
Diagnosis and Treatment of Peripheral and Cranial Nerve Tumors with Expert Recommendations: An EUropean Network for RAre CANcers (EURACAN) Initiative
 , , , ,
, , , ,  and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Methods
3. Epidemiology and Clinical Features
4. Genetic Tumor Syndromes Correlated with Cranial and Peripheral Nerve Sheath Tumors
5. Pathology and Molecular Markers
6. Imaging
7. Surgery
8. Radiotherapy
9. Medical Treatments
9.1. Vestibular Schwannomas
9.2. Plexiform Neurofibromas
9.3. Malignant Nerve Sheath Tumors
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data availability Statement
Conflicts of Interest
References
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- The WHO Classification of Tumours Editorial Board. WHO Classification of Tumours Soft Tissue and Bone Tumours, 5th ed.; IARC Press: Lyon, France, 2020. [Google Scholar]
- Ostrom, Q.T.; Cioffi, G.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2014–2018. Neuro-Oncology 2021, 23 (Suppl. S3), iii1–iii105. [Google Scholar] [CrossRef] [PubMed]
- Brainin, M.; Barnes, M.; Baron, J.C.; Gilhus, N.E.; Hughes, R.; Selmaj, K.; Waldemar, G.; Guideline Standards Subcommittee of the EFNS Scientific Committee. Guidance for the preparation of neurological management guidelines by EFNS scientific task forces—Revised recommendations 2004. Eur. J. Neurol. 2004, 11, 577–581. [Google Scholar] [CrossRef] [PubMed]
- Casadei, G.P.; Komori, T.; Scheithauer, B.W.; Miller, G.M.; Parisi, J.E.; Kelly, P.J. Intracranial parenchymal schwannoma. A clinico-pathological and neuroimaging study of nine cases. J. Neurosurg. 1993, 79, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Voltaggio, L.; Murray, R.; Lasota, J.; Miettinen, M. Gastric schwannoma: A clinicopathologic study of 51 cases and critical review of the literature. Hum. Pathol. 2012, 43, 650–659. [Google Scholar] [CrossRef] [Green Version]
- Babu, R.; Sharma, R.; Bagley, J.H.; Hatef, J.; Friedman, A.H.; Adamson, C. Vestibular schwannomas in the modern era: Epidemiology, treatment trends, and disparities in management. J. Neurosurg. 2013, 119, 121–130. [Google Scholar] [CrossRef] [Green Version]
- Evans, D.G.R. Neurofibromatosis type 2 (NF2): A clinical and molecular review. Orphanet J. Rare Dis. 2009, 4, 16. [Google Scholar] [CrossRef] [Green Version]
- Andersen, J.F.; Nilsen, K.S.; Vassbotn, F.S.; Møller, P.; Myrseth, E.; Lund-Johansen, M.; Goplen, F.K. Predictors of Vertigo in Patients with Untreated Vestibular Schwannoma. Otol. Neurotol. 2015, 36, 647–652. [Google Scholar] [CrossRef]
- Woodruff, J.M.; Selig, A.M.; Crowley, K.; Allen, P.W. Schwannoma (Neurilemoma) with Malignant Transformation A Rare, Distinctive Peripheral Nerve Tumor. Am. J. Surg. Pathol. 1994, 18, 882–895. [Google Scholar] [CrossRef]
- McMenamin, M.E.; Fletcher, C.D.M. Expanding the Spectrum of Malignant Change in Schwannomas: Epithelioid malignant change, epithelioid malignant peripheral nerve sheath tumor, and epithelioid angiosarcoma: A study of 17 cases. Am. J. Surg. Pathol. 2001, 25, 13–25. [Google Scholar] [CrossRef]
- Carter, J.M.; O’Hara, C.; Dundas, G.; Gilchrist, D.; Collins, M.S.; Eaton, K.; Judkins, A.R.; Biegel, J.A.; Folpe, A.L. Epithelioid Malignant Peripheral Nerve Sheath Tumor Arising in a Schwannoma, in a Patient With “Neuroblastoma-like” Schwannomatosis and a Novel Germline SMARCB1 Mutation. Am. J. Surg. Pathol. 2012, 36, 154–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perry, A.; Reuss, D.E.; Rodriguez, F. Neurofibroma. In WHO Classification of Tumours Series, 5th ed.; IARC Press: Lyon, France, 2020; Volume 3, pp. 232–236. [Google Scholar]
- Wolkenstein, P.; Zeller, J.; Revuz, J.; Ecosse, E.; Leplège, A. Quality-of-Life Impairment in Neurofibromatosis Type 1: A cross-sectional study of 128 cases. Arch. Dermatol. 2001, 137, 1421–1425. [Google Scholar] [CrossRef] [PubMed]
- Meyer, A.; Billings, S.D. What’s new in nerve sheath tumors. Virchows Arch. 2020, 476, 65–80. [Google Scholar] [CrossRef]
- Collins-Sawaragi, Y.C.; Ferner, R.; Vassallo, G.; De Agrò, G.; Eccles, S.; Cadwgan, J.; Hargrave, D.; Hupton, E.; Eelloo, J.; Lunt, L.; et al. Location, symptoms, and management of plexiform neurofibromas in 127 children with neurofibromatosis 1, attending the National Complex Neurofibromatosis 1 service, 2018–2019. Am. J. Med. Genet. Part A 2022, 188, 1723–1727. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, M.M.; Antonescu, C.R.; Fletcher, C.D.; Kim, A.; Lazar, A.J.; Quezado, M.M.; Reilly, K.M.; Stemmer-Rachamimov, A.; Stewart, D.R.; Viskochil, D.; et al. Histopathologic evaluation of atypical neurofibromatous tumors and their transformation into malignant peripheral nerve sheath tumor in patients with neurofibromatosis 1—A consensus overview. Hum. Pathol. 2017, 67, 1–10. [Google Scholar] [CrossRef]
- Stewart, D.R.; Korf, B.R.; Nathanson, K.L.; Stevenson, D.A.; Yohay, K. Care of adults with neurofibromatosis type 1: A clinical practice resource of the American College of Medical Genetics and Genomics (ACMG). Anesth. Analg. 2018, 20, 671–682. [Google Scholar] [CrossRef] [Green Version]
- Bs, J.E.B.; Peterson, C.R.; Dhakal, S.; Giampoli, E.J.; Constine, L.S. Malignant peripheral nerve sheath tumors (MPNST): A SEER analysis of incidence across the age spectrum and therapeutic interventions in the pediatric population. Pediatr. Blood Cancer 2014, 61, 1955–1960. [Google Scholar] [CrossRef]
- Somatilaka, B.N.; Sadek, A.; McKay, R.M.; Le, L.Q. Malignant peripheral nerve sheath tumor: Models, biology, and translation. Oncogene 2022, 41, 2405–2421. [Google Scholar] [CrossRef]
- Ferner, R.E.; Gutmann, D.H. International consensus statement on malignant peripheral nerve sheath tumors in neurofibroma-tosis. Cancer Res. 2002, 62, 1573–1577. [Google Scholar]
- Evans, D.G.R.; Baser, M.E.; McGaughran, J.; Sharif, S.; Howard, E.; Moran, A. Malignant peripheral nerve sheath tumours in neurofibromatosis 1. J. Med. Genet. 2002, 39, 311–314. [Google Scholar] [CrossRef] [Green Version]
- Foley, K.M.; Woodruff, J.M.; Ellis, F.T.; Posner, J.B. Radiation-induced malignant and atypical peripheral nerve sheath tumors. Ann. Neurol. 1980, 7, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Carli, M.; Ferrari, A.; Mattke, A.; Zanetti, I.; Casanova, M.; Bisogno, G.; Cecchetto, G.; Alaggio, R.; De Sio, L.; Koscielniak, E.; et al. Pediatric Malignant Peripheral Nerve Sheath Tumor: The Italian and German Soft Tissue Sarcoma Cooperative Group. J. Clin. Oncol. 2005, 23, 8422–8430. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.W.; Hirose, T.; Scheithauer, B.W.; Schild, S.; Gunderson, L.L. Malignant peripheral nerve sheath tumor: Analysis of treatment outcome. Int. J. Radiat. Oncol. 1998, 42, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Stucky, C.-C.H.; Johnson, K.N.; Gray, R.J.; Pockaj, B.A.; Ocal, I.T.; Rose, P.S.; Wasif, N. Malignant Peripheral Nerve Sheath Tumors (MPNST): The Mayo Clinic Experience. Ann. Surg. Oncol. 2011, 19, 878–885. [Google Scholar] [CrossRef]
- Valentin, T.; Le Cesne, A.; Ray-Coquard, I.; Italiano, A.; Decanter, G.; Bompas, E.; Isambert, N.; Thariat, J.; Linassier, C.; Bertucci, F.; et al. Management and prognosis of malignant peripheral nerve sheath tumors: The experience of the French Sarcoma Group (GSF-GETO). Eur. J. Cancer 2016, 56, 77–84. [Google Scholar] [CrossRef]
- Torres-Mora, J.; Dry, S.; Li, X.; Binder, S.; Amin, M.; Folpe, A.L. Malignant Melanotic Schwannian Tumor: A clinicopathologic, im-munohistochemical, and gene expression profiling study of 40 cases, with a proposal for the reclassification of “melanotic schwannoma”. Am. J. Surg. Pathol. 2014, 38, 94–105. [Google Scholar] [CrossRef]
- Carney, J.A. Psammomatous melanotic schwannoma. A distinctive, heritable tumor with special associations, including cardiac myxoma and the Cushing syndrome. Am. J. Surg. Pathol. 1990, 14, 206–222. [Google Scholar] [CrossRef]
- Zhang, H.-Y.; Yang, G.-H.; Chen, H.-J.; Wei, B.; Ke, Q.; Guo, H.; Ye, L.; Bu, H.; Yang, K.; Zhang, Y.-H. Clinicopathological, immunohistochemical, and ultrastructural study of 13 cases of melanotic schwannoma. Chin. Med. J. 2005, 118, 1451–1461. [Google Scholar]
- Wang, L.; Zehir, A.; Sadowska, J.; Zhou, N.; Rosenblum, M.; Busam, K.; Agaram, N.; Travis, W.; Arcila, M.; Dogan, S.; et al. Consistent copy number changes and recurrentPRKAR1Amutations distinguish Melanotic Schwannomas from Melanomas: SNP-array and next generation sequencing analysis. Genes Chromosom. Cancer 2015, 54, 463–471. [Google Scholar] [CrossRef]
- Chen, Y.-Y.; Yen, H.-H.; Soon, M.-S. Solitary gastric melanotic schwannoma: Sonographic findings. J. Clin. Ultrasound 2007, 35, 52–54. [Google Scholar] [CrossRef]
- Chetty, R.; Vajpeyi, R.; Penwick, J.L. Psammomatous melanotic schwannoma presenting as colonic polyps. Virchows Arch. 2007, 451, 717–720. [Google Scholar] [CrossRef]
- Lindholm, K.; Moran, C.A. Primary mediastinal melanotic schwannian tumors: A clinicopathological and immunohistochemical study of 5 cases. Ann. Diagn. Pathol. 2018, 37, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Khoo, M.; Pressney, I.; Hargunani, R.; Tirabosco, R. Melanotic schwannoma: An 11-year case series. Skelet. Radiol. 2016, 45, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Lenartowicz, K.A.; Goyal, A.; Mauermann, M.L.; Wilson, T.J.; Spinner, R.J. Clinical Features, Natural History, and Outcomes of Intraneural Perineuriomas: A Systematic Review of the Literature. World Neurosurg. 2021, 154, 120–131.e8. [Google Scholar] [CrossRef] [PubMed]
- Almefty, R.; Webber, B.L.; Arnautović, K.I. Intraneural perineurioma of the third cranial nerve: Occurrence and identification: Case report. J. Neurosurg. 2006, 104, 824–827. [Google Scholar] [CrossRef] [Green Version]
- Christoforidis, M.; Buhl, R.; Paulus, W.; Sepehrnia, A. intraneural perineurioma of the viiith cranial nerve: Case report. Neurosurgery 2007, 61, E652. [Google Scholar] [CrossRef]
- Giannini, C.; Scheithauer, B.W.; Steinberg, J.; Cosgrove, T.J. Intraventricular Perineurioma: Case Report. Neurosurgery 1998, 43, 1478–1481. [Google Scholar] [CrossRef]
- Mauermann, M.L.; Amrami, K.K.; Kuntz, N.L.; Spinner, R.J.; Dyck, P.J.; Bosch, E.P.; Engelstad, J.; Felmlee, J.P.; Dyck, P.J. Longitudinal study of intraneural perineurioma--a benign, focal hypertrophic neuropathy of youth. Brain 2009, 132, 2265–2276. [Google Scholar] [CrossRef] [Green Version]
- Boyanton, B.L., Jr.; Jones, J.K.; Shenaq, S.M.; Hicks, M.J.; Bhattacharjee, M.B. Intraneural Perineurioma: A Systematic Review with Illustrative Cases. Arch. Pathol. Lab. Med. 2007, 131, 1382–1392. [Google Scholar] [CrossRef]
- Wilson, T.J.; Howe, B.M.; Stewart, S.A.; Spinner, R.J.; Amrami, K.K. Clinicoradiological features of intraneural perineuriomas obviate the need for tissue diagnosis. J. Neurosurg. 2018, 129, 1034–1040. [Google Scholar] [CrossRef] [Green Version]
- Pendleton, C.; Lenartowicz, K.A.; Howe, B.M.; Spinner, R.J. Limb Undergrowth in Intraneural Perineuriomas: An Under-Recognized Association. World Neurosurg. 2020, 141, e670–e676. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, I.-M.; Ströbel, P.; Thiha, A.; Sohns, J.M.; Mühlfeld, C.; Küffer, S.; Felmerer, G.; Stepniewski, A.; Pauli, S.; Agaimy, A. Soft tissue perineurioma and other unusual tumors in a patient with neurofibromatosis type 1. Int. J. Clin. Exp. Pathol. 2013, 6, 3003–3008. [Google Scholar] [PubMed]
- Al-Adnani, M. Soft Tissue Perineurioma in a Child with Neurofibromatosis Type 1: A Case Report and Review of the Literature. Pediatr. Dev. Pathol. 2017, 20, 444–448. [Google Scholar] [CrossRef] [PubMed]
- White, B.; Belzberg, A.; Ahlawat, S.; Blakeley, J.; Rodriguez, F.J. Intraneural perineurioma in neurofibromatosis type 2 with molecular analysis. Clin. Neuropathol. 2020, 39, 167–171. [Google Scholar] [CrossRef] [PubMed]
- Pendleton, C.; Spinner, R.J.; Dyck, P.J.B.; Mauermann, M.L.; Ladak, A.; Restrepo, C.E.; Baheti, S.; Klein, C.J. Association of intraneural perineurioma with neurofibromatosis type 2. Acta Neurochir. 2020, 162, 1891–1897. [Google Scholar] [CrossRef]
- Hornick, J.L.; Bundock, E.A.; Fletcher, C.D.M. Hybrid Schwannoma/Perineurioma: Clinicopathologic analysis of 42 distinctive benign nerve sheath tumors. Am. J. Surg. Pathol. 2009, 33, 1554–1561. [Google Scholar] [CrossRef]
- Michal, M.; Kazakov, D.V.; Belousova, I.; Bisceglia, M.; Zamecnik, M.; Mukensnabl, P. A benign neoplasm with histopathological features of both schwannoma and retiform perineurioma (benign schwannoma-perineurioma): A report of six cases of a distinctive soft tissue tumor with a predilection for the fingers. Virchows Arch. 2004, 445, 347–353. [Google Scholar] [CrossRef]
- Goyal-Honavar, A.; Gupta, A.; Chacko, G.; Chacko, A.G. Trigeminal hybrid nerve sheath tumor—A case report and literature review. Br. J. Neurosurg. 2021, 1–4. [Google Scholar] [CrossRef]
- Las Heras, F.L.; Martuza, R.; Caruso, P.; Rincon, S.; Stemmer-Rachamimov, A. 24-Year-Old Woman with An Internal Auditory Canal Mass. Hybrid peripheral nerve sheath tumor with schwannoma/perineurioma components. Brain Pathol. 2013, 23, 361–362. [Google Scholar] [CrossRef]
- Matsuo, S.; Higaki, R.; Matsukado, K. Microsurgical Resection of Hybrid Nerve Sheath Tumor Involving the Orbit and Lateral Wall of the Cavernous Sinus: 2-Dimensional Operative Video. Oper. Neurosurg. 2021, 21, E551. [Google Scholar] [CrossRef]
- Harder, A.; Wesemann, M.; Hagel, C.; Schittenhelm, J.; Fischer, S.; Tatagiba, M.; Nagel, C.; Jeibmann, A.; Bohring, A.; Mautner, V.-F.; et al. Hybrid Neurofibroma/Schwannoma is Overrepresented Among Schwannomatosis and Neurofibromatosis Patients. Am. J. Surg. Pathol. 2012, 36, 702–709. [Google Scholar] [CrossRef] [PubMed]
- MacCollin, M.; Chiocca, E.A.; Evans, G.; Friedman, J.; Horvitz, R.; Jaramillo, D.; Lev, M.; Mautner, V.F.; Niimura, M.; Plotkin, S.R.; et al. Diagnostic criteria for schwannomatosis. Neurology 2005, 64, 1838–1845. [Google Scholar] [CrossRef] [PubMed]
- Kacerovska, D.; Michal, M.; Kuroda, N.; Tanaka, A.; Sima, R.; Denisjuk, N.; Kreuzberg, B.; Ricarova, R.; Kazakov, D.V. Hybrid Peripheral Nerve Sheath Tumors, Including a Malignant Variant in Type 1 Neurofibromatosis. Am. J. Dermatopathol. 2013, 35, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Inatomi, Y.; Ito, T.; Nagae, K.; Yamada, Y.; Kiyomatsu, M.; Nakano-Nakamura, M.; Uchi, H.; Oda, Y.; Furue, M. Hybrid perineurioma-neurofibroma in a patient with neurofibromatosis type 1, clinically mimicking malignant peripheral nerve sheath tumor. Eur. J. Dermatol. 2014, 24, 412–413. [Google Scholar] [CrossRef]
- Engelhard, H.H.; Villano, J.L.; Porter, K.R.; Stewart, A.K.; Barua, M.; Ii, F.G.B.; Newton, H.B.; Takashima, H.; Takebayashi, T.; Yoshimoto, M.; et al. Clinical presentation, histology, and treatment in 430 patients with primary tumors of the spinal cord, spinal meninges, or cauda equina. J. Neurosurg. Spine 2010, 13, 67–77. [Google Scholar] [CrossRef]
- Palmisciano, P.; Sagoo, N.S.; Haider, A.S.; Ogasawara, C.; Ogasawara, M.; Bin Alamer, O.; Heidari, K.S.; Raj, K.M.; Scalia, G.; Umana, G.E.; et al. Primary Paraganglioma of the Spine: A Systematic Review of Clinical Features and Surgical Management in Cauda Equina versus Non–Cauda Equina Lesions. World Neurosurg. 2022, 161, 190–197.e20. [Google Scholar] [CrossRef]
- Shtaya, A.; Iorga, R.; Hettige, S.; Bridges, L.R.; Stapleton, S.; Johnston, F.G. Paraganglioma of the cauda equina: A tertiary centre experience and scoping review of the current literature. Neurosurg. Rev. 2021, 45, 103–118. [Google Scholar] [CrossRef]
- Blades, D.A.; Hardy, R.W.; Cohen, M. Cervical paraganglioma with subsequent intracranial and intraspinal metastases. J. Neurosurg. 1991, 75, 320–323. [Google Scholar] [CrossRef]
- Sachani, H.; Tripathi, M.; Madhusudan, K.S.; Semalti, K.; Shanker, S.; ArunRaj, S.T.; Bal, C. Thoracic Extradural Paraganglioma Localized on 68Ga-DOTANOC PET/CT. Clin. Nucl. Med. 2021, 46, e471–e472. [Google Scholar] [CrossRef]
- Gelabert-González, M. Paragangliomas of the lumbar region. Report of two cases and review of the literature. J. Neurosurg. Spine 2005, 2, 354–365. [Google Scholar] [CrossRef] [Green Version]
- Adriani, K.S.; Stenvers, D.J.; Imanse, J.G. Pearls & Oy-sters: Lumbar paraganglioma: Can you see it in the eyes? Neurology 2012, 78, e27–e28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moran, C.; Rush, W.; Mena, H. Primary spinal paragangliomas: A clinicopathological and immunohistochemical study of 30 cases. Histopathology 1997, 31, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Roche, P.H.; Figarella-Branger, D.; Regis, J.; Peragut, J.C. Cauda equina paraganglioma with subsequent intracranial and intraspinal metastases. Acta Neurochir. 1996, 138, 475–479. [Google Scholar] [CrossRef]
- Thines, L.; Lejeune, J.P.; Ruchoux, M.M.; Assaker, R. Management of delayed intracranial and intraspinal metastases of intradural spinal paragangliomas. Acta Neurochir. 2006, 148, 63–66. [Google Scholar] [CrossRef] [PubMed]
- Thomson, N.; Pacak, K.; Schmidt, M.H.; Palmer, C.A.; Salzman, K.L.; Champine, M.; Schiffman, J.D.; Cohen, A.L. Leptomeningeal dissemination of a low-grade lumbar paraganglioma: Case report. J. Neurosurg. Spine 2017, 26, 501–506. [Google Scholar] [CrossRef] [Green Version]
- Sonneland, P.R.; Scheithauer, B.W.; LeChago, J.; Crawford, B.G.; Onofrio, B.M. Paraganglioma of the cauda equina region. Clinico-pathologic study of 31 cases with special reference to immunocytology and ultrastructure. Cancer 1986, 58, 1720–1735. [Google Scholar] [CrossRef]
- Easton, D.F.; Ponder, M.A.; Huson, S.M.; Ponder, B.A. An analysis of variation in expression of neurofibromatosis (NF) type 1 (NF1): Evidence for modifying genes. Am. J. Hum. Genet. 1993, 53, 305–313. [Google Scholar]
- Jordan, J.T.; Plotkin, S.R. Neurofibromatoses. Hematol. Oncol. Clin. N. Am. 2022, 36, 253–267. [Google Scholar] [CrossRef]
- Legius, E.; Messiaen, L.; Wolkenstein, P.; Pancza, P.; Avery, R.A.; Berman, Y.; Blakeley, J.; Babovic-Vuksanovic, D.; Cunha, K.S.; Ferner, R.; et al. Revised diagnostic criteria for neurofibromatosis type 1 and Legius syndrome: An international consensus recommendation. Anesth. Analg. 2021, 23, 1506–1513. [Google Scholar] [CrossRef]
- Messiaen, L. Multidiscipilinary Approach to Neurofibromatosis 1. In Molecular Diagnosis of NF1; Tadini, G., Legius, E., Brems, H., Eds.; Springer: Cham, Switzerland, 2020. [Google Scholar]
- Carton, C.; Evans, D.G.; Blanco, I.; Friedrich, R.E.; Ferner, R.E.; Farschtschi, S.; Salvador, H.; Azizi, A.A.; Mautner, V.; Röhl, C.; et al. ERN GENTURIS tumour surveillance guidelines for individuals with neurofibromatosis type 1. Eclinicalmedicine 2023, 56, 101818. [Google Scholar] [CrossRef]
- Plotkin, S.R.; Messiaen, L.; Legius, E.; Pancza, P.; Avery, R.A.; Blakeley, J.O.; Babovic-Vuksanovic, D.; Ferner, R.; Fisher, M.J.; Friedman, J.M.; et al. Updated diagnostic criteria and nomenclature for neurofibromatosis type 2 and schwannomatosis: An international consensus recommendation. Anesth. Analg. 2022, 24, 1967–1977. [Google Scholar] [CrossRef] [PubMed]
- Burns, R.; Niendorf, K.; Steinberg, K.; Mueller, A.; Ly, I.; Jordan, J.T.; Plotkin, S.R.; Hicks, S.R. Genetic testing to gain diagnostic clarity in neurofibromatosis type 2 and schwannomatosis. Am. J. Med. Genet. Part A 2022, 188, 2413–2420. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.G.; Hartley, C.L.; Smith, P.T.; King, A.T.; Bowers, N.L.; Tobi, S.; Wallace, A.J.; Perry, M.; Anup, R.; Lloyd, S.K.W.; et al. Incidence of mosaicism in 1055 de novo NF2 cases: Much higher than previous estimates with high utility of next-generation sequencing. Anesth. Analg. 2020, 22, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.J.; Bowers, N.L.; Banks, C.; Coates-Brown, R.; Morris, K.A.; Ewans, L.; Wilson, M.; Pinner, J.; Bhaskar, S.S.; Cammarata-Scalisi, F.; et al. A deep intronic SMARCB1 variant associated with schwannomatosis. Clin. Genet. 2020, 97, 376–377. [Google Scholar] [CrossRef]
- Nonaka, D.; Chiriboga, L.; Rubin, B.P. Sox10: A Pan-Schwannian and Melanocytic Marker. Am. J. Surg. Pathol. 2008, 32, 1291–1298. [Google Scholar] [CrossRef] [PubMed]
- Karamchandani, J.R.; Nielsen, T.O.; van de Rijn, M.; West, R.B. Sox10 and S100 in the Diagnosis of Soft-tissue Neoplasms. Appl. Immunohistochem. Mol. Morphol. 2012, 20, 445–450. [Google Scholar] [CrossRef] [Green Version]
- Jo, V.Y.; Fletcher, C.D. SMARCB1/INI1 Loss in Epithelioid Schwannoma: A Clinicopathologic and Immunohistochemical Study of 65 Cases. Am. J. Surg. Pathol. 2017, 41, 1013–1022. [Google Scholar] [CrossRef]
- Schaefer, I.-M.; Dong, F.; Garcia, E.P.; Fletcher, C.D.M.; Jo, V.Y. Recurrent SMARCB1 Inactivation in Epithelioid Malignant Peripheral Nerve Sheath Tumors. Am. J. Surg. Pathol. 2019, 43, 835–843. [Google Scholar] [CrossRef]
- Colman, S.D.; Williams, C.A.; Wallace, M.R. Benign neurofibromas in type 1 neurofibromatosis (NF1) show somatic deletions of the NF1 gene. Nat. Genet. 1995, 11, 90–92. [Google Scholar] [CrossRef]
- Klein, C.J.; Wu, Y.; Jentoft, M.E.; Mer, G.; Spinner, R.J.; Dyck, P.J.B.; Mauermann, M.L. Genomic analysis reveals frequentTRAF7mutations in intraneural perineuriomas. Ann. Neurol. 2017, 81, 316–321. [Google Scholar] [CrossRef] [Green Version]
- Carter, J.M.; Wu, Y.; Blessing, M.M.; Folpe, A.L.; Thorland, E.C.; Spinner, R.J.; Jentoft, M.E.; Wang, C.; Baheti, S.; Niu, Z.; et al. Recurrent Genomic Alterations in Soft Tissue Perineuriomas. Am. J. Surg. Pathol. 2018, 42, 1708–1714. [Google Scholar] [CrossRef] [PubMed]
- Brock, J.E.; Perez-Atayde, A.R.; Kozakewich, H.P.W.; Richkind, K.E.; Fletcher, J.A.; Vargas, S.O. Cytogenetic Aberrations in Perineurioma: Variation with subtype. Am. J. Surg. Pathol. 2005, 29, 1164–1169. [Google Scholar] [CrossRef] [PubMed]
- Ronellenfitsch, M.W.; Harter, P.N.; Kirchner, M.; Heining, C.; Hutter, B.; Gieldon, L.; Schittenhelm, J.; Schuhmann, M.U.; Tatagiba, M.; Marquardt, G.; et al. Targetable ERBB2 mutations identified in neurofibroma/schwannoma hybrid nerve sheath tumors. J. Clin. Investig. 2020, 130, 2488–2495. [Google Scholar] [CrossRef]
- Lee, W.; Teckie, S.; Wiesner, T.; Ran, L.; Granada, C.N.P.; Lin, M.; Zhu, S.; Cao, Z.; Liang, Y.; Sboner, A.; et al. PRC2 is recurrently inactivated through EED or SUZ12 loss in malignant peripheral nerve sheath tumors. Nat. Genet. 2014, 46, 1227–1232. [Google Scholar] [CrossRef] [Green Version]
- De Raedt, T.; Beert, E.; Pasmant, E.; Luscan, A.; Brems, H.; Ortonne, N.; Helin, K.; Hornick, J.L.; Mautner, V.; Kehrer-Sawatzki, H.; et al. PRC2 loss amplifies Ras-driven transcription and confers sensitivity to BRD4-based therapies. Nature 2014, 514, 247–251. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Wang, Y.; Jones, S.; Sausen, M.; McMahon, K.; Sharma, R.; Wang, Q.; Belzberg, A.J.; Chaichana, K.; Gallia, G.L.; et al. Somatic mutations of SUZ12 in malignant peripheral nerve sheath tumors. Nat. Genet. 2014, 46, 1170–1172. [Google Scholar] [CrossRef] [Green Version]
- Pemov, A.; Hansen, N.F.; Sindiri, S.; Patidar, R.; Higham, C.S.; Dombi, E.; Miettinen, M.M.; Fetsch, P.; Brems, H.; Chandrasekharappa, S.C.; et al. Low mutation burden and frequent loss of CDKN2A/B and SMARCA2, but not PRC2, define premalignant neurofibromatosis type 1–associated atypical neurofibromas. Neuro-Oncology 2019, 21, 981–992. [Google Scholar] [CrossRef]
- Prieto-Granada, C.N.; Wiesner, T.; Messina, J.L.; Jungbluth, A.A.; Chi, P.; Antonescu, C.R. Loss of H3K27me3 Expression Is a Highly Sensitive Marker for Sporadic and Radiation-induced MPNST. Am. J. Surg. Pathol. 2016, 40, 479–489. [Google Scholar] [CrossRef] [Green Version]
- Schaefer, I.-M.; Fletcher, C.D.; Hornick, J.L. Loss of H3K27 trimethylation distinguishes malignant peripheral nerve sheath tumors from histologic mimics. Mod. Pathol. 2016, 29, 4–13. [Google Scholar] [CrossRef] [Green Version]
- Bockmayr, M.; Körner, M.; Schweizer, L.; Schüller, U. Cauda equina paragangliomas express HOXB13. Neuropathol. Appl. Neurobiol. 2021, 47, 889–890. [Google Scholar] [CrossRef]
- Haller, F.; Moskalev, E.A.; Faucz, F.R.; Barthelmeß, S.; Wiemann, S.; Bieg, M.; Assié, G.; Bertherat, J.; Schaefer, I.-M.; Otto, C.; et al. Aberrant DNA hypermethylation of SDHC: A novel mechanism of tumor development in Carney triad. Endocrine-Related Cancer 2014, 21, 567–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Letouzé, E.; Martinelli, C.; Loriot, C.; Burnichon, N.; Abermil, N.; Ottolenghi, C.; Janin, M.; Menara, M.; Nguyen, A.T.; Benit, P.; et al. SDH Mutations Establish a Hypermethylator Phenotype in Paraganglioma. Cancer Cell 2013, 23, 739–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, F.O.; Cartwright, M.S.; Alter, K.E.; Visser, L.H.; Hobson-Webb, L.D.; Padua, L.; Strakowski, J.A.; Preston, D.C.; Boon, A.J.; Axer, H.; et al. Indications for neuromuscular ultrasound: Expert opinion and review of the literature. Clin. Neurophysiol. 2018, 129, 2658–2679. [Google Scholar] [CrossRef] [PubMed]
- Hung, E.H.Y.; Griffith, J.F.; Ng, A.W.H.; Lee, R.K.L.; Lau, D.T.Y.; Leung, J.C.S. Ultrasound of Musculoskeletal Soft-Tissue Tumors Superficial to the Investing Fascia. Am. J. Roentgenol. 2014, 202, W532–W540. [Google Scholar] [CrossRef] [PubMed]
- Tøttrup, M.; Eriksen, J.D.; Hellfritzsch, M.B.; Sørensen, F.B.; Baad-Hansen, T. Diagnostic accuracy of ultrasound-guided core biopsy of peripheral nerve sheath tumors. J. Clin. Ultrasound 2020, 48, 134–138. [Google Scholar] [CrossRef] [PubMed]
- Pendleton, C.; Spinner, R.J. Image-guided percutaneous biopsy of peripheral nerve tumors of indeterminate nature: Risks and benefits. Acta Neurochir. 2020, 162, 1425–1429. [Google Scholar] [CrossRef]
- Grover, S.B.; Kundra, R.; Grover, H.; Gupta, V.; Gupta, R. Imaging diagnosis of plexiform neurofibroma- unravelling the confounding features: A report of two cases. Radiol. Case Rep. 2021, 16, 2824–2833. [Google Scholar] [CrossRef]
- Bruscella, S.; Alfieri, A.; de Bellis, A.; Rolando, F.; Covelli, E.M.; Manfredonia, L.; Orabona, P.; de Marinis, P. Malignant Intracerebral Nerve Sheath Tumor Presenting with Intratumoral Hemorrhage. World Neurosurg. 2021, 145, 370–375. [Google Scholar] [CrossRef]
- Tanaka, M.; Shibui, S.; Nomura, K.; Nakanishi, Y.; Hasegawa, T.; Hirose, T.; Le Fèvre, C.; Castelli, J.; Perrin, C.; Hénaux, P.; et al. Malignant intracerebral nerve sheath tumor with intratumoral calcification. J. Neurosurg. 2000, 92, 338–341. [Google Scholar] [CrossRef]
- Agarwal, A.; Chandra, A.; Jaipal, U.; Bagarhatta, M.; Mendiratta, K.; Goyal, A.; Kumar, R.; Mangalhara, N. Can imaging be the new yardstick for diagnosing peripheral neuropathy?—A comparison between high resolution ultrasound and MR neurography with an approach to diagnosis. Insights Imaging 2019, 10, 104–113. [Google Scholar] [CrossRef]
- Noebauer-Huhmann, I.M.; Weber, M.-A.; Lalam, R.K.; Trattnig, S.; Bohndorf, K.; Vanhoenacker, F.; Tagliafico, A.; Van Rijswijk, C.; Vilanova, J.C.; Afonso, P.D.; et al. Soft Tissue Tumors in Adults: ESSR-Approved Guidelines for Diagnostic Imaging. Semin. Musculoskelet. Radiol. 2015, 19, 475–482. [Google Scholar] [CrossRef] [Green Version]
- Laffan, E.E.; Ngan, B.-Y.; Navarro, O.M. Pediatric Soft-Tissue Tumors and Pseudotumors: MR Imaging Features with Pathologic Correlation: Part 2. Tumors of Fibroblastic/Myofibroblastic, So-called Fibrohistiocytic, Muscular, Lymphomatous, Neurogenic, Hair Matrix, and Uncertain Origin. Radiographics 2009, 29, e36. [Google Scholar] [CrossRef] [Green Version]
- Ahlawat, S.; Chhabra, A.; Blakely, J. Magnetic Resonance Neurography of Peripheral Nerve Tumors and Tumorlike Conditions. Neuroimaging Clin. N. Am. 2014, 24, 171–192. [Google Scholar] [CrossRef] [PubMed]
- Fisher, S.; Karri, S.; Ramzi, A.; Sharma, R.; Chhabra, A.; Soldatos, T. Advanced MR Imaging of Peripheral Nerve Sheath Tumors Including Diffusion Imaging. Semin. Musculoskelet. Radiol. 2015, 19, 179–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chhabra, A.; Deshmukh, S.D.; Lutz, A.M.; Fritz, J.; Andreisek, G.; Sneag, D.B.; Subhawong, T.; Singer, A.D.; Wong, P.K.; Thakur, U.; et al. Neuropathy Score Reporting and Data System: A Reporting Guideline for MRI of Peripheral Neuropathy with a Multicenter Validation Study. Am. J. Roentgenol. 2022, 219, 279–291. [Google Scholar] [CrossRef]
- Chhabra, A.; Deshmukh, S.D.; Lutz, A.M.; Fritz, J.; Sneag, D.B.; Mogharrabi, B.; Guirguis, M.; Andreisek, G.; Xi, Y.; Ahlawat, S. Neuropathy Score Reporting and Data System (NS-RADS): MRI Reporting Guideline of Peripheral Neuropathy Explained and Reviewed. Skelet. Radiol. 2022, 51, 1909–1922. [Google Scholar] [CrossRef] [PubMed]
- Broski, S.M.; Johnson, G.; Howe, B.M.; Nathan, M.A.; Wenger, D.E.; Spinner, R.J.; Amrami, K.K. Evaluation of 18F-FDG PET and MRI in differentiating benign and malignant peripheral nerve sheath tumors. Skelet. Radiol. 2016, 45, 1097–1105. [Google Scholar] [CrossRef]
- Raad, R.A.; Lala, S.; Allen, J.C.; Babb, J.; Mitchell, C.W.; Franceschi, A.M.; Yohay, K.; Friedman, K.P. Comparison of hybrid 18F-fluorodeoxyglucose positron emission tomography/magnetic resonance imaging and positron emission tomography/computed tomography for evaluation of peripheral nerve sheath tumors in patients with neurofibromatosis type 1. World J. Nucl. Med. 2018, 17, 241–248. [Google Scholar] [CrossRef]
- Reinert, C.P.; Schuhmann, M.U.; Bender, B.; Gugel, I.; la Fougère, C.; Schäfer, J.; Gatidis, S. Comprehensive anatomical and functional imaging in patients with type I neurofibromatosis using simultaneous FDG-PET/MRI. Eur. J. Nucl. Med. 2019, 46, 776–787. [Google Scholar] [CrossRef]
- Bredella, M.A.; Torriani, M.; Hornicek, F.; Ouellette, H.A.; Plamer, W.E.; Williams, Z.; Fischman, A.J.; Plotkin, S.R. Value of PET in the Assessment of Patients with Neurofibromatosis Type 1. Am. J. Roentgenol. 2007, 189, 928–935. [Google Scholar] [CrossRef]
- Li, C.-S.; Huang, G.-S.; Wu, H.-D.; Chen, W.-T.; Shih, L.-S.; Lii, J.-M.; Duh, S.-J.; Chen, R.-C.; Tu, H.-Y.; Chan, W.P. Differentiation of soft tissue benign and malignant peripheral nerve sheath tumors with magnetic resonance imaging. Clin. Imaging 2008, 32, 121–127. [Google Scholar] [CrossRef]
- Mautner, V.F.; Hartmann, M.; Kluwe, L.; Friedrich, R.E.; Fünsterer, C. MRI growth patterns of plexiform neurofibromas in patients with neurofibromatosis type 1. Neuroradiology 2006, 48, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Razek, A.A.K.A.; Ashmalla, G.A. Assessment of paraspinal neurogenic tumors with diffusion-weighted MR imaging. Eur. Spine J. 2018, 27, 841–846. [Google Scholar] [CrossRef] [PubMed]
- Fayad, L.M.; Wang, X.; Blakeley, J.O.; Durand, D.J.; Jacobs, M.A.; Demehri, S.; Subhawong, T.K.; Soldatos, T.; Barker, P.B. Characterization of Peripheral Nerve Sheath Tumors with 3T Proton MR Spectroscopy. Am. J. Neuroradiol. 2014, 35, 1035–1041. [Google Scholar] [CrossRef] [PubMed]
- Ogose, A.; Hotta, T.; Morita, T.; Higuchi, T.; Umezu, H.; Imaizumi, S.; Hatano, H.; Kawashima, H.; Gu, W.; Endo, N. Diagnosis of Peripheral Nerve Sheath Tumors around the Pelvis. Jpn. J. Clin. Oncol. 2004, 34, 405–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, S.-S.; Zager, E.L.; Coyne, T.M.; Nangunoori, R.; Kneeland, J.B.; Nathanson, K. Hybrid peripheral nerve sheath tumor: Case report. J. Neurosurg. 2012, 117, 897–901. [Google Scholar] [CrossRef] [Green Version]
- Koeller, K.K.; Shih, R.Y. Intradural Extramedullary Spinal Neoplasms: Radiologic-Pathologic Correlation. RadioGraphics 2019, 39, 468–490. [Google Scholar] [CrossRef]
- Nguyen, R.; Dombi, E.; Widemann, B.C.; Solomon, J.; Fuensterer, C.; Kluwe, L.; Friedman, J.M.; Mautner, V.-F. Growth dynamics of plexiform neurofibromas: A retrospective cohort study of 201 patients with neurofibromatosis 1. Orphanet J. Rare Dis. 2012, 7, 75. [Google Scholar] [CrossRef] [Green Version]
- Rubino, F.; Eichberg, D.G.; Shah, A.H.; Luther, E.M.; Lu, V.M.; Saad, A.G.; Kahn, D.; Komotar, R.J.; Ivan, M.E. When “Peripheral” Becomes “Central”: Primary and Secondary Malignant Intracerebral Nerve Sheath Tumor: A Case Report and a Systematic Review. Neurosurgery 2021, 88, 1074–1087. [Google Scholar] [CrossRef]
- Le Fèvre, C.; Castelli, J.; Perrin, C.; Hénaux, P.L.; Noël, G. Tumeurs malignes des gaines nerveuses périphériques intracérébrales métastatiques: À propos de deux cas et revue exhaustive des cas de la littérature. Cancer/Radiothérapie 2016, 20, 119–132. [Google Scholar] [CrossRef]
- Mackel, C.E.; Medeiros, I.; Moore, B.E.; Zhao, Q.; Jha, R. Intracranial Malignant Peripheral Nerve Sheath Tumors Not Associated with a Cranial Nerve: Systematic Review and Illustrative Case. World Neurosurg. 2021, 156, 76–91. [Google Scholar] [CrossRef]
- Kozić, D.; Nagulić, M.; Samardzić, M.; Ostojić, J.; Rasulić, L.; Cvetković-Dozić, D. Intrapontine malignant nerve sheath tumor: MRI and MRS features. Acta Neurol. Belg. 2008, 10, 67–71. [Google Scholar]
- Ishimaru, H.; Morikawa, M.; Iwanaga, S.; Kaminogo, M.; Ochi, M.; Hayashi, K. Differentiation between high-grade glioma and metastatic brain tumor using single-voxel proton MR spectroscopy. Eur. Radiol. 2001, 11, 1784–1791. [Google Scholar] [CrossRef]
- Sundgren, P.; Annertz, M.; Englund, E.; Strömblad, L.G.; Holtås, Ś. Paragangliomas of the spinal canal. Neuroradiology 1999, 41, 788–794. [Google Scholar] [CrossRef] [PubMed]
- Makhdoomi, R.; Nayil, K.; Santosh, V. Primary Spinal Paragangliomas: A Review. Neurosurg. Q. 2009, 19, 196–199. [Google Scholar] [CrossRef]
- Sharma, A.; Gaikwad, S.B.; Goyal, M.; Mishra, N.K.; Sharma, M.C. Calcified filum terminale paraganglioma causing superficial siderosis. Am. J. Roentgenol. 1998, 170, 1650–1652. [Google Scholar] [CrossRef] [Green Version]
- Schweizer, L.; Thierfelder, F.; Thomas, C.; Soschinski, P.; Suwala, A.; Stichel, D.; Wefers, A.K.; Wessels, L.; Misch, M.; Kim, H.-Y.; et al. Molecular characterization of CNS paragangliomas identifies cauda equina paragangliomas as a distinct tumor entity. Acta Neuropathol. 2020, 140, 893–906. [Google Scholar] [CrossRef]
- Klekamp, J.; Samii, M. Surgery of Spinal Nerve Sheath Tumors with Special Reference to Neurofibromatosis. Neurosurgery 1998, 42, 279–289. [Google Scholar] [CrossRef]
- Robla-Costales, J.; Rodríguez-Aceves, C.; Martínez-Benia, F.; Socolovsky, M. State of the Art and Advances in Peripheral Nerve Surgery. Adv. Tech. Stand. Neurosurg. 2022, 45, 245–283. [Google Scholar] [CrossRef]
- Zipfel, J.; Al-Hariri, M.; Gugel, I.; Grimm, A.; Steger, V.; Ladurner, R.; Krimmel, M.; Tatagiba, M.; Schuhmann, M. Surgical Management of Sporadic Peripheral Nerve Schwannomas in Adults: Indications and Outcome in a Single Center Cohort. Cancers 2021, 13, 1017. [Google Scholar] [CrossRef]
- Pedro, M.T.; Antoniadis, G.; Scheuerle, A.; Pham, M.; Wirtz, C.R.; Koenig, R.W. Intraoperative high-resolution ultrasound and contrast-enhanced ultrasound of peripheral nerve tumors and tumorlike lesions. Neurosurg. Focus 2015, 39, E5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gugel, I.; Grimm, F.; Tatagiba, M.; Schuhmann, M.U.; Zipfel, J. Management of neurofibromatosis type 2 and schwannomatosis associated peripheral and intraspinal schwannomas: Influence of surgery, genetics, and localization. J. Neuro-Oncol. 2022, 159, 271–279. [Google Scholar] [CrossRef]
- Siqueira, M.G.; Socolovsky, M.; Martins, R.S.; Robla-Costales, J.; Di Masi, G.; Heise, C.O.; Cosamalón, J.G. Surgical treatment of typical peripheral schwannomas: The risk of new postoperative deficits. Acta Neurochir. 2013, 155, 1745–1749. [Google Scholar] [CrossRef] [PubMed]
- Germano, I.M.; Sheehan, J.; Parish, J.; Atkins, T.; Asher, A.; Hadjipanayis, C.G.; Burri, S.H.; Green, S.; Olson, J.J. Congress of Neurological Surgeons Systematic Review and Evidence-Based Guidelines on the Role of Radiosurgery and Radiation Therapy in the Management of Patients with Vestibular Schwannomas. Neurosurgery 2018, 82, E49–E51. [Google Scholar] [CrossRef]
- Paldor, I.; Chen, A.S.; Kaye, A.H. Growth rate of vestibular schwannoma. J. Clin. Neurosci. 2016, 32, 1–8. [Google Scholar] [CrossRef]
- Hunter, J.B.; Francis, D.O.; O’Connell, B.P.; Kabagambe, E.K.; Bennett, M.L.; Wanna, G.B.; Rivas, A.; Thompson, R.C.; Haynes, D.S. Single Institutional Experience with Observing 564 Vestibular Schwannomas: Factors Associated with Tumor Growth. Otol. Neurotol. 2016, 37, 1630–1636. [Google Scholar] [CrossRef] [Green Version]
- Tveiten, O.V.; Carlson, M.L.; Goplen, F.; Vassbotn, F.; Link, M.J.; Lund-Johansen, M. Long-term Auditory Symptoms in Patients with Sporadic Vestibular Schwannoma: An International Cross-Sectional Study. Neurosurgery 2015, 77, 218–227. [Google Scholar] [CrossRef] [PubMed]
- Goldbrunner, R.; Weller, M.; Regis, J.; Lund-Johansen, M.; Stavrinou, P.; Reuss, D.; Evans, D.G.; Lefranc, F.; Sallabanda, K.; Falini, A.; et al. EANO guideline on the diagnosis and treatment of vestibular schwannoma. Neuro-Oncology 2019, 22, 31–45. [Google Scholar] [CrossRef]
- Hadjipanayis, C.G.; Carlson, M.L.; Link, M.J.; Rayan, T.A.; Parish, J.; Atkins, T.; Asher, A.L.; Dunn, I.F.; Corrales, C.E.; Van Gompel, J.J.; et al. Congress of Neurological Surgeons Systematic Review and Evidence-Based Guidelines on Surgical Resection for the Treatment of Patients with Vestibular Schwannomas. Neurosurgery 2018, 82, E40–E43. [Google Scholar] [CrossRef] [Green Version]
- Ferrari, A.; Casanova, M.; Bisogno, G.; Mattke, A.; Meazza, C.; Gandola, L.; Sotti, G.; Cecchetto, G.; Harms, D.; Koscielniak, E.; et al. Clear cell sarcoma of tendons and aponeuroses in pediatric patients: A report from the Italian and German Soft Tissue Sarcoma Cooperative Group. Cancer 2002, 94, 3269–3276. [Google Scholar] [CrossRef]
- Gachiani, J.; Kim, D.; Nelson, A.; Kline, D. Surgical management of malignant peripheral nerve sheath tumors. Neurosurg. Focus 2007, 22, 1–8. [Google Scholar] [CrossRef]
- Kahn, J.; Gillespie, A.; Tsokos, M.; Ondos, J.; Dombi, E.; Camphausen, K.; Widemann, B.C.; Kaushal, A. Radiation Therapy in Management of Sporadic and Neurofibromatosis Type 1-Associated Malignant Peripheral Nerve Sheath Tumors. Front. Oncol. 2014, 4, 324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, E.; Coert, J.H.; Flucke, U.E.; Slooff, W.-B.M.; Ho, V.K.; van der Graaf, W.T.; van Dalen, T.; van de Sande, M.A.; van Houdt, W.J.; Grünhagen, D.J.; et al. A nationwide cohort study on treatment and survival in patients with malignant peripheral nerve sheath tumours. Eur. J. Cancer 2020, 124, 77–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spunt, S.L.; Million, L.; Chi, Y.-Y.; Anderson, J.; Tian, J.; Hibbitts, E.; Coffin, C.; McCarville, M.B.; Randall, R.L.; Parham, D.M.; et al. A risk-based treatment strategy for non-rhabdomyosarcoma soft-tissue sarcomas in patients younger than 30 years (ARST0332): A Children’s Oncology Group prospective study. Lancet Oncol. 2020, 21, 145–161. [Google Scholar] [CrossRef] [PubMed]
- Myrseth, E.; Møller, P.; Pedersen, P.-H.; Lund-Johansen, M. Vestibular schwannoma: Surgery or gamma knife radiosurgery? A prospective, nonrandomized study. Neurosurgery 2009, 64, 654–663. [Google Scholar] [CrossRef]
- Régis, J.; Pellet, W.; Delsanti, C.; Dufour, H.; Roche, P.H.; Thomassin, J.M.; Zanaret, M.; Peragut, J.C. Functional outcome after gamma knife surgery or microsurgery for vestibular schwannomas. J. Neurosurg. 2002, 97, 1091–1100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karpinos, M.; Teh, B.S.; Zeck, O.; Carpenter, L.; Phan, C.; Mai, W.-Y.; Lu, H.H.; Chiu, J.; Butler, E.; Gormley, W.B.; et al. Treatment of acoustic neuroma: Stereotactic radiosurgery vs. microsurgery. Int. J. Radiat. Oncol. 2002, 54, 1410–1421. [Google Scholar] [CrossRef]
- Pollock, B.E.; Driscoll, C.L.; Foote, R.L.; Link, M.J.; Gorman, D.A.; Bauch, C.D.; Mandrekar, J.N.; Krecke, K.N.; Johnson, C.H. Patient Outcomes After Vestibular Schwannoma Management: A Prospective Comparison of Microsurgical Resection and Stereotactic Radiosurgery. Neurosurgery 2006, 59, 77–85. [Google Scholar] [CrossRef]
- Carlson, M.L.; Vivas, E.X.; McCracken, D.J.; Sweeney, A.D.; Neff, B.A.; Shepard, N.T.; Olson, J.J. Congress of Neurological Surgeons Systematic Review and Evidence-Based Guidelines on Hearing Preservation Outcomes in Patients with Sporadic Vestibular Schwannomas. Neurosurgery 2018, 82, E35–E39. [Google Scholar] [CrossRef] [Green Version]
- Tsao, M.N.; Sahgal, A.; Xu, W.; De Salles, A.; Hayashi, M.; Levivier, M.; Ma, L.; Martinez, R.; Régis, J.; Ryu, S.; et al. Stereotactic radiosurgery for vestibular schwannoma: International Stereotactic Radiosurgery Society (ISRS) Practice Guideline. J. Radiosurg. SBRT 2017, 5, 5–24. [Google Scholar]
- Huy, P.T.B. Radiotherapy for glomus jugulare paraganglioma. Eur. Ann. Otorhinolaryngol. Head Neck Dis. 2014, 131, 223–226. [Google Scholar] [CrossRef] [Green Version]
- Plotkin, S.R.; Merker, V.L.; Halpin, C.; Jennings, D.; McKenna, M.J.; Harris, G.J.; Barker, F.G.I., 2nd. Bevacizumab for Progressive Vestibular Schwannoma in Neurofibromatosis Type 2: A retrospective review of 31 patients. Otol. Neurotol. 2012, 33, 1046–1052. [Google Scholar] [CrossRef] [PubMed]
- Blakeley, J.O.; Ye, X.; Duda, D.G.; Halpin, C.F.; Bergner, A.L.; Muzikansky, A.; Merker, V.; Gerstner, E.R.; Fayad, L.M.; Ahlawat, S.; et al. Efficacy and Biomarker Study of Bevacizumab for Hearing Loss Resulting from Neurofibromatosis Type 2–Associated Vestibular Schwannomas. J. Clin. Oncol. 2016, 34, 1669–1675. [Google Scholar] [CrossRef] [PubMed]
- Robertson, K.A.; Nalepa, G.; Yang, F.-C.; Bowers, D.C.; Ho, C.Y.; Hutchins, G.D.; Croop, J.M.; Vik, T.A.; Denne, S.C.; Parada, L.F.; et al. Imatinib mesylate for plexiform neurofibromas in patients with neurofibromatosis type 1: A phase 2 trial. Lancet Oncol. 2012, 13, 1218–1224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Widemann, B.C.; Dombi, E.; Gillespie, A.; Wolters, P.L.; Belasco, J.; Goldman, S.; Korf, B.R.; Solomon, J.; Martin, S.; Salzer, W.; et al. Phase 2 randomized, flexible crossover, double-blinded, placebo-controlled trial of the farnesyltransferase inhibitor tipifarnib in children and young adults with neurofibromatosis type 1 and progressive plexiform neurofibromas. Neuro-Oncology 2014, 16, 707–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Widemann, B.C.; Babovic-Vuksanovic, D.; Dombi, E.; Wolters, P.L.; Goldman, S.; Martin, S.; Goodwin, A.; Goodspeed, W.; Kieran, M.W.; Cohen, B.; et al. Phase II trial of pirfenidone in children and young adults with neurofibromatosis type 1 and progressive plexiform neurofibromas. Pediatr. Blood Cancer 2014, 61, 1598–1602. [Google Scholar] [CrossRef] [PubMed]
- Weiss, B.; Widemann, B.C.; Wolters, P.; Dombi, E.; Vinks, A.A.; Cantor, A.; Korf, B.; Perentesis, J.; Gutmann, D.H.; Schorry, E.; et al. Sirolimus for non-progressive NF1-associated plexiform neurofibromas: An NF clinical trials consortium phase II study. Pediatr. Blood Cancer 2013, 61, 982–986. [Google Scholar] [CrossRef] [PubMed]
- Weiss, B.; Widemann, B.C.; Wolters, P.; Dombi, E.; Vinks, A.; Cantor, A.; Perentesis, J.; Schorry, E.; Ullrich, N.; Gutmann, D.H.; et al. Sirolimus for progressive neurofibromatosis type 1-associated plexiform neurofibromas: A Neurofibromatosis Clinical Trials Consortium phase II study. Neuro-Oncology 2015, 17, 596–603. [Google Scholar] [CrossRef] [Green Version]
- Jakacki, R.I.; Dombi, E.; Steinberg, S.M.; Goldman, S.; Kieran, M.W.; Ullrich, N.J.; Pollack, I.F.; Goodwin, A.; Manley, P.E.; Fangusaro, J.; et al. Phase II trial of pegylated interferon alfa-2b in young patients with neurofibromatosis type 1 and unresectable plexiform neurofibromas. Neuro-Oncology 2017, 19, 289–297. [Google Scholar] [CrossRef] [Green Version]
- Zehou, O.; Ferkal, S.; Brugieres, P.; Barbarot, S.; Bastuji-Garin, S.; Combemale, P.; Valeyrie-Allanore, L.; Sbidian, E.; Wolkenstein, P. Absence of Efficacy of Everolimus in Neurofibromatosis 1-Related Plexiform Neurofibromas: Results from a Phase 2a Trial. J. Investig. Dermatol. 2019, 139, 718–720. [Google Scholar] [CrossRef] [Green Version]
- Gross, A.M.; Dombi, E.; Widemann, B.C. Current status of MEK inhibitors in the treatment of plexiform neurofibromas. Child’s Nerv. Syst. 2020, 36, 2443–2452. [Google Scholar] [CrossRef]
- Dombi, E.; Baldwin, A.; Marcus, L.J.; Fisher, M.J.; Weiss, B.; Kim, A.; Whitcomb, P.; Martin, S.; Aschbacher-Smith, L.E.; Rizvi, T.A.; et al. Activity of Selumetinib in Neurofibromatosis Type 1–Related Plexiform Neurofibromas. N. Engl. J. Med. 2016, 375, 2550–2560. [Google Scholar] [CrossRef] [PubMed]
- Gross, A.M.; Wolters, P.L.; Dombi, E.; Baldwin, A.; Whitcomb, P.; Fisher, M.J.; Weiss, B.; Kim, A.; Bornhorst, M.; Shah, A.C.; et al. Selumetinib in Children with Inoperable Plexiform Neurofibromas. N. Engl. J. Med. 2020, 382, 1430–1442. [Google Scholar] [CrossRef] [PubMed]
- Gross, A.M.; Glassberg, B.; Wolters, P.L.; Dombi, E.; Baldwin, A.; Fisher, M.J.; Kim, A.R.; Bornhorst, M.; Weiss, B.D.; Blakeley, J.O.; et al. Selumetinib in children with neurofibromatosis type 1 and asymptomatic inoperable plexiform neurofibroma at risk for developing tumor-related morbidity. Neuro-Oncology 2022, 24, 1978–1988. [Google Scholar] [CrossRef]
- Jackson, S.; Baker, E.H.; Gross, A.M.; Whitcomb, P.; Baldwin, A.; Derdak, J.; Tibery, C.; Desanto, J.; Carbonell, A.; Yohay, K.; et al. The MEK inhibitor selumetinib reduces spinal neurofibroma burden in patients with NF1 and plexiform neurofibromas. Neuro-Oncol. Adv. 2020, 2, vdaa095. [Google Scholar] [CrossRef]
- Coyne, G.H.O.; Gross, A.M.; Dombi, E.; Tibery, C.; Carbonell, A.; Takebe, N.; Derdak, J.; Pichard, D.; Srivastava, A.K.; Herrick, W.; et al. Phase II trial of the MEK 1/2 inhibitor selumetinib (AZD6244, ARRY-142886 Hydrogen Sulfate) in adults with neurofibromatosis type 1 (NF1) and inoperable plexiform neurofibromas (PN). J. Clin. Oncol. 2020, 38, 3612. [Google Scholar] [CrossRef]
- Santo, V.E.; Passos, J.; Nzwalo, H.; Carvalho, I.; Santos, F.; Martins, C.; Salgado, L.; Silva, C.E.; Vinhais, S.; Vilares, M.; et al. Selumetinib for plexiform neurofibromas in neurofibromatosis type 1: A single-institution experience. J. Neuro-Oncol. 2020, 147, 459–463. [Google Scholar] [CrossRef]
- Weiss, B.D.; Wolters, P.L.; Plotkin, S.R.; Widemann, B.C.; Tonsgard, J.H.; Blakeley, J.; Allen, J.C.; Schorry, E.; Korf, B.; Robison, N.J.; et al. NF106: A Neurofibromatosis Clinical Trials Consortium Phase II Trial of the MEK Inhibitor Mirdametinib (PD-0325901) in Adolescents and Adults with NF1-Related Plexiform Neurofibromas. J. Clin. Oncol. 2021, 39, 797–806. [Google Scholar] [CrossRef]
- McCowage, G.B.; Mueller, S.; Pratilas, C.A.; Hargrave, D.R.; Moertel, C.L.; Whitlock, J.; Fox, E.; Hingorani, P.; Russo, M.W.; Dasgupta, K.; et al. Trametinib in pediatric patients with neurofibromatosis type 1 (NF-1)–associated plexiform neurofibroma: A phase I/IIa study. J. Clin. Oncol. 2018, 36, 10504. [Google Scholar] [CrossRef]
- Wang, D.; Ge, L.; Guo, Z.; Li, Y.; Zhu, B.; Wang, W.; Wei, C.; Li, Q.; Wang, Z. Efficacy and Safety of Trametinib in Neurofibromatosis Type 1-Associated Plexiform Neurofibroma and Low-Grade Glioma: A Systematic Review and Meta-Analysis. Pharmaceuticals 2022, 15, 956. [Google Scholar] [CrossRef]
- Mueller, S.; Reddy, A.T.; Dombi, E.; Allen, J.; Packer, R.; Clapp, W.; Goldman, S.; Schorry, E.; Tonsgard, J.; Blakeley, J.; et al. MEK inhibitor binimetinib shows clinical activity in children with neurofibromatosis type 1-associated plexiform neurofibromas: A report from PNOC and the NF clinical trials consortium. Neuro Oncol. 2020, 22 (Suppl. 3), iii420–iii421. [Google Scholar] [CrossRef]
- Fisher, M.J.; Shih, C.-S.; Rhodes, S.D.; Armstrong, A.E.; Wolters, P.L.; Dombi, E.; Zhang, C.; Angus, S.P.; Johnson, G.L.; Packer, R.J.; et al. Cabozantinib for neurofibromatosis type 1–related plexiform neurofibromas: A phase 2 trial. Nat. Med. 2021, 27, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Nutakki, K.; Varni, J.W.; Swigonski, N.L. PedsQL Neurofibromatosis Type 1 Module for children, adolescents and young adults: Feasibility, reliability, and validity. J. Neuro-Oncol. 2018, 137, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Pollard, K.; Calizo, A.; Pratilas, C.A. Activation of Receptor Tyrosine Kinases Mediates Acquired Resistance to MEK Inhibition in Malignant Peripheral Nerve Sheath Tumors. Cancer Res 2021, 81, 747–762. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Tang, X.; Liang, H.; Yang, R.; Yan, T.; Guo, W. Prognosis and risk factors for malignant peripheral nerve sheath tumor: A systematic review and meta-analysis. World J. Surg. Oncol. 2020, 18, 257. [Google Scholar] [CrossRef]
- Yan, P.; Huang, R.; Hu, P.; Liu, F.; Zhu, X.; Hu, P.; Yin, H.; Zhang, J.; Meng, T.; Huang, Z. Nomograms for predicting the overall and cause-specific survival in patients with malignant peripheral nerve sheath tumor: A population-based study. J. Neuro-Oncol. 2019, 143, 495–503. [Google Scholar] [CrossRef]
- Judson, I.; Verweij, J.; Gelderblom, H.; Hartmann, J.T.; Schöffski, P.; Blay, J.-Y.; Kerst, J.M.; Sufliarsky, J.; Whelan, J.; Hohenberger, P.; et al. Doxorubicin alone versus intensified doxorubicin plus ifosfamide for first-line treatment of advanced or metastatic soft-tissue sarcoma: A randomised controlled phase 3 trial. Lancet Oncol. 2014, 15, 415–423. [Google Scholar] [CrossRef]
- Skotheim, R.I.; Kallioniemi, A.; Bjerkhagen, B.; Mertens, F.; Brekke, H.R.; Monni, O.; Mousses, S.; Mandahl, N.; Sœter, G.; Nesland, J.M.; et al. Topoisomerase-IIα Is Upregulated in Malignant Peripheral Nerve Sheath Tumors and Associated with Clinical Outcome. J. Clin. Oncol. 2003, 21, 4586–4591, Erratum in J. Clin. Oncol. 2004, 22, 969. [Google Scholar] [CrossRef]
- Higham, C.S.; Steinberg, S.M.; Dombi, E.; Perry, A.; Helman, L.J.; Schuetze, S.M.; Ludwig, J.A.; Staddon, A.; Milhem, M.; Rushing, D.; et al. SARC006: Phase II Trial of Chemotherapy in Sporadic and Neurofibromatosis Type 1 Associated Chemotherapy-Naive Malignant Peripheral Nerve Sheath Tumors. Sarcoma 2017, 2017, 8685638. [Google Scholar] [CrossRef] [Green Version]
- Steins, M.B.; Serve, H.; Zühlsdorf, M.; Senninger, N.; Semik, M.; Berdel, W.E. Carboplatin/etoposide induces remission of metasta-sised malignant peripheral nerve tumours (malignant schwannoma) refractory to first-line therapy. Oncol Rep. 2002, 9, 627–630. [Google Scholar] [PubMed]
- Leu, K.M.; Ostruszka, L.J.; Shewach, D.; Zalupski, M.; Sondak, V.; Biermann, J.S.; Lee, J.S.-J.; Couwlier, C.; Palazzolo, K.; Baker, L.H. Laboratory and Clinical Evidence of Synergistic Cytotoxicity of Sequential Treament with Gemcitabine Followed by Docetaxel in the Treatment of Sarcoma. J. Clin. Oncol. 2004, 22, 1706–1712. [Google Scholar] [CrossRef]
- Takahashi, M.; Komine, K.; Imai, H.; Okada, Y.; Saijo, K.; Takahashi, M.; Shirota, H.; Ohori, H.; Takahashi, S.; Chiba, N.; et al. Efficacy and safety of gemcitabine plus docetaxel in Japanese patients with unresectable or recurrent bone and soft tissue sarcoma: Results from a single-institutional analysis. PLoS ONE 2017, 12, e0176972. [Google Scholar] [CrossRef] [PubMed]
- Moretti, V.M.; Crawford, E.A.; Staddon, A.P.; Lackman, R.D.; Ogilvie, C.M. Early Outcomes for Malignant Peripheral Nerve Sheath Tumor Treated with Chemotherapy. Am. J. Clin. Oncol. 2011, 34, 417–421. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, H.G.; Rostad, S.; Ross, J.S.; Ali, S.M.; Millis, S.Z. Genomic Profiling in Patients with Malignant Peripheral Nerve Sheath Tumors Reveals Multiple Pathways with Targetable Mutations. J. Natl. Compr. Cancer Netw. 2018, 16, 967–974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagabushan, S.; Lau, L.M.S.; Barahona, P.; Wong, M.; Sherstyuk, A.; Marshall, G.M.; Tyrrell, V.; Wegner, E.A.; Ekert, P.G.; Cowley, M.J.; et al. Efficacy of MEK inhibition in a recurrent malignant peripheral nerve sheath tumor. NPJ Precis. Oncol. 2021, 5, 9. [Google Scholar] [CrossRef]
- Grit, J.L.; Pridgeon, M.G.; Essenburg, C.J.; Wolfrum, E.; Madaj, Z.B.; Turner, L.; Wulfkuhle, J.; Iii, E.F.P.; Graveel, C.R.; Steensma, M.R. Kinome Profiling of NF1-Related MPNSTs in Response to Kinase Inhibition and Doxorubicin Reveals Therapeutic Vulnerabilities. Genes 2020, 11, 331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maki, R.G.; D’Adamo, D.R.; Keohan, M.L.; Saulle, M.; Schuetze, S.M.; Undevia, S.D.; Livingston, M.B.; Cooney, M.M.; Hensley, M.L.; Mita, M.M.; et al. Phase II Study of Sorafenib in Patients with Metastatic or Recurrent Sarcomas. J. Clin. Oncol. 2009, 27, 3133–3140. [Google Scholar] [CrossRef] [Green Version]
- González-Muñoz, T.; Kim, A.; Ratner, N.; Peinado, H. The Need for New Treatments Targeting MPNST: The Potential of Strategies Combining MEK Inhibitors with Antiangiogenic Agents. Clin. Cancer Res. 2022, 28, 3185–3195. [Google Scholar] [CrossRef]
- Widemann, B.C.; Lu, Y.; Reinke, D.; Okuno, S.H.; Meyer, C.F.; Cote, G.M.; Chugh, R.; Milhem, M.; Hirbe, A.C.; Kim, A.; et al. Targeting Sporadic and Neurofibromatosis Type 1 (NF1) Related Refractory Malignant Peripheral Nerve Sheath Tumors (MPNST) in a Phase II Study of Everolimus in Combination with Bevacizumab (SARC016). Sarcoma 2019, 2019, 7656747. [Google Scholar] [CrossRef] [Green Version]
- Kim, A.; Lu, Y.; Okuno, S.H.; Reinke, D.; Maertens, O.; Perentesis, J.; Basu, M.; Wolters, P.L.; De Raedt, T.; Chawla, S.; et al. Targeting Refractory Sarcomas and Malignant Peripheral Nerve Sheath Tumors in a Phase I/II Study of Sirolimus in Combination with Ganetespib (SARC023). Sarcoma 2020, 2020, 5784876. [Google Scholar] [CrossRef] [Green Version]
- Chugh, R.; Wathen, J.K.; Maki, R.G.; Benjamin, R.S.; Patel, S.R.; Myers, P.A.; Priebat, D.A.; Reinke, D.K.; Thomas, D.G.; Keohan, M.L.; et al. Phase II Multicenter Trial of Imatinib in 10 Histologic Subtypes of Sarcoma Using a Bayesian Hierarchical Statistical Model. J. Clin. Oncol. 2009, 27, 3148–3153. [Google Scholar] [CrossRef]
- Schuetze, S.M.; Wathen, J.K.; Lucas, D.R.; Choy, E.; Samuels, B.L.; Staddon, A.P.; Ganjoo, K.N.; von Mehren, M.; Chow, W.A.; Loeb, D.M.; et al. SARC009: Phase 2 study of dasatinib in patients with previously treated, high-grade, advanced sarcoma. Cancer 2015, 122, 868–874. [Google Scholar] [CrossRef] [PubMed]
- Manji, G.A.; Van Tine, B.A.; Lee, S.M.; Raufi, A.G.; Pellicciotta, I.; Hirbe, A.C.; Pradhan, J.; Chen, A.; Rabadan, R.; Schwartz, G.K. A Phase I Study of the Combination of Pexidartinib and Sirolimus to Target Tumor-Associated Macrophages in Unresectable Sarcoma and Malignant Peripheral Nerve Sheath Tumors. Clin. Cancer Res. 2021, 27, 5519–5527. [Google Scholar] [CrossRef] [PubMed]
- Nishida, Y.; Urakawa, H.; Nakayama, R.; Kobayashi, E.; Ozaki, T.; Ae, K.; Matsumoto, Y.; Tsuchiya, H.; Goto, T.; Hiraga, H.; et al. PhaseIIclinical trial of pazopanib for patients with unresectable or metastatic malignant peripheral nerve sheath tumors. Int. J. Cancer 2021, 148, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Albritton, K.H.; Rankin, C.; Coffin, C.M.; Ratner, N.; Budd, G.T.; Schuetze, S.M.; Randall, R.L.; DeClue, J.E.; Borden, E.C. Phase II study of erlotinib in metastatic or unresectable malignant peripheral nerve sheath tumors (MPNST). J. Clin. Oncol. 2006, 24, 9518. [Google Scholar] [CrossRef]
- Dickson, M.A.; Mahoney, M.R.; Tap, W.D.; D’Angelo, S.P.; Keohan, M.L.; Van Tine, B.A.; Agulnik, M.; Horvath, L.E.; Nair, J.S.; Schwartz, G.K. Phase II study of MLN8237 (Alisertib) in advanced/metastatic sarcoma. Ann. Oncol. 2016, 27, 1855–1860. [Google Scholar] [CrossRef]
- Al-Ezzi, E.; Gounder, M.; Watson, G.; Mazzocca, A.; D’Angelo, S.P.; Bravetti, J.; Wang, H.; Razak, A.A.; Vincenzi, B. Selinexor, a First in Class, Nuclear Export Inhibitor for the Treatment of Advanced Malignant Peripheral Nerve Sheath Tumor. Oncologist 2021, 26, e710–e714. [Google Scholar] [CrossRef]
- Kobayashi, H.; Makise, N.; Shinozaki-Ushiku, A.; Zhang, L.; Ishibashi, Y.; Ikegami, M.; Tsuda, Y.; Kohsaka, S.; Ushiku, T.; Oda, K.; et al. Dramatic response to entrectinib in a patient with malignant peripheral nerve sheath tumor harboring novel SNRNP70-NTRK3 fusion gene. Genes Chromosom. Cancer 2022, 62, 47–51. [Google Scholar] [CrossRef]
- Özdemir, B.C.; Bohanes, P.; Bisig, B.; Missiaglia, E.; Tsantoulis, P.; Coukos, G.; Montemurro, M.; Homicsko, K.; Michielin, O. Deep Response to Anti-PD-1 Therapy of Metastatic Neurofibromatosis Type 1-Associated Malignant Peripheral Nerve Sheath Tumor wth CD274/PD-L1 Amplification. JCO Precis. Oncol. 2019, 3, 1–6. [Google Scholar] [CrossRef]
- Davis, L.E.; Nicholls, L.A.; Babiker, H.M.; Liau, J.; Mahadevan, D. PD-1 Inhibition Achieves a Complete Metabolic Response in a Patient with Malignant Peripheral Nerve Sheath Tumor. Cancer Immunol. Res. 2019, 7, 1396–1400. [Google Scholar] [CrossRef]


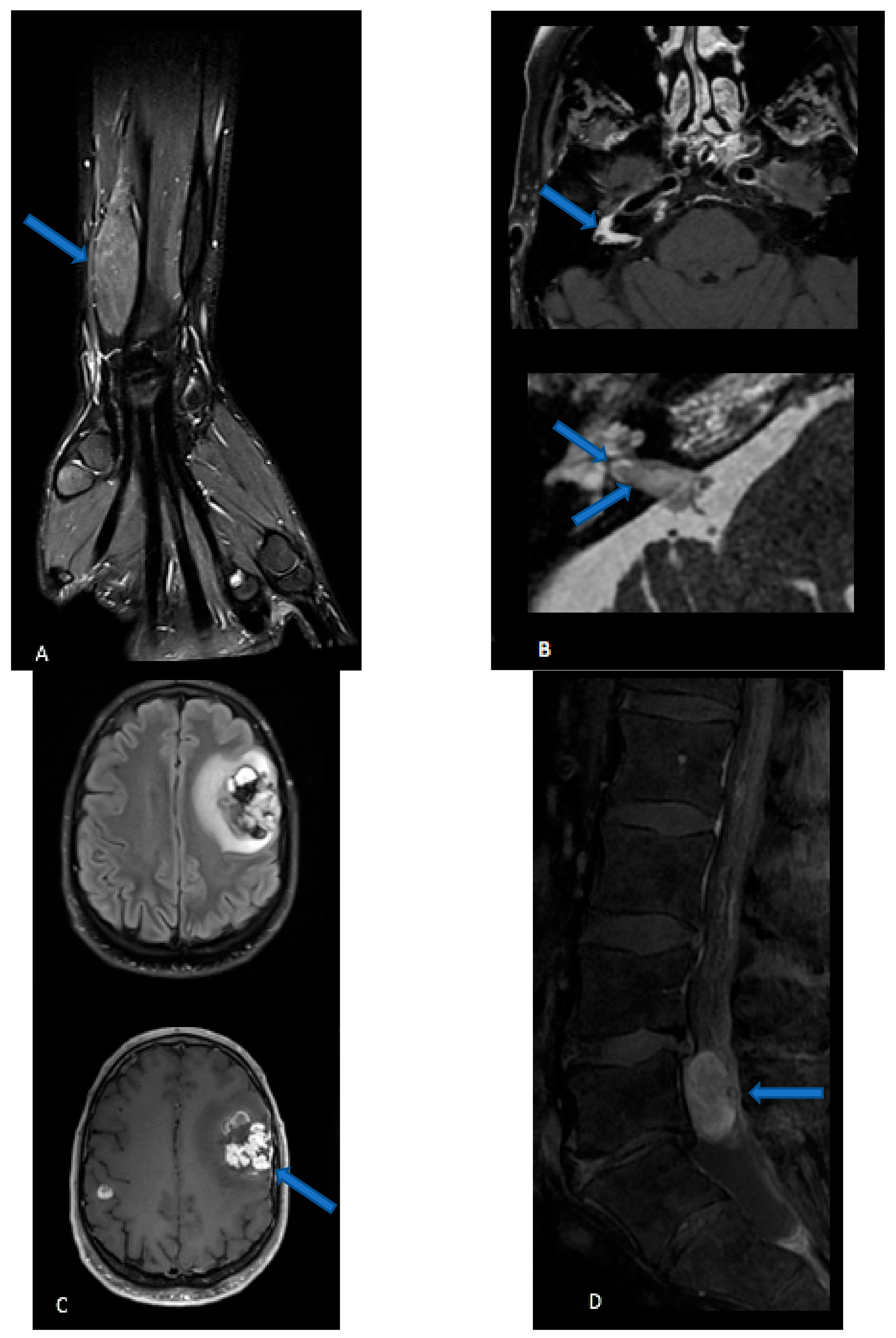{kind=link}
| Tumor Type | Estimated Incidence | Age | Sex | Location | Clinical Presentation |
|---|---|---|---|---|---|
| Schwannoma | 1.09 per 100,000/year | All age groups are affected, with a peak incidence in the 4th–6th decades | No sex predilection |
|
|
| Neurofibroma (Localized, diffuse, plexiform subtype) | 5.3% of all benign soft tissue tumors | All age groups are equally affected | No sex predilection |
|
|
| Perineurioma (Intraneural and soft tissue subtypes) | 1% of nerve sheath and soft tissue neoplasms, respectively (>50 cases of intraneural perineuriomas and >300 cases of soft tissue perineuriomas have been described) |
| Slight prevalence for female (M:F ratio 1:2) |
|
|
| Hybrid nerve sheath tumor | Very rare | Over a wide age range, with a peak in young adults | Equal sex distribution |
|
|
| Malignant peripheral nerve sheath tumour (MPNST) (epithelioid and perineural subtypes) | 2–10% of soft tissue sarcomas. Epithelioid MPNST is particularly rare (~5% of all MPNST) | Sporadic MPNST occurs most commonly in patients aged 20–50 years. MPNSTs in children are usually associated with NF1 | Similar sex distribution (M:F 1.2:1) |
|
|
| Malignant melanotic nerve sheath tumour (MMNST) | Very rare | Median age of 22.5 years in patients with Carney complex and 33.2 years in patients with sporadic presentation | No sex predilection |
|
|
| Neuroendocrine tumour (previously paraganglioma) | Very rare | 40–60 years | No sex predilection |
|
|
| Tumor Type | Malignancy * | Essential Diagnostic Criteria |
|---|---|---|
| Schwannoma | Benign | Extensive S100 and SOX10 expression; Verocay bodies; hyalinized blood vessels; loss of INI1 expression (epithelioid schwannoma) of mosaic pattern of INI1 expression (syndrome-associated schwannoma) |
| Neurofibroma | Benign | Infiltrative, low-cellularity spindle cell neoplasm associated with a variably myxoid to collagenous stroma and a mixed cell population |
| Perineurioma | Benign | Slender spindle cells with bipolar cytoplasmic processes in a storiform and/or whorled architecture or pseudo-onion bulb pattern positive for at least one perineurial antigen and negative for S100 |
| Hybrid nerve sheath tumors | Benign | Intermingled features of two types of benign nerve sheath tumors; appropriate immunohistochemical staining for each component |
| MPNST | Malignant | For patients with NF1, a histopathological consistent malignant spindle cell tumor is sufficient. For sporadic tumors, additional focal S100 or SOX10 expression and association with a peripheral nerve and no (X;18)(p11.2;q11.2) translocation (oncogenic SS18-SSX1 fusion) of association with a peripheral nerve and immunohistochemical/molecular proof of PRC2 inactivation or methylation profile of MPNST |
| Epithelioid MPNST | Malignant | Epithelioid cells with prominent nucleoli showing diffuse expression of S100, absence of melanocytic markers, and SMARCB1 loss |
| Malignant melanotic nerve sheath tumor | Malignant | Fascicular to sheet-like proliferation of variably pigmented, relatively uniform plump spindled cells with coexpression of S100/SOX10 and melanocytic markers or PRKAR1A mutation Desirable: origin from paraspinal or visceral autonomic nerves |
| Cauda equina neuroendocrine tumor | Benign | Well-demarcated tumor of the cauda equina with a nested pattern and synaptophysin or chromogranin expression |
| Tumor Type | Impact of Gross Total Resection | Consequences of Residual Tumor | Indications for Radiotherapy |
|---|---|---|---|
| Schwannoma |
|
|
|
| Vestibular schwannoma(VS) |
|
|
|
| Neurofibroma |
|
|
|
| Perineurioma |
|
|
|
| Hybrid nerve sheath tumors |
|
|
|
| MPNST |
|
|
|
| Cauda equina neuroendocrine tumor |
|
|
|
| Class of Evidence | Level of Recommendation | |
|---|---|---|
| Resection is recommended to obtain a histological and molecular diagnosis. | II | B |
| Gross total resection is recommended as therapy of first choice when feasible in PNST. When risk of neurological sequelae from surgery is high, detailed informed preoperative counseling by a surgeon experienced in performing such surgery is important. | III | B |
| Use of intraoperative electrophysiological monitoring is mandatory to preserve nerve functioning during surgery of PNST. | III | B |
| Intraoperative high-resolution ultrasound and/or use of a microscope are recommended to achieve complete resection by intracapsular dissection and preserving the attached functional nerve fibers. | IV | Good practice point |
| Observaton with serial MRI and audiological monitoring without any tumor-directed treatment is considered appropriate for incidental, asymptomatic vestibular schwannomas | III | C |
| Surgery is considered the primary treatment to reduce mass effect in vestibular schwannomas | II | B |
| To improve the rate of functional preservation, intraoperative monitoring is mandatory for surgery of vestibular schwannomas and should include somatosensoric evoked potentials, monitoring of the facial nerve comprising direct electrical stimulation and free-running electromyography, and brainstem auditory evoked responses | III | B |
| The frequency of surveillance imaging with MRI should be based on the extent of resection (GTR vs. non-GTR) and tumor aggressiveness, and the duration should be up to 5 years in PNST | IV | Good practice point |
| Annual MRI is recommended for 5 years in patients with untreated, incidental schwannomas, as well as conservatively treated, irradiated, and incompletely resected vestibular schwannomas. Thereafter, the follow-up intervals can be increased | IV | Good practice point |
| Repeated surgery in patients with local tumor progression or recurrence of PNST should be considered | IV | Good practice point |
| Radiotherapy may be omitted in benign tumors after complete resection (e.g., schwannomas) | II | B |
| SRS is superior over microsurgery for patients with vestibular schwannomas < 3 cm in terms of preserving facial nerve and hearing function | II | B |
| SRS should be delivered with a dose of 11–14 Gy at the margin in vestibular schwannomas and 11–12 Gy when the risk of hearing loss is a critical issue | III | C |
| Radiotherapy is not recommended for NF-related plexiform neurofibromas given the theoretic risk of secondary malignancy in a tumor-suppressor syndrome | IV | Good practice point |
| Perineurioma/hybrid nerve sheath tumors with high risk of neurological sequelae after surgery less than 15 mm in size may be treated with a single fraction radiosurgery with 12–14 Gy, while fractionated stereotactic radiotherapy with 1.8 Gy to 50.4 Gy may be considered for larger tumors | IV | C |
| Radiotherapy (55.8 Gy in 1.8 Gy) should be considered in MPNST after incomplete resection or greater than 5 cm, and pre-operatively in case of unresectable MPNST to improve local control and/or increase the potential of an R1 or even R0 resection | IV | C |
| Final dose of adjuvant radiotherapy after surgery of MPNST may be determined by the resection status: a boost of 10.8 Gy (R1 resection; cumulative dose 55.8 Gy) or 19.8 Gy (R2 resection; cumulative dose 64.8 Gy) should be considered if necessary | IV | C |
| If not operable, cauda equine neuroendocrine tumors may be treated with a radiation dose depending on the treatment intent (curative vs. palliative symptomatic) | IV | C |
| Consider bevacizumab in patients with multiple rapidly enlarging tumours, who are inoperable (e.g., bilateral vestibular schwannomas) | II | B |
| In patients with plexiform neurofibromas or MPNSTs, systemic treatments should be considered especially when complete resection is not feasible | II | B |
| Consider targeted therapy with MEK inhibitor selumetinib in children ≥ 2 years with NF1 and inoperable and symptomatic plexiform neurofibromas | I | B |
| Consider targeted therapy with MEK inhibitor selumetinib NF1 adult patients with unresectable, symptomatic, and/or progressive plexiform neurofibromas | II | B |
| Anthracycline-based treatment is the first-line therapy for unresectable, locally advanced, or metastatic MPNST | II | B |
| The topoisomerase II inhibitor etoposide, alone or in association with ifosfamide, could be considered in case of progression of MPNST following anthracycline-based therapy | III | C |
| Other cytotoxic chemotherapy regimens, including gemcitabine plus docetaxel, may be considered when anthracycline and etoposide-based therapy fails to control MPNST | IV | C |
| Sequencing to identify molecular targets to direct potential targeted therapy may be performed for incompletely resected or recurrent tumors that have exhausted treatment options | III | C |
| In patients with recurrent plexiform neurofibromas or MPNST who are no longer eligible for local treatments, enrollment in clinical trials might be warranted, particularly in patients with a good performance status. | II | C |
| Study | Phase | N° of Patients | Treatment | Outcome Measure |
|---|---|---|---|---|
| Cutaneous neurofibroma | ||||
| NCT04730583 | 1 | 20 |
| Primary:
|
| NCT05199376 CryoNF1 | NA | 30 |
| Primary:
|
| NCT05005845 | 2 | 168 |
| Primary:
|
| NCT02728388 | 2 | 30 |
| Primary:
|
| Atypical/plexiform neurofibroma | ||||
| NCT04750928 | 1/2 | 50 |
| Primary:
|
| NCT04954001 | 1/2 | 160 |
| Primary:
|
| NCT05309668 (SPRINKLE) | 1/2 | 44 |
| Primary:
|
| NCT03326388 (INSPECT) | 1/2 | 30 |
| Primary:
|
| NCT02390752 | 1/2 | 81 |
| Primary:
|
| NCT03363217 | 2 | 150 (including also LGG and HGG MAPK/ERK mutated) |
| Primary:
|
| Malignant peripheral nerve sheath tumor | ||||
| NCT02584647 | 1/2 | 43 |
| Primary:
|
| NCT05107037 | 1 | 120 |
| Primary:
|
| NCT05011019 (Chinese patients) | 1/2 | 192 |
| Primary:
|
| NCT02700230 | 1 | 30 |
| Primary:
|
| NCT05245500 | 1/2 | 339 (including also solid tumors with MTAP deletion) |
| Primary:
|
| NCT04917042 | 2 | 24 |
| Primary:
|
| NCT04897321 | 1 | 32 (including also B7-H3+ solid tumors) |
| Primary:
|
| NCT04872543 | 2 | 25 |
| Primary:
|
| NCT04465643 | 2 | 18 |
| Primary:
|
| NCT04420975 | 1 | 20 (including also other soft tissue tumors) |
| Primary:
|
| NCT04222413 | 1 | 54 (including also other advanced or metastatic solid tumors) |
| Primary:
|
| NCT03618381 | 1 | 36 (including also other soft tissue tumors) |
| Primary:
|
| NCT03611868 | 1/2 | 224 (including also metastatic melanoma or advanced solid tumors) |
| Primary:
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pellerino, A.; Verdijk, R.M.; Nichelli, L.; Andratschke, N.H.; Idbaih, A.; Goldbrunner, R. Diagnosis and Treatment of Peripheral and Cranial Nerve Tumors with Expert Recommendations: An EUropean Network for RAre CANcers (EURACAN) Initiative. Cancers 2023, 15, 1930. https://doi.org/10.3390/cancers15071930
Pellerino A, Verdijk RM, Nichelli L, Andratschke NH, Idbaih A, Goldbrunner R. Diagnosis and Treatment of Peripheral and Cranial Nerve Tumors with Expert Recommendations: An EUropean Network for RAre CANcers (EURACAN) Initiative. Cancers. 2023; 15(7):1930. https://doi.org/10.3390/cancers15071930
Chicago/Turabian StylePellerino, Alessia, Robert M. Verdijk, Lucia Nichelli, Nicolaus H. Andratschke, Ahmed Idbaih, and Roland Goldbrunner. 2023. "Diagnosis and Treatment of Peripheral and Cranial Nerve Tumors with Expert Recommendations: An EUropean Network for RAre CANcers (EURACAN) Initiative" Cancers 15, no. 7: 1930. https://doi.org/10.3390/cancers15071930