
Aberrant HMGA2 Expression Sustains Genome Instability That Promotes Metastasis and Therapeutic Resistance in Colorectal Cancer

Abstract
:Simple Summary
Abstract
1. Introduction
1.1. Colorectal Cancer
1.2. CRC Pathogenesis
1.3. Genome Instability in CRC
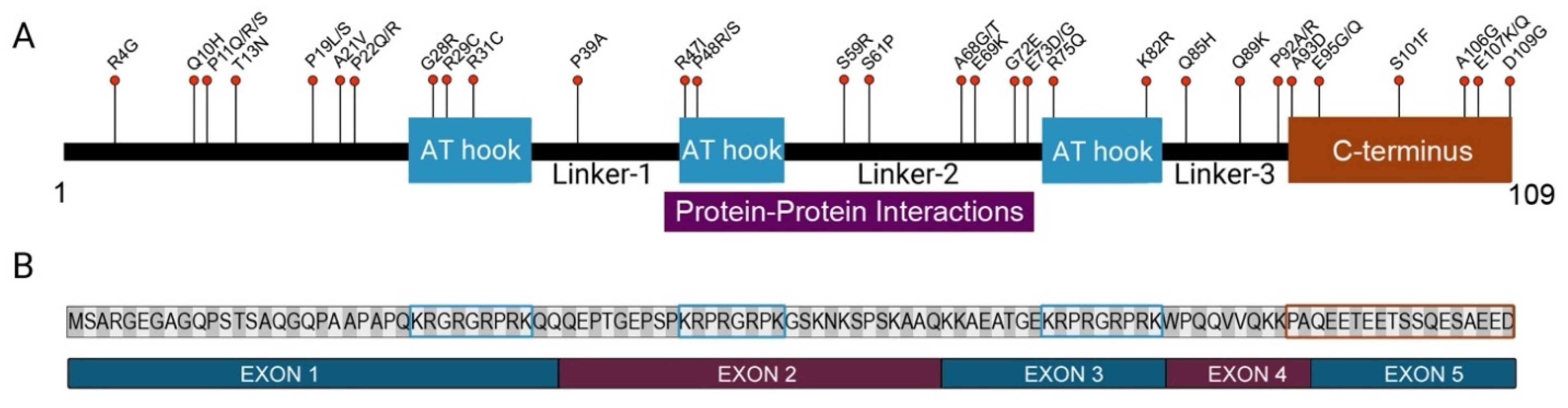
1.4. HMGA2 and CRC
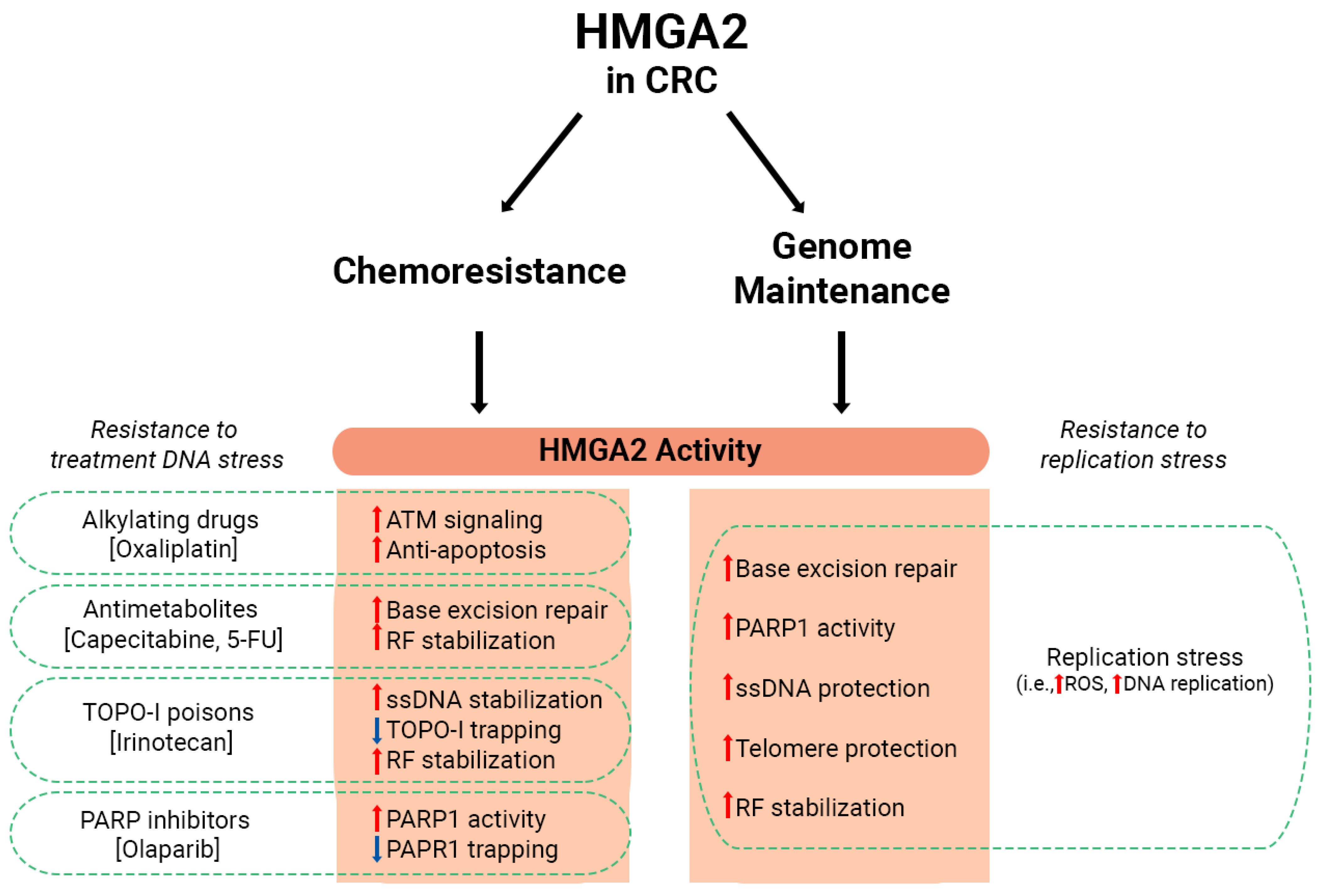
1.5. The Role of HMGA2 in Genome Maintenance
1.6. Aberrant HMGA2 Expression Promotes CRC Metastasis
1.7. HMGA2 Promotes Chemoresistance in CRC
2. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Xi, Y.; Xu, P. Global colorectal cancer burden in 2020 and projections to 2040. Transl. Oncol. 2021, 14, 101174. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Dekker, E.; Tanis, P.J.; Vleugels, J.L.A.; Kasi, P.M.; Wallace, M.B. Colorectal cancer. Lancet 2019, 394, 1467–1480. [Google Scholar] [CrossRef]
- Guren, M.G. The global challenge of colorectal cancer. Lancet Gastroenterol. Hepatol. 2019, 4, 894–895. [Google Scholar] [CrossRef] [Green Version]
- Brenner, D.R.; Weir, H.K.; Demers, A.A.; Ellison, L.F.; Louzado, C.; Shaw, A.; Turner, D.; Woods, R.R.; Smith, L.M.; Canadian Cancer Statistics Advisory, C. Projected estimates of cancer in Canada in 2020. CMAJ 2020, 192, E199–E205. [Google Scholar] [CrossRef] [Green Version]
- Canadian Cancer Society, Statistics Canada and the Public Health Agency of Canada. Canadian Cancer Statistics 2021. HPCDP J. 2021, 41, 57. [Google Scholar]
- Colorectal Cancer: Statistics—American Society of Clinical Oncology (ASCO). Available online: https://www.cancer.net/cancer-types/colorectal-cancer/statistics (accessed on 15 August 2022).
- Hewitson, P.; Glasziou, P.; Watson, E.; Towler, B.; Irwig, L. Cochrane systematic review of colorectal cancer screening using the fecal occult blood test (hemoccult): An update. Am. J. Gastroenterol. 2008, 103, 1541–1549. [Google Scholar] [CrossRef]
- Elmunzer, B.J.; Hayward, R.A.; Schoenfeld, P.S.; Saini, S.D.; Deshpande, A.; Waljee, A.K. Effect of flexible sigmoidoscopy-based screening on incidence and mortality of colorectal cancer: A systematic review and meta-analysis of randomized controlled trials. PLoS Med. 2012, 9, e1001352. [Google Scholar] [CrossRef]
- Hardcastle, J.D.; Chamberlain, J.O.; Robinson, M.H.; Moss, S.M.; Amar, S.S.; Balfour, T.W.; James, P.D.; Mangham, C.M. Randomised controlled trial of faecal-occult-blood screening for colorectal cancer. Lancet 1996, 348, 1472–1477. [Google Scholar] [CrossRef] [PubMed]
- Kronborg, O.; Jorgensen, O.D.; Fenger, C.; Rasmussen, M. Randomized study of biennial screening with a faecal occult blood test: Results after nine screening rounds. Scand. J. Gastroenterol. 2004, 39, 846–851. [Google Scholar] [CrossRef] [PubMed]
- Ferlitsch, M.; Moss, A.; Hassan, C.; Bhandari, P.; Dumonceau, J.M.; Paspatis, G.; Jover, R.; Langner, C.; Bronzwaer, M.; Nalankilli, K.; et al. Colorectal polypectomy and endoscopic mucosal resection (EMR): European Society of Gastrointestinal Endoscopy (ESGE) Clinical Guideline. Endoscopy 2017, 49, 270–297. [Google Scholar] [CrossRef] [Green Version]
- Siegel, R.L.; Miller, K.D.; Goding Sauer, A.; Fedewa, S.A.; Butterly, L.F.; Anderson, J.C.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 145–164. [Google Scholar] [CrossRef] [Green Version]
- Araghi, M.; Arnold, M.; Rutherford, M.J.; Guren, M.G.; Cabasag, C.J.; Bardot, A.; Ferlay, J.; Tervonen, H.; Shack, L.; Woods, R.R.; et al. Colon and rectal cancer survival in seven high-income countries 2010–2014: Variation by age and stage at diagnosis (the ICBP SURVMARK-2 project). Gut 2021, 70, 114–126. [Google Scholar] [CrossRef] [PubMed]
- Bryan, S.; Masoud, H.; Weir, H.K.; Woods, R.; Lockwood, G.; Smith, L.; Brierley, J.; Gospodarowicz, M.; Badets, N. Cancer in Canada: Stage at diagnosis. Health Rep. 2018, 29, 21–25. [Google Scholar]
- Wingfield, S.A.; Heflin, M.T. Cancer Screening in Older Adults. Clin. Geriatr. Med. 2016, 32, 17–33. [Google Scholar] [CrossRef]
- Munkholm, P. Review article: The incidence and prevalence of colorectal cancer in inflammatory bowel disease. Aliment. Pharmacol. Ther. 2003, 18 (Suppl. 2), 1–5. [Google Scholar] [CrossRef] [PubMed]
- Binefa, G.; Rodriguez-Moranta, F.; Teule, A.; Medina-Hayas, M. Colorectal cancer: From prevention to personalized medicine. World J. Gastroenterol. 2014, 20, 6786–6808. [Google Scholar] [CrossRef]
- Al-Sukhni, W.; Aronson, M.; Gallinger, S. Hereditary colorectal cancer syndromes: Familial adenomatous polyposis and lynch syndrome. Surg. Clin. N. Am. 2008, 88, 819–844. [Google Scholar] [CrossRef]
- Vogelstein, B.; Fearon, E.R.; Hamilton, S.R.; Kern, S.E.; Preisinger, A.C.; Leppert, M.; Nakamura, Y.; White, R.; Smits, A.M.; Bos, J.L. Genetic alterations during colorectal-tumor development. N. Engl. J. Med. 1988, 319, 525–532. [Google Scholar] [CrossRef] [Green Version]
- Canadian Cancer Statistics Advisory Committee. Canadian Cancer Statistics: A 2018 Special Report on Cancer Incidence by Stage; Canadian Cancer Statistics Advisory Committee: Toronto, ON, Canada, 2018. [Google Scholar]
- Syngal, S.; Brand, R.E.; Church, J.M.; Giardiello, F.M.; Hampel, H.L.; Burt, R.W.; Randall, W. ACG clinical guideline: Genetic testing and management of hereditary gastrointestinal cancer syndromes. Am. J. Gastroenterol. 2015, 110, 223–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samadder, N.J.; Baffy, N.; Giridhar, K.V.; Couch, F.J.; Riegert-Johnson, D. Hereditary Cancer Syndromes-A Primer on Diagnosis and Management, Part 2: Gastrointestinal Cancer Syndromes. Mayo Clin. Proc. 2019, 94, 1099–1116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hampel, H.; Frankel, W.L.; Martin, E.; Arnold, M.; Khanduja, K.; Kuebler, P.; Nakagawa, H.; Sotamaa, K.; Prior, T.W.; Westman, J.; et al. Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer). N. Engl. J. Med. 2005, 352, 1851–1860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonadona, V.; Bonaiti, B.; Olschwang, S.; Grandjouan, S.; Huiart, L.; Longy, M.; Guimbaud, R.; Buecher, B.; Bignon, Y.J.; Caron, O.; et al. Cancer risks associated with germline mutations in MLH1, MSH2, and MSH6 genes in Lynch syndrome. JAMA 2011, 305, 2304–2310. [Google Scholar] [CrossRef] [Green Version]
- Tutlewska, K.; Lubinski, J.; Kurzawski, G. Germline deletions in the EPCAM gene as a cause of Lynch syndrome—Literature review. Hered. Cancer Clin. Pract. 2013, 11, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas, N. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [Green Version]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reynies, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef]
- Munemitsu, S.; Albert, I.; Souza, B.; Rubinfeld, B.; Polakis, P. Regulation of intracellular beta-catenin levels by the adenomatous polyposis coli (APC) tumor-suppressor protein. Proc. Natl. Acad. Sci. USA 1995, 92, 3046–3050. [Google Scholar] [CrossRef] [Green Version]
- Kolligs, F.T. Diagnostics and Epidemiology of Colorectal Cancer. Visc. Med. 2016, 32, 158–164. [Google Scholar] [CrossRef] [Green Version]
- Jones, S.; Chen, W.D.; Parmigiani, G.; Diehl, F.; Beerenwinkel, N.; Antal, T.; Traulsen, A.; Nowak, M.A.; Siegel, C.; Velculescu, V.E.; et al. Comparative lesion sequencing provides insights into tumor evolution. Proc. Natl. Acad. Sci. USA 2008, 105, 4283–4288. [Google Scholar] [CrossRef] [Green Version]
- Stryker, S.J.; Wolff, B.G.; Culp, C.E.; Libbe, S.D.; Ilstrup, D.M.; MacCarty, R.L. Natural history of untreated colonic polyps. Gastroenterology 1987, 93, 1009–1013. [Google Scholar] [CrossRef]
- Cho, K.R.; Vogelstein, B. Genetic alterations in the adenoma—Carcinoma sequence. Cancer 1992, 70, 1727–1731. [Google Scholar] [CrossRef] [PubMed]
- Hisamuddin, I.M.; Yang, V.W. Molecular Genetics of Colorectal Cancer: An Overview. Curr. Colorectal. Cancer Rep. 2006, 2, 53–59. [Google Scholar] [CrossRef] [Green Version]
- Rao, C.V.; Yamada, H.Y. Genomic instability and colon carcinogenesis: From the perspective of genes. Front. Oncol. 2013, 3, 130. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.; Mishra, L.; Deng, C.X. The role of TGF-beta/SMAD4 signaling in cancer. Int. J. Biol. Sci. 2018, 14, 111–123. [Google Scholar] [CrossRef] [Green Version]
- Barber, T.D.; McManus, K.; Yuen, K.W.; Reis, M.; Parmigiani, G.; Shen, D.; Barrett, I.; Nouhi, Y.; Spencer, F.; Markowitz, S.; et al. Chromatid cohesion defects may underlie chromosome instability in human colorectal cancers. Proc. Natl. Acad. Sci. USA 2008, 105, 3443–3448. [Google Scholar] [CrossRef] [Green Version]
- Palmer, M.C.L.; Neudorf, N.M.; Farrell, A.C.; Razi, T.; Lichtensztejn, Z.; McManus, K.J. The F-box protein, FBXO7 is required to maintain chromosome stability in humans. Hum. Mol. Genet. 2021, 31, 1471–1486. [Google Scholar] [CrossRef]
- Thompson, L.L.; Baergen, A.K.; Lichtensztejn, Z.; McManus, K.J. Reduced SKP1 Expression Induces Chromosome Instability through Aberrant Cyclin E1 Protein Turnover. Cancers 2020, 12, 531. [Google Scholar] [CrossRef] [Green Version]
- Jeusset, L.M.; Guppy, B.J.; Lichtensztejn, Z.; McDonald, D.; McManus, K.J. Reduced USP22 Expression Impairs Mitotic Removal of H2B Monoubiquitination, Alters Chromatin Compaction and Induces Chromosome Instability That May Promote Oncogenesis. Cancers 2021, 13, 1043. [Google Scholar] [CrossRef]
- Kuipers, E.J.; Grady, W.M.; Lieberman, D.; Seufferlein, T.; Sung, J.J.; Boelens, P.G.; van de Velde, C.J.; Watanabe, T. Colorectal cancer. Nat. Rev. Dis. Prim. 2015, 1, 15065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noffsinger, A.E. Serrated polyps and colorectal cancer: New pathway to malignancy. Annu. Rev. Pathol. 2009, 4, 343–364. [Google Scholar] [CrossRef] [PubMed]
- Szylberg, L.; Janiczek, M.; Popiel, A.; Marszalek, A. Serrated polyps and their alternative pathway to the colorectal cancer: A systematic review. Gastroenterol. Res. Pract. 2015, 2015, 573814. [Google Scholar] [CrossRef]
- Hahn, M.M.; de Voer, R.M.; Hoogerbrugge, N.; Ligtenberg, M.J.; Kuiper, R.P.; van Kessel, A.G. The genetic heterogeneity of colorectal cancer predisposition—Guidelines for gene discovery. Cell. Oncol. 2016, 39, 491–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baran, B.; Mert Ozupek, N.; Yerli Tetik, N.; Acar, E.; Bekcioglu, O.; Baskin, Y. Difference between Left-Sided and Right-Sided Colorectal Cancer: A Focused Review of Literature. Gastroenterol. Res. 2018, 11, 264–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, G.H.; Malietzis, G.; Askari, A.; Bernardo, D.; Al-Hassi, H.O.; Clark, S.K. Is right-sided colon cancer different to left-sided colorectal cancer?—A systematic review. Eur. J. Surg. Oncol. 2015, 41, 300–308. [Google Scholar] [CrossRef]
- Li, F.Y.; Lai, M.D. Colorectal cancer, one entity or three. J. Zhejiang Univ. Sci. B 2009, 10, 219–229. [Google Scholar] [CrossRef] [Green Version]
- Bufill, J.A. Colorectal cancer: Evidence for distinct genetic categories based on proximal or distal tumor location. Ann. Intern. Med. 1990, 113, 779–788. [Google Scholar] [CrossRef] [PubMed]
- Iacopetta, B. Are there two sides to colorectal cancer? Int. J. Cancer 2002, 101, 403–408. [Google Scholar] [CrossRef]
- Donaldson, G.P.; Lee, S.M.; Mazmanian, S.K. Gut biogeography of the bacterial microbiota. Nat. Rev. Microbiol. 2016, 14, 20–32. [Google Scholar] [CrossRef] [Green Version]
- Flynn, K.J.; Ruffin, M.T.t.; Turgeon, D.K.; Schloss, P.D. Spatial Variation of the Native Colon Microbiota in Healthy Adults. Cancer Prev. Res. (Phila) 2018, 11, 393–402. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Geigl, J.B.; Obenauf, A.C.; Schwarzbraun, T.; Speicher, M.R. Defining ‘chromosomal instability’. Trends Genet. 2008, 24, 64–69. [Google Scholar] [CrossRef]
- Sieber, O.M.; Heinimann, K.; Tomlinson, I.P. Genomic instability—The engine of tumorigenesis? Nat. Rev. Cancer 2003, 3, 701–708. [Google Scholar] [CrossRef]
- Lengauer, C.; Kinzler, K.W.; Vogelstein, B. Genetic instability in colorectal cancers. Nature 1997, 386, 623–627. [Google Scholar] [CrossRef] [PubMed]
- Lepage, C.C.; Morden, C.R.; Palmer, M.C.L.; Nachtigal, M.W.; McManus, K.J. Detecting Chromosome Instability in Cancer: Approaches to Resolve Cell-To-Cell Heterogeneity. Cancers 2019, 11, 226. [Google Scholar] [CrossRef] [Green Version]
- Giardiello, F.M.; Allen, J.I.; Axilbund, J.E.; Boland, C.R.; Burke, C.A.; Burt, R.W.; Church, J.M.; Dominitz, J.A.; Johnson, D.A.; Kaltenbach, T.; et al. Guidelines on genetic evaluation and management of Lynch syndrome: A consensus statement by the US Multi-Society Task Force on colorectal cancer. Gastroenterology 2014, 147, 502–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markowitz, S.D.; Bertagnolli, M.M. Molecular origins of cancer: Molecular basis of colorectal cancer. N. Engl. J. Med. 2009, 361, 2449–2460. [Google Scholar] [CrossRef] [Green Version]
- Nazemalhosseini Mojarad, E.; Kuppen, P.J.; Aghdaei, H.A.; Zali, M.R. The CpG island methylator phenotype (CIMP) in colorectal cancer. Gastroenterol. Hepatol. Bed Bench 2013, 6, 120–128. [Google Scholar]
- Orr, B.C.; Compton, D.A. A double-edged sword: How oncogenes and tumor suppressor genes can contribute to chromosomal instability. Front. Oncol. 2013, 3, 164. [Google Scholar] [CrossRef] [Green Version]
- Heng, J.; Heng, H.H. Two-phased evolution: Genome chaos-mediated information creation and maintenance. Prog. Biophys. Mol. Biol. 2021, 165, 29–42. [Google Scholar] [CrossRef]
- Watkins, T.B.K.; Lim, E.L.; Petkovic, M.; Elizalde, S.; Birkbak, N.J.; Wilson, G.A.; Moore, D.A.; Gronroos, E.; Rowan, A.; Dewhurst, S.M.; et al. Pervasive chromosomal instability and karyotype order in tumour evolution. Nature 2020, 587, 126–132. [Google Scholar] [CrossRef]
- Baker, D.J.; Jin, F.; Jeganathan, K.B.; van Deursen, J.M. Whole chromosome instability caused by Bub1 insufficiency drives tumorigenesis through tumor suppressor gene loss of heterozygosity. Cancer Cell 2009, 16, 475–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salgueiro, L.; Buccitelli, C.; Rowald, K.; Somogyi, K.; Kandala, S.; Korbel, J.O.; Sotillo, R. Acquisition of chromosome instability is a mechanism to evade oncogene addiction. EMBO Mol. Med. 2020, 12, e10941. [Google Scholar] [CrossRef]
- Jusino, S.; Fernandez-Padin, F.M.; Saavedra, H.I. Centrosome aberrations and chromosome instability contribute to tumorigenesis and intra-tumor heterogeneity. J. Cancer Metastasis Treat. 2018, 4, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, S.L.; Compton, D.A. Chromosome missegregation in human cells arises through specific types of kinetochore-microtubule attachment errors. Proc. Natl. Acad. Sci. USA 2011, 108, 17974–17978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dumont, M.; Gamba, R.; Gestraud, P.; Klaasen, S.; Worrall, J.T.; De Vries, S.G.; Boudreau, V.; Salinas-Luypaert, C.; Maddox, P.S.; Lens, S.M.; et al. Human chromosome-specific aneuploidy is influenced by DNA-dependent centromeric features. EMBO J. 2020, 39, e102924. [Google Scholar] [CrossRef]
- Worrall, J.T.; Tamura, N.; Mazzagatti, A.; Shaikh, N.; van Lingen, T.; Bakker, B.; Spierings, D.C.J.; Vladimirou, E.; Foijer, F.; McClelland, S.E. Non-Random Mis-Segregation of Human Chromosomes. Cell Rep. 2018, 23, 3366–3380. [Google Scholar] [CrossRef]
- Bakhoum, S.F.; Kabeche, L.; Murnane, J.P.; Zaki, B.I.; Compton, D.A. DNA-damage response during mitosis induces whole-chromosome missegregation. Cancer Discov. 2014, 4, 1281–1289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laughney, A.M.; Elizalde, S.; Genovese, G.; Bakhoum, S.F. Dynamics of Tumor Heterogeneity Derived from Clonal Karyotypic Evolution. Cell Rep. 2015, 12, 809–820. [Google Scholar] [CrossRef] [Green Version]
- Waclaw, B.; Bozic, I.; Pittman, M.E.; Hruban, R.H.; Vogelstein, B.; Nowak, M.A. A spatial model predicts that dispersal and cell turnover limit intratumour heterogeneity. Nature 2015, 525, 261–264. [Google Scholar] [CrossRef] [Green Version]
- Hoevenaar, W.H.M.; Janssen, A.; Quirindongo, A.I.; Ma, H.; Klaasen, S.J.; Teixeira, A.; van Gerwen, B.; Lansu, N.; Morsink, F.H.M.; Offerhaus, G.J.A.; et al. Degree and site of chromosomal instability define its oncogenic potential. Nat. Commun. 2020, 11, 1501. [Google Scholar] [CrossRef] [Green Version]
- Gao, C.; Su, Y.; Koeman, J.; Haak, E.; Dykema, K.; Essenberg, C.; Hudson, E.; Petillo, D.; Khoo, S.K.; Vande Woude, G.F. Chromosome instability drives phenotypic switching to metastasis. Proc. Natl. Acad. Sci. USA 2016, 113, 14793–14798. [Google Scholar] [CrossRef] [Green Version]
- Bakhoum, S.F.; Ngo, B.; Laughney, A.M.; Cavallo, J.A.; Murphy, C.J.; Ly, P.; Shah, P.; Sriram, R.K.; Watkins, T.B.K.; Taunk, N.K.; et al. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 2018, 553, 467–472. [Google Scholar] [CrossRef] [Green Version]
- Sottoriva, A.; Kang, H.; Ma, Z.; Graham, T.A.; Salomon, M.P.; Zhao, J.; Marjoram, P.; Siegmund, K.; Press, M.F.; Shibata, D.; et al. A Big Bang model of human colorectal tumor growth. Nat. Genet. 2015, 47, 209–216. [Google Scholar] [CrossRef] [Green Version]
- Young, A.R.; Narita, M. Oncogenic HMGA2: Short or small? Genes Dev. 2007, 21, 1005–1009. [Google Scholar] [CrossRef] [Green Version]
- Hammond, S.M.; Sharpless, N.E. HMGA2, microRNAs, and stem cell aging. Cell 2008, 135, 1013–1016. [Google Scholar] [CrossRef] [Green Version]
- Chau, K.Y.; Patel, U.A.; Lee, K.L.; Lam, H.Y.; Crane-Robinson, C. The gene for the human architectural transcription factor HMGI-C consists of five exons each coding for a distinct functional element. Nucleic Acids Res. 1995, 23, 4262–4266. [Google Scholar] [CrossRef] [Green Version]
- Reeves, R. Structure and function of the HMGI(Y) family of architectural transcription factors. Environ. Health Perspect. 2000, 108 (Suppl. 5), 803–809. [Google Scholar] [CrossRef]
- Pfannkuche, K.; Summer, H.; Li, O.; Hescheler, J.; Droge, P. The high mobility group protein HMGA2: A co-regulator of chromatin structure and pluripotency in stem cells? Stem Cell Rev. Rep. 2009, 5, 224–230. [Google Scholar] [CrossRef]
- Fedele, M.; Fidanza, V.; Battista, S.; Pentimalli, F.; Klein-Szanto, A.J.; Visone, R.; De Martino, I.; Curcio, A.; Morisco, C.; Del Vecchio, L.; et al. Haploinsufficiency of the Hmga1 gene causes cardiac hypertrophy and myelo-lymphoproliferative disorders in mice. Cancer Res. 2006, 66, 2536–2543. [Google Scholar] [CrossRef] [Green Version]
- Foti, D.; Chiefari, E.; Fedele, M.; Iuliano, R.; Brunetti, L.; Paonessa, F.; Manfioletti, G.; Barbetti, F.; Brunetti, A.; Croce, C.M.; et al. Lack of the architectural factor HMGA1 causes insulin resistance and diabetes in humans and mice. Nat. Med. 2005, 11, 765–773. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Benson, K.F.; Ashar, H.R.; Chada, K. Mutation responsible for the mouse pygmy phenotype in the developmentally regulated factor HMGI-C. Nature 1995, 376, 771–774. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.O.; Li, J.; Davis, B.W.; Upadhyay, S.; Al Muhisen, H.M.; Suva, L.J.; Clement, T.M.; Andersson, L. Hmga2 deficiency is associated with allometric growth retardation, infertility, and behavioral abnormalities in mice. G3 (Bethesda) 2022, 12, jkab417. [Google Scholar] [CrossRef] [PubMed]
- Cattaruzzi, G.; Altamura, S.; Tessari, M.A.; Rustighi, A.; Giancotti, V.; Pucillo, C.; Manfioletti, G. The second AT-hook of the architectural transcription factor HMGA2 is determinant for nuclear localization and function. Nucleic Acids Res. 2007, 35, 1751–1760. [Google Scholar] [CrossRef] [Green Version]
- Noro, B.; Licheri, B.; Sgarra, R.; Rustighi, A.; Tessari, M.A.; Chau, K.Y.; Ono, S.J.; Giancotti, V.; Manfioletti, G. Molecular dissection of the architectural transcription factor HMGA2. Biochemistry 2003, 42, 4569–4577. [Google Scholar] [CrossRef]
- Sgarra, R.; Maurizio, E.; Zammitti, S.; Lo Sardo, A.; Giancotti, V.; Manfioletti, G. Macroscopic differences in HMGA oncoproteins post-translational modifications: C-terminal phosphorylation of HMGA2 affects its DNA binding properties. J. Proteome Res. 2009, 8, 2978–2989. [Google Scholar] [CrossRef]
- Sgarra, R.; Rustighi, A.; Tessari, M.A.; Di Bernardo, J.; Altamura, S.; Fusco, A.; Manfioletti, G.; Giancotti, V. Nuclear phosphoproteins HMGA and their relationship with chromatin structure and cancer. FEBS Lett. 2004, 574, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Sgarra, R.; Tessari, M.A.; Di Bernardo, J.; Rustighi, A.; Zago, P.; Liberatori, S.; Armini, A.; Bini, L.; Giancotti, V.; Manfioletti, G. Discovering high mobility group A molecular partners in tumour cells. Proteomics 2005, 5, 1494–1506. [Google Scholar] [CrossRef]
- Pentimalli, F.; Palmieri, D.; Pacelli, R.; Garbi, C.; Cesari, R.; Martin, E.; Pierantoni, G.M.; Chieffi, P.; Croce, C.M.; Costanzo, V.; et al. HMGA1 protein is a novel target of the ATM kinase. Eur. J. Cancer 2008, 44, 2668–2679. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, D.; Valentino, T.; D’Angelo, D.; De Martino, I.; Postiglione, I.; Pacelli, R.; Croce, C.M.; Fedele, M.; Fusco, A. HMGA proteins promote ATM expression and enhance cancer cell resistance to genotoxic agents. Oncogene 2011, 30, 3024–3035. [Google Scholar] [CrossRef] [Green Version]
- Hombach-Klonisch, S.; Kalantari, F.; Medapati, M.R.; Natarajan, S.; Krishnan, S.N.; Kumar-Kanojia, A.; Thanasupawat, T.; Begum, F.; Xu, F.Y.; Hatch, G.M.; et al. HMGA2 as a functional antagonist of PARP1 inhibitors in tumor cells. Mol. Oncol. 2019, 13, 153–170. [Google Scholar] [CrossRef] [Green Version]
- UniProt, C. UniProt: The Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 2023, 51, D523–D531. [Google Scholar] [CrossRef]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 76, 7–20. [Google Scholar] [CrossRef] [Green Version]
- Vaser, R.; Adusumalli, S.; Leng, S.N.; Sikic, M.; Ng, P.C. SIFT missense predictions for genomes. Nat. Protoc. 2016, 11, 1–9. [Google Scholar] [CrossRef]
- Reeves, R. Molecular biology of HMGA proteins: Hubs of nuclear function. Gene 2001, 277, 63–81. [Google Scholar] [CrossRef] [PubMed]
- Catez, F.; Yang, H.; Tracey, K.J.; Reeves, R.; Misteli, T.; Bustin, M. Network of dynamic interactions between histone H1 and high-mobility-group proteins in chromatin. Mol. Cell. Biol. 2004, 24, 4321–4328. [Google Scholar] [CrossRef] [Green Version]
- Reeves, R. Nuclear functions of the HMG proteins. Biochim. Biophys. Acta 2010, 1799, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Li, O.; Li, J.; Droge, P. DNA architectural factor and proto-oncogene HMGA2 regulates key developmental genes in pluripotent human embryonic stem cells. FEBS Lett. 2007, 581, 3533–3537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Lim, H.H.; Tjokro, N.O.; Sathiyanathan, P.; Natarajan, S.; Chew, T.W.; Klonisch, T.; Goodman, S.D.; Surana, U.; Droge, P. Chaperoning HMGA2 protein protects stalled replication forks in stem and cancer cells. Cell Rep. 2014, 6, 684–697. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, S.M.; Droge, P. Oncofetal HMGA2 attenuates genotoxic damage induced by topoisomerase II target compounds through the regulation of local DNA topology. Mol. Oncol. 2019, 13, 2062–2078. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, S.M.; Ramani, P.D.; Wong, S.Q.R.; Zhao, X.; Ivanyi-Nagy, R.; Leong, T.C.; Chua, C.; Li, Z.; Hentze, H.; Tan, I.B.; et al. The chromatin structuring protein HMGA2 influences human subtelomere stability and cancer chemosensitivity. PLoS ONE 2019, 14, e0215696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Bacolla, A.; Wang, G.; Vasquez, K.M. Non-B DNA structure-induced genetic instability and evolution. Cell. Mol. Life Sci. 2010, 67, 43–62. [Google Scholar] [CrossRef] [Green Version]
- Summer, H.; Li, O.; Bao, Q.; Zhan, L.; Peter, S.; Sathiyanathan, P.; Henderson, D.; Klonisch, T.; Goodman, S.D.; Droge, P. HMGA2 exhibits dRP/AP site cleavage activity and protects cancer cells from DNA-damage-induced cytotoxicity during chemotherapy. Nucleic Acids Res. 2009, 37, 4371–4384. [Google Scholar] [CrossRef]
- Lindahl, T.; Barnes, D.E. Repair of endogenous DNA damage. Cold Spring Harb. Symp. Quant. Biol. 2000, 65, 127–133. [Google Scholar] [CrossRef]
- Natarajan, S.; Hombach-Klonisch, S.; Droge, P.; Klonisch, T. HMGA2 inhibits apoptosis through interaction with ATR-CHK1 signaling complex in human cancer cells. Neoplasia 2013, 15, 263–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hombach-Klonisch, S.; Natarajan, S.; Thanasupawat, T.; Medapati, M.; Pathak, A.; Ghavami, S.; Klonisch, T. Mechanisms of therapeutic resistance in cancer (stem) cells with emphasis on thyroid cancer cells. Front. Endocrinol. 2014, 5, 37. [Google Scholar] [CrossRef] [Green Version]
- Danescu, A.; Herrero Gonzalez, S.; Di Cristofano, A.; Mai, S.; Hombach-Klonisch, S. Three-dimensional nuclear telomere architecture changes during endometrial carcinoma development. Genes Chromosomes Cancer 2013, 52, 716–732. [Google Scholar] [CrossRef] [Green Version]
- Wark, L.; Danescu, A.; Natarajan, S.; Zhu, X.; Cheng, S.Y.; Hombach-Klonisch, S.; Mai, S.; Klonisch, T. Three-dimensional telomere dynamics in follicular thyroid cancer. Thyroid 2014, 24, 296–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Brenes, I.A.; Wodarz, D.; Komarova, N.L. Quantifying replicative senescence as a tumor suppressor pathway and a target for cancer therapy. Sci. Rep. 2015, 5, 17660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, A.Y.; Lin, H.H.; Kuo, C.Y.; Shih, H.M.; Wang, C.C.; Yen, Y.; Ann, D.K. High-mobility group A2 protein modulates hTERT transcription to promote tumorigenesis. Mol. Cell. Biol. 2011, 31, 2605–2617. [Google Scholar] [CrossRef] [Green Version]
- Natarajan, S.; Begum, F.; Gim, J.; Wark, L.; Henderson, D.; Davie, J.R.; Hombach-Klonisch, S.; Klonisch, T. High Mobility Group A2 protects cancer cells against telomere dysfunction. Oncotarget 2016, 7, 12761–12782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Lange, T. Shelterin: The protein complex that shapes and safeguards human telomeres. Genes Dev. 2005, 19, 2100–2110. [Google Scholar] [CrossRef] [Green Version]
- Zechiedrich, E.L.; Osheroff, N. Eukaryotic topoisomerases recognize nucleic acid topology by preferentially interacting with DNA crossovers. EMBO J. 1990, 9, 4555–4562. [Google Scholar] [CrossRef]
- Peter, S.; Yu, H.; Ivanyi-Nagy, R.; Droge, P. Cell-based high-throughput compound screening reveals functional interaction between oncofetal HMGA2 and topoisomerase I. Nucleic Acids Res. 2016, 44, e162. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Zhao, Z.; Xu, C.; Zhou, Z.; Zhu, Z.; You, T. HMGA2 induces transcription factor Slug expression to promote epithelial-to-mesenchymal transition and contributes to colon cancer progression. Cancer Lett. 2014, 355, 130–140. [Google Scholar] [CrossRef]
- Zhao, X.; Peter, S.; Droge, P.; Yan, J. Oncofetal HMGA2 effectively curbs unconstrained (+) and (−) DNA supercoiling. Sci. Rep. 2017, 7, 8440. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Liu, X.; Li, A.Y.; Chen, L.; Lai, L.; Lin, H.H.; Hu, S.; Yao, L.; Peng, J.; Loera, S.; et al. Overexpression of HMGA2 promotes metastasis and impacts survival of colorectal cancers. Clin. Cancer Res. 2011, 17, 2570–2580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellarin, I.; Arnoldo, L.; Costantini, S.; Pegoraro, S.; Ros, G.; Penzo, C.; Triolo, G.; Demarchi, F.; Sgarra, R.; Vindigni, A.; et al. The Architectural Chromatin Factor High Mobility Group A1 Enhances DNA Ligase IV Activity Influencing DNA Repair. PLoS ONE 2016, 11, e0164258. [Google Scholar] [CrossRef] [Green Version]
- Li, A.Y.; Boo, L.M.; Wang, S.Y.; Lin, H.H.; Wang, C.C.; Yen, Y.; Chen, B.P.; Chen, D.J.; Ann, D.K. Suppression of nonhomologous end joining repair by overexpression of HMGA2. Cancer Res. 2009, 69, 5699–5706. [Google Scholar] [CrossRef] [Green Version]
- Thanos, D.; Maniatis, T. The high mobility group protein HMG I(Y) is required for NF-kappa B-dependent virus induction of the human IFN-beta gene. Cell 1992, 71, 777–789. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zeng, K.; Xu, M.; Liu, X.; Hu, X.; Xu, T.; He, B.; Pan, Y.; Sun, H.; Wang, S. P53-induced miR-1249 inhibits tumor growth, metastasis, and angiogenesis by targeting VEGFA and HMGA2. Cell Death Dis. 2019, 10, 131. [Google Scholar] [CrossRef] [Green Version]
- Krahn, N.; Meier, M.; To, V.; Booy, E.P.; McEleney, K.; O’Neil, J.D.; McKenna, S.A.; Patel, T.R.; Stetefeld, J. Nanoscale Assembly of High-Mobility Group AT-Hook 2 Protein with DNA Replication Fork. Biophys. J. 2017, 113, 2609–2620. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Hu, L.; Wang, J.; Li, X.; Sahengbieke, S.; Wu, J.; Lai, M. HMGA2 promotes intestinal tumorigenesis by facilitating MDM2-mediated ubiquitination and degradation of p53. J. Pathol. 2018, 246, 508–518. [Google Scholar] [CrossRef]
- Wu, J.; Wang, Y.; Xu, X.; Cao, H.; Sahengbieke, S.; Sheng, H.; Huang, Q.; Lai, M. Transcriptional activation of FN1 and IL11 by HMGA2 promotes the malignant behavior of colorectal cancer. Carcinogenesis 2016, 37, 511–521. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.; Mou, Y.; Shi, X.; Liu, T.; Chen, Z.; Zuo, X. Circular RNA 100146 Promotes Colorectal Cancer Progression by the MicroRNA 149/HMGA2 Axis. Mol. Cell. Biol. 2021, 41, e00445-20. [Google Scholar] [CrossRef]
- Ye, J.; Liu, J.; Tang, T.; Xin, L.; Bao, X.; Yan, Y. LINC00963 affects the development of colorectal cancer via MiR-532-3p/HMGA2 axis. Cancer Cell Int. 2021, 21, 87. [Google Scholar] [CrossRef] [PubMed]
- Mansoori, B.; Mohammadi, A.; Naghizadeh, S.; Gjerstorff, M.; Shanehbandi, D.; Shirjang, S.; Najafi, S.; Holmskov, U.; Khaze, V.; Duijf, P.H.G.; et al. miR-330 suppresses EMT and induces apoptosis by downregulating HMGA2 in human colorectal cancer. J. Cell. Physiol. 2020, 235, 920–931. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Zou, Q.; He, H.; Liang, Y.; Lei, M.; Zhou, Q.; Fan, D.; Shen, L. Long non-coding RNA PCAT6 targets miR-204 to modulate the chemoresistance of colorectal cancer cells to 5-fluorouracil-based treatment through HMGA2 signaling. Cancer Med. 2019, 8, 2484–2495. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Liang, Y.; Shen, L.; Shen, L. MicroRNA-204 modulates colorectal cancer cell sensitivity in response to 5-fluorouracil-based treatment by targeting high mobility group protein A2. Biol. Open 2016, 5, 563–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, R.X.; Chen, X.; Xia, L.P.; Zhang, J.X.; Pan, Z.Z.; Ma, X.D.; Han, K.; Chen, J.W.; Judde, J.G.; Deas, O.; et al. N(6)-methyladenosine modification of circNSUN2 facilitates cytoplasmic export and stabilizes HMGA2 to promote colorectal liver metastasis. Nat. Commun. 2019, 10, 4695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, C.; Lin, Y.; Mao, Y.; Huang, Z.; Liu, A.Y.; Ma, H.; Yu, D.; Maitikabili, A.; Xiao, H.; Zhang, C.; et al. MicroRNA-543 suppresses colorectal cancer growth and metastasis by targeting KRAS, MTA1 and HMGA2. Oncotarget 2016, 7, 21825–21839. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.C.; Wang, G.P.; Yin, L.M.; Li, M.; Wu, L.L. Increasing miR-150 and lowering HMGA2 inhibit proliferation and cycle progression of colon cancer in SW480 cells. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 6793–6800. [Google Scholar] [CrossRef]
- Yu, F.Y.; Tu, Y.; Deng, Y.; Guo, C.; Ning, J.; Zhu, Y.; Lv, X.; Ye, H. MiR-4500 is epigenetically downregulated in colorectal cancer and functions as a novel tumor suppressor by regulating HMGA2. Cancer Biol. Ther. 2016, 17, 1149–1157. [Google Scholar] [CrossRef] [Green Version]
- Lu, W.; Huang, Z.; Wang, J.; Liu, H. Long non-coding RNA DANCR accelerates colorectal cancer progression via regulating the miR-185-5p/HMGA2 axis. J. Biochem. 2022, 171, 389–398. [Google Scholar] [CrossRef]
- Deng, X.; Kong, F.; Li, S.; Jiang, H.; Dong, L.; Xu, X.; Zhang, X.; Yuan, H.; Xu, Y.; Chu, Y.; et al. A KLF4/PiHL/EZH2/HMGA2 regulatory axis and its function in promoting oxaliplatin-resistance of colorectal cancer. Cell Death Dis. 2021, 12, 485. [Google Scholar] [CrossRef]
- Mizuno, R.; Kawada, K.; Sakai, Y. The Molecular Basis and Therapeutic Potential of Let-7 MicroRNAs against Colorectal Cancer. Can. J. Gastroenterol. Hepatol. 2018, 2018, 5769591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canadian Cancer Society. Chemotherapy for Colorectal Cancer. Available online: https://cancer.ca/en/cancer-information/cancer-types/colorectal/treatment/chemotherapy (accessed on 4 February 2023).
- Hammond, W.A.; Swaika, A.; Mody, K. Pharmacologic resistance in colorectal cancer: A review. Ther. Adv. Med. Oncol. 2016, 8, 57–84. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Wang, Y.; Deng, H.; Liu, C.; Wu, J.; Lai, M. HMGA2 enhances 5-fluorouracil chemoresistance in colorectal cancer via the Dvl2/Wnt pathway. Oncotarget 2018, 9, 9963–9974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pommier, Y.; Sun, Y.; Huang, S.N.; Nitiss, J.L. Roles of eukaryotic topoisomerases in transcription, replication and genomic stability. Nat. Rev. Mol. Cell. Biol. 2016, 17, 703–721. [Google Scholar] [CrossRef] [PubMed]
- Koster, D.A.; Palle, K.; Bot, E.S.; Bjornsti, M.A.; Dekker, N.H. Antitumour drugs impede DNA uncoiling by topoisomerase I. Nature 2007, 448, 213–217. [Google Scholar] [CrossRef]
- Vaughn, C.M.; Selby, C.P.; Yang, Y.; Hsu, D.S.; Sancar, A. Genome-wide single-nucleotide resolution of oxaliplatin-DNA adduct repair in drug-sensitive and -resistant colorectal cancer cell lines. J. Biol. Chem. 2020, 295, 7584–7594. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Variant ID | Amino Acid Position A | Amino Acid Change B | Predicted Clinical Significance C | |
|---|---|---|---|---|
| PolyPhen-2 [95] | SIFT [96] | |||
| rs1305472211 | 3 | A > T | Benign | Tolerated |
| rs1345615964 | 4 | R > G | Benign | Deleterious |
| rs986654581 | 5 | G > A | Benign | Tolerated |
| rs760771175 | 5 | G > S | Benign | Tolerated |
| rs1240728249 | 10 | Q > H | Possibly damaging | Deleterious |
| rs570487984 | 11 | P > Q | Benign | Deleterious |
| rs570487984 | 11 | P > R | Benign | Deleterious |
| rs764301435 | 11 | P > S | Benign | Deleterious |
| rs1192097965 | 12 | S > P | Benign | Tolerated |
| rs1397468624 | 13 | T > I | Benign | Tolerated |
| rs1397468624 | 13 | T > N | Benign | Deleterious |
| rs1385684307 | 19 | P > L | Benign | Deleterious |
| rs1172122751 | 19 | P > S | Benign | Deleterious |
| rs1455452771 | 21 | A > V | Benign | Deleterious |
| rs934404193 | 22 | P > Q | Benign | Deleterious |
| rs934404193 | 22 | P > R | Benign | Deleterious |
| rs1316442498 | 22 | P > S | Benign | Tolerated |
| rs1441432033 | 23 | A > E | Benign | Tolerated |
| rs7968829 | 28 | G > R | Probably damaging | Tolerated |
| rs1232428321 | 29 | R > C | Benign | Deleterious |
| rs1414030211 | 29 | R > P | Benign | Tolerated |
| rs914226102 | 31 | R > C | Probably damaging | Deleterious |
| rs1356918678 | 38 | E > K | Benign | Tolerated |
| rs759194451 | 39 | P > A | Probably damaging | Tolerated |
| rs1457257561 | 41 | G > D | Benign | Tolerated |
| rs1565699476 | 41 | G > S | Benign | Tolerated |
| rs974285143 | 47 | R > I | Probably damaging | Deleterious |
| rs762404803 | 48 | P > R | Probably damaging | Deleterious |
| rs994288858 | 48 | P > S | Probably damaging | Deleterious |
| rs1416276943 | 59 | S > R | Possibly damaging | Deleterious |
| rs374963072 | 61 | S > P | Probably damaging | Tolerated |
| rs192426102 | 68 | A > G | Probably damaging | Deleterious |
| rs756682360 | 68 | A > T | Probably damaging | Tolerated |
| rs79011113 | 69 | E > K | Possibly damaging | Tolerated |
| rs745772022 | 70 | A > T | Benign | Tolerated |
| rs1483054103 | 71 | T > I | Benign | Tolerated |
| rs779988617 | 72 | G > E | Probably damaging | Deleterious |
| rs768584493 | 73 | E > D | Possibly damaging | Tolerated |
| rs1187672823 | 73 | E > G | Probably damaging | Tolerated |
| rs1376518929 | 75 | R > Q | Probably damaging | Tolerated |
| rs1433416843 | 82 | K > R | Probably damaging | Deleterious |
| rs537621666 | 85 | Q > H | Probably damaging | Tolerated |
| rs1176973487 | 87 | V > A | Benign | Tolerated |
| rs1257572569 | 89 | Q > K | Possibly damaging | Tolerated |
| rs151017786 | 92 | P > A | Probably damaging | Tolerated |
| rs1251832694 | 92 | P > R | Probably damaging | Tolerated |
| rs1384638234 | 93 | A > D | Possibly damaging | Tolerated |
| rs1316622465 | 95 | E > G | Probably damaging | Deleterious |
| rs755573046 | 95 | E > Q | Probably damaging | Tolerated |
| rs1405500903 | 96 | E > D | Benign | Tolerated |
| rs1272332080 | 99 | E > G | Benign | Tolerated |
| rs1363332064 | 99 | E > K | Benign | Tolerated |
| rs1302947401 | 101 | S > F | Probably damaging | Tolerated |
| rs528046066 | 106 | A > G | Probably damaging | Tolerated |
| rs745605177 | 107 | E > K | Possibly damaging | Deleterious |
| rs745605177 | 107 | E > Q | Probably damaging | Deleterious |
| rs1180385418 | 109 | D > G | Probably damaging | Deleterious |
| ncRNA | Effect | Reference |
|---|---|---|
| circRNA 100146 | Promoter Inhibits miRNA 149, which inversely regulates HMGA2 expression | [127] |
| miRNA 149 | Inhibitor Suppresses HMGA2 expression | [127] |
| LINC00963 | Promoter Inhibits miRNA 532-3p, which inversely regulates HMGA2 expression | [128] |
| miRNA 532-3p | Inhibitor Suppresses HMGA2 expression | [128] |
| miRNA 330 | Inhibitor Suppresses HMGA2 expression | [129] |
| lncRNA PCAT6 | Promoter Inhibits miRNA 204, which inversely regulates HMGA2 expression Reduced sensitivity to 5-fluorouracil-based chemotherapies | [130] |
| miRNA 204 | Inhibitor Negatively regulates HMGA2 expression | [131] |
| circRNA NSUN2 | Promoter Creates an RNA-protein complex by interacting with IGF2BP2 and HMGA2 | [132] |
| miRNA 543 | Inhibitor Regulates KRAS, MTA1 and HMGA2 expression | [133] |
| miRNA 150 | Inhibitor Downregulates HMGA2 expression, which reduces Cyclin A levels, leading to cell cycle arrest | [134] |
| miRNA 4500 | Inhibitor Suppresses HMGA2 expression | [135] |
| miRNA 185-5p | Inhibitor Suppresses HMGA2 expression | [136] |
| lncRNA DANCR | Promoter Acts as a molecular sponge for miRNA 185-5p, thus upregulating HMGA2 expression | [136] |
| P53 inHibiting LncRNA (PiHL) | Promoter Reduces Oxaliplatin sensitivity | [137] |
| Let-7 miRNAs | Inhibitor Reduced Let-7 miRNAs abort tumor suppression by Let-7 targets (e.g., HMGA2) | [138] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Campos Gudiño, R.; McManus, K.J.; Hombach-Klonisch, S. Aberrant HMGA2 Expression Sustains Genome Instability That Promotes Metastasis and Therapeutic Resistance in Colorectal Cancer. Cancers 2023, 15, 1735. https://doi.org/10.3390/cancers15061735
Campos Gudiño R, McManus KJ, Hombach-Klonisch S. Aberrant HMGA2 Expression Sustains Genome Instability That Promotes Metastasis and Therapeutic Resistance in Colorectal Cancer. Cancers. 2023; 15(6):1735. https://doi.org/10.3390/cancers15061735
Chicago/Turabian StyleCampos Gudiño, Rubi, Kirk J. McManus, and Sabine Hombach-Klonisch. 2023. "Aberrant HMGA2 Expression Sustains Genome Instability That Promotes Metastasis and Therapeutic Resistance in Colorectal Cancer" Cancers 15, no. 6: 1735. https://doi.org/10.3390/cancers15061735