
Senescent Stromal Cells in the Tumor Microenvironment: Victims or Accomplices?

{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
- Which types of stromal cells undergo senescence in the TME and their corresponding biological changes?
- Which signals do tumor cells transmit to and receive from the senescent stromal cells?
- What is the role of infiltrated senescent immune cells in tumor development and progression?
- What is the role of senescent stromal cells to develop therapy resistance in the TME?
2. The Discovery of Senescent Stroma
2.1. Biomarkers of Senescent Cells
2.2. Evidence of the Occurrence of Cellular Senescence in Tumor Stroma
3. Formation of Senescent Stroma
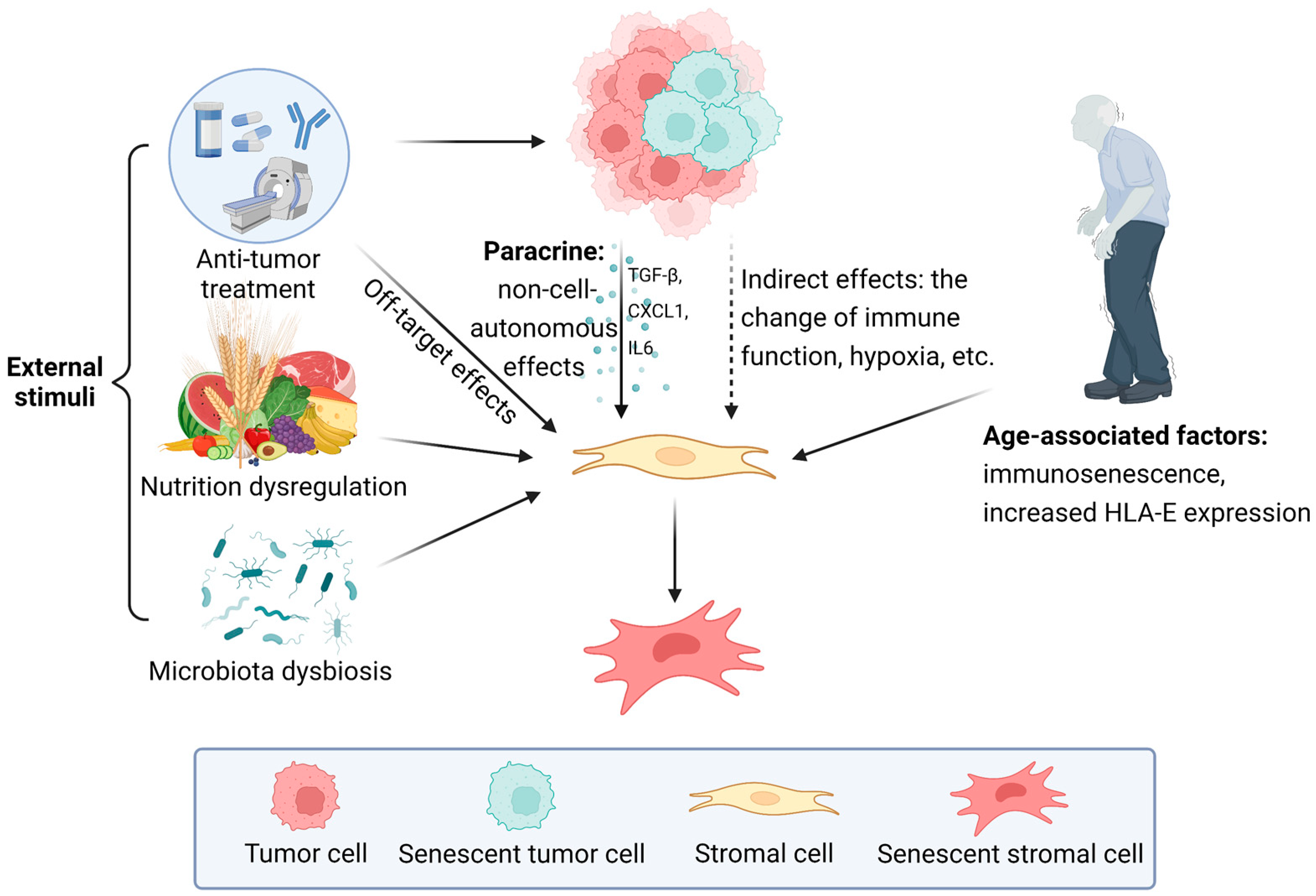
3.1. The Influence of External Stimuli
3.2. The Induction from Senescent Cells in TME
3.3. Age-Associated Accumulation of Senescent Cells
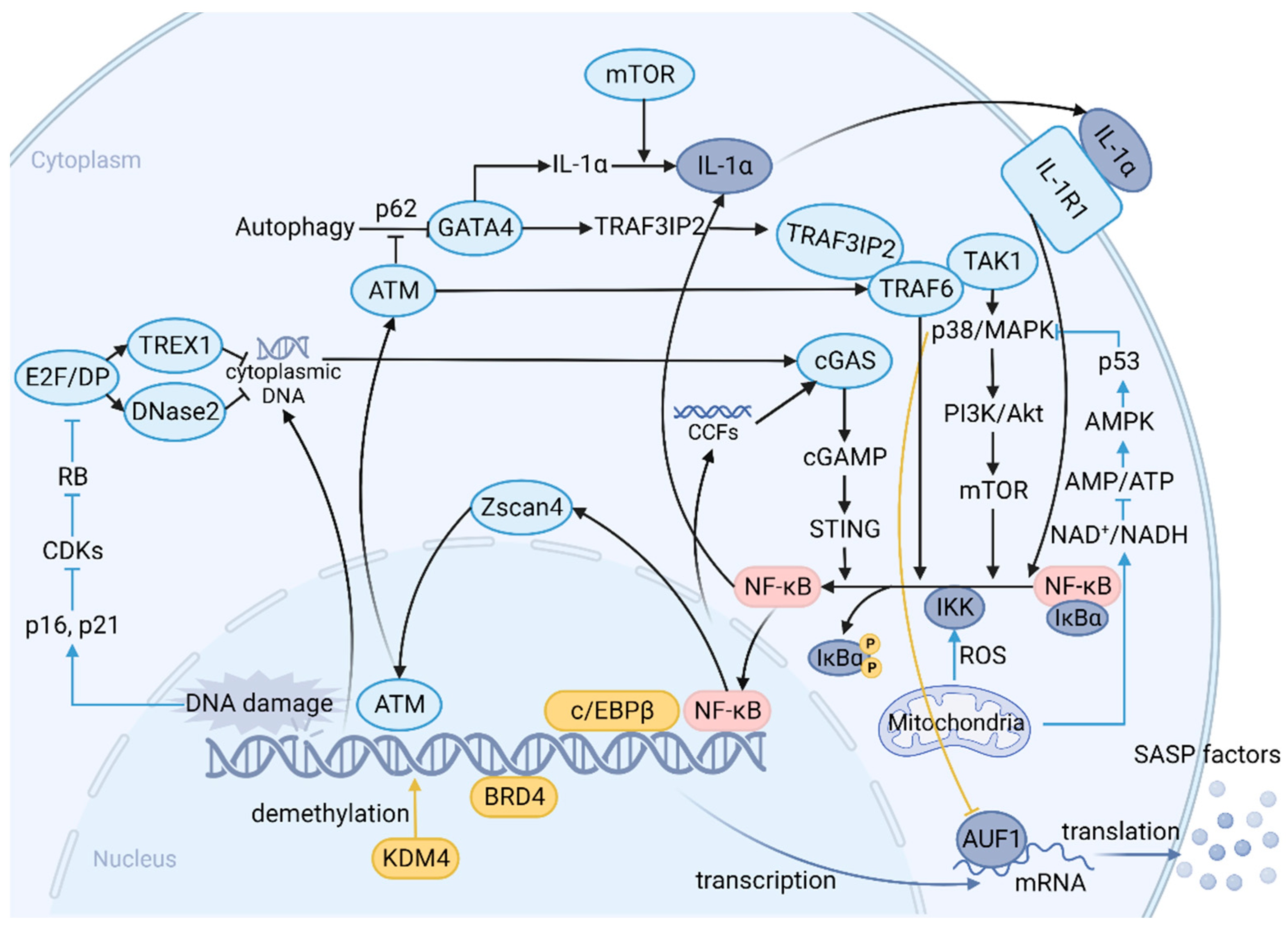
4. SASP: A Function Arm of Cellular Senescence and a Complex Network of Signaling Pathways
5. How Senescent Stroma Promotes Tumor Progression
5.1. Epithelial–Mesenchymal Transition (EMT)
5.2. Cancer Stem Cells (CSCs) and Cancer Stemness
5.3. Angiogenesis
5.4. ECM Remodeling and MMPs
6. Distant Metastasis: Distinct Niches Shaped by Senescent Stroma
7. Senescent Stroma and Immune Infiltration in the TME
7.1. Inflammation Induced by SASP
7.2. Immunosuppressive Cells: Assistant to the Progression of Malignancy
7.3. Senescent T Cells
8. Tumor Therapy Resistance Promoted by Senescent Stroma
9. Prospects
10. Conclusions
10.1. BOX1 CAFs, Myofibroblasts, and Senescent Fibroblasts: Unresolved Dispute
10.2. BOX2 “One-Two Punch” Therapy
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef] [PubMed]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Espín, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef] [PubMed]
- Di Micco, R.; Krizhanovsky, V.; Baker, D.; d’Adda di Fagagna, F. Cellular senescence in ageing: From mechanisms to therapeutic opportunities. Nat. Rev. Mol. Cell Biol. 2021, 22, 75–95. [Google Scholar] [CrossRef]
- Shmulevich, R.; Krizhanovsky, V. Cell Senescence, DNA Damage, and Metabolism. Antioxid. Redox Signal. 2021, 34, 324–334. [Google Scholar] [CrossRef]
- Galanos, P.; Vougas, K.; Walter, D.; Polyzos, A.; Maya-Mendoza, A.; Haagensen, E.J.; Kokkalis, A.; Roumelioti, F.-M.; Gagos, S.; Tzetis, M.; et al. Chronic p53-independent p21 expression causes genomic instability by deregulating replication licensing. Nat. Cell Biol. 2016, 18, 777–789. [Google Scholar] [CrossRef]
- Milanovic, M.; Fan, D.N.Y.; Belenki, D.; Däbritz, J.H.M.; Zhao, Z.; Yu, Y.; Dörr, J.R.; Dimitrova, L.; Lenze, D.; Monteiro Barbosa, I.A.; et al. Senescence-associated reprogramming promotes cancer stemness. Nature 2018, 553, 96–100. [Google Scholar] [CrossRef] [Green Version]
- Campisi, J. Aging, cellular senescence, and cancer. Annu. Rev. Physiol. 2013, 75, 685–705. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Lankhorst, L.; Bernards, R. Exploiting senescence for the treatment of cancer. Nat. Rev. Cancer 2022, 22, 340–355. [Google Scholar] [CrossRef]
- Sharpless, N.E.; Sherr, C.J. Forging a signature of in vivo senescence. Nat. Rev. Cancer 2015, 15, 397–408. [Google Scholar] [CrossRef]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic ras Provokes Premature Cell Senescence Associated with Accumulation of p53 and p16INK4a. Cell 1997, 88, 593–602. [Google Scholar] [CrossRef] [Green Version]
- Lin, A.W.; Barradas, M.; Stone, J.C.; van Aelst, L.; Serrano, M.; Lowe, S.W. Premature senescence involving p53 and p16 is activated in response to constitutive MEK/MAPK mitogenic signaling. Genes Dev. 1998, 12, 3008–3019. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Woods, D.; McMahon, M.; Bishop, J.M. Senescence of human fibroblasts induced by oncogenic Raf. Genes Dev. 1998, 12, 2997–3007. [Google Scholar] [CrossRef] [Green Version]
- Dimri, G.P.; Itahana, K.; Acosta, M.; Campisi, J. Regulation of a Senescence Checkpoint Response by the E2F1 Transcription Factor and p14 ARF Tumor Suppressor. Mol. Cell. Biol. 2000, 20, 273–285. [Google Scholar] [CrossRef] [Green Version]
- Childs, B.G.; Gluscevic, M.; Baker, D.J.; Laberge, R.-M.; Marquess, D.; Dananberg, J.; van Deursen, J.M. Senescent cells: An emerging target for diseases of ageing. Nat. Rev. Drug Discov. 2017, 16, 718–735. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; de Oliveira, R.L.; Wang, C.; Neto, J.M.F.; Mainardi, S.; Evers, B.; Lieftink, C.; Morris, B.; Jochems, F.; Willemsen, L.; et al. High-Throughput Functional Genetic and Compound Screens Identify Targets for Senescence Induction in Cancer. Cell Rep. 2017, 21, 773–783. [Google Scholar] [CrossRef] [Green Version]
- Demirci, D.; Dayanc, B.; Mazi, F.A.; Senturk, S. The Jekyll and Hyde of Cellular Senescence in Cancer. Cells 2021, 10, 208. [Google Scholar] [CrossRef]
- Coppé, J.-P.; Desprez, P.-Y.; Krtolica, A.; Campisi, J. The Senescence-Associated Secretory Phenotype: The Dark Side of Tumor Suppression. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 99–118. [Google Scholar] [CrossRef] [Green Version]
- Shelton, D.N.; Chang, E.; Whittier, P.S.; Choi, D.; Funk, W.D. Microarray analysis of replicative senescence. Curr. Biol. 1999, 9, 939–945. [Google Scholar] [CrossRef] [Green Version]
- Coppé, J.-P.; Patil, C.K.; Rodier, F.; Sun, Y.; Muñoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.-Y.; Campisi, J. Senescence-Associated Secretory Phenotypes Reveal Cell-Nonautonomous Functions of Oncogenic RAS and the p53 Tumor Suppressor. PLoS Biol. 2008, 6, e301. [Google Scholar] [CrossRef]
- Faget, D.V.; Ren, Q.; Stewart, S.A. Unmasking senescence: Context-dependent effects of SASP in cancer. Nat. Rev. Cancer 2019, 19, 439–453. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paget, S. The distribution of secondary growths in cancer of the breast. Cancer Metastasis Rev. 1989, 8, 98–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maman, S.; Witz, I.P. A history of exploring cancer in context. Nat. Rev. Cancer 2018, 18, 359–376. [Google Scholar] [CrossRef]
- Jin, M.-Z.; Jin, W.-L. The updated landscape of tumor microenvironment and drug repurposing. Signal Transduct. Target. Ther. 2020, 5, 166. [Google Scholar] [CrossRef]
- Krtolica, A.; Parrinello, S.; Lockett, S.; Desprez, P.-Y.; Campisi, J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: A link between cancer and aging. Proc. Natl. Acad. Sci. USA 2001, 98, 12072–12077. [Google Scholar] [CrossRef] [Green Version]
- Gabai, Y.; Assouline, B.; Ben-Porath, I. Senescent stromal cells: Roles in the tumor microenvironment. Trends Cancer 2022, 9, 28–41. [Google Scholar] [CrossRef]
- Muñoz-Espín, D.; Cañamero, M.; Maraver, A.; Gómez-López, G.; Contreras, J.; Murillo-Cuesta, S.; Rodríguez-Baeza, A.; Varela-Nieto, I.; Ruberte, J.; Collado, M.; et al. Programmed Cell Senescence during Mammalian Embryonic Development. Cell 2013, 155, 1104–1118. [Google Scholar] [CrossRef] [Green Version]
- Krizhanovsky, V.; Yon, M.; Dickins, R.A.; Hearn, S.; Simon, J.; Miething, C.; Yee, H.; Zender, L.; Lowe, S.W. Senescence of Activated Stellate Cells Limits Liver Fibrosis. Cell 2008, 134, 657–667. [Google Scholar] [CrossRef] [Green Version]
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018, 28, 436–453. [Google Scholar] [CrossRef]
- Kurz, D.J.; Decary, S.; Hong, Y.; Erusalimsky, J.D. Senescence-associated (beta)-galactosidase reflects an increase in lysosomal mass during replicative ageing of human endothelial cells. J. Cell Sci. 2000, 113 Pt 20, 3613–3622. [Google Scholar] [CrossRef] [PubMed]
- Evangelou, K.; Lougiakis, N.; Rizou, S.V.; Kotsinas, A.; Kletsas, D.; Muñoz-Espín, D.; Kastrinakis, N.G.; Pouli, N.; Marakos, P.; Townsend, P.; et al. Robust, universal biomarker assay to detect senescent cells in biological specimens. Aging Cell 2017, 16, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Pazolli, E.; Luo, X.; Brehm, S.; Carbery, K.; Chung, J.-J.; Prior, J.L.; Doherty, J.; Demehri, S.; Salavaggione, L.; Piwnica-Worms, D.; et al. Senescent Stromal-Derived Osteopontin Promotes Preneoplastic Cell Growth. Cancer Res. 2009, 69, 1230–1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burd, C.E.; Sorrentino, J.A.; Clark, K.S.; Darr, D.B.; Krishnamurthy, J.; Deal, A.M.; Bardeesy, N.; Castrillon, D.H.; Beach, D.H.; Sharpless, N.E. Monitoring Tumorigenesis and Senescence In Vivo with a p16INK4a-Luciferase Model. Cell 2013, 152, 340–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okuma, A.; Hanyu, A.; Watanabe, S.; Hara, E. p16(Ink4a) and p21(Cip1/Waf1) promote tumour growth by enhancing myeloid-derived suppressor cells chemotaxis. Nat. Commun. 2017, 8, 2050. [Google Scholar] [CrossRef] [Green Version]
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.P.; Athineos, D.; Kang, T.-W.; Lasitschka, F.; Andrulis, M.; et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nature 2013, 15, 978–990. [Google Scholar] [CrossRef]
- Montes, C.L.; Chapoval, A.I.; Nelson, J.; Orhue, V.; Zhang, X.; Schulze, D.H.; Strome, S.E.; Gastman, B.R. Tumor-Induced Senescent T Cells with Suppressor Function: A Potential Form of Tumor Immune Evasion. Cancer Res. 2008, 68, 870–879. [Google Scholar] [CrossRef] [Green Version]
- Golomb, L.; Sagiv, A.; Pateras, I.S.; Maly, A.; Krizhanovsky, V.; Gorgoulis, V.G.; Oren, M.; Ben-Yehuda, A. Age-associated inflammation connects RAS-induced senescence to stem cell dysfunction and epidermal malignancy. Cell Death Differ. 2015, 22, 1764–1774. [Google Scholar] [CrossRef] [Green Version]
- Tokarsky-Amiel, R.; Azazmeh, N.; Helman, A.; Stein, Y.; Hassan, A.; Maly, A.; Ben-Porath, I. Dynamics of Senescent Cell Formation and Retention Revealed by p14ARF Induction in the Epidermis. Cancer Res. 2013, 73, 2829–2839. [Google Scholar] [CrossRef] [Green Version]
- Yoshimoto, S.; Loo, T.M.; Atarashi, K.; Kanda, H.; Sato, S.; Oyadomari, S.; Iwakura, Y.; Oshima, K.; Morita, H.; Hattori, M.; et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 2013, 499, 97–101. [Google Scholar] [CrossRef]
- Chiesa-Vottero, A.G.; Malpica, A.; Deavers, M.T.; Broaddus, R.; Nuovo, G.J.; Silva, E.G. Immunohistochemical Overexpression of p16 and p53 in Uterine Serous Carcinoma and Ovarian High-grade Serous Carcinoma. Int. J. Gynecol. Pathol. 2007, 26, 328–333. [Google Scholar] [CrossRef]
- Yemelyanova, A.; Ji, H.; Shih, I.M.; Wang, T.L.; Wu, L.S.; Ronnett, B.M. Utility of p16 expression for distinction of uterine serous carcinomas from endometrial endometrioid and endocervical adenocarcinomas: Immunohistochemical analysis of 201 cases. Am. J. Surg. Pathol. 2009, 33, 1504–1514. [Google Scholar] [CrossRef]
- Moritani, S.; Ichihara, S.; Hasegawa, M.; Iwakoshi, A.; Murakami, S.; Sato, T.; Okamoto, T.; Mori, Y.; Kuhara, H.; Silverberg, S.G. Stromal p16 expression differentiates endometrial polyp from endometrial hyperplasia. Virchows Arch. 2012, 461, 141–148. [Google Scholar] [CrossRef]
- Yoon, G.; Koh, C.W.; Yoon, N.; Kim, J.-Y.; Kim, H.-S. Stromal p16 expression is significantly increased in endometrial carcinoma. Oncotarget 2016, 8, 4826–4836. [Google Scholar] [CrossRef] [Green Version]
- Yoon, N.; Yoon, G.; Park, C.K.; Kim, H.-S. Stromal p16 expression is significantly increased in malignant ovarian neoplasms. Oncotarget 2016, 7, 64665–64673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, T.; Do, S.-I.; NA, K.; Kim, G.; Jeong, Y.I.; Kim, Y.W.; Kim, H.-S. Stromal p16 Overexpression in Gastric-type Mucinous Carcinoma of the Uterine Cervix. Anticancer Res. 2018, 38, 3551–3558. [Google Scholar] [CrossRef] [Green Version]
- Kihara, A.; Amano, Y.; Yoshimoto, T.; Matsubara, D.; Fukushima, N.; Fujiwara, H.; Niki, T. Stromal p16 Expression Helps Distinguish Atypical Polypoid Adenomyoma From Myoinvasive Endometrioid Carcinoma of the Uterus. Am. J. Surg. Pathol. 2019, 43, 1526–1535. [Google Scholar] [CrossRef]
- Bohn, O.L.; Fuertes-Camilo, M.; Navarro, L.; Saldivar, J.; Sanchez-Sosa, S. p16INK4a expression in basal-like breast carcinoma. Int. J. Clin. Exp. Pathol. 2010, 3, 600–607. [Google Scholar]
- Harbhajanka, A.; Lamzabi, I.; Bitterman, P.; Reddy, V.B.; Ghai, R.; Gattuso, P. Correlation of p16 Expression on Cancer and Stromal Cells With Clinicopathologic and Immunohistochemical Features of Lobular Breast Carcinoma. Appl. Immunohistochem. Mol. Morphol. 2019, 27, 658–662. [Google Scholar] [CrossRef]
- Na, K.; Sung, J.-Y.; Kim, H.-S. Stromal p16 Overexpression in Adult Granulosa Cell Tumors of the Ovary. Anticancer Res. 2017, 37, 2437–2444. [Google Scholar] [CrossRef] [Green Version]
- Ye, S.-W.; Wang, Y.; Valmori, D.; Ayyoub, M.; Han, Y.; Xu, X.-L.; Zhao, A.-L.; Qu, L.; Gnjatic, S.; Ritter, G.; et al. Ex-Vivo Analysis of CD8+ T Cells Infiltrating Colorectal Tumors Identifies a Major Effector-Memory Subset with Low Perforin Content. J. Clin. Immunol. 2006, 26, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Hwang, H.J.; Lee, Y.-R.; Kang, D.; Lee, H.C.; Seo, H.R.; Ryu, J.-K.; Kim, Y.-N.; Ko, Y.-G.; Park, H.J.; Lee, J.-S. Endothelial cells under therapy-induced senescence secrete CXCL11, which increases aggressiveness of breast cancer cells. Cancer Lett. 2020, 490, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.-G.; Jeon, B.-M.; Yun, Y.-J.; Cui, C.-H.; Kim, S.-C. Ginsenoside Rh2 Ameliorates Doxorubicin-Induced Senescence Bystander Effect in Breast Carcinoma Cell MDA-MB-231 and Normal Epithelial Cell MCF-10A. Int. J. Mol. Sci. 2019, 20, 1244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.-J.; Park, I.H.; Rhee, W.J.; Kim, H.S.; Shin, J.-S. HMGB1 modulates the balance between senescence and apoptosis in response to genotoxic stress. FASEB J. 2019, 33, 10942–10953. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-J.; Park, I.H.; Kwak, M.S.; Rhee, W.J.; Kim, S.H.; Shin, J.-S. HMGB1 orchestrates STING-mediated senescence via TRIM30α modulation in cancer cells. Cell Death Discov. 2021, 7, 28. [Google Scholar] [CrossRef]
- Miyake, T.; Shimada, M.; Matsumoto, Y.; Okino, A. DNA Damage Response After Ionizing Radiation Exposure in Skin Keratinocytes Derived from Human-Induced Pluripotent Stem Cells. Int. J. Radiat. Oncol. 2019, 105, 193–205. [Google Scholar] [CrossRef]
- Meng, J.; Li, Y.; Wan, C.; Sun, Y.; Dai, X.; Huang, J.; Hu, Y.; Gao, Y.; Wu, B.; Zhang, Z.; et al. Targeting senescence-like fibroblasts radiosensitizes non–small cell lung cancer and reduces radiation-induced pulmonary fibrosis. J. Clin. Investig. 2021, 6, e146334. [Google Scholar] [CrossRef]
- Benadjaoud, M.A.; Soysouvanh, F.; Tarlet, G.; Paget, V.; Buard, V.; de Andrade, H.S.; Morilla, I.; Dos Santos, M.; Bertho, A.; L’Homme, B.; et al. Deciphering the Dynamic Molecular Program of Radiation-Induced Endothelial Senescence. Int. J. Radiat. Oncol. 2021, 112, 975–985. [Google Scholar] [CrossRef]
- Prise, K.M.; O’Sullivan, J.M. Radiation-induced bystander signalling in cancer therapy. Nat. Rev. Cancer 2009, 9, 351–360. [Google Scholar] [CrossRef]
- Gupta, K.; Burns, T.C. Radiation-Induced Alterations in the Recurrent Glioblastoma Microenvironment: Therapeutic Implications. Front. Oncol. 2018, 8, 503. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.Y.; Souroullas, G.P.; Diekman, B.O.; Krishnamurthy, J.; Hall, B.M.; Sorrentino, J.A.; Parker, J.S.; Sessions, G.A.; Gudkov, A.V.; Sharpless, N.E. Cells exhibiting strong p16(INK4a) promoter activation in vivo display features of senescence. Proc. Natl. Acad. Sci. USA 2019, 116, 2603–2611. [Google Scholar] [CrossRef] [Green Version]
- Fane, M.; Weeraratna, A.T. How the ageing microenvironment influences tumour progression. Nat. Rev. Cancer 2019, 20, 89–106. [Google Scholar] [CrossRef]
- Boyajian, J.L.; Ghebretatios, M.; Schaly, S.; Islam, P.; Prakash, S. Microbiome and Human Aging: Probiotic and Prebiotic Potentials in Longevity, Skin Health and Cellular Senescence. Nutrients 2021, 13, 4550. [Google Scholar] [CrossRef]
- Yang, G.; Rosen, D.G.; Zhang, Z.; Bast, R.C.; Mills, G.B.; Colacino, J.A.; Mercado-Uribe, I.; Liu, J. The chemokine growth-regulated oncogene 1 (Gro-1) links RAS signaling to the senescence of stromal fibroblasts and ovarian tumorigenesis. Proc. Natl. Acad. Sci. USA 2006, 103, 16472–16477. [Google Scholar] [CrossRef] [Green Version]
- Lugo, R.; Gabasa, M.; Andriani, F.; Puig, M.; Facchinetti, F.; Ramírez, J.; Gómez-Caro, A.; Pastorino, U.; Fuster, G.; Almendros, I.; et al. Heterotypic paracrine signaling drives fibroblast senescence and tumor progression of large cell carcinoma of the lung. Oncotarget 2016, 7, 82324–82337. [Google Scholar] [CrossRef] [Green Version]
- Hassona, Y.; Cirillo, N.; Lim, K.P.; Herman, A.; Mellone, M.; Thomas, G.J.; Pitiyage, G.N.; Parkinson, E.; Prime, S.S. Progression of genotype-specific oral cancer leads to senescence of cancer-associated fibroblasts and is mediated by oxidative stress and TGF-β. Carcinogenesis 2013, 34, 1286–1295. [Google Scholar] [CrossRef] [Green Version]
- Cai, L.; Xu, S.; Piao, C.; Qiu, S.; Li, H.; Du, J. Adiponectin induces CXCL1 secretion from cancer cells and promotes tumor angiogenesis by inducing stromal fibroblast senescence. Mol. Carcinog. 2015, 55, 1796–1806. [Google Scholar] [CrossRef]
- Kim, E.K.; Moon, S.; Kim, D.K.; Zhang, X.; Kim, J. CXCL1 induces senescence of cancer-associated fibroblasts via autocrine loops in oral squamous cell carcinoma. PLoS ONE 2018, 13, e0188847. [Google Scholar] [CrossRef]
- Lian, J.; Yue, Y.; Yu, W.; Zhang, Y. Immunosenescence: A key player in cancer development. J. Hematol. Oncol. 2020, 13, 151. [Google Scholar] [CrossRef]
- Ye, J.; Huang, X.; Hsueh, E.C.; Zhang, Q.; Ma, C.; Zhang, Y.; Varvares, M.A.; Hoft, D.F.; Peng, G. Human regulatory T cells induce T-lymphocyte senescence. Blood 2012, 120, 2021–2031. [Google Scholar] [CrossRef]
- Liu, X.; Mo, W.; Ye, J.; Li, L.; Zhang, Y.; Hsueh, E.C.; Hoft, D.F.; Peng, G. Regulatory T cells trigger effector T cell DNA damage and senescence caused by metabolic competition. Nat. Commun. 2018, 9, 249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Yang, L.; Yue, D.; Cao, L.; Li, L.; Wang, D.; Ping, Y.; Shen, Z.; Zheng, Y.; Wang, L.; et al. Macrophage-derived CCL22 promotes an immunosuppressive tumor microenvironment via IL-8 in malignant pleural effusion. Cancer Lett. 2019, 452, 244–253. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Ma, C.; Hsueh, E.C.; Dou, J.; Mo, W.; Liu, S.; Han, B.; Huang, Y.; Zhang, Y.; A Varvares, M.; et al. TLR 8 signaling enhances tumor immunity by preventing tumor-induced T-cell senescence. EMBO Mol. Med. 2014, 6, 1294–1311. [Google Scholar] [CrossRef] [PubMed]
- Desdín-Micó, G.; Soto-Heredero, G.; Aranda, J.F.; Oller, J.; Carrasco, E.; Gabandé-Rodríguez, E.; Blanco, E.M.; Alfranca, A.; Cussó, L.; Desco, M.; et al. T cells with dysfunctional mitochondria induce multimorbidity and premature senescence. Science 2020, 368, 1371–1376. [Google Scholar] [CrossRef]
- Yousefzadeh, M.J.; Flores, R.R.; Zhu, Y.; Schmiechen, Z.C.; Brooks, R.W.; Trussoni, C.E.; Cui, Y.; Angelini, L.; Lee, K.-A.; McGowan, S.J.; et al. An aged immune system drives senescence and ageing of solid organs. Nature 2021, 594, 100–105. [Google Scholar] [CrossRef]
- Burton, D.G.; Stolzing, A. Cellular senescence: Immunosurveillance and future immunotherapy. Ageing Res. Rev. 2018, 43, 17–25. [Google Scholar] [CrossRef] [Green Version]
- Pereira, B.I.; Devine, O.P.; Vukmanovic-Stejic, M.; Chambers, E.S.; Subramanian, P.; Patel, N.; Virasami, A.; Sebire, N.J.; Kinsler, V.; Valdovinos, A.; et al. Senescent cells evade immune clearance via HLA-E-mediated NK and CD8+ T cell inhibition. Nat. Commun. 2019, 10, 2387. [Google Scholar] [CrossRef] [Green Version]
- Di Matteo, S.; Avanzini, M.A.; Pelizzo, G.; Calcaterra, V.; Croce, S.; Spaggiari, G.M.; Theuer, C.; Zuccotti, G.; Moretta, L.; Pelosi, A.; et al. Neuroblastoma Tumor-Associated Mesenchymal Stromal Cells Regulate the Cytolytic Functions of NK Cells. Cancers 2022, 15, 19. [Google Scholar] [CrossRef]
- Waaijer, M.E.C.; Goldeck, D.; Gunn, D.A.; van Heemst, D.; Westendorp, R.G.J.; Pawelec, G.; Maier, A.B. Are skin senescence and immunosenescence linked within individuals? Aging Cell 2019, 18, e12956. [Google Scholar] [CrossRef] [Green Version]
- Lau, L.; Porciuncula, A.; Yu, A.; Iwakura, Y.; David, G. Uncoupling the Senescence-Associated Secretory Phenotype from Cell Cycle Exit via Interleukin-1 Inactivation Unveils Its Protumorigenic Role. Mol. Cell. Biol. 2019, 39, e00586-18. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Fu, D.; Xu, Q.; Cong, X.; Wu, C.; Zhong, X.; Ma, Y.; Lv, Z.; Chen, F.; Han, L.; et al. The senescence-associated secretory phenotype is potentiated by feedforward regulatory mechanisms involving Zscan4 and TAK1. Nat. Commun. 2018, 9, 1723. [Google Scholar] [CrossRef] [Green Version]
- Nelson, G.; Kucheryavenko, O.; Wordsworth, J.; von Zglinicki, T. The senescent bystander effect is caused by ROS-activated NF-κB signalling. Mech. Ageing Dev. 2018, 170, 30–36. [Google Scholar] [CrossRef]
- Farfariello, V.; Gordienko, D.V.; Mesilmany, L.; Touil, Y.; Germain, E.; Fliniaux, I.; Desruelles, E.; Gkika, D.; Roudbaraki, M.; Shapovalov, G.; et al. TRPC3 shapes the ER-mitochondria Ca2+ transfer characterizing tumour-promoting senescence. Nat. Commun. 2022, 13, 956. [Google Scholar] [CrossRef]
- Glück, S.; Guey, B.; Gulen, M.F.; Wolter, K.; Kang, T.-W.; Schmacke, N.A.; Bridgeman, A.; Rehwinkel, J.; Zender, L.; Ablasser, A. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nature 2017, 19, 1061–1070. [Google Scholar] [CrossRef]
- Flanagan, K.C.; Alspach, E.; Pazolli, E.; Parajuli, S.; Ren, Q.; Arthur, L.L.; Tapia, R.; Stewart, S.A. c-Myb and C/EBPβ regulate OPN and other senescence-associated secretory phenotype factors. Oncotarget 2017, 9, 21–36. [Google Scholar] [CrossRef] [Green Version]
- Callender, L.A.; Carroll, E.C.; Beal, R.W.J.; Chambers, E.S.; Nourshargh, S.; Akbar, A.N.; Henson, S.M. Human CD8+ EMRA T cells display a senescence-associated secretory phenotype regulated by p38 MAPK. Aging Cell 2017, 17, e12675. [Google Scholar] [CrossRef]
- Bent, E.H.; Gilbert, L.A.; Hemann, M.T. A senescence secretory switch mediated by PI3K/AKT/mTOR activation controls chemoprotective endothelial secretory responses. Genes Dev. 2016, 30, 1811–1821. [Google Scholar] [CrossRef] [Green Version]
- Alspach, E.; Flanagan, K.C.; Luo, X.; Ruhland, M.K.; Huang, H.; Pazolli, E.; Donlin, M.J.; Marsh, T.; Piwnica-Worms, D.; Monahan, J.; et al. p38MAPK Plays a Crucial Role in Stromal-Mediated Tumorigenesis. Cancer Discov. 2014, 4, 716–729. [Google Scholar] [CrossRef] [Green Version]
- Laberge, R.-M.; Sun, Y.; Orjalo, A.V.; Patil, C.K.; Freund, A.; Zhou, L.; Curran, S.C.; Davalos, A.R.; Wilson-Edell, K.A.; Liu, S.; et al. MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat. Cell Biol. 2015, 17, 1049–1061. [Google Scholar] [CrossRef]
- Kang, C.; Xu, Q.; Martin, T.D.; Li, M.Z.; DeMaria, M.; Aron, L.; Lu, T.; Yankner, B.A.; Campisi, J.; Elledge, S.J. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science 2015, 349, aaa5612. [Google Scholar] [CrossRef] [Green Version]
- Loo, T.M.; Kamachi, F.; Watanabe, Y.; Yoshimoto, S.; Kanda, H.; Arai, Y.; Nakajima-Takagi, Y.; Iwama, A.; Koga, T.; Sugimoto, Y.; et al. Gut Microbiota Promotes Obesity-Associated Liver Cancer through PGE2-Mediated Suppression of Antitumor Immunity. Cancer Discov. 2017, 7, 522–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, A.; Loo, T.M.; Okada, R.; Kamachi, F.; Watanabe, Y.; Wakita, M.; Watanabe, S.; Kawamoto, S.; Miyata, K.; Barber, G.N.; et al. Downregulation of cytoplasmic DNases is implicated in cytoplasmic DNA accumulation and SASP in senescent cells. Nat. Commun. 2018, 9, 1249. [Google Scholar] [CrossRef] [PubMed]
- Hoare, M.; Ito, Y.; Kang, T.-W.; Weekes, M.; Matheson, N.; Patten, D.; Shetty, S.; Parry, A.; Menon, S.; Salama, R.; et al. NOTCH1 mediates a switch between two distinct secretomes during senescence. Nat. Cell Biol. 2016, 18, 979–992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tasdemir, N.; Banito, A.; Roe, J.-S.; Alonso-Curbelo, D.; Camiolo, M.; Tschaharganeh, D.F.; Huang, C.-H.; Aksoy, O.; Bolden, J.E.; Chen, C.-C.; et al. BRD4 Connects Enhancer Remodeling to Senescence Immune Surveillance. Cancer Discov. 2016, 6, 612–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Long, Q.; Wu, S.; Xu, Q.; Song, S.; Han, L.; Qian, M.; Ren, X.; Liu, H.; Jiang, J.; et al. KDM4 orchestrates epigenomic remodeling of senescent cells and potentiates the senescence-associated secretory phenotype. Nat. Aging 2021, 1, 454–472. [Google Scholar] [CrossRef]
- Han, L.; Long, Q.; Li, S.; Xu, Q.; Zhang, B.; Dou, X.; Qian, M.; Jiramongkol, Y.; Guo, J.; Cao, L.; et al. Senescent Stromal Cells Promote Cancer Resistance through SIRT1 Loss-Potentiated Overproduction of Small Extracellular Vesicles. Cancer Res. 2020, 80, 3383–3398. [Google Scholar] [CrossRef]
- Borghesan, M.; Fafián-Labora, J.; Eleftheriadou, O.; Carpintero-Fernández, P.; Paez-Ribes, M.; Vizcay-Barrena, G.; Swisa, A.; Kolodkin-Gal, D.; Ximénez-Embún, P.; Lowe, R.; et al. Small Extracellular Vesicles Are Key Regulators of Non-cell Autonomous Intercellular Communication in Senescence via the Interferon Protein IFITM3. Cell Rep. 2019, 27, 3956–3971. [Google Scholar] [CrossRef] [Green Version]
- Fafián-Labora, J.A.; O’Loghlen, A. NF-κB/IKK activation by small extracellular vesicles within the SASP. Aging Cell 2021, 20, e13426. [Google Scholar] [CrossRef]
- Guan, X.; LaPak, K.M.; Hennessey, R.C.; Yu, C.Y.; Shakya, R.; Zhang, J.; Burd, C.E. Stromal Senescence By Prolonged CDK4/6 Inhibition Potentiates Tumor Growth. Mol Cancer Res. 2017, 15, 237–249. [Google Scholar] [CrossRef] [Green Version]
- Pardella, E.; Pranzini, E.; Nesi, I.; Parri, M.; Spatafora, P.; Torre, E.; Muccilli, A.; Castiglione, F.; Fambrini, M.; Sorbi, F.; et al. Therapy-Induced Stromal Senescence Promoting Aggressiveness of Prostate and Ovarian Cancer. Cells 2022, 11, 4026. [Google Scholar] [CrossRef]
- Kanehira, M.; Fujiwara, T.; Nakajima, S.; Okitsu, Y.; Onishi, Y.; Fukuhara, N.; Ichinohasama, R.; Okada, Y.; Harigae, H. A Lysophosphatidic Acid Receptors 1 and 3 Axis Governs Cellular Senescence of Mesenchymal Stromal Cells and Promotes Growth and Vascularization of Multiple Myeloma. Stem Cells 2016, 35, 739–753. [Google Scholar] [CrossRef]
- Stoddart, A.; Wang, J.; Fernald, A.A.; Davis, E.M.; Johnson, C.R.; Hu, C.; Cheng, J.X.; McNerney, M.E.; Le Beau, M.M. Cytotoxic Therapy-Induced Effects on Both Hematopoietic and Marrow Stromal Cells Promotes Therapy-Related Myeloid Neoplasms. Blood Cancer Discov. 2020, 1, 32–47. [Google Scholar]
- Kutyna, M.M.; Kok, C.H.; Lim, Y.; Tran, E.N.H.; Campbell, D.; Paton, S.; Thompson-Peach, C.; Lim, K.; Cakouros, D.; Arthur, A.; et al. A senescence stress secretome is a hallmark of therapy-related myeloid neoplasm stromal tissue occurring soon after cytotoxic exposure. Leukemia 2022, 36, 2678–2689. [Google Scholar] [CrossRef]
- Zhou, P.; Xia, C.; Wang, T.; Dong, Y.; Weng, Q.; Liu, X.; Geng, Y.; Wang, J.; Du, J. Senescent bone marrow microenvironment promotes Nras-mutant leukemia. J. Mol. Cell Biol. 2020, 13, 72–74. [Google Scholar] [CrossRef]
- Mikuła-Pietrasik, J.; Uruski, P.; Matuszkiewicz, K.; Szubert, S.; Moszyński, R.; Szpurek, D.; Sajdak, S.; Tykarski, A.; Książek, K. Ovarian cancer-derived ascitic fluids induce a senescence-dependent pro-cancerogenic phenotype in normal peritoneal mesothelial cells. Cell. Oncol. 2016, 39, 473–481. [Google Scholar] [CrossRef]
- Chen, F.; Long, Q.; Fu, D.; Zhu, D.; Ji, Y.; Han, L.; Zhang, B.; Xu, Q.; Liu, B.; Li, Y.; et al. Targeting SPINK1 in the damaged tumour microenvironment alleviates therapeutic resistance. Nat. Commun. 2018, 9, 4315. [Google Scholar] [CrossRef] [Green Version]
- Xu, Q.; Long, Q.; Zhu, D.; Fu, D.; Zhang, B.; Han, L.; Qian, M.; Guo, J.; Xu, J.; Cao, L.; et al. Targeting amphiregulin (AREG) derived from senescent stromal cells diminishes cancer resistance and averts programmed cell death 1 ligand (PD-L1)-mediated immunosuppression. Aging Cell 2019, 18, e13027. [Google Scholar] [CrossRef] [Green Version]
- Bracken, C.P.; Goodall, G.J. The many regulators of epithelial−mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2021, 23, 89–90. [Google Scholar] [CrossRef]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2018, 20, 69–84. [Google Scholar] [CrossRef]
- Taki, M.; Abiko, K.; Ukita, M.; Murakami, R.; Yamanoi, K.; Yamaguchi, K.; Hamanishi, J.; Baba, T.; Matsumura, N.; Mandai, M. Tumor Immune Microenvironment during Epithelial–Mesenchymal Transition. Clin. Cancer Res. 2021, 27, 4669–4679. [Google Scholar] [CrossRef]
- Mikuła-Pietrasik, J.; Sosińska, P.; Maksin, K.; Kucińska, M.; Piotrowska, H.; Murias, M.; Woźniak, A.; Szpurek, D.; Książek, K. Colorectal cancer-promoting activity of the senescent peritoneal mesothelium. Oncotarget 2015, 6, 29178–29195. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, P.T.; Kanno, K.; Pham, Q.T.; Kikuchi, Y.; Kakimoto, M.; Kobayashi, T.; Otani, Y.; Kishikawa, N.; Miyauchi, M.; Arihiro, K.; et al. Senescent hepatic stellate cells caused by deoxycholic acid modulates malignant behavior of hepatocellular carcinoma. J. Cancer Res. Clin. Oncol. 2020, 146, 3255–3268. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Montero, P.; Londoño-Vallejo, A.; Vernot, J.-P. Senescence-associated IL-6 and IL-8 cytokines induce a self- and cross-reinforced senescence/inflammatory milieu strengthening tumorigenic capabilities in the MCF-7 breast cancer cell line. Cell Commun. Signal. 2017, 15, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Campisi, J.; Higano, C.; Beer, T.M.; Porter, P.; Coleman, I.; True, L.; Nelson, P.S. Treatment-induced damage to the tumor microenvironment promotes prostate cancer therapy resistance through WNT16B. Nat. Med. 2012, 18, 1359–1368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Long, Q.; Fu, Q.; Xu, Q.; Da Fu, D.; Li, Y.; Gao, L.; Guo, J.; Zhang, X.; Lam, E.W.-F.; et al. Targeting epiregulin in the treatment-damaged tumor microenvironment restrains therapeutic resistance. Oncogene 2022, 41, 4941–4959. [Google Scholar] [CrossRef]
- Xu, J.; Lamouille, S.; Derynck, R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef]
- Plaks, V.; Kong, N.; Werb, Z. The Cancer Stem Cell Niche: How Essential Is the Niche in Regulating Stemness of Tumor Cells? Cell Stem Cell 2015, 16, 225–238. [Google Scholar] [CrossRef] [Green Version]
- Kreso, A.; Dick, J.E. Evolution of the Cancer Stem Cell Model. Cell Stem Cell 2014, 14, 275–291. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Finkelstein, D.; Gao, C.; Shi, L.; Wang, Y.; López-Terrada, D.; Wang, K.; Utley, S.; Pounds, S.; Neale, G.; et al. Multi-organ Mapping of Cancer Risk. Cell 2016, 166, 1132–1146.e7. [Google Scholar] [CrossRef] [Green Version]
- Quail, D.F.; Taylor, M.J.; Postovit, L.M. Microenvironmental Regulation of Cancer Stem Cell Phenotypes. Curr. Stem Cell Res. Ther. 2012, 7, 197–216. [Google Scholar] [CrossRef]
- Batlle, E.; Clevers, H. Cancer stem cells revisited. Nat. Med. 2017, 23, 1124–1134. [Google Scholar] [CrossRef]
- Prasetyanti, P.R.; Medema, J.P. Intra-tumor heterogeneity from a cancer stem cell perspective. Mol. Cancer 2017, 16, 41. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.-Y.; Kang, J.W.; Song, X.; Kim, B.K.; Yoo, Y.D.; Kwon, Y.T.; Lee, Y.J. Role of the IL-6-JAK1-STAT3-Oct-4 pathway in the conversion of non-stem cancer cells into cancer stem-like cells. Cell. Signal. 2013, 25, 961–969. [Google Scholar] [CrossRef] [Green Version]
- Korkaya, H.; Kim, G.-I.; Davis, A.; Malik, F.; Henry, N.L.; Ithimakin, S.; Quraishi, A.A.; Tawakkol, N.; D’Angelo, R.; Paulson, A.K.; et al. Activation of an IL6 Inflammatory Loop Mediates Trastuzumab Resistance in HER2+ Breast Cancer by Expanding the Cancer Stem Cell Population. Mol. Cell 2012, 47, 570–584. [Google Scholar] [CrossRef] [Green Version]
- Ospina-Muñoz, N.; Vernot, J.-P. Partial acquisition of stemness properties in tumorspheres obtained from interleukin-8-treated MCF-7 cells. Tumor Biol. 2020, 42, 1010428320979438. [Google Scholar] [CrossRef]
- Ma, X.; Chen, J.; Liu, J.; Xu, B.; Liang, X.; Yang, X.; Feng, Y.; Liang, X.; Liu, J. IL-8/CXCR2 mediates tropism of human bone marrow-derived mesenchymal stem cells toward CD133(+) /CD44(+) Colon cancer stem cells. J. Cell. Physiol. 2021, 236, 3114–3128. [Google Scholar] [CrossRef]
- Kim, B.; Seo, Y.; Kwon, J.; Shin, Y.; Kim, S.; Park, S.J.; Park, J.J.; Cheon, J.H.; Kim, W.H.; Kim, T.I. IL-6 and IL-8, secreted by myofibroblasts in the tumor microenvironment, activate HES1 to expand the cancer stem cell population in early colorectal tumor. Mol. Carcinog. 2021, 60, 188–200. [Google Scholar] [CrossRef]
- Ritschka, B.; Storer, M.; Mas, A.; Heinzmann, F.; Ortells, M.C.; Morton, J.P.; Sansom, O.J.; Zender, L.; Keyes, W.M. The senescence-associated secretory phenotype induces cellular plasticity and tissue regeneration. Genes Dev. 2017, 31, 172–183. [Google Scholar] [CrossRef] [Green Version]
- Milanovic, M.; Yu, Y.; Schmitt, C.A. The Senescence–Stemness Alliance—A Cancer-Hijacked Regeneration Principle. Trends Cell Biol. 2018, 28, 1049–1061. [Google Scholar] [CrossRef]
- Alimirah, F.; Pulido, T.; Valdovinos, A.; Alptekin, S.; Chang, E.; Jones, E.; Diaz, D.A.; Flores, J.; Velarde, M.C.; Demaria, M.; et al. Cellular Senescence Promotes Skin Carcinogenesis through p38MAPK and p44/42MAPK Signaling. Cancer Res. 2020, 80, 3606–3619. [Google Scholar] [CrossRef]
- Jiang, X.; Wang, J.; Deng, X.; Xiong, F.; Zhang, S.; Gong, Z.; Li, X.; Cao, K.; Deng, H.; He, Y.; et al. The role of microenvironment in tumor angiogenesis. J. Exp. Clin. Cancer Res. 2020, 39, 1745–1770. [Google Scholar] [CrossRef] [PubMed]
- Lugano, R.; Ramachandran, M.; Dimberg, A. Tumor angiogenesis: Causes, consequences, challenges and opportunities. Cell. Mol. Life Sci. 2020, 77, 1745–1770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Folkman, J. Patterns and Emerging Mechanisms of the Angiogenic Switch during Tumorigenesis. Cell 1996, 86, 353–364. [Google Scholar] [CrossRef] [Green Version]
- Mikuła-Pietrasik, J.; Sosińska, P.; Naumowicz, E.; Maksin, K.; Piotrowska, H.; Woźniak, A.; Szpurek, D.; Książek, K. Senescent peritoneal mesothelium induces a pro-angiogenic phenotype in ovarian cancer cells in vitro and in a mouse xenograft model in vivo. Clin. Exp. Metastasis 2015, 33, 15–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rutecki, S.; Szulc, P.; Pakuła, M.; Uruski, P.; Radziemski, A.; Naumowicz, E.; Moszyński, R.; Tykarski, A.; Mikuła-Pietrasik, J.; Książek, K. Pro-cancerogenic effects of spontaneous and drug-induced senescence of ovarian cancer cells in vitro and in vivo: A comparative analysis. J. Ovarian Res. 2022, 15, 87. [Google Scholar] [CrossRef]
- Ramello, M.C.; Boari, J.T.; Canale, F.P.; A Mena, H.; Negrotto, S.; Gastman, B.; Gruppi, A.; Rodríguez, E.V.A.; Montes, C.L. Tumor-induced senescent T cells promote the secretion of pro-inflammatory cytokines and angiogenic factors by human monocytes/macrophages through a mechanism that involves Tim-3 and CD40L. Cell Death Dis. 2014, 5, e1507. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Hong, Y.; Kim, M. Structural and Functional Changes and Possible Molecular Mechanisms in Aged Skin. Int. J. Mol. Sci. 2021, 22, 12489. [Google Scholar] [CrossRef]
- Malaquin, N.; Vercamer, C.; Bouali, F.; Martien, S.; Deruy, E.; Wernert, N.; Chwastyniak, M.; Pinet, F.; Abbadie, C.; Pourtier, A. Senescent Fibroblasts Enhance Early Skin Carcinogenic Events via a Paracrine MMP-PAR-1 Axis. PLoS ONE 2013, 8, e63607. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Hornsby, P.J. Senescent Human Fibroblasts Increase the Early Growth of Xenograft Tumors via Matrix Metalloproteinase Secretion. Cancer Res. 2007, 67, 3117–3126. [Google Scholar] [CrossRef] [Green Version]
- Papadopoulou, A.; Kletsas, D. Human lung fibroblasts prematurely senescent after exposure to ionizing radiation enhance the growth of malignant lung epithelial cells in vitro and in vivo. Int. J. Oncol. 2011, 39, 989–999. [Google Scholar]
- Hassona, Y.; Cirillo, N.; Heesom, K.; Parkinson, E.K.; Prime, S.S. Senescent cancer-associated fibroblasts secrete active MMP-2 that promotes keratinocyte dis-cohesion and invasion. Br. J. Cancer 2014, 111, 1230–1237. [Google Scholar] [CrossRef] [Green Version]
- Laplane, L.; Duluc, D.; Larmonier, N.; Pradeu, T.; Bikfalvi, A. The Multiple Layers of the Tumor Environment. Trends Cancer 2018, 4, 802–809. [Google Scholar] [CrossRef]
- Laplane, L.; Duluc, D.; Bikfalvi, A.; Larmonier, N.; Pradeu, T. Beyond the tumour microenvironment. Int. J. Cancer 2019, 145, 2611–2618. [Google Scholar] [CrossRef] [Green Version]
- Mikuła-Pietrasik, J.; Uruski, P.; Sosińska, P.; Maksin, K.; Piotrowska-Kempisty, H.; Kucińska, M.; Murias, M.; Szubert, S.; Woźniak, A.; Szpurek, D.; et al. Senescent peritoneal mesothelium creates a niche for ovarian cancer metastases. Cell Death Dis. 2016, 7, e2565. [Google Scholar] [CrossRef] [Green Version]
- Luo, X.; Fu, Y.; Loza, A.J.; Murali, B.; Leahy, K.M.; Ruhland, M.K.; Gang, M.; Su, X.; Zamani, A.; Shi, Y.; et al. Stromal-Initiated Changes in the Bone Promote Metastatic Niche Development. Cell Rep. 2015, 14, 82–92. [Google Scholar] [CrossRef] [Green Version]
- Fane, M.E.; Chhabra, Y.; Alicea, G.M.; Maranto, D.A.; Douglass, S.M.; Webster, M.R.; Rebecca, V.W.; Marino, G.E.; Almeida, F.; Ecker, B.L.; et al. Stromal changes in the aged lung induce an emergence from melanoma dormancy. Nature 2022, 606, 396–405. [Google Scholar] [CrossRef]
- Jin, J.; Li, L.; Fu, C. Age-induced changes in lung microenvironment: From melanoma dormancy to outgrowth. Signal Transduct. Target. Ther. 2023, 8, 33. [Google Scholar] [CrossRef]
- Hui, L.; Chen, Y. Tumor microenvironment: Sanctuary of the devil. Cancer Lett. 2015, 368, 7–13. [Google Scholar] [CrossRef]
- Gajewski, T.F.; Schreiber, H.; Fu, Y.-X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013, 14, 1014–1022. [Google Scholar] [CrossRef] [Green Version]
- Zinger, A.; Cho, W.C.; Ben-Yehuda, A. Cancer and Aging—The Inflammatory Connection. Aging Dis. 2017, 8, 611–627. [Google Scholar] [CrossRef] [Green Version]
- Olivieri, F.; Prattichizzo, F.; Grillari, J.; Balistreri, C.R. Cellular Senescence and Inflammaging in Age-Related Diseases. Mediat. Inflamm. 2018, 2018, 9076485. [Google Scholar] [CrossRef] [PubMed]
- Rathinam, V.A.; Fitzgerald, K.A. Inflammasome Complexes: Emerging Mechanisms and Effector Functions. Cell 2016, 165, 792–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wieland, E.; Rodriguez-Vita, J.; Liebler, S.S.; Mogler, C.; Moll, I.; Herberich, S.E.; Espinet, E.; Herpel, E.; Menuchin, A.; Chang-Claude, J.; et al. Endothelial Notch1 Activity Facilitates Metastasis. Cancer Cell 2017, 31, 355–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lagnado, A.; Leslie, J.; Ruchaud-Sparagano, M.; Victorelli, S.; Hirsova, P.; Ogrodnik, M.; Collins, A.L.; Vizioli, M.G.; Habiballa, L.; Saretzki, G.; et al. Neutrophils induce paracrine telomere dysfunction and senescence in ROS-dependent manner. EMBO J. 2021, 40, e106048. [Google Scholar] [CrossRef]
- Xu, M.; Tchkonia, T.; Ding, H.; Ogrodnik, M.; Lubbers, E.R.; Pirtskhalava, T.; White, T.A.; Johnson, K.O.; Stout, M.B.; Mezera, V.; et al. JAK inhibition alleviates the cellular senescence-associated secretory phenotype and frailty in old age. Proc. Natl. Acad. Sci. USA 2015, 112, E6301–E6310. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Xu, J.; Lan, H. Tumor-associated macrophages in tumor metastasis: Biological roles and clinical therapeutic applications. J. Hematol. Oncol. 2019, 12, 76. [Google Scholar] [CrossRef]
- Ruhland, M.K.; Loza, A.J.; Capietto, A.-H.; Luo, X.; Knolhoff, B.L.; Flanagan, K.C.; Belt, B.A.; Alspach, E.; Leahy, K.; Luo, J.; et al. Stromal senescence establishes an immunosuppressive microenvironment that drives tumorigenesis. Nat. Commun. 2016, 7, 11762. [Google Scholar] [CrossRef] [Green Version]
- Eggert, T.; Wolter, K.; Ji, J.; Ma, C.; Yevsa, T.; Klotz, S.; Medina-Echeverz, J.; Longerich, T.; Forgues, M.; Reisinger, F.; et al. Distinct Functions of Senescence-Associated Immune Responses in Liver Tumor Surveillance and Tumor Progression. Cancer Cell 2016, 30, 533–547. [Google Scholar] [CrossRef] [Green Version]
- Lujambio, A.; Akkari, L.; Simon, J.; Grace, D.; Tschaharganeh, D.F.; Bolden, J.E.; Zhao, Z.; Thapar, V.; Joyce, J.A.; Krizhanovsky, V.; et al. Non-Cell-Autonomous Tumor Suppression by p53. Cell 2013, 153, 449–460. [Google Scholar] [CrossRef] [Green Version]
- Henson, S.M.; Macaulay, R.; Riddell, N.E.; Nunn, C.J.; Akbar, A.N. Blockade of PD-1 or p38 MAP kinase signaling enhances senescent human CD8+T-cell proliferation by distinct pathways. Eur. J. Immunol. 2015, 45, 1441–1451. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Wang, B.; Song, R.; Hao, Y.; Wang, D.; Li, Y.; Jiang, Y.; Xu, L.; Ma, Y.; Zheng, H.; et al. T-cell Immunoglobulin and ITIM Domain Contributes to CD8+ T-cell Immunosenescence. Aging Cell 2018, 17, e12716. [Google Scholar] [CrossRef]
- Huff, W.X.; Kwon, J.H.; Henriquez, M.; Fetcko, K.; Dey, M. The Evolving Role of CD8+CD28− Immunosenescent T Cells in Cancer Immunology. Int. J. Mol. Sci. 2019, 20, 2810. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Hartman, C.L.; Li, L.; Albert, C.J.; Si, F.; Gao, A.; Huang, L.; Zhao, Y.; Lin, W.; Hsueh, E.C.; et al. Reprogramming lipid metabolism prevents effector T cell senescence and enhances tumor immunotherapy. Sci. Transl. Med. 2021, 13, eaaz6314. [Google Scholar] [CrossRef]
- Gao, A.; Liu, X.; Lin, W.; Wang, J.; Wang, S.; Si, F.; Huang, L.; Zhao, Y.; Sun, Y.; Peng, G. Tumor-derived ILT4 induces T cell senescence and suppresses tumor immunity. J. Immunother. Cancer 2021, 9, e001536. [Google Scholar] [CrossRef]
- Pereira, B.I.; De Maeyer, R.P.H.; Covre, L.P.; Nehar-Belaid, D.; Lanna, A.; Ward, S.; Marches, R.; Chambers, E.S.; Gomes, D.C.O.; Riddell, N.E.; et al. Sestrins induce natural killer function in senescent-like CD8+ T cells. Nat. Immunol. 2020, 21, 684–694. [Google Scholar] [CrossRef]
- Hubackova, S.; Krejcikova, K.; Bartek, J.; Hodny, Z. IL1- and TGFβ-Nox4 signaling, oxidative stress and DNA damage response are shared features of replicative, oncogene-induced, and drug-induced paracrine ‘Bystander senescence’. Aging 2012, 4, 932–951. [Google Scholar] [CrossRef]
- Mikuła-Pietrasik, J.; Sosińska, P.; Janus, J.; Rubiś, B.; Brewińska-Olchowik, M.; Piwocka, K.; Książek, K. Bystander senescence in human peritoneal mesothelium and fibroblasts is related to thrombospondin-1-dependent activation of transforming growth factor-β1. Int. J. Biochem. Cell Biol. 2013, 45, 2087–2096. [Google Scholar] [CrossRef]
- Shafqat, S.; Chicas, E.A.; Shafqat, A.; Hashmi, S.K. The Achilles’ heel of cancer survivors: Fundamentals of accelerated cellular senescence. J. Clin. Investig. 2022, 132. [Google Scholar] [CrossRef]
- Kamdje, E.A.A.H.N.; Etet, P.F.S.; Simo, R.T.; Vecchio, L.; Lukong, K.E.; Krampera, M. Emerging data supporting stromal cell therapeutic potential in cancer: Reprogramming stromal cells of the tumor microenvironment for anti-cancer effects. Cancer Biol. Med. 2020, 17, 828–841. [Google Scholar] [CrossRef]
- Biffi, G.; Tuveson, D.A. Diversity and Biology of Cancer-Associated Fibroblasts. Physiol. Rev. 2021, 101, 147–176. [Google Scholar] [CrossRef]
- De Wever, O.; Demetter, P.; Mareel, M.; Bracke, M. Stromal myofibroblasts are drivers of invasive cancer growth. Int. J. Cancer 2008, 123, 2229–2238. [Google Scholar] [CrossRef] [PubMed]
- Fujita, H.; Ohuchida, K.; Mizumoto, K.; Nakata, K.; Yu, J.; Kayashima, T.; Cui, L.; Manabe, T.; Ohtsuka, T.; Tanaka, M. α-Smooth Muscle Actin Expressing Stroma Promotes an Aggressive Tumor Biology in Pancreatic Ductal Adenocarcinoma. Pancreas 2010, 39, 1254–1262. [Google Scholar] [CrossRef] [PubMed]
- Marsh, D.; Suchak, K.; A Moutasim, K.; Vallath, S.; Hopper, C.; Jerjes, W.; Upile, T.; Kalavrezos, N.; Violette, S.M.; Weinreb, P.H.; et al. Stromal features are predictive of disease mortality in oral cancer patients. J. Pathol. 2011, 223, 470–481. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, M.; Ogawa, T.; Zhang, X.; Hanamura, N.; Kashikura, Y.; Takamura, M.; Yoneda, M.; Shiraishi, T. Role of stromal myofibroblasts in invasive breast cancer: Stromal expression of alpha-smooth muscle actin correlates with worse clinical outcome. Breast Cancer 2010, 19, 170–176. [Google Scholar] [CrossRef]
- Underwood, T.J.; Hayden, A.L.; Derouet, M.; Garcia, E.; Noble, F.; White, M.; Thirdborough, S.; Mead, A.; Clemons, N.; Mellone, M.; et al. Cancer-associated fibroblasts predict poor outcome and promote periostin-dependent invasion in oesophageal adenocarcinoma. J. Pathol. 2015, 235, 466–477. [Google Scholar] [CrossRef] [Green Version]
- Mellone, M.; Hanley, C.J.; Thirdborough, S.; Mellows, T.; Garcia, E.; Woo, J.; Tod, J.; Frampton, S.; Jenei, V.; Moutasim, K.A.; et al. Induction of fibroblast senescence generates a non-fibrogenic myofibroblast phenotype that differentially impacts on cancer prognosis. Aging 2016, 9, 114–132. [Google Scholar] [CrossRef] [Green Version]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [Green Version]
- Lord, C.J.; Tutt, A.N.; Ashworth, A. Synthetic Lethality and Cancer Therapy: Lessons Learned from the Development of PARP Inhibitors. Annu. Rev. Med. 2015, 66, 455–470. [Google Scholar] [CrossRef]
- Lopez, J.S.; Banerji, U. Combine and conquer: Challenges for targeted therapy combinations in early phase trials. Nat. Rev. Clin. Oncol. 2016, 14, 57–66. [Google Scholar] [CrossRef]
- Wang, L.; de Oliveira, R.L.; Huijberts, S.; Bosdriesz, E.; Pencheva, N.; Brunen, D.; Bosma, A.; Song, J.-Y.; Zevenhoven, J.; Vries, G.T.L.-D.; et al. An Acquired Vulnerability of Drug-Resistant Melanoma with Therapeutic Potential. Cell 2018, 173, 1413–1425.e14. [Google Scholar] [CrossRef] [Green Version]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ye, M.; Huang, X.; Wu, Q.; Liu, F. Senescent Stromal Cells in the Tumor Microenvironment: Victims or Accomplices? Cancers 2023, 15, 1927. https://doi.org/10.3390/cancers15071927
Ye M, Huang X, Wu Q, Liu F. Senescent Stromal Cells in the Tumor Microenvironment: Victims or Accomplices? Cancers. 2023; 15(7):1927. https://doi.org/10.3390/cancers15071927
Chicago/Turabian StyleYe, Minghan, Xinyi Huang, Qianju Wu, and Fei Liu. 2023. "Senescent Stromal Cells in the Tumor Microenvironment: Victims or Accomplices?" Cancers 15, no. 7: 1927. https://doi.org/10.3390/cancers15071927