 5548–5560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apostolova, S.N.; Toshkova, R.A.; Momchilova, A.B.; Tzoneva, R.D. Statins and Alkylphospholipids as New Anticancer Agents Targeting Lipid Metabolism. Anticancer. Agents Med. Chem. 2016, 16, 1512–1522. [Google Scholar] [CrossRef] [PubMed]
- Longo, J.; van Leeuwen, J.E.; Elbaz, M.; Branchard, E.; Penn, L.Z. Statins as Anticancer Agents in the Era of Precision Medicine. Clin. Cancer Res. 2020, 26, 5791–5800. [Google Scholar] [CrossRef] [PubMed]
- Knox, J.J.; Siu, L.L.; Chen, E.; Dimitroulakos, J.; Kamel-Reid, S.; Moore, M.J.; Chin, S.; Irish, J.; LaFramboise, S.; Oza, A.M. A Phase I Trial of Prolonged Administration of Lovastatin in Patients with Recurrent or Metastatic Squamous Cell Carcinoma of the Head and Neck or of the Cervix. Eur. J. Cancer 2005, 41, 523–530. [Google Scholar] [CrossRef]
- Kornblau, S.M.; Banker, D.E.; Stirewalt, D.; Shen, D.; Lemker, E.; Verstovsek, S.; Estrov, Z.; Faderl, S.; Cortes, J.; Beran, M.; et al. Blockade of Adaptive Defensive Changes in Cholesterol Uptake and Synthesis in AML by the Addition of Pravastatin to Idarubicin + High-Dose Ara-C: A Phase 1 Study. Blood 2007, 109, 2999–3006. [Google Scholar] [CrossRef]
- Hus, M.; Grzasko, N.; Szostek, M.; Pluta, A.; Helbig, G.; Woszczyk, D.; Adamczyk-Cioch, M.; Jawniak, D.; Legiec, W.; Morawska, M.; et al. Thalidomide, Dexamethasone and Lovastatin with Autologous Stem Cell Transplantation as a Salvage Immunomodulatory Therapy in Patients with Relapsed and Refractory Multiple Myeloma. Ann. Hematol. 2011, 90, 1161–1166. [Google Scholar] [CrossRef] [Green Version]
- Goss, G.D.; Jonker, D.J.; Laurie, S.A.; Weberpals, J.I.; Oza, A.M.; Spaans, J.N.; la Porte, C.; Dimitroulakos, J. A Phase I Study of High-Dose Rosuvastatin with Standard Dose Erlotinib in Patients with Advanced Solid Malignancies. J. Transl. Med. 2016, 14, 83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kant, S.; Kesarwani, P.; Guastella, A.R.; Kumar, P.; Graham, S.F.; Buelow, K.L.; Nakano, I.; Chinnaiyan, P. Perhexiline Demonstrates FYN-Mediated Antitumor Activity in Glioblastoma. Mol. Cancer Ther. 2020, 19, 1415–1422. [Google Scholar] [CrossRef]
- Ren, X.-R.; Wang, J.; Osada, T.; Mook, R.A.; Morse, M.A.; Barak, L.S.; Lyerly, H.K.; Chen, W. Perhexiline Promotes HER3 Ablation through Receptor Internalization and Inhibits Tumor Growth. Breast Cancer Res. 2015, 17, 20. [Google Scholar] [CrossRef] [Green Version]
- Binienda, A.; Ziolkowska, S.; Pluciennik, E. The Anticancer Properties of Silibinin: Its Molecular Mechanism and Therapeutic Effect in Breast Cancer. Anticancer. Agents Med. Chem. 2020, 20, 1787–1796. [Google Scholar] [CrossRef]
- Brovkovych, V.; Izhar, Y.; Danes, J.M.; Dubrovskyi, O.; Sakallioglu, I.T.; Morrow, L.M.; Atilla-Gokcumen, G.E.; Frasor, J. Fatostatin Induces Pro- and Anti-Apoptotic Lipid Accumulation in Breast Cancer. Oncogenesis 2018, 7, 66. [Google Scholar] [CrossRef] [Green Version]
- Gholkar, A.A.; Cheung, K.; Williams, K.J.; Lo, Y.-C.; Hamideh, S.A.; Nnebe, C.; Khuu, C.; Bensinger, S.J.; Torres, J.Z. Fatostatin Inhibits Cancer Cell Proliferation by Affecting Mitotic Microtubule Spindle Assembly and Cell Division. J. Biol. Chem. 2016, 291, 17001–17008. [Google Scholar] [CrossRef] [Green Version]
- Williams, K.J.; Argus, J.P.; Zhu, Y.; Wilks, M.Q.; Marbois, B.N.; York, A.G.; Kidani, Y.; Pourzia, A.L.; Akhavan, D.; Lisiero, D.N.; et al. An Essential Requirement for the SCAP/SREBP Signaling Axis to Protect Cancer Cells from Lipotoxicity. Cancer Res. 2013, 73, 2850–2862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, M.; Su, L.; Yuan, Y.-C.; Li, H.; Chow, W.A. Nelfinavir and Nelfinavir Analogs Block Site-2 Protease Cleavage to Inhibit Castration-Resistant Prostate Cancer. Sci. Rep. 2015, 5, 9698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heuer, T.S.; Ventura, R.; Mordec, K.; Lai, J.; Fridlib, M.; Buckley, D.; Kemble, G. FASN Inhibition and Taxane Treatment Combine to Enhance Anti-Tumor Efficacy in Diverse Xenograft Tumor Models through Disruption of Tubulin Palmitoylation and Microtubule Organization and FASN Inhibition-Mediated Effects on Oncogenic Signaling and Gene Expression. EBioMedicine 2017, 16, 51–62. [Google Scholar] [CrossRef] [Green Version]
- Tan, W.; Zhong, Z.; Wang, S.; Suo, Z.; Yang, X.; Hu, X.; Wang, Y. Berberine Regulated Lipid Metabolism in the Presence of C75, Compound C, and TOFA in Breast Cancer Cell Line MCF-7. Evid.-Based Complement. Altern. Med. 2015, 2015, 396035. [Google Scholar] [CrossRef] [Green Version]
- Potze, L.; di Franco, S.; Grandela, C.; Pras-Raves, M.L.; Picavet, D.I.; van Veen, H.A.; van Lenthe, H.; Mullauer, F.B.; van der Wel, N.N.; Luyf, A.; et al. Betulinic Acid Induces a Novel Cell Death Pathway That Depends on Cardiolipin Modification. Oncogene 2016, 35, 427–437. [Google Scholar] [CrossRef]
- Zhang, I.; Cui, Y.; Amiri, A.; Ding, Y.; Campbell, R.E.; Maysinger, D. Pharmacological Inhibition of Lipid Droplet Formation Enhances the Effectiveness of Curcumin in Glioblastoma. Eur. J. Pharm. Biopharm. 2016, 100, 66–76. [Google Scholar] [CrossRef] [Green Version]
- Dang, C.V. MYC, Metabolism, Cell Growth, and Tumorigenesis. Cold Spring Harb. Perspect. Med. 2013, 3, a014217. [Google Scholar] [CrossRef] [Green Version]
- Sradhanjali, S.; Reddy, M.M. Inhibition of Pyruvate Dehydrogenase Kinase as a Therapeutic Strategy against Cancer. Curr. Top. Med. Chem. 2018, 18, 444–453. [Google Scholar] [CrossRef]
- Ren, P.; Yue, M.; Xiao, D.; Xiu, R.; Gan, L.; Liu, H.; Qing, G. ATF4 and N-Myc Coordinate Glutamine Metabolism in MYCN -Amplified Neuroblastoma Cells through ASCT2 Activation. J. Pathol. 2015, 235, 90–100. [Google Scholar] [CrossRef]
- Borah, N.A.; Reddy, M.M. Aurora Kinase B Inhibition: A Potential Therapeutic Strategy for Cancer. Molecules 2021, 26, 1981. [Google Scholar] [CrossRef] [PubMed]
- Borah, N.A.; Sradhanjali, S.; Barik, M.R.; Jha, A.; Tripathy, D.; Kaliki, S.; Rath, S.; Raghav, S.K.; Patnaik, S.; Mittal, R.; et al. Aurora Kinase B Expression, Its Regulation and Therapeutic Targeting in Human Retinoblastoma. Investig. Opthalmol. Vis. Sci. 2021, 62, 16. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, C.; Wang, X.; Briggs, M.R.; Admon, A.; Wu, J.; Hua, X.; Goldstein, J.L.; Brown, M.S. SREBP-1, a Basic-Helix-Loop-Helix-Leucine Zipper Protein That Controls Transcription of the Low Density Lipoprotein Receptor Gene. Cell 1993, 75, 187–197. [Google Scholar] [CrossRef]
- Horton, J.D.; Shah, N.A.; Warrington, J.A.; Anderson, N.N.; Park, S.W.; Brown, M.S.; Goldstein, J.L. Combined Analysis of Oligonucleotide Microarray Data from Transgenic and Knockout Mice Identifies Direct SREBP Target Genes. Proc. Natl. Acad. Sci. USA 2003, 100, 12027–12032. [Google Scholar] [CrossRef] [Green Version]
- Horton, J.D.; Goldstein, J.L.; Brown, M.S. SREBPs: Activators of the Complete Program of Cholesterol and Fatty Acid Synthesis in the Liver. J. Clin. Investig. 2002, 109, 1125–1131. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.-C.; Li, X.; Liu, J.; Lin, J.; Chung, L.W.K. Activation of Androgen Receptor, Lipogenesis, and Oxidative Stress Converged by SREBP-1 Is Responsible for Regulating Growth and Progression of Prostate Cancer Cells. Mol. Cancer Res. 2012, 10, 133–142. [Google Scholar] [CrossRef] [Green Version]
- Guo, D.; Reinitz, F.; Youssef, M.; Hong, C.; Nathanson, D.; Akhavan, D.; Kuga, D.; Amzajerdi, A.N.; Soto, H.; Zhu, S.; et al. An LXR Agonist Promotes Glioblastoma Cell Death through Inhibition of an EGFR/AKT/SREBP-1/LDLR–Dependent Pathway. Cancer Discov. 2011, 1, 442–456. [Google Scholar] [CrossRef] [Green Version]
- Bao, J.; Zhu, L.; Zhu, Q.; Su, J.; Liu, M.; Huang, W. SREBP-1 Is an Independent Prognostic Marker and Promotes Invasion and Migration in Breast Cancer. Oncol. Lett. 2016, 12, 2409–2416. [Google Scholar] [CrossRef] [Green Version]
- Cheng, C.; Ru, P.; Geng, F.; Liu, J.; Yoo, J.Y.; Wu, X.; Cheng, X.; Euthine, V.; Hu, P.; Guo, J.Y.; et al. Glucose-Mediated N-Glycosylation of SCAP Is Essential for SREBP-1 Activation and Tumor Growth. Cancer Cell 2015, 28, 569–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hua, X.; Yokoyama, C.; Wu, J.; Briggs, M.R.; Brown, M.S.; Goldstein, J.L.; Wang, X. SREBP-2, a Second Basic-Helix-Loop-Helix-Leucine Zipper Protein That Stimulates Transcription by Binding to a Sterol Regulatory Element. Proc. Natl. Acad. Sci. USA 1993, 90, 11603–11607. [Google Scholar] [CrossRef] [Green Version]
- Hu, Z.; Cheng, C.; Wang, Y.; Chen, T.; Tu, J.; Niu, C.; Xing, R.; Wang, Y.; Xu, Y. Synergistic Effect of Statins and Abiraterone Acetate on the Growth Inhibition of Neuroblastoma via Targeting Androgen Receptor. Front Oncol 2021, 11, 595285. [Google Scholar] [CrossRef]
- Wu, Y.; Chen, K.; Liu, X.; Huang, L.; Zhao, D.; Li, L.; Gao, M.; Pei, D.; Wang, C.; Liu, X. Srebp-1 Interacts with c-Myc to Enhance Somatic Cell Reprogramming. Stem Cells 2016, 34, 83–92. [Google Scholar] [CrossRef]
- Shroff, E.H.; Eberlin, L.S.; Dang, V.M.; Gouw, A.M.; Gabay, M.; Adam, S.J.; Bellovin, D.I.; Tran, P.T.; Philbrick, W.M.; Garcia-Ocana, A.; et al. MYC Oncogene Overexpression Drives Renal Cell Carcinoma in a Mouse Model through Glutamine Metabolism. Proc. Natl. Acad. Sci. USA 2015, 112, 6539–6544. [Google Scholar] [CrossRef] [Green Version]
- Gouw, A.M.; Eberlin, L.S.; Margulis, K.; Sullivan, D.K.; Toal, G.G.; Tong, L.; Zare, R.N.; Felsher, D.W. Oncogene KRAS Activates Fatty Acid Synthase, Resulting in Specific ERK and Lipid Signatures Associated with Lung Adenocarcinoma. Proc. Natl. Acad. Sci. USA 2017, 114, 4300–4305. [Google Scholar] [CrossRef] [Green Version]
- Kondo, A.; Yamamoto, S.; Nakaki, R.; Shimamura, T.; Hamakubo, T.; Sakai, J.; Kodama, T.; Yoshida, T.; Aburatani, H.; Osawa, T. Extracellular Acidic PH Activates the Sterol Regulatory Element-Binding Protein 2 to Promote Tumor Progression. Cell Rep. 2017, 18, 2228–2242. [Google Scholar] [CrossRef] [Green Version]
- Carroll, P.A.; Diolaiti, D.; McFerrin, L.; Gu, H.; Djukovic, D.; Du, J.; Cheng, P.F.; Anderson, S.; Ulrich, M.; Hurley, J.B.; et al. Deregulated Myc Requires MondoA/Mlx for Metabolic Reprogramming and Tumorigenesis. Cancer Cell 2015, 27, 271–285. [Google Scholar] [CrossRef] [Green Version]
- Praz, V. The Eukaryotic Promoter Database, EPD: New Entry Types and Links to Gene Expression Data. Nucleic Acids Res. 2002, 30, 322–324. [Google Scholar] [CrossRef]
- Singh, K.B.; Hahm, E.-R.; Kim, S.-H.; Wendell, S.G.; Singh, S.V. A Novel Metabolic Function of Myc in Regulation of Fatty Acid Synthesis in Prostate Cancer. Oncogene 2021, 40, 592–602. [Google Scholar] [CrossRef]
- Morelli, E.; Fulciniti, M.; Samur, M.K.; Ribeiro, C.; Wert-Lamas, L.; Gulla, A.; Aktas Samur, A.; Federico, C.; Scionti, F.; Yao, Y.; et al. Targeting Myeloma Cell Metab.olism Via Disruption of the Lnc-17-92 Transcriptional Program: Druggable New Vulnerability in Multiple Myeloma. Blood 2019, 134, 317. [Google Scholar] [CrossRef]
- Rugolo, F.; Bazan, N.G.; Calandria, J.; Jun, B.; Raschellà, G.; Melino, G.; Agostini, M. The Expression of ELOVL4, Repressed by MYCN, Defines Neuroblastoma Patients with Good Outcome. Oncogene 2021, 40, 5741–5751. [Google Scholar] [CrossRef]
- Qin, X.-Y.; Su, T.; Yu, W.; Kojima, S. Lipid Desaturation-Associated Endoplasmic Reticulum Stress Regulates MYCN Gene Expression in Hepatocellular Carcinoma Cells. Cell Death Dis. 2020, 11, 66. [Google Scholar] [CrossRef] [Green Version]
- Qin, X.-Y.; Gailhouste, L. Non-Genomic Control of Dynamic MYCN Gene Expression in Liver Cancer. Front. Oncol. 2021, 10, 618515. [Google Scholar] [CrossRef]
- Tsai, W.-C.; Hsu, S.-D.; Hsu, C.-S.; Lai, T.-C.; Chen, S.-J.; Shen, R.; Huang, Y.; Chen, H.-C.; Lee, C.-H.; Tsai, T.-F.; et al. MicroRNA-122 Plays a Critical Role in Liver Homeostasis and Hepatocarcinogenesis. J. Clin. Investig. 2012, 122, 2884–2897. [Google Scholar] [CrossRef] [Green Version]
- Horie, T.; Ono, K.; Horiguchi, M.; Nishi, H.; Nakamura, T.; Nagao, K.; Kinoshita, M.; Kuwabara, Y.; Marusawa, H.; Iwanaga, Y.; et al. MicroRNA-33 Encoded by an Intron of Sterol Regulatory Element-Binding Protein 2 (Srebp2) Regulates HDL in Vivo. Proc. Natl. Acad. Sci. USA 2010, 107, 17321–17326. [Google Scholar] [CrossRef] [Green Version]
- Rayner, K.J.; Suárez, Y.; Dávalos, A.; Parathath, S.; Fitzgerald, M.L.; Tamehiro, N.; Fisher, E.A.; Moore, K.J.; Fernández-Hernando, C. MiR-33 Contributes to the Regulation of Cholesterol Homeostasis. Science 2010, 328, 1570–1573. [Google Scholar] [CrossRef] [Green Version]
- Horie, T.; Nishino, T.; Baba, O.; Kuwabara, Y.; Nakao, T.; Nishiga, M.; Usami, S.; Izuhara, M.; Sowa, N.; Yahagi, N.; et al. MicroRNA-33 Regulates Sterol Regulatory Element-Binding Protein 1 Expression in Mice. Nat. Commun. 2013, 4, 2883. [Google Scholar] [CrossRef] [Green Version]
- Shirasaki, T.; Honda, M.; Shimakami, T.; Horii, R.; Yamashita, T.; Sakai, Y.; Sakai, A.; Okada, H.; Watanabe, R.; Murakami, S.; et al. MicroRNA-27a Regulates Lipid Metabolism and Inhibits Hepatitis C Virus Replication in Human Hepatoma Cells. J. Virol. 2013, 87, 5270–5286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iliopoulos, D.; Drosatos, K.; Hiyama, Y.; Goldberg, I.J.; Zannis, V.I. MicroRNA-370 Controls the Expression of MicroRNA-122 and Cpt1α and Affects Lipid Metabolism. J. Lipid Res. 2010, 51, 1513–1523. [Google Scholar] [CrossRef] [Green Version]
- Guo, S.; Zhang, Y.; Wang, S.; Yang, T.; Ma, B.; Li, X.; Zhang, Y.; Jiang, X. LncRNA PCA3 Promotes Antimony-Induced Lipid Metabolic Disorder in Prostate Cancer by Targeting MIR-132-3 P/SREBP1 Signaling. Toxicol. Lett. 2021, 348, 50–58. [Google Scholar] [CrossRef]
- Guo, L.; Lu, J.; Gao, J.; Li, M.; Wang, H.; Zhan, X. The Function of SNHG7/MiR-449a/ACSL1 Axis in Thyroid Cancer. J. Cell Biochem. 2020, 121, 4034–4042. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Talapatra, J.; Reddy, M.M. Lipid Metabolic Reprogramming in Embryonal Neoplasms with MYCN Amplification. Cancers 2023, 15, 2144. https://doi.org/10.3390/cancers15072144
Talapatra J, Reddy MM. Lipid Metabolic Reprogramming in Embryonal Neoplasms with MYCN Amplification. Cancers. 2023; 15(7):2144. https://doi.org/10.3390/cancers15072144
Chicago/Turabian StyleTalapatra, Jyotirmayee, and Mamatha M. Reddy. 2023. "Lipid Metabolic Reprogramming in Embryonal Neoplasms with MYCN Amplification" Cancers 15, no. 7: 2144. https://doi.org/10.3390/cancers15072144