

The Role of Different Immunocompetent Cell Populations in the Pathogenesis of Head and Neck Cancer—Regulatory Mechanisms of Pro- and Anti-Cancer Activity and Their Impact on Immunotherapy

Abstract
:Simple Summary
Abstract
1. Introduction
1.1. Emerging Key Immune Cells in the Immune Evasion of Cancer—Tumour Infiltrating Lymphocytes (TILs) and Tumour-Associated Immunocompetent Cells in the TME
1.1.1. Tumour Infiltrating Lymphocytes (TILs)
1.1.2. Tumour-Associated Immunocompetent Cells in the Tumour Microenvironment (TME)
Antigen-Presenting Cells (APCs)—Dendritic Cells (DCs)
Regulatory T Cells (CD4+CD25+Foxp3+Tregs)
Tumour-Associated Macrophages (TAMs/MФ)
Myeloid-Derived Suppressor Cells (MDSCs)
Tumour-Associated Neutrophils (TANs)
Carcinoma-Associated Fibroblasts (CAFs)
Natural Killer Cells (NK)
T Helper 17 Cells (Th17)
HPV Infection as a Pivotal Modulator of the Immune Response in HNSCC
2. Materials and Methods
3. Results
3.1. Immunoediting and Immune Surveillance, Biological Mechanisms of Tumour Immune Escape
3.1.1. Immunoediting and Immune Surveillance
3.1.2. Biological Mechanisms of Tumour Immune Escape
Deficiency in the Tumour Antigen Release from the Tumour Cells (TM)
Disturbances in the Maturation and Function of Dendritic Cells (DCs)
Defects in Immunocompetent Cell Priming and Activation of T cells in Local Lymph Nodes
Disturbed Trafficking of T Cells to the Tumour Microenvironment (TME) and Defect in the Recognition of Tumour Cells by Immunocompetent Cells
Lack of T Cell Mediated Immune Response and Deficiency in the Killing of Tumour Cells
3.2. Current Perspectives and Future Prospects of Immunotherapeutic Strategies for Overcoming Immune Escape of HNSCC
3.2.1. HNSCC Immune Checkpoints Receptors and Their Targeting by Immunotherapy (Immune Checkpoint Inhibitors, ICIs)
PD-1/PD-L1 Inhibitors
CTLA-4 Inhibitors
ICI Combination Therapy
Other Checkpoint Modulator Inhibitors
3.2.2. Therapeutic Strategies for TME-Directed Therapy—Targeting TAMs, MDSCs, Tregs, CAFs and NK Cells
Strategies for TAMs
Strategies for MDSCs
Strategies for CD4+CD25+Foxp3+Tregs
Strategies for CAFs
Strategies for NK Cells
3.2.3. Adoptive Cell Therapy (ADP) and CAR-T Cell Therapy in Patients with HNSCC
3.2.4. Therapeutic Vaccines in Patients with HNSCC
3.2.5. Immunotherapy Based on Nanotechnology in Patients with HNSCC
3.3. Summary of Key Recommendations for HNSCC Immunotherapy According to the Society for Immunotherapy of Cancer (SITC), The National Comprehensive Cancer Network (NCCN) and the American Society of Clinical Oncology (ASCO)
- Integration of immunotherapy with PD-1 inhibitors in the treatment of relapsed/metastatic head and neck squamous cell cancer (R/M HNSCC):
- Frontline therapy: The FDA approved pembrolizumab (Keytruda, Merck), the anti-programmed cell death protein (PD-1) monoclonal antibody, for the first-line treatment of patients with unresectable R/M HNSCC;
- Pembrolizumab has been approved for mono immunotherapy in HNSCC patients with naïve R/M HNSCC, whose tumours express a PD-L1 biomarker with a combined positive score, CPS ≥ 1 confirmed by IHC staining (or positivity for PD-L1 is ≥1% tumour proportion score, TPS), as defined by the FDA-approved test;
- Pembrolizumab has been approved for immunotherapy in combination with platinum and fluorouracil (FU) in HNSCC patients with R/M HNSCC, whose tumours express a PD-L1 biomarker with a combined positive score, CPS < 1 confirmed by IHC staining), as defined by the FDA-approved test;
- Pembrolizumab has been approved for immunotherapy in combination with platinum and fluorouracil (FU) in all naïve R/M HNSCC patients, regardless of PD-L1 biomarker specifications;
- The FDA also expanded the use of the PD-L1 IHC 22C3 pharmDx kit for selecting patients with HNSCC for pembrolizumab treatment as a single immunotherapeutic agent.
- Second-line therapy: The FDA approved two immunotherapeutic agents, the anti-programmed cell death protein (PD-1) monoclonal antibodies, nivolumab (Opdivo, Bristol-Myers Squibb) and pembrolizumab (Keytruda, Merck), for the treatment of patients with R/M HNSCC unresponsive to platinum-based treatment:
- Pembrolizumab or nivolumab monotherapy may be proposed for the treatment of patients with R/M HNSCC without a clinical response to six months of platinum-based chemotherapy (if TPS ≥ 50);
- Option: best supportive care if not eligible for the treatment of patients with R/M HNSCC without a clinical response to six months of platinum-based chemotherapy and prior immunotherapy;
- Pembrolizumab or nivolumab monotherapy may be proposed for the treatment of patients with any PD-L1 CPS status and platinum-refractory R/M HNSCC.
- HPV status and the use of immunotherapy in HNSCC: HPV status (based on the p16 biomarker IHC overexpression) should be taken into account in the selection of therapy, but it has no influence on the decisions for patients with R/M HNSCC treatment with the use of immunotherapy.
- Evaluation of immune treatment response and further treatment recommendations for patients with advanced HNSCC:
- Initial clinical follow-up after one month of treatment with assessment of immune-related symptoms and adverse events (AEs);
- Each subsequent assessment of immune-related symptoms and AEs should be evaluated at least monthly;
- For initial assessment, a baseline clinical exam of the patient with imaging via CT or PET-CT scan following, should be performed. In monitoring patients for signs of response, patient evaluation (via radiographic imaging) should occur every three months. If radiographic progression is observed early in treatment, and the patient is clinically stable, treatment continuation until progression is confirmed on a second scan is recommended;
- If clinical response after treatment and six months of maintenance immunotherapy is observed, continue treatment for at least two years or until disease progression or toxicity;
- If disease progression on or after treatment with a PD-1 inhibitor is observed, enrolment in a clinical trial, treat with palliative radiotherapy and/or chemotherapy is recommended;
- Combination therapy (notably chemotherapy + immunotherapy IO) for rapidly growing disease due to the need for an enhanced response rate is recommended;
- In the first line setting for patients with R/M HNSCC with rapid/symptomatic progression, whose tumours express the PD-L1 biomarker, extreme chemotherapy regimen is recommended/Option: TPeX regimen or pembrolizumab + chemotherapy (FDA);
- In the first line setting for patients with R/M HNSCC with rapid/symptomatic progression, but without PD-L1 assessment, pembrolizumab + chemotherapy (FDA) or extreme chemotherapy regimen are recommended/Option: TPeX regimen;
- TMB (tumour mutational burden) testing if CPS is not available or in patients with rare tumours; TMB ≥ 10 interpreted as high; correlating with a clinical benefit to PD1 inhibitors
3.4. Latest Immunotherapies for HNSCC
4. Conclusions and Future Directions
Supplementary Materials
Funding
Data Availability Statement
Conflicts of Interest
References
- Yan, K.; Agrawal, N.; Gooi, Z. Head and Neck Masses. Med. Clin. N. Am. 2018, 102, 1013–1025. [Google Scholar] [CrossRef]
- El-Naggar, A.K.; Chan, C.J.; Grandis, J.R.; Takata, T.; Slootweg, P.J. WHO Classification of Head and Neck Tumours, 4th ed.; IARC: Lyon, France, 2017; ISBN 9789283224389. [Google Scholar]
- Seeburg, D.P.; Baer, A.H.; Aygun, N. Imaging of Patients with Head and Neck Cancer: From Staging to Surveillance. Oral Maxillofac. Surg. Clin. N. Am. 2018, 30, 421–433. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Gormley, M.; Creaney, G.; Schache, A.; Ingarfield, K.; Conway, D.I. Reviewing the epidemiology of head and neck cancer: Definitions, trends and risk factors. Br. Dent. J. 2022, 233, 780–786. [Google Scholar] [CrossRef]
- Chow, L.Q.M. Head and Neck Cancer. N. Engl. J. Med. 2020, 382, 60–72. [Google Scholar] [CrossRef]
- Fakhry, C.; Westra, W.H.; Wang, S.J.; van Zante, A.; Zhang, Y.; Rettig, E.; Yin, L.X.; Ryan, W.R.; Ha, P.K.; Wentz, A.; et al. The prognostic role of sex, race, and human papillomavirus in oropharyngeal and nonoropharyngeal head and neck squamous cell cancer. Cancer 2017, 123, 1566–1575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bravi, F.; Lee, Y.A.; Hashibe, M.; Boffetta, P.; Conway, D.I.; Ferraroni, M.; La Vecchia, C.; Edefonti, V. INHANCE Consortium investigators. Lessons learned from the INHANCE consortium: An overview of recent results on head and neck cancer. Oral Dis. 2021, 27, 73–93. [Google Scholar] [CrossRef]
- Ang, K.K.; Harris, J.; Wheeler, R.; Weber, R.; Rosenthal, D.I.; Nguyen-Tân, P.F.; Westra, W.H.; Chung, C.H.; Jordan, R.C.; Lu, C.; et al. Human papillomavirus and survival of patients with oropharyngeal cancer. N. Engl. J. Med. 2010, 363, 24–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freitag, J.; Wald, T.; Kuhnt, T.; Gradistanac, T.; Kolb, M.; Dietz, A.; Wiegand, S.; Wichmann, G. Extracapsular extension of neck nodes and absence of human papillomavirus 16-DNA are predictors of impaired survival in p16-positive oropharyngeal squamous cell carcinoma. Cancer 2020, 126, 1856–1872. [Google Scholar] [CrossRef] [Green Version]
- Leemans, C.R.; Snijders, P.J.F.; Brakenhoff, R.H. The molecular landscape of head and neck cancer. Nat. Rev. Cancer 2018, 18, 269–282. [Google Scholar] [CrossRef] [PubMed]
- Du, E.; Mazul, A.L.; Farquhar, D.; Brennan, P.; Anantharaman, D.; Abedi-Ardekani, B.; Weissler, M.C.; Hayes, D.N.; Olshan, A.F.; Zevallos, J.P. Long-term Survival in Head and Neck Cancer: Impact of Site, Stage, Smoking, and Human Papillomavirus Status. Laryngoscope 2019, 129, 2506–2513. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Sun, X.; Chen, Z.; Du, J.; Wu, Y. Head and Neck Squamous Cell Carcinoma: Risk Factors, Molecular Alterations, Immunology and Peptide Vaccines. Int. J. Pept. Res. Ther. 2022, 28, 19. [Google Scholar] [CrossRef] [PubMed]
- Miranda-Galvis, M.; Loveless, R.; Kowalski, L.P.; Teng, Y. Impacts of Environmental Factors on Head and Neck Cancer Pathogenesis and Progression. Cells 2021, 10, 389. [Google Scholar] [CrossRef]
- Nichols, A.C.; Lang, P.; Prisman, E.; Berthelet, E.; Tran, E.; Hamilton, S.; Wu, J.; Fung, K.; de Almeida, J.R.; Bayley, A.; et al. Treatment de-escalation for HPV-associated oropharyngeal squamous cell carcinoma with radiotherapy vs. trans-oral surgery (ORATOR2): Study protocol for a randomized phase II trial. BMC Cancer 2020, 20, 125. [Google Scholar] [CrossRef] [Green Version]
- Saada-Bouzid, E.; Peyrade, F.; Guigay, J. Molecular genetics of head and neck squamous cell carcinoma. Curr. Opin. Oncol. 2019, 31, 131–137. [Google Scholar] [CrossRef]
- Zhang, Y.X.; Koneva, L.A.; Virani, S.; Arthur, A.E.; Virani, A.; Hall, P.B.; Warden, C.D.; Carey, T.E.; Chepeha, D.B.; Prince, M.E.; et al. Subtypes of HPV-Positive Head and Neck Cancers Are Associated with HPV Characteristics, Copy Number Alterations, PIK3CA Mutation, and Pathway Signatures. Clin. Cancer Res. 2016, 22, 4735–4745. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, M.S.; Sougnez, C.; Lichtenstein, L.; Cibulskis, K.; Lander, E.; Gabriel, S.B.; Getz, G. Comprehensive genomic characterization of head and neck squamous cell carcinomas. The Cancer Genome Atlas Network. Nature 2015, 517, 576–582. [Google Scholar] [CrossRef] [Green Version]
- Zanoni, D.K.; Patel, S.G.; Shah, J.P. Changes in the 8th Edition of the American Joint Committee on Cancer (AJCC) Staging of Head and Neck Cancer: Rationale and Implications. Curr. Oncol. Rep. 2019, 21, 52. [Google Scholar] [CrossRef]
- Fialova, A.; Koucky, V.; Hajduskova, M.; Hladikova, K.; Spisek, R. Immunological Network in Head and Neck Squamous Cell Carcinoma-A Prognostic Tool Beyond HPV Status. Front. Oncol. 2020, 10, 1701. [Google Scholar] [CrossRef]
- Taberna, M.; Mena, M.; Pavon, M.A.; Alemany, L.; Gillison, M.L.; Mesia, R. Human papillomavirus-related oropharyngeal cancer. Ann. Oncol. 2017, 28, 2386–2398. [Google Scholar] [CrossRef]
- de Freitas, A.C.; de Oliveira, T.H.A.; Barros, M.R.; Venuti, A. hrHPV E5 oncoprotein: Immune evasion and related immunotherapies. J. Exp. Clin. Cancer Res. 2017, 36, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pal, A.; Kundu, R. Human Papillomavirus E6 and E7: The Cervical Cancer Hallmarks and Targets for Therapy. Front. Microbiol. 2020, 10, 3116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canning, M.; Guo, G.; Yu, M.; Myint, C.; Groves, M.W.; Byrd, J.K.; Cui, Y. Heterogeneity of the Head and Neck Squamous Cell Carcinoma Immune Landscape and Its Impact on Immunotherapy. Front. Cell Dev. Biol. 2019, 7, 52. [Google Scholar] [CrossRef]
- Seiwert, T.Y.; Zuo, Z.X.; Keck, M.K.; Khattri, A.; Pedamallu, C.S.; Stricker, T.; Brown, C.; Pugh, T.J.; Stojanov, P.; Cho, J.; et al. Integrative and Comparative Genomic Analysis of HPV-Positive and HPV-Negative Head and Neck Squamous Cell Carcinomas. Clin. Cancer Res. 2015, 21, 632–641. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.M.; Shi, C.J.; Xia, R.H.; Wang, L.Z.; Tian, Z.; Ye, W.M.; Liu, L.; Liu, S.L.; Zhang, C.Y.; Hu, Y.H.; et al. Analysis of Immunological Characteristics and Genomic Alterations in HPV-Positive Oropharyngeal Squamous Cell Carcinoma Based on PD-L1 Expression. Front. Immunol. 2022, 12, 5948. [Google Scholar] [CrossRef] [PubMed]
- Wuerdemann, N.; Gultekin, S.E.; Putz, K.; Wittekindt, C.; Huebbers, C.U.; Sharma, S.J.; Eckel, H.; Schubotz, A.B.; Gattenlohner, S.; Buttner, R.; et al. PD-L1 Expression and a High Tumor Infiltrate of CD8+Lymphocytes Predict Outcome in Patients with Oropharyngeal Squamous Cells Carcinoma. Int. J. Mol. Sci. 2020, 21, 5228. [Google Scholar] [CrossRef]
- Tang, H.; Zhou, X.; Ye, Y.; Zhou, Y.; Wu, C.Y.; Xu, Y. The different role of PD-L1 in head and neck squamous cell carcinomas: A meta-analysis. Path. Res. Prac. 2020, 216, 152768. [Google Scholar] [CrossRef]
- Schoenfeld, J.D.; Gjini, E.; Rodig, S.J.; Tishler, R.B.; Rawal, B.; Catalano, P.J.; Uppaluri, R.; Haddad, R.I.; Hanna, G.J.; Chau, N.G.; et al. Evaluating the PD-1 Axis and Immune Effector Cell Infiltration in Oropharyngeal Squamous Cell Carcinoma. Int. J. Radiat. Oncol. Biol. Phys. 2018, 102, 137–145. [Google Scholar] [CrossRef]
- Tosi, A.; Parisatto, B.; Menegaldo, A.; Spinato, G.; Guido, M.; Del Mistro, A.; Bussani, R.; Zanconati, F.; Tofanelli, M.; Tirelli, G.; et al. The immune microenvironment of HPV-positive and HPV-negative oropharyngeal squamous cell carcinoma: A multiparametric quantitative and spatial analysis unveils a rationale to target treatment-naive tumors with immune checkpoint inhibitors. J. Exp. Clin. Cancer Res. 2022, 41, 279. [Google Scholar] [CrossRef]
- Amin, M.B.; Greene, F.L.; Edge, S.B.; Compton, C.C.; Gershenwald, J.E.; Brookland, R.K.; Meyer, L.; Gress, D.M.; Byrd, D.R.; Winchester, D.P. The eighth edition AJCC cancer staging manual: Continuing to build a bridge from a population-based to a more “personalized” approach to cancer staging. CA Cancer J. Clin. 2017, 67, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Deng, S.Z.; Wang, S.M.; Shi, X.K.; Zhou, H.M. Microenvironment in Oral Potentially Malignant Disorders: Multi-Dimensional Characteristics and Mechanisms of Carcinogenesis. Int. J. Mol. Sci. 2022, 23, 8940. [Google Scholar] [CrossRef] [PubMed]
- Noel, G.; Fontsa, M.L.; Willard-Gallo, K. The impact of tumor cell metabolism on T cell-mediated immune responses and immuno-metabolic biomarkers in cancer. Semin. Cancer Biol. 2018, 52, 66–74. [Google Scholar] [CrossRef]
- Farlow, J.L.; Brenner, J.C.; Lei, Y.L.; Chinn, S.B. Immune deserts in head and neck squamous cell carcinoma: A review of challenges and opportunities for modulating the tumor immune microenvironment. Oral Oncol. 2021, 120, 105420. [Google Scholar] [CrossRef]
- Giraldo, N.A.; Sanchez-Salas, R.; Peske, J.D.; Vano, Y.; Becht, E.; Petitprez, F.; Validire, P.; Ingels, A.; Cathelineau, X.; Fridman, W.H.; et al. The clinical role of the TME in solid cancer. Br. J. Cancer 2019, 120, 45–53. [Google Scholar] [CrossRef]
- Sathasivam, H.P.; Kist, R.; Sloan, P.; Thomson, P.; Nugent, M.; Alexander, J.; Haider, S.; Robinson, M. Predicting the clinical outcome of oral potentially malignant disorders using transcriptomic-based molecular pathology. Br. J. Cancer 2021, 125, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Wen, L.; Mu, W.; Lu, H.; Wang, X.; Fang, J.; Jia, Y.; Li, Q.; Wang, D.; Wen, S.; Guo, J.; et al. Porphyromonas gingivalis Promotes Oral Squamous Cell Carcinoma Progression in an Immune Microenvironment. J. Dent. Res. 2020, 99, 666–675. [Google Scholar] [CrossRef]
- Hinshaw, D.C.; Shevde, L.A. The Tumor Microenvironment Innately Modulates Cancer Progression. Cancer Res. 2019, 79, 4557–4566. [Google Scholar] [CrossRef] [Green Version]
- Pereira, D.; Martins, D.; Mendes, F. Immunotherapy in Head and Neck Cancer When, How, and Why? Biomedicines 2022, 10, 2151. [Google Scholar] [CrossRef]
- Stern, P.L.; Dalianis, T. Oropharyngeal Squamous Cell Carcinoma Treatment in the Era of Immune Checkpoint Inhibitors. Viruses 2021, 13, 1234. [Google Scholar] [CrossRef]
- Society for Immunotherapy of Cancer. Cancer Immunotherapy Guidelines. 2017. Available online: https://www.sitcancer.org/research/cancer-immunotherapy-guidelines (accessed on 31 January 2023).
- The National Comprehensive Cancer Network® (NCCN®); Clinical Practice Guidelines in Oncology (NCCN Guidelines®). Head and Neck Cancers; NCCN Evidence BlocksTM (Version 1.2023, 01/30/23). Patents for Current List of Applicable Patents. Head and Neck Cancers Version 1.2023. Available online: www.nccn.org (accessed on 30 January 2023).
- Forster, M.D.; Devlin, M.J. Immune Checkpoint Inhibition in Head and Neck Cancer. Front. Oncol. 2018, 8, 310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poulose, J.V.; Kainickal, C.T. Immune checkpoint inhibitors in head and neck squamous cell carcinoma: A systematic review of phase-3 clinical trials. World J. Clin. Oncol. 2022, 13, 388–411. [Google Scholar] [CrossRef] [PubMed]
- Ghanizada, M.; Jakobsen, K.K.; Gronhoj, C.; von Buchwald, C. The effects of checkpoint inhibition on head and neck squamous cell carcinoma: A systematic review. Oral Oncol. 2019, 90, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Dogan, V.; Rieckmann, T.; Munscher, A.; Busch, C.J. Current studies of immunotherapy in head and neck cancer. Clin. Otolaryngol. 2018, 43, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Economopoulou, P.; Kotsantis, I.; Psyrri, A. Tumor Microenvironment and Immunotherapy Response in Head and Neck Cancer. Cancers 2020, 12, 3377. [Google Scholar] [CrossRef] [PubMed]
- Jumaniyazova, E.; Lokhonina, A.; Dzhalilova, D.; Kosyreva, A.; Fatkhudinov, T. Immune Cells in Head-and-Neck Tumor Microenvironments. J. Pers. Med. 2022, 12, 1521. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.M.Y.; Krinsky, A.L.; Woolaver, R.A.; Wang, X.G.; Chen, Z.G.; Wang, J.H. Tumor immune microenvironment in head and neck cancers. Mol. Carcinog. 2020, 59, 766–774. [Google Scholar] [CrossRef]
- Elmusrati, A.; Wang, J.; Wang, C.Y. Tumor microenvironment and immune evasion in head and neck squamous cell carcinoma. Int. J. Oral Sci. 2021, 13, 24. [Google Scholar] [CrossRef]
- Boulter, L.; Bullock, E.; Mabruk, Z.; Brunton, V.G. The fibrotic and immune microenvironments as targetable drivers of metastasis. Br. J. Cancer 2021, 124, 27–36. [Google Scholar] [CrossRef]
- Hussain, S.; Peng, B.; Cherian, M.; Song, J.W.; Ahirwar, D.K.; Ganju, R.K. The Roles of Stroma-Derived Chemokine in Different Stages of Cancer Metastases. Front. Immunol. 2020, 11, 598532. [Google Scholar] [CrossRef]
- Naleskina, L.A.; Kunska, L.M.; Chekhun, V.F. Modern views on the role of main components of stroma and tumor microinvironment in invasion, migration and metastasis. Exp. Oncol. 2020, 42, 252–262. [Google Scholar] [CrossRef] [PubMed]
- Hendry, S.; Salgado, R.; Gevaert, T.; Russell, P.A.; John, T.; Thapa, B.; Christie, M.; van de Vijver, K.; Estrada, M.V.; Gonzalez-Ericsson, P.I.; et al. Assessing Tumor-Infiltrating Lymphocytes in Solid Tumors: A Practical Review for Pathologists and Proposal for a Standardized Method from the International Immuno-Oncology Biomarkers Working Group: Part 2: TILs in Melanoma, Gastrointestinal Tract Carcinomas, Non-Small Cell Lung Carcinoma and Mesothelioma, Endometrial and Ovarian Carcinomas, Squamous Cell Carcinoma of the Head and Neck, Genitourinary Carcinomas, and Primary Brain Tumors. Adv. Anat. Pathol. 2017, 24, 311–335. [Google Scholar] [CrossRef] [PubMed]
- Furgiuele, S.; Descamps, G.; Lechien, J.R.; Dequanter, D.; Journe, F.; Saussez, S. Immunoscore Combining CD8, FoxP3, and CD68-Positive Cells Density and Distribution Predicts the Prognosis of Head and Neck Cancer Patients. Cells 2022, 11, 2050. [Google Scholar] [CrossRef]
- Lei, X.; Lei, Y.; Li, J.K.; Du, W.X.; Li, R.G.; Yang, J.; Li, J.; Li, F.; Tan, H.B. Immune cells within the tumor microenvironment: Biological functions and roles in cancer immunotherapy. Cancer Lett. 2020, 470, 126–133. [Google Scholar] [CrossRef]
- De Meulenaere, A.; Vermassen, T.; Aspeslagh, S.; Vandecasteele, K.; Rottey, S.; Ferdinande, L. TILs in Head and Neck Cancer: Ready for Clinical Implementation and Why (Not)? Head Neck Pathol. 2017, 11, 354–363. [Google Scholar] [CrossRef] [Green Version]
- Gajewski, T.F.; Schreiber, H.; Fu, Y.X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013, 14, 1014–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crespo, J.; Sun, H.Y.; Welling, T.H.; Tian, Z.G.; Zou, W.P. T cell anergy, exhaustion, senescence, and stemness in the tumor microenvironment. Curr. Opin. Immunol. 2013, 25, 214–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchan, S.L.; Rogel, A.; Al-Shamkhani, A. The immunobiology of CD27 and OX40 and their potential as targets for cancer immunotherapy. Blood 2018, 131, 39–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandal, R.; Senbabaoglu, Y.; Desrichard, A.; Havel, J.J.; Dalin, M.G.; Riaz, N.; Lee, K.W.; Ganly, I.; Hakimi, A.A.; Chan, T.A.; et al. The head and neck cancer immune landscape and its immunotherapeutic implications. JCI Insight 2016, 1, e89829. [Google Scholar] [CrossRef] [Green Version]
- Borsetto, D.; Tomasoni, M.; Payne, K.; Polesel, J.; Deganello, A.; Bossi, P.; Tysome, J.R.; Masterson, L.; Tirelli, G.; Tofanelli, M.; et al. Prognostic Significance of CD4+and CD8+Tumor-Infiltrating Lymphocytes in Head and Neck Squamous Cell Carcinoma: A Meta-Analysis. Cancers 2021, 13, 781. [Google Scholar] [CrossRef]
- de Ruiter, E.J.; Ooft, M.L.; Devriese, L.A.; Willems, S.M. The prognostic role of tumor infiltrating T-lymphocytes in squamous cell carcinoma of the head and neck: A systematic review and meta-analysis. Oncoimmunology 2017, 6, e1356148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Kempen, P.M.W.; Noorlag, R.; Swartz, J.E.; Bovenschen, N.; Braunius, W.W.; Vermeulen, J.F.; Van Cann, E.M.; Grolman, W.; Willems, S.M. Oropharyngeal squamous cell carcinomas differentially express granzyme inhibitors. Cancer Immunol. Immunother. 2016, 65, 575–585. [Google Scholar] [CrossRef] [Green Version]
- Welters, M.J.P.; Ma, W.B.; Santegoets, S.; Goedemans, R.; Ehsan, I.; Jordanova, E.S.; van Ham, V.J.; van Unen, V.; Koning, F.; van Egmond, S.I.; et al. Intratumoral HPV16-Specific T Cells Constitute a Type I-Oriented Tumor Microenvironment to Improve Survival in HPV16-Driven Oropharyngeal Cancer. Clin. Cancer Res. 2018, 24, 634–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landin, D.; Ahrlund-Richter, A.; Mirzaie, L.; Mints, M.; Nasman, A.; Kolev, A.; Marklund, L.; Dalianis, T.; Munck-Wikland, E.; Ramqvist, T. Immune related proteins and tumor infiltratingCD8+lymphocytes in hypopharyngeal cancer in relation to human papillomavirus (HPV) and clinical outcome. Head Neck-J. Sci. Spec. 2020, 42, 3206–3217. [Google Scholar] [CrossRef]
- Hu, C.Y.; Tian, S.; Lin, L.; Zhang, J.H.; Ding, H. Prognostic and clinicopathological significance of PD-L1 and tumor infiltrating lymphocytes in hypopharyngeal squamous cell carcinoma. Oral Oncol. 2020, 102, 104560. [Google Scholar] [CrossRef] [PubMed]
- Ono, T.; Azuma, K.; Kawahara, A.; Akiba, J.; Kakuma, T.; Chitose, S.; Umeno, H. Pre-treatment CD8(+) tumour-infiltrating lymphocyte density predicts distant metastasis after definitive treatment in patients with stage III/IV hypopharyngeal squamous cell carcinoma. Clin. Otolaryngol. 2018, 43, 1312–1320. [Google Scholar] [CrossRef]
- Balermpas, P.; Michel, Y.; Wagenblast, J.; Seitz, O.; Weiss, C.; Rodel, F.; Rodel, C.; Fokas, E. Tumour-infiltrating lymphocytes predict response to definitive chemoradiotherapy in head and neck cancer. Br. J. Cancer 2014, 110, 501–509. [Google Scholar] [CrossRef]
- Kim, H.R.; Ha, S.J.; Hong, M.H.; Heo, S.J.; Koh, Y.W.; Choi, E.C.; Kim, E.K.; Pyo, K.H.; Jung, I.; Seo, D.; et al. PD-L1 expression on immune cells, but not on tumor cells, is a favorable prognostic factor for head and neck cancer patients. Sci. Rep. 2016, 6, 36956. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, N.; Bellile, E.; Thomas, D.; McHugh, J.; Rozek, L.; Virani, S.; Peterson, L.; Carey, T.E.; Walline, H.; Moyer, J.; et al. Tumor infiltrating lymphocytes and survival in patients with head and neck squamous cell carcinoma. Head Neck-J. Sci. Spec. 2016, 38, 1074–1084. [Google Scholar] [CrossRef] [Green Version]
- Oguejiofor, K.; Hall, J.; Slater, C.; Betts, G.; Hall, G.; Slevin, N.; Dovedi, S.; Stern, P.L.; West, C.M.L. Stromal infiltration of CD8 T cells is associated with improved clinical outcome in HPV-positive oropharyngeal squamous carcinoma. Br. J. Cancer 2015, 113, 886–893. [Google Scholar] [CrossRef]
- Hsu, J.M.; Xia, W.; Hsu, Y.H.; Chan, L.C.; Yu, W.H.; Cha, J.H.; Chen, C.T.; Liao, H.W.; Kuo, C.W.; Khoo, K.-H.; et al. STT3-dependent PD-L1 accumulation on cancer stem cells promotes immune evasion. Nat. Commun. 2018, 9, 1908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mansour, F.A.; Al-Mazrou, A.; Al-Mohanna, F.; Al-Alwan, M.; Ghebeh, H. PD-L1 is overexpressed on breast cancer stem cells through notch3/mTOR axis. Oncoimmunology 2020, 9, 1729299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, F.; Zhang, T.; Deng, S.C.; Wei, J.C.; Yang, P.; Wang, Q.; Chen, Z.P.; Li, W.L.; Chen, H.C.; Hu, H.; et al. PD-L1 promotes colorectal cancer stem cell expansion by activating HMGA1-dependent signaling pathways. Cancer Lett. 2019, 450, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Li, M.O.; Wolf, N.; Raulet, D.H.; Akkari, L.; Pittet, M.J.; Rodriguez, P.C.; Kaplan, R.N.; Munitz, A.; Zhang, Z.M.; Cheng, S.J.; et al. Innate immune cells in the tumor microenvironment. Cancer Cell 2021, 39, 725–729. [Google Scholar] [CrossRef] [PubMed]
- Kazemi, M.H.; Sadri, M.; Najafi, A.; Rahimi, A.; Baghernejadan, Z.; Khorramdelazad, H.; Falak, R. Tumor-infiltrating lymphocytes for treatment of solid tumors: It takes two to tango? Front. Immunol. 2022, 13, 1018962. [Google Scholar] [CrossRef]
- Ruhle, A.; Todorovic, J.; Spohn, S.S.K.; Gkika, E.; Becker, C.; Knopf, A.; Zamboglou, C.; Sprave, T.; Werner, M.; Grosu, A.L.; et al. Prognostic value of tumor-infiltrating immune cells and immune checkpoints in elderly head-and-neck squamous cell carcinoma patients undergoing definitive (chemo)radiotherapy. Radiat. Oncol. 2022, 17, 181. [Google Scholar] [CrossRef]
- Wondergem, N.E.; Nauta, I.H.; Muijlwijk, T.; Leemans, C.R.; van de Ven, R. The Immune Microenvironment in Head and Neck Squamous Cell Carcinoma: On Subsets and Subsites. Curr. Oncol. Rep. 2020, 22, 81. [Google Scholar] [CrossRef]
- Plesca, I.; Muller, L.; Bottcher, J.P.; Medyouf, H.; Wehner, R.; Schmitz, M. Tumor-associated human dendritic cell subsets: Phenotype, functional orientation, and clinical relevance. Eur. J. Immunol. 2022, 52, 1750–1758. [Google Scholar] [CrossRef]
- Kim, C.W.; Kim, K.D.; Lee, H.K. The role of dendritic cells in tumor microenvironments and their uses as therapeutic targets. BMB Rep. 2021, 54, 31–43. [Google Scholar] [CrossRef]
- Veglia, F.; Gabrilovich, D.I. Dendritic cells in cancer: The role revisited. Curr. Opin. Immunol. 2017, 45, 43–51. [Google Scholar] [CrossRef] [Green Version]
- Guillerey, C.; Huntington, N.D.; Smyth, M.J. Targeting natural killer cells in cancer immunotherapy. Nat. Immunol. 2016, 17, 1025–1036. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Zheng, X.; Gao, W.; Wang, B.; Wu, Y. Tumor microenvironment and immune-related therapies of head and neck squamous cell carcinoma. Mol. Ther. Oncolytics 2021, 20, 342–351. [Google Scholar] [CrossRef] [PubMed]
- Michielsen, A.J.; Hogan, A.E.; Marry, J.; Tosetto, M.; Cox, F.; Hyland, J.M.; Sheahan, K.D.; O’Donoghue, D.P.; Mulcahy, H.E.; Ryan, E.J.; et al. Tumour Tissue Microenvironment Can Inhibit Dendritic Cell Maturation in Colorectal Cancer. PLoS ONE 2011, 6, e27944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krempski, J.; Karyampudi, L.; Behrens, M.D.; Erskine, C.L.; Hartmann, L.; Dong, H.D.; Goode, E.L.; Kalli, K.R.; Knutson, K.L. Tumor-Infiltrating Programmed Death Receptor-1(+) Dendritic Cells Mediate Immune Suppression in Ovarian Cancer. J. Immunol. 2011, 186, 6905–6913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kießler, M.; Plesca, I.; Sommer, U.; Wehner, R.; Wilczkowski, F.; Müller, L.; Tunger, A.; Lai, X.; Rentsch, A.; Peuker, K.; et al. Tumor-infiltrating plasmacytoid dendritic cells are associated with survival in human colon cancer. J. Immunother. Cancer 2021, 9, e001813. [Google Scholar] [CrossRef]
- Han, N.N.; Zhang, Z.; Liu, S.W.; Ow, A.; Ruan, M.; Yang, W.J.; Zhang, C.P. Increased tumor-infiltrating plasmacytoid dendritic cells predicts poor prognosis in oral squamous cell carcinoma. Arch. Oral Biol. 2017, 78, 129–134. [Google Scholar] [CrossRef]
- Han, N.N.; Zhang, Z.; Jv, H.Y.; Hu, J.Z.; Ruan, M.; Zhang, C.P. Culture supernatants of oral cancer cells induce impaired IFN-alpha production of pDCs partly through the down-regulation of TLR-9 expression. Arch. Oral Biol. 2018, 93, 141–148. [Google Scholar] [CrossRef]
- Han, N.N.; Li, X.; Wang, Y.P.; Wang, L.; Zhang, C.Y.; Zhang, Z.; Ruan, M.; Zhang, C.P. Increased tumor-infiltrating plasmacytoid dendritic cells promote cancer cell proliferation and invasion via TNF-alpha/NF-kappa B/CXCR-4 pathway in oral squamous cell carcinoma. J. Cancer 2021, 12, 3045–3056. [Google Scholar] [CrossRef]
- Marmonti, E.; Oliva-Ramirez, J.; Haymaker, C. Dendritic Cells: The Long and Evolving Road towards Successful Targetability in Cancer. Cells 2022, 11, 3028. [Google Scholar] [CrossRef]
- Bol, K.F.; Schreibelt, G.; Rabold, K.; Wculek, S.K.; Schwarze, J.K.; Dzionek, A.; Teijeira, A.; Kandalaft, L.E.; Romero, P.; Coukos, G.; et al. The clinical application of cancer immunotherapy based on naturally circulating dendritic cells. J. Immunother. Cancer 2019, 7, 109. [Google Scholar] [CrossRef] [Green Version]
- Carenza, C.; Calcaterra, F.; Oriolo, F.; Di Vito, C.; Ubezio, M.; Della Porta, M.G.; Mavilio, D.; Della Bella, S. Costimulatory Molecules and Immune Checkpoints Are Differentially Expressed on Different Subsets of Dendritic Cells. Front. Immunol. 2019, 10, 1325. [Google Scholar] [CrossRef] [PubMed]
- Zou, S.L.; Tong, Q.Y.; Liu, B.W.; Huang, W.; Tian, Y.; Fu, X.H. Targeting STAT3 in Cancer Immunotherapy. Mol. Cancer 2020, 19, 145. [Google Scholar] [CrossRef]
- Thuring, M.; Knuchel, R.; Picchetta, L.; Keller, D.; Schmidli, T.S.; Provenzano, M. The Prognostic Value of Indoleamine-2,3-Dioxygenase Gene Expression in Urine of Prostate Cancer Patients Undergoing Radical Prostatectomy as First Treatment of Choice. Front. Immunol. 2020, 11, 1244. [Google Scholar] [CrossRef] [PubMed]
- Gomes, B.; Driessens, G.; Bartlett, D.; Cai, D.Y.; Cauwenberghs, S.; Crosignani, S.; Dalvie, D.; Denies, S.; Dillon, C.P.; Fantin, V.R.; et al. Characterization of the Selective lndoleamine 2,3-Dioxygenase-1 (IDO1) Catalytic Inhibitor EOS200271/PF-06840003 Supports IDO1 as a Critical Resistance Mechanism to PD-(L)1 Blockade Therapy. Mol. Cancer Ther. 2018, 17, 2530–2542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Naour, J.; Galluzzi, L.; Zitvogel, L.; Kroemer, G.; Vacchelli, E. Trial watch: IDO inhibitors in cancer therapy. Oncoimmunology 2020, 9, 1777625. [Google Scholar] [CrossRef]
- Di, S.M.; Zhou, M.; Pan, Z.Y.; Sun, R.X.; Chen, M.H.; Jiang, H.; Shi, B.Z.; Luo, H.; Li, Z.H. Combined Adjuvant of Poly I:C Improves Antitumor Effects of CAR-T Cells. Front. Oncol. 2019, 9, 241. [Google Scholar] [CrossRef]
- Sato-Kaneko, F.; Yao, S.Y.; Ahmadi, A.; Zhang, S.S.; Hosoya, T.; Kaneda, M.M.; Varner, J.A.; Pu, M.; Messer, K.S.; Guiducci, C.; et al. Combination immunotherapy with TLR agonists and checkpoint inhibitors suppresses head and neck cancer. JCI Insight 2017, 2, e93397. [Google Scholar] [CrossRef]
- Woo, S.R.; Fuertes, M.B.; Corrales, L.; Spranger, S.; Furdyna, M.J.; Leung, M.Y.K.; Duggan, R.; Wang, Y.; Barber, G.N.; Fitzgerald, K.A.; et al. STING-Dependent Cytosolic DNA Sensing Mediates Innate Immune Recognition of Immunogenic Tumors. Immunity 2014, 41, 830–842. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Diaz, A.; Shin, D.S.; Moreno, B.H.; Saco, J.; Escuin-Ordinas, H.; Rodriguez, G.A.; Zaretsky, J.M.; Sun, L.; Hugo, W.; Wang, X.Y.; et al. Interferon Receptor Signaling Pathways Regulating PD-L1 and PD-L2 Expression. Cell Rep. 2017, 19, 1189–1201. [Google Scholar] [CrossRef] [Green Version]
- Li, T.J.; Cheng, H.; Yuan, H.; Xu, Q.M.; Shu, C.; Zhang, Y.F.; Xu, P.B.; Tan, J.S.; Rui, Y.C.; Li, P.W.; et al. Antitumor Activity of cGAMP via Stimulation of cGAS-cGAMPSTING-IRF3 Mediated Innate Immune Response. Sci. Rep. 2016, 6, 19049. [Google Scholar] [CrossRef] [Green Version]
- Marcus, A.; Mao, A.J.; Lensink-Vasan, M.; Wang, L.; Vance, R.E.; Raulet, D.H. Tumor-Derived cGAMP Triggers a STING-Mediated Interferon Response in Non-tumor Cells to Activate the NK Cell Response. Immunity 2018, 49, 754–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, C.H.A.; Zundell, J.A.; Ranatunga, S.; Lin, C.; Nefedova, Y.; Del Valle, J.R.; Hu, C.C.A. Agonist-Mediated Activation of STING Induces Apoptosis in Malignant B Cells. Cancer Res. 2016, 76, 2137–2152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.X.; Ye, S.B.; Ni, J.J.; Cai, T.T.; Liu, Y.N.; Huang, D.J.; Mai, H.Q.; Chen, Q.Y.; He, J.; Zhang, X.S.; et al. STING signaling remodels the tumor microenvironment by antagonizing myeloid-derived suppressor cell expansion. Cell Death Differ. 2019, 26, 2314–2328. [Google Scholar] [CrossRef] [PubMed]
- Amouzegar, A.; Chelvanambi, M.; Filderman, J.N.; Storkus, W.J.; Luke, J.J. STING Agonists as Cancer Therapeutics. Cancers 2021, 13, 2695. [Google Scholar] [CrossRef] [PubMed]
- Kalantari, T.; Ciric, B.; Kamali-Sarvestani, E.; Rostami, A. Bone marrow dendritic cells deficient for CD40 and IL-23p19 are tolerogenic in vitro. Iran. J. Basic Med. Sci. 2020, 23, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Djureinovic, D.; Wang, M.N.; Kluger, H.M. Agonistic CD40 Antibodies in Cancer Treatment. Cancers 2021, 13, 1302. [Google Scholar] [CrossRef]
- Mangsbo, S.M.; Broos, S.; Fletcher, E.; Veitonmaki, N.; Furebring, C.; Dahlen, E.; Norlen, P.; Lindstedt, M.; Totterman, T.H.; Ellmark, P. The Human Agonistic CD40 Antibody ADC-1013 Eradicates Bladder Tumors and Generates T-cell-Dependent Tumor Immunity. Clin. Cancer Res. 2015, 21, 1115–1126. [Google Scholar] [CrossRef] [Green Version]
- Byrne, K.T.; Betts, C.B.; Mick, R.; Sivagnanam, S.; Bajor, D.L.; Laheru, D.A.; Chiorean, E.G.; O’Hara, M.H.; Liudahl, S.M.; Newcomb, C.; et al. Neoadjuvant Selicrelumab, an Agonist CD40 Antibody, Induces Changes in the Tumor Microenvironment in Patients with Resectable Pancreatic Cancer. Clin. Cancer Res. 2021, 27, 4574–4586. [Google Scholar] [CrossRef]
- Morrison, A.H.; Diamond, M.S.; Hay, C.A.; Byrne, K.T.; Vonderheide, R.H. Sufficiency of CD40 activation and immune checkpoint blockade for T cell priming and tumor immunity. Proc. Natl. Acad. Sci. USA 2020, 117, 8022–8031. [Google Scholar] [CrossRef]
- Vonderheide, R.H. CD40 Agonist Antibodies in Cancer Immunotherapy. Annu. Rev. Med. 2020, 71, 47–58. [Google Scholar] [CrossRef] [Green Version]
- Rech, A.J.; Dada, H.; Kotzin, J.J.; Henao-Mejia, J.; Minn, A.J.; Victor, C.T.S.; Vonderheide, R.H. Radiotherapy and CD40 Activation Separately Augment Immunity to Checkpoint Blockade in Cancer. Cancer Res. 2018, 78, 4282–4291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ngiow, S.F.; Young, A.; Blake, S.J.; Hill, G.R.; Yagita, H.; Teng, M.W.L.; Korman, A.J.; Smyth, M.J. Agonistic CD40 mAb-Driven IL12 Reverses Resistance to Anti-PD1 in a T-cell-Rich Tumor. Cancer Res. 2016, 76, 6266–6277. [Google Scholar] [CrossRef] [Green Version]
- Sanborn, R.G.N.; O’Hara, M. Phase 1 study of the CD40 agonist monoclonal antibody (mAb); CDX-1140 alone and in combination with CDX-301 (rhFLT3L) in patients with advanced cancers. In 34th Annual Meeting & Pre-Conference Programs of the Society for Immunotherapy of Cancer (SITC 2019): Part 2: National Harbour, MD, USA. P827. J. Immunother. Cancer 2019, 7, 283. [Google Scholar] [CrossRef] [Green Version]
- Xavier, F.C.A.; Silva, J.C.; Rodini, C.O.; Rodrigues, M.F.S.D. Mechanisms of immune evasion by head and neck cancer stem cells. Front. Oral Health 2022, 3, 957310. [Google Scholar] [CrossRef] [PubMed]
- Scott, E.N.; Gocher, A.M.; Workman, C.J.; Vignali, D.A.A. Regulatory T Cells: Barriers of Immune Infiltration Into the Tumor Microenvironment. Front. Immunol. 2021, 12, 2282. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.J.; Guo, J.H.; Jia, R. Treg: A Promising Immunotherapeutic Target in Oral Diseases. Front. Immunol. 2021, 12, 667862. [Google Scholar] [CrossRef] [PubMed]
- Mair, F.; Erickson, J.R.; Frutoso, M.; Konecny, A.J.; Greene, E.; Voillet, V.; Maurice, N.J.; Rongvaux, A.; Dixon, D.; Barber, B.; et al. Extricating human tumour immune alterations from tissue inflammation. Nature 2022, 605, 728–735. [Google Scholar] [CrossRef]
- Wang, H.C.; Chan, L.P.; Cho, S.F. Targeting the Immune Microenvironment in the Treatment of Head and Neck Squamous Cell Carcinoma. Front. Oncol. 2019, 9, 1084. [Google Scholar] [CrossRef] [Green Version]
- Peltanova, B.; Raudenska, M.; Masarik, M. Effect of tumor microenvironment on pathogenesis of the head and neck squamous cell carcinoma: A systematic review. Mol. Cancer 2019, 18, 63. [Google Scholar] [CrossRef] [Green Version]
- Sasidharan, N.V.; Elkord, E. Immune checkpoint inhibitors in cancer therapy: A focus on T-regulatory cells. Immunol. Cell Biol. 2018, 96, 21. [Google Scholar] [CrossRef]
- Bruni, D.; Angell, H.K.; Galon, J. The immune contexture and Immunoscore in cancer prognosis and therapeutic efficacy. Nat. Rev. Cancer 2020, 20, 662–680. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.Q.; McMichael, E.L.; Shayan, G.; Li, J.; Chen, K.; Srivastava, R.; Kane, L.P.; Lu, B.F.; Ferris, R.L. Novel Effector Phenotype of Tim-3(+) Regulatory T Cells Leads to Enhanced Suppressive Function in Head and Neck Cancer Patients. Clin. Cancer Res. 2018, 24, 4529–4538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oweida, A.J.; Darragh, L.; Phan, A.; Binder, D.; Bhatia, S.; Mueller, A.; Van Court, B.; Milner, D.; Raben, D.; Woessner, R.; et al. STAT3 Modulation of Regulatory T Cells in Response to Radiation Therapy in Head and Neck Cancer. J. Natl. Cancer Inst. 2019, 111, 1339–1349. [Google Scholar] [CrossRef] [PubMed]
- Niu, M.L.; Combs, S.E.; Linge, A.; Krause, M.; Baumann, M.; Lohaus, F.; Ebert, N.; Tinhofer, I.; Budach, V.; von der Grun, J.; et al. Comparison of the composition of lymphocyte subpopulations in non-relapse and relapse patients with squamous cell carcinoma of the head and neck before, during radiochemotherapy and in the follow-up period: A multicenter prospective study of the German Cancer Consortium Radiation Oncology Group (DKTK-ROG). Radiat. Oncol. 2021, 16, 141. [Google Scholar] [CrossRef] [PubMed]
- Echarti, A.; Hecht, M.; Buttner-Herold, M.; Haderlein, M.; Hartmann, A.; Fietkau, R.; Distel, L. CD8+and Regulatory T cells Differentiate Tumor Immune Phenotypes and Predict Survival in Locally Advanced Head and Neck Cancer. Cancers 2019, 11, 1398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ihara, F.; Sakurai, D.; Horinaka, A.; Makita, Y.; Fujikawa, A.; Sakurai, T.; Yamasaki, K.; Kunii, N.; Motohashi, S.; Nakayama, T.; et al. CD45RA (-)Foxp3(high) regulatory T cells have a negative impact on the clinical outcome of head and neck squamous cell carcinoma. Cancer Immunol. Immunother. 2017, 66, 1275–1285. [Google Scholar] [CrossRef]
- Hur, J.Y.; Ku, B.M.; Park, S.; Jung, H.A.; Lee, S.H.; Ahn, M.J. Prognostic value of FOXP3+ regulatory T cells for patients with locally advanced oropharyngeal squamous cell carcinoma. PLoS ONE 2022, 17, e0274830. [Google Scholar] [CrossRef]
- Punt, S.; Dronkers, E.A.C.; Welters, M.J.P.; Goedemans, R.; Koljenovic, S.; Bloemena, E.; Snijders, P.J.F.; Gorter, A.; van der Burg, S.H.; de Jong, R.J.B.; et al. A beneficial tumor microenvironment in oropharyngeal squamous cell carcinoma is characterized by a high T cell and low IL-17(+) cell frequency. Cancer Immunol. Immunother. 2016, 65, 393–403. [Google Scholar] [CrossRef] [Green Version]
- Norouzian, M.; Mehdipour, F.; Ashraf, M.J.; Khademi, B.; Ghaderi, A. Regulatory and effector T cell subsets in tumor-draining lymph nodes of patients with squamous cell carcinoma of head and neck. BMC Immunol. 2022, 23, 56. [Google Scholar] [CrossRef]
- Shang, B.; Liu, Y.; Jiang, S.J. Prognostic value of tumor-infiltrating FoxP3(+) regulatory T cells in cancers: A systematic review and meta-analysis. Sci. Rep. 2015, 5, 15179. [Google Scholar] [CrossRef] [Green Version]
- Song, J.J.; Zhao, S.J.; Fang, J.; Ma, D.; Liu, X.Q.; Chen, X.B.; Wang, Y.; Cheng, B.; Wang, Z. Foxp3 overexpression in tumor cells predicts poor survival in oral squamous cell carcinoma. BMC Cancer 2016, 16, 530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chuckran, C.A.; Cillo, A.R.; Moskovitz, J.; Overacre-Delgoffe, A.; Somasundaram, A.S.; Shan, F.; Magnon, G.C.; Kunning, S.R.; Abecassis, I.; Zureikat, A.H.; et al. Prevalence of intratumoral regulatory T cells expressing neuropilin-1 is associated with poorer outcomes in patients with cancer. Sci. Transl. Med. 2021, 13, eabf8495. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.; Kim, J.A.; Kim, Y.J.; Lee, H.W.; Kim, C.H.; Haam, S.; Kim, Y.S. A Neuropilin-1 Antagonist Exerts Antitumor Immunity by Inhibiting the Suppressive Function of Intratumoral Regulatory T Cells. Cancer Immunol. Res. 2020, 8, 46–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seminerio, I.; Descamps, G.; Dupont, S.; de Marrez, L.; Laigle, J.A.; Lechien, J.R.; Kindt, N.; Journe, F.; Saussez, S. Infiltration of FoxP3+Regulatory T Cells is a Strong and Independent Prognostic Factor in Head and Neck Squamous Cell Carcinoma. Cancers 2019, 11, 227. [Google Scholar] [CrossRef] [Green Version]
- Vari, F.; Arpon, D.; Keane, C.; Hertzberg, M.S.; Talaulikar, D.; Jain, S.; Cui, Q.Y.; Han, E.; Tobin, J.; Bird, R.; et al. Immune evasion via PD-1/PD-L1 on NK cells and monocyte/macrophages is more prominent in Hodgkin lymphoma than DLBCL. Blood 2018, 131, 1809–1819. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.; He, H.Y.; Liu, H.; Li, R.C.; Chen, Y.F.; Qi, Y.Y.; Jiang, Q.; Chen, L.L.; Zhang, P.P.; Zhang, H.; et al. Tumour-associated macrophages-derived CXCL8 determines immune evasion through autonomous PD-L1 expression in gastric cancer. Gut 2019, 68, 1764–1773. [Google Scholar] [CrossRef]
- Cassetta, L.; Pollard, J.W. Targeting macrophages: Therapeutic approaches in cancer. Nat. Rev. Drug Discov. 2018, 17, 887–904. [Google Scholar] [CrossRef]
- Hu, B.Y.; Wang, Z.W.; Zeng, H.; Qi, Y.Y.; Chen, Y.F.; Wang, T.; Wang, J.J.; Chang, Y.; Bai, Q.; Xia, Y.; et al. Blockade of DC-SIGNthorn Tumor-Associated Macrophages Reactivates Antitumor Immunity and Improves Immunotherapy in Muscle-Invasive Bladder Cancer. Cancer Res. 2020, 80, 1707–1719. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef]
- Chamoto, K.; Chowdhury, P.S.; Kumar, A.; Sonomura, K.; Matsuda, F.; Fagarasan, S.; Honjo, T. Mitochondrial activation chemicals synergize with surface receptor PD-1 blockade for T cell-dependent antitumor activity. Proc. Natl. Acad. Sci. USA 2017, 114, E761–E770. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; He, M.Y.; Zhu, L.F.; Yang, C.C.; Zhou, M.L.; Wang, Q.; Zhang, W.; Zheng, Y.Y.; Wang, D.M.; Xu, Z.Q.; et al. Tumor- associated macrophages correlate with the clinicopathological features and poor outcomes via inducing epithelial to mesenchymal transition in oral squamous cell carcinoma. J. Exp. Clin. Cancer Res. 2016, 35, 12. [Google Scholar] [CrossRef] [Green Version]
- Seminerio, I.; Kindt, N.; Descamps, G.; Bellier, J.; Lechien, J.R.; Mat, Q.; Pottier, C.; Journé, F.; Saussez, S. High infiltration of CD68+ macrophages is associated with poor prognoses of head and neck squamous cell carcinoma patients and is influenced by human papillomavirus. Oncotarget 2018, 9, 11046–11059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, M.; Buttner-Herold, M.; Hyckel, P.; Moebius, P.; Distel, L.; Ries, J.; Amann, K.; Neukam, F.W.; Wehrhan, F. Small oral squamous cell carcinomas with nodal lymphogenic metastasis show increased infiltration of M2 polarized macrophages—An immunohistochemical analysis. J. Cranio-Maxillofac. Surg. 2014, 42, 1087–1094. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.Z.; Yi, M.; Niu, M.K.; Mei, Q.; Wu, K.M. Myeloid-derived suppressor cells: An emerging target for anticancer immunotherapy. Mol. Cancer 2022, 21, 184. [Google Scholar] [CrossRef]
- Mastio, J.; Condamine, T.; Dominguez, G.; Kossenkov, A.V.; Donthireddy, L.; Veglia, F.; Lin, C.; Wang, F.; Fu, S.Y.; Zhou, J.; et al. Identification of monocyte-like precursors of granulocytes in cancer as a mechanism for accumulation of PMN-MDSCs. J. Exp. Med. 2019, 216, 2150–2169. [Google Scholar] [CrossRef] [Green Version]
- Dysthe, M.; Parihar, R. Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. In Tumor Microenvironment; Hematopoietic Cells—Part A; Springer: Berlin/Heidelberg, Germany, 2020; Volume 1224, pp. 117–140. [Google Scholar] [CrossRef]
- Park, M.J.; Lee, S.H.; Kim, E.K.; Lee, E.J.; Baek, J.A.; Park, S.H.; Kwok, S.K.; Cho, M.L. Interleukin-10 produced by myeloid-derived suppressor cells is critical for the induction of Tregs and attenuation of rheumatoid inflammation in mice. Sci. Rep. 2018, 8, 3753. [Google Scholar] [CrossRef] [Green Version]
- Kumar, V.; Patel, S.; Tcyganov, E.; Gabrilovich, D.I. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol. 2016, 37, 208–220. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.J.; Wei, G.W.; Cheng, W.A.; Dong, Z.Y.; Sun, H.; Lee, V.Y.; Cha, S.C.; Smith, D.L.; Kwak, L.W.; Qin, H. Targeting myeloid-derived suppressor cells for cancer immunotherapy. Cancer Immunol. Immunoth. 2018, 67, 1181–1195. [Google Scholar] [CrossRef]
- Bronte, V.; Brandau, S.; Chen, S.H.; Colombo, M.P.; Frey, A.B.; Greten, T.F.; Mandruzzato, S.; Murray, P.J.; Ochoa, A.; Ostrand-Rosenberg, S.; et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat. Commun. 2016, 7, 12150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, S.L.; Yang, Z.Z.; Hao, X.; Tang, W.X.; Ma, W.; Zong, H. Roles of HMGB1 in regulating myeloid-derived suppressor cells in the tumor microenvironment. Biomark. Res. 2020, 8, 21. [Google Scholar] [CrossRef] [PubMed]
- Younis, R.H.; Han, K.L.; Webb, T.J. Human Head and Neck Squamous Cell Carcinoma-Associated Semaphorin 4D Induces Expansion of Myeloid-Derived Suppressor Cells. J. Immunol. 2016, 196, 1419–1429. [Google Scholar] [CrossRef] [Green Version]
- Jayakumar, A.; Bothwell, A.L.M. Functional Diversity of Myeloid-Derived Suppressor Cells: The Multitasking Hydra of Cancer. J. Immunol. 2019, 203, 1095–1103. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.; Liang, F.; Thompson, E.A.; Vono, M.; Ols, S.; Lindgren, G.; Hassett, K.; Salter, H.; Ciaramella, G.; Lore, K. Rhesus Macaque Myeloid-Derived Suppressor Cells Demonstrate T Cell Inhibitory Functions and Are Transiently Increased after Vaccination. J. Immunol. 2018, 200, 286–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostrand-Rosenberg, S.; Fenselau, C. Myeloid-Derived Suppressor Cells: Immune-Suppressive Cells That Impair Antitumor Immunity and Are Sculpted by Their Environment. J. Immunol. 2018, 200, 422–431. [Google Scholar] [CrossRef] [Green Version]
- Peng, D.J.; Tanikawa, T.; Li, W.; Zhao, L.L.; Vatan, L.; Szeliga, W.; Wan, S.S.; Wei, S.; Wang, Y.; Liu, Y.; et al. Myeloid-Derived Suppressor Cells Endow Stem-like Qualities to Breast Cancer Cells through IL6/STAT3 and NO/NOTCH Cross-talk Signaling. Cancer Res. 2016, 76, 3156–3165. [Google Scholar] [CrossRef] [Green Version]
- Netherby, C.S.; Messmer, M.N.; Burkard-Mandel, L.; Colligan, S.; Miller, A.; Gomez, E.C.; Wang, J.M.; Nemeth, M.J.; Abrams, S.I. The Granulocyte Progenitor Stage Is a Key Target of IRF8-Mediated Regulation of Myeloid-Derived Suppressor Cell Production. J. Immunol. 2017, 198, 4129–4139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yaddanapudi, K.; Rendon, B.E.; Lamont, G.; Kim, E.J.; Al Rayyan, N.; Richie, J.; Albeituni, S.; Waigel, S.; Wise, A.; Mitchell, R.A. MIF Is Necessary for Late-Stage Melanoma Patient MDSC Immune Suppression and Differentiation. Cancer Immunol. Res. 2016, 4, 101–112. [Google Scholar] [CrossRef] [Green Version]
- Al-Khami, A.A.; Zheng, L.Q.; Del Valle, L.; Hossain, F.; Wyczechowska, D.; Zabaleta, J.; Sanchez, M.D.; Dean, M.J.; Rodriguez, P.C.; Ochoa, A.C. Exogenous lipid uptake induces metabolic and functional reprogramming of tumor-associated myeloid-derived suppressor cells. Oncoimmunology 2017, 6, e1344804. [Google Scholar] [CrossRef] [Green Version]
- Mojsilovic, S.; Mojsilovic, S.S.; Bjelica, S.; Santibanez, J.F. Transforming growth factor-beta1 and myeloid-derived suppressor cells: A cancerous partnership. Dev. Dyn. 2022, 251, 105–124. [Google Scholar] [CrossRef]
- Li, J.; Wang, L.; Chen, X.; Li, L.; Li, Y.; Ping, Y.; Huang, L.; Yue, D.; Zhang, Z.; Wang, F.; et al. CD39/CD73 upregulation on myeloid-derived suppressor cells via TGF-β-mTOR-HIF-1 signaling in patients with non-small cell lung cancer. Oncoimmunology 2017, 6, e1320011. [Google Scholar] [CrossRef] [Green Version]
- Wu, P.; Wu, D.; Ni, C.; Ye, J.; Chen, W.; Hu, G.; Wang, Z.; Wang, C.; Zhang, Z.; Xia, W.; et al. gamma delta T17 Cells Promote the Accumulation and Expansion of Myeloid-Derived Suppressor Cells in Human Colorectal Cancer. Immunity 2014, 40, 785–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.B.; He, T.; Liu, J.K.; Tai, J.J.; Wang, B.; Chen, Z.Y.; Quan, Z.X. Pan-cancer analysis reveals tumor-associated macrophage communication in the tumor microenvironment. Exp. Hematol. Oncol. 2021, 10, 31. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.S.; Cui, Y.D.; Yang, J.; Wang, S.J. Epithelial-mesenchymal transition: When tumor cells meet myeloid-derived suppressor cells. Biochim. Biophys. Acta Rev. Cancer 2021, 1876, 188564. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.S.; Ha, S.Y.; Kim, H.M.; Ahn, S.M.; Kang, M.S.; Kim, K.M.; Choi, M.G.; Lee, J.H.; Sohn, T.S.; Bae, J.M.; et al. The prognostic effects of tumor infiltrating regulatory T cells and myeloid derived suppressor cells assessed by multicolor flow cytometry in gastric cancer patients. Oncotarget 2016, 7, 7940–7951. [Google Scholar] [CrossRef] [Green Version]
- Chevalier, M.F.; Trabanelli, S.; Racle, J.; Salome, B.; Cesson, V.; Gharbi, D.; Bohner, P.; Domingos-Pereira, S.; Dartiguenave, F.; Fritschi, A.S.; et al. ILC2-modulated T cell-to-MDSC balance is associated with bladder cancer recurrence. J. Clin. Investig. 2017, 127, 2916–2929. [Google Scholar] [CrossRef] [Green Version]
- Safarzadeh, E.; Orangi, M.; Mohammadi, H.; Babaie, F.; Baradaran, B. Myeloid-derived suppressor cells: Important contributors to tumor progression and metastasis. J. Cell. Physiol. 2018, 233, 3024–3036. [Google Scholar] [CrossRef]
- Safarzadeh, E.; Hashemzadeh, S.; Duijf, P.H.G.; Mansoori, B.; Khaze, V.; Mohammadi, A.; Kazemi, T.; Yousefi, M.; Asadi, M.; Mohammadi, H.; et al. Circulating myeloid-derived suppressor cells: An independent prognostic factor in patients with breast cancer. J. Cell. Physiol. 2019, 234, 3515–3525. [Google Scholar] [CrossRef]
- Qiu, Y.Q.; Chen, T.; Hu, R.; Zhu, R.Y.; Li, C.J.; Ruan, Y.C.; Xie, X.L.; Li, Y.H. Next frontier in tumor immunotherapy: Macrophage-mediated immune evasion. Biomark. Res. 2021, 9, 72. [Google Scholar] [CrossRef]
- Zhu, S.L.; Yi, M.; Wu, Y.Z.; Dong, B.; Wu, K.M. Roles of tumor-associated macrophages in tumor progression: Implications on therapeutic strategies (vol 10, 60, 2021). Exp. Hematol. Oncol. 2022, 11, 4. [Google Scholar] [CrossRef]
- Gao, X.D.; Sui, H.S.; Zhao, S.; Gao, X.M.; Su, Y.P.; Qu, P. Immunotherapy Targeting Myeloid-Derived Suppressor Cells (MDSCs) in Tumor Microenvironment. Front. Immunol. 2021, 11, 585214. [Google Scholar] [CrossRef]
- Sui, H.S.; Dongye, S.Y.; Liu, X.C.; Xu, X.H.; Wang, L.; Jin, C.Q.; Yao, M.H.; Gong, Z.Q.; Jiang, D.; Zhang, K.X.; et al. Immunotherapy of targeting MDSCs in tumor microenvironment. Front. Immunol. 2022, 13, 990463. [Google Scholar] [CrossRef]
- Lang, S.; Bruderek, K.; Kaspar, C.; Höing, B.; Kanaan, O.; Dominas, N.; Hussain, T.; Droege, F.; Eyth, C.; Hadaschik, B.; et al. Clinical Relevance and Suppressive Capacity of Human Myeloid-Derived Suppressor Cell Subsets. Clin. Cancer Res. 2018, 24, 4834–4844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenkins, R.W.; Barbie, D.A.; Flaherty, K.T. Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer 2018, 118, 9–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Cicco, P.; Ercolano, G.; Ianaro, A. The New Era of Cancer Immunotherapy: Targeting Myeloid-Derived Suppressor Cells to Overcome Immune Evasion. Front. Immunol. 2020, 11, 1680. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.; Sharabi, A. Targeting myeloid-derived suppressor cells to enhance natural killer cell-based immunotherapy. Pharmacol. Ther. 2022, 235, 108114. [Google Scholar] [CrossRef]
- Zalfa, C.; Paust, S. Natural Killer Cell Interactions With Myeloid Derived Suppressor Cells in the Tumor Microenvironment and Implications for Cancer Immunotherapy. Front. Immunol. 2021, 12, 633205. [Google Scholar] [CrossRef]
- Carnevalli, L.S.; Ghadially, H.; Barry, S.T. Therapeutic Approaches Targeting the Natural Killer-Myeloid Cell Axis in the Tumor Microenvironment. Front. Immunol. 2021, 12, 633685. [Google Scholar] [CrossRef]
- Hou, A.H.; Hou, K.Y.; Huang, Q.B.; Lei, Y.J.; Chen, W.L. Targeting Myeloid-Derived Suppressor Cell, a Promising Strategy to Overcome Resistance to Immune Checkpoint Inhibitors. Front. Immunol. 2020, 11, 783. [Google Scholar] [CrossRef]
- Lei, Q.Y.; Wang, D.; Sun, K.; Wang, L.P.; Zhang, Y. Resistance Mechanisms of Anti-PD1/PDL1 Therapy in Solid Tumors. Front. Cell Dev. Biol. 2020, 8, 672. [Google Scholar] [CrossRef]
- Powell, D.R.; Huttenlocher, A. Neutrophils in the Tumor Microenvironment. Trends Immunol. 2016, 37, 41–52. [Google Scholar] [CrossRef] [Green Version]
- Shaul, M.E.; Fridlender, Z.G. Neutrophils as active regulators of the immune system in the tumor microenvironment. J. Leukoc. Biol. 2017, 102, 343–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosales, C.; Lowell, C.A.; Schnoor, M.; Uribe-Querol, E. Neutrophils: Their Role in Innate and Adaptive Immunity 2017. J. Immunol. Res. 2017, 2017, 9748345. [Google Scholar] [CrossRef] [PubMed]
- Chadwick, J.W.; Fine, N.; Khoury, W.; Tasevski, N.; Sun, C.X.; Boroumand, P.; Klip, A.; Glogauer, M. Tissue-specific murine neutrophil activation states in health and inflammation. J. Leukoc. Biol. 2021, 110, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Oveisi, M.; Shifman, H.; Fine, N.; Sun, C.X.; Glogauer, N.; Senadheera, D.; Glogauer, M. Novel Assay To Characterize Neutrophil Responses to Oral Biofilms. Infect. Immun. 2019, 87, e00790-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lakschevitz, F.S.; Hassanpour, S.; Rubin, A.; Fine, N.; Sun, C.X.; Glogauer, M. Identification of neutrophil surface marker changes in health and inflammation using high-throughput screening flow cytometry. Exp. Cell Res. 2016, 342, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Moses, K.; Brandau, S. Human neutrophils: Their role in cancer and relation to myeloid-derived suppressor cells. Semin. Immunol. 2016, 28, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Mishalian, I.; Granot, Z.; Fridlender, Z.G. The diversity of circulating neutrophils in cancer. Immunobiology 2017, 222, 82–88. [Google Scholar] [CrossRef]
- Jablonska, E.; Slodczyk, B.; Wawrusiewicz-Kurylonek, N.; Dziemianczyk-Pakiela, D.; Garley, M.; Ratajczak-Wrona, W.; Borys, J.; Kretowski, A. Comparison of B-Cell Activating Factor Expression in Neutrophils in Patients with Potentially Malignant Disorders and Patients with Cancer in the Same Site. Clin. Lab. 2016, 62, 1507–1514. [Google Scholar] [CrossRef]
- Bouma, G.; Ancliff, P.J.; Thrasher, A.J.; Burns, S.O. Recent advances in the understanding of genetic defects of neutrophil number and function. Br. J. Haematol. 2010, 151, 312–326. [Google Scholar] [CrossRef]
- Mishalian, I.; Bayuh, R.; Levy, L.; Zolotarov, L.; Michaeli, J.; Fridlender, Z.G. Tumor-associated neutrophils (TAN) develop pro-tumorigenic properties during tumor progression. Cancer Immunol. Immunoth. 2013, 62, 1745–1756. [Google Scholar] [CrossRef]
- Dumitru, C.A.; Lang, S.; Brandau, S. Modulation of neutrophil granulocytes in the tumor microenvironment: Mechanisms and consequences for tumor progression. Semin. Cancer Biol. 2013, 23, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Demkow, U. Neutrophil Extracellular Traps (NETs) in Cancer Invasion, Evasion and Metastasis. Cancers 2021, 13, 4495. [Google Scholar] [CrossRef] [PubMed]
- Masucci, M.T.; Minopoli, M.; Del Vecchio, S.; Carriero, M.V. The Emerging Role of Neutrophil Extracellular Traps (NETs) in Tumor Progression and Metastasis. Front. Immunol. 2020, 11, 1749. [Google Scholar] [CrossRef]
- Hu, W.X.; Lee, S.M.L.; Bazhin, A.V.; Guba, M.; Werner, J.; Niess, H. Neutrophil extracellular traps facilitate cancer metastasis: Cellular mechanisms and therapeutic strategies. J. Cancer Res. Clin. Oncol. 2022. [Google Scholar] [CrossRef] [PubMed]
- De Meo, M.L.; Spicer, J.D. The role of neutrophil extracellular traps in cancer progression and metastasis. Semin. Immunol. 2021, 57, 101595. [Google Scholar] [CrossRef]
- Long, W.; Chen, J.J.; Gao, C.; Lin, Z.; Xie, X.B.; Dai, H.L. Brief review on the roles of neutrophils in cancer development. J. Leukoc. Biol. 2021, 109, 407–413. [Google Scholar] [CrossRef]
- Ponzetta, A.; Mantovani, A.; Jaillon, S. Dissecting neutrophil complexity in cancer. Emerg. Top. Life Sci. 2017, 1, 457–470. [Google Scholar] [CrossRef] [PubMed]
- Kaltenmeier, C.; Simmons, R.L.; Tohme, S.; Yazdani, H.O. Neutrophil Extracellular Traps (NETs) in Cancer Metastasis. Cancers 2021, 13, 6131. [Google Scholar] [CrossRef]
- Gershkovitz, M.; Caspi, Y.; Fainsod-Levi, T.; Katz, B.; Michaeli, J.; Khawaled, S.; Lev, S.; Polyansky, L.; Shaul, M.E.; Sionov, R.V.; et al. TRPM2 Mediates Neutrophil Killing of Disseminated Tumor Cells. Cancer Res. 2018, 78, 2680–2690. [Google Scholar] [CrossRef] [Green Version]
- Tolle, F.; Umansky, V.; Utikal, J.; Kreis, S.; Brechard, S. Neutrophils in Tumorigenesis: Missing Targets for Successful Next Generation Cancer Therapies? Int. J. Mol. Sci. 2021, 22, 6744. [Google Scholar] [CrossRef]
- Sun, B.W.; Qin, W.T.; Song, M.M.; Liu, L.; Yu, Y.; Qi, X.X.; Sun, H. Neutrophil Suppresses Tumor Cell Proliferation via Fas/Fas Ligand Pathway Mediated Cell Cycle Arrested. Int. J. Biol. Sci. 2018, 14, 2103–2113. [Google Scholar] [CrossRef] [PubMed]
- Moonen, C.G.J.; Hirschfeld, J.; Cheng, L.L.; Chapple, I.L.C.; Loos, B.G.; Nicu, E.A. Oral Neutrophils Characterized: Chemotactic, Phagocytic, and Neutrophil Extracellular Trap (NET) Formation Properties. Front. Immunol. 2019, 10, 635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uribe-Querol, E.; Rosales, C. Neutrophils in Cancer: Two Sides of the Same Coin. J. Immunol. Res. 2015, 2015, 983698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lecot, P.; Sarabi, M.; Abrantes, M.P.; Mussard, J.; Koenderman, L.; Caux, C.; Bendriss-Vermare, N.; Michallet, M.C. Neutrophil Heterogeneity in Cancer: From Biology to Therapies. Front. Immunol. 2019, 10, 2155. [Google Scholar] [CrossRef] [Green Version]
- Grecian, R.; Whyte, M.K.B.; Walmsley, S.R. The role of neutrophils in cancer. Br. Med. Bull. 2018, 128, 5–14. [Google Scholar] [CrossRef] [Green Version]
- Rakic, A.; Beaudry, P.; Mahoney, D.J. The complex interplay between neutrophils and cancer. Cell Tissue Res. 2018, 371, 517–529. [Google Scholar] [CrossRef]
- Hurt, B.; Schulick, R.; Edil, B.; El Kasmi, K.C.; Barnett, C. Cancer-promoting mechanisms of tumor-associated neutrophils. Am. J. Surg. 2017, 214, 938–944. [Google Scholar] [CrossRef]
- Demers, M.; Wong, S.L.; Martinod, K.; Gallant, M.; Cabral, J.E.; Wang, Y.M.; Wagner, D.D. Priming of neutrophils toward NETosis promotes tumor growth. Oncoimmunology 2016, 5, e1134073. [Google Scholar] [CrossRef] [Green Version]
- He, M.; Peng, A.P.; Huang, X.Z.; Shi, D.C.; Wang, J.C.; Zhao, Q.Y.; Lin, H.B.; Kuang, D.M.; Ke, P.F.; Lao, X.M. Peritumoral stromal neutrophils are essential for c-Met-elicited metastasis in human hepatocellular carcinoma. Oncoimmunology 2016, 5, e1219828. [Google Scholar] [CrossRef] [Green Version]
- Swierczak, A.; Mouchemore, K.A.; Hamilton, J.A.; Anderson, R.L. Neutrophils: Important contributors to tumor progression and metastasis. Cancer Metastasis Rev. 2015, 34, 735–751. [Google Scholar] [CrossRef]
- Zhang, Y.T.; Liu, G.Q.; Sun, M.M.; Lu, X. Targeting and exploitation of tumor-associated neutrophils to enhance immunotherapy and drug delivery for cancer treatment. Cancer Biol. Med. 2020, 17, 32–43. [Google Scholar] [CrossRef]
- Condamine, T.; Dominguez, G.A.; Youn, J.I.; Kossenkov, A.V.; Mony, S.; Alicea-Torres, K.; Tcyganov, E.; Hashimoto, A.; Nefedova, Y.; Lin, C.; et al. Lectin-type oxidized LDL receptor-1 distinguishes population of human polymorphonuclear myeloid-derived suppressor cells in cancer patients. Sci. Immunol. 2016, 1, aaf8943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veglia, F.; Tyurin, V.A.; Blasi, M.; De Leo, A.; Kossenkov, A.V.; Donthireddy, L.; To, T.K.J.; Schug, Z.; Basu, S.; Wang, F.; et al. Fatty acid transport protein 2 reprograms neutrophils in cancer. Nature 2019, 569, 73–78. [Google Scholar] [CrossRef]
- Zhao, Y.; Rahmy, S.; Liu, Z.M.; Zhang, C.; Lu, X. Rational targeting of immunosuppressive neutrophils in cancer. Pharmacol. Ther. 2020, 212, 107556. [Google Scholar] [CrossRef]
- Jamieson, T.; Clarke, M.; Steele, C.W.; Samuel, M.S.; Neumann, J.; Jung, A.; Huels, D.; Olson, M.F.; Das, S.; Nibbs, R.J.; et al. Inhibition of CXCR2 profoundly suppresses inflammation-driven and spontaneous tumorigenesis. J. Clin. Investig. 2012, 122, 3127–3144. [Google Scholar] [CrossRef] [PubMed]
- Cupp, M.A.; Cariolou, M.; Tzoulaki, I.; Aune, D.; Evangelou, E.; Berlanga-Taylor, A.J. Neutrophil to lymphocyte ratio and cancer prognosis: An umbrella review of systematic reviews and meta-analyses of observational studies. BMC Med. 2020, 18, 360. [Google Scholar] [CrossRef]
- Howard, R.; Kanetsky, P.A.; Egan, K.M. Exploring the prognostic value of the neutrophil-to-lymphocyte ratio in cancer. Sci. Rep. 2019, 9, 19673. [Google Scholar] [CrossRef] [Green Version]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [Green Version]
- Kanzaki, R.; Pietras, K. Heterogeneity of cancer-associated fibroblasts: Opportunities for precision medicine. Cancer Sci. 2020, 111, 2708–2717. [Google Scholar] [CrossRef]
- Mao, X.Q.; Xu, J.; Wang, W.; Liang, C.; Hua, J.; Liu, J.; Zhang, B.; Meng, Q.C.; Yu, X.J.; Shi, S. Crosstalk between cancer-associated fibroblasts and immune cells in the tumor microenvironment: New findings and future perspectives. Mol. Cancer 2021, 20, 131. [Google Scholar] [CrossRef] [PubMed]
- Jia, C.C.; Wang, G.Y.; Wang, T.T.; Fu, B.S.; Zhang, Y.C.; Huang, L.; Deng, Y.N.; Chen, G.Z.; Wu, X.C.; Chen, J.N.; et al. Cancer-associated Fibroblasts induce epithelial-mesenchymal transition via the Transglutaminase 2-dependent IL-6/IL6R/STAT3 axis in Hepatocellular Carcinoma. Int. J. Biol. Sci. 2020, 16, 2542–2558. [Google Scholar] [CrossRef]
- Custodio, M.; Biddle, A.; Tavassoli, M. Portrait of a CAF: The story of cancer-associated fibroblasts in head and neck cancer. Oral Oncol. 2020, 110, 104972. [Google Scholar] [CrossRef]
- Wei, L.Y.; Lee, J.J.; Yeh, C.Y.; Yang, C.J.; Kok, S.H.; Ko, J.Y.; Tsai, F.C.; Chia, J.S. Reciprocal activation of cancer-associated fibroblasts and oral squamous carcinoma cells through CXCL1. Oral Oncol. 2019, 88, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Yamao, T.; Yamashita, Y.; Yamamura, K.; Nakao, Y.; Tsukamoto, M.; Nakagawa, S.; Okabe, H.; Hayashi, H.; Imai, K.; Baba, H. Cellular Senescence, Represented by Expression of Caveolin-1, in Cancer-Associated Fibroblasts Promotes Tumor Invasion in Pancreatic Cancer. Ann. Surg. Oncol. 2019, 26, 1552–1559. [Google Scholar] [CrossRef]
- Miyai, Y.; Esaki, N.; Takahashi, M.; Enomoto, A. Cancer-associated fibroblasts that restrain cancer progression: Hypotheses and perspectives. Cancer Sci. 2020, 111, 1047–1057. [Google Scholar] [CrossRef] [Green Version]
- Mizutani, Y.; Kobayashi, H.; Iida, T.; Asai, N.; Masamune, A.; Hara, A.; Esaki, N.; Ushida, K.; Mii, S.; Shiraki, Y.; et al. Meflin-Positive Cancer-Associated Fibroblasts Inhibit Pancreatic Carcinogenesis. Cancer Res. 2019, 79, 5367–5381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ichihara, R.; Shiraki, Y.; Mizutani, Y.; Iida, T.; Miyai, Y.; Esaki, N.; Kato, A.; Mii, S.; Ando, R.; Hayashi, M.; et al. Matrix remodeling-associated protein 8 is a marker of a subset of cancer-associated fibroblasts in pancreatic cancer. Pathol. Int. 2022, 72, 161–175. [Google Scholar] [CrossRef] [PubMed]
- Neuzillet, C.; Tijeras-Raballand, A.; Ragulan, C.; Cros, J.; Patil, Y.; Martinet, M.; Erkan, M.; Kleeff, J.; Wilson, J.; Apte, M.; et al. Inter- and intra-tumoural heterogeneity in cancer-associated fibroblasts of human pancreatic ductal adenocarcinoma. J. Pathol. 2019, 248, 51–65. [Google Scholar] [CrossRef] [Green Version]
- Gunaydin, G. CAFs Interacting With TAMs in Tumor Microenvironment to Enhance Tumorigenesis and Immune Evasion. Front. Oncol. 2021, 11, 668349. [Google Scholar] [CrossRef]
- Shiraki, Y.; Mii, S.; Esaki, N.; Enomoto, A. Possible disease-protective roles of fibroblasts in cancer and fibrosis and their therapeutic application. Nagoya J. Med. Sci. 2022, 84, 484–496. [Google Scholar] [CrossRef]
- Chen, P.Y.; Wei, W.F.; Wu, H.Z.; Fan, L.S.; Wang, W. Cancer-Associated Fibroblast Heterogeneity: A Factor That Cannot Be Ignored in Immune Microenvironment Remodeling. Front. Immunol. 2021, 12, 671595. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; McAndrews, K.M.; Kalluri, R. Clinical and therapeutic relevance of cancer-associated fibroblasts. Nat. Rev. Clin. Oncol. 2021, 18, 792–804. [Google Scholar] [CrossRef]
- Ji, Q.; Zhou, L.H.; Sui, H.; Yang, L.; Wu, X.N.; Song, Q.; Jia, R.; Li, R.X.; Sun, J.; Wang, Z.Y.; et al. Primary tumors release ITGBL1-rich extracellular vesicles to promote distal metastatic tumor growth through fibroblast-niche formation. Nat. Commun. 2020, 11, 1211. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.L.; Wang, W.; Lan, X.L.; Zeng, Z.C.; Liang, Y.S.; Yan, Y.R.; Song, F.Y.; Wang, F.F.; Zhu, X.H.; Liao, W.J.; et al. CAFs secreted exosomes promote metastasis and chemotherapy resistance by enhancing cell stemness and epithelial-mesenchymal transition in colorectal cancer. Mol. Cancer 2019, 18, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.F.; Sun, Y.Q.; Yu, W.W.; Yan, Y.; Qiao, M.; Jiang, R.Q.; Guan, W.B.; Wang, L.F. Downregulation of miRNA-214 in cancer-associated fibroblasts contributes to migration and invasion of gastric cancer cells through targeting FGF9 and inducing EMT. J. Exp. Clin. Cancer Res. 2019, 38, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, X.; Yan, M.; Wang, X.; Xu, Q.; Wang, X.N.; Zhu, X.Q.; Shi, J.B.; Li, Z.H.; Zhang, J.J.; Chen, W.T. Cancer-associated Fibroblast-derived IL-6 Promotes Head and Neck Cancer Progression via the Osteopontin-NF-kappa B Signaling Pathway. Theranostics 2018, 8, 921–940. [Google Scholar] [CrossRef]
- Kumar, D.; New, J.; Vishwakarma, V.; Joshi, R.; Enders, J.; Lin, F.C.; Dasari, S.; Gutierrez, W.R.; Leef, G.; Ponnurangam, S.; et al. Cancer-Associated Fibroblasts Drive Glycolysis in a Targetable Signaling Loop Implicated in Head and Neck Squamous Cell Carcinoma Progression. Cancer Res. 2018, 78, 3769–3782. [Google Scholar] [CrossRef] [Green Version]
- Yu, B.B.; Wu, K.L.; Wang, X.; Zhang, J.J.; Wang, L.Z.; Jiang, Y.Y.; Zhu, X.Q.; Chen, W.T.; Yan, M. Periostin secreted by cancer-associated fibroblasts promotes cancer stemness in head and neck cancer by activating protein tyrosine kinase 7. Cell Death Dis. 2018, 9, 1082. [Google Scholar] [CrossRef] [Green Version]
- Principe, S.; Mejia-Guerrero, S.; Ignatchenko, V.; Sinha, A.; Ignatchenko, A.; Shi, W.; Pereira, K.; Su, S.S.; Huang, S.H.; O’Sullivan, B.; et al. Proteomic Analysis of Cancer-Associated Fibroblasts Reveals a Paracrine Role for MFAP5 in Human Oral Tongue Squamous Cell Carcinoma. J. Proteome Res. 2018, 17, 2045–2059. [Google Scholar] [CrossRef]
- Huang, Y.H.; Chang, C.Y.; Kuo, Y.Z.; Fang, W.Y.; Kao, H.Y.; Tsai, S.T.; Wu, L.W. Cancer-associated fibroblast-derived interleukin-1 beta activates protumor C-C motif chemokine ligand 22 signaling in head and neck cancer. Cancer Sci. 2019, 110, 2783–2793. [Google Scholar] [CrossRef] [Green Version]
- Glentis, A.; Oertle, P.; Mariani, P.; Chikina, A.; El Marjou, F.; Attieh, Y.; Zaccarini, F.; Lae, M.; Loew, D.; Dingli, F.; et al. Cancer-associated fibroblasts induce metalloprotease-independent cancer cell invasion of the basement membrane. Nat. Commun. 2017, 8, 924. [Google Scholar] [CrossRef] [Green Version]
- Liao, Z.H.; Tan, Z.W.; Zhu, P.C.; Tan, N.S. Cancer-associated fibroblasts in tumor microenvironment—Accomplices in tumor malignancy. Cell. Immunol. 2019, 343, 103729. [Google Scholar] [CrossRef] [PubMed]
- Iida, T.; Mizutani, Y.; Esaki, N.; Ponik, S.M.; Burkel, B.M.; Weng, L.; Kuwata, K.; Masamune, A.; Ishihara, S.; Haga, H.; et al. Pharmacologic conversion of cancer-associated fibroblasts from a protumor phenotype to an antitumor phenotype improves the sensitivity of pancreatic cancer to chemotherapeutics. Oncogene 2022, 41, 2764–2777. [Google Scholar] [CrossRef]
- Su, S.C.; Chen, J.N.; Yao, H.R.; Liu, J.; Yu, S.B.; Lao, L.Y.; Wang, M.H.; Luo, M.L.; Xing, Y.; Chen, F.; et al. CD10(+) GPR77(+) Cancer-Associated Fibroblasts Promote Cancer Formation and Chemoresistance by Sustaining Cancer Stemness. Cell 2018, 172, 841–856. [Google Scholar] [CrossRef]
- Tao, L.L.; Huang, G.C.; Wang, R.; Pan, Y.; He, Z.Y.; Chu, X.Y.; Song, H.Z.; Chen, L.B. Cancer-associated fibroblasts treated with cisplatin facilitates chemoresistance of lung adenocarcinoma through IL-11/IL-11R/STAT3 signaling pathway. Sci. Rep. 2016, 6, 38408. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Li, X.J.; Zeng, C.; Liu, C.L.; Hao, Q.; Li, W.N.; Zhang, K.; Zhang, W.Q.; Wang, S.N.; Zhao, H.D.; et al. CD63(+)Cancer-Associated Fibroblasts Confer Tamoxifen Resistance to Breast Cancer Cells through Exosomal miR-22. Adv. Sci. 2020, 7, 2002518. [Google Scholar] [CrossRef]
- Zhang, H.Y.; Deng, T.; Liu, R.; Ning, T.; Yang, H.O.; Liu, D.Y.; Zhang, Q.M.; Lin, D.; Ge, S.H.; Bai, M.; et al. CAF secreted miR-522 suppresses ferroptosis and promotes acquired chemo-resistance in gastric cancer. Mol. Cancer 2020, 19, 43. [Google Scholar] [CrossRef] [Green Version]
- Zhai, J.; Shen, J.J.; Xie, G.P.; Wu, J.Q.; He, M.F.; Gao, L.L.; Zhang, Y.F.; Yao, X.Q.; Shen, L.Z. Cancer-associated fibroblasts-derived IL-8 mediates resistance to cisplatin in human gastric cancer. Cancer Lett. 2019, 454, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Richards, K.E.; Zeleniak, A.E.; Fishel, M.L.; Wu, J.; Littlepage, L.E.; Hill, R. Cancer-associated fibroblast exosomes regulate survival and proliferation of pancreatic cancer cells. Oncogene 2017, 36, 1770–1778. [Google Scholar] [CrossRef] [Green Version]
- Fiori, M.E.; Di Franco, S.; Villanova, L.; Bianca, P.; Stassi, G.; De Maria, R. Cancer-associated fibroblasts as abettors of tumor progression at the crossroads of EMT and therapy resistance. Mol. Cancer 2019, 18, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ford, K.; Hanley, C.J.; Mellone, M.; Szyndralewiez, C.; Heitz, F.; Wiesel, P.; Wood, O.; Machado, M.; Lopez, M.A.; Ganesan, A.P.; et al. NOX4 Inhibition Potentiates Immunotherapy by Overcoming Cancer-Associated Fibroblast-Mediated CD8 T-cell Exclusion from Tumors. Cancer Res. 2020, 80, 1846–1860. [Google Scholar] [CrossRef] [Green Version]
- Schmid, P.; Rugo, H.S.; Adams, S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Dieras, V.; Henschel, V.; Molinero, L.; Chui, S.Y.; et al. Atezolizumab plus nab-paclitaxel as first-line treatment for unresectable, locally advanced or metastatic triple-negative breast cancer (IMpassion130): Updated efficacy results from a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2020, 21, 44–59. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.K.; Zhou, B.; Zhuang, X.M.; Zhuang, P.L.; Zhang, D.M.; Chen, W.L. Cancer-associated fibroblasts confer cisplatin resistance of tongue cancer via autophagy activation. Biomed. Pharmacother. 2018, 97, 1341–1348. [Google Scholar] [CrossRef]
- Steinbichler, T.B.; Metzler, V.; Pritz, C.; Riechelmann, H.; Dudas, J. Tumor-associated fibroblast-conditioned medium induces CDDP resistance in HNSCC cells. Oncotarget 2016, 7, 2508–2518. [Google Scholar] [CrossRef] [Green Version]
- Ayuso, J.M.; Vitek, R.; Swick, A.D.; Skala, M.C.; Wisinski, K.B.; Kimple, R.J.; Lambert, P.F.; Beebe, D.J. Effects of culture method on response to EGFR therapy in head and neck squamous cell carcinoma cells. Sci. Rep. 2019, 9, 12480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cirillo, N.; Hassona, Y.; Celentano, A.; Lim, K.P.; Manchella, S.; Parkinson, E.K.; Prime, S.S. Cancer-associated fibroblasts regulate keratinocyte cell-cell adhesion via TGF-β-dependent pathways in genotype-specific oral cancer. Carcinogenesis 2017, 38, 76–85. [Google Scholar] [CrossRef] [Green Version]
- Habif, G.; Crinier, A.; Andre, P.; Vivier, E.; Narni-Mancinelli, E. Targeting natural killer cells in solid tumors. Cell. Mol. Immunol. 2019, 16, 415–422. [Google Scholar] [CrossRef]
- Stabile, H.; Fionda, C.; Gismondi, A.; Santoni, A. Role of Distinct natural Killer Cell Subsets in Anticancer Response. Front. Immunol. 2017, 8, 293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michel, T.; Poli, A.; Cuapio, A.; Briquemont, B.; Iserentant, G.; Ollert, M.; Zimmer, J. Human CD56(bright) NK Cells: An Update. J. Immunol. 2016, 196, 2923–2931. [Google Scholar] [CrossRef] [Green Version]
- Sojka, D.K.; Tian, Z.G.; Yokoyama, W.M. Tissue-resident natural killer cells and their potential diversity. Semin. Immunol. 2014, 26, 127–131. [Google Scholar] [CrossRef] [Green Version]
- Lugthart, G.; Melsen, J.E.; Vervat, C.; van Ostaijen-ten Dam, M.M.; Corver, W.E.; Roelen, D.L.; van Bergen, J.; van Tol, M.J.D.; Lankester, A.C.; Schilham, M.W. Human Lymphoid Tissues Harbor a Distinct CD69(+)CXCR6(+) NK Cell Population. J. Immunol. 2016, 197, 78–84. [Google Scholar] [CrossRef] [Green Version]
- Melsen, J.E.; Lugthart, G.; Lankester, A.C.; Schilham, M.W. Human Circulating and Tissue-Resident CD56(bright) Natural Killer Cell Populations. Front. Immunol. 2016, 7, 262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glasner, A.; Levi, A.; Enk, J.; Isaacson, B.; Viukov, S.; Orlanski, S.; Scope, A.; Neuman, T.; Enk, C.D.; Hanna, J.H.; et al. NKp46 Receptor-Mediated Interferon-gamma Production by Natural Killer Cells Increases Fibronectin 1 to Alter Tumor Architecture and Control Metastasis. Immunity 2018, 48, 107–119. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Wang, X.; Jin, T.; Tian, Y.; Dai, C.; Widarma, C.; Song, R.; Xu, F. Immune checkpoint molecules in natural killer cells as potential targets for cancer immunotherapy. Signal Transduct. Target. Ther. 2020, 5, 250. [Google Scholar] [CrossRef]
- Santos, E.M.; de Matos, F.R.; de Morais, E.F.; Galvao, H.C.; Freitas, R.D. Evaluation of Cd8+and natural killer cells defense in oral and oropharyngeal squamous cell carcinoma. J. Cranio-Maxillofac. Surg. 2019, 47, 676–681. [Google Scholar] [CrossRef] [PubMed]
- He, X.H.; Xiao, Y.L.; Liu, S.; Deng, R.Y.; Li, Z.M.; Zhu, X.Y. Predicting the Immune Microenvironment and Prognosis with a NETosis-Related lncRNA Signature in Head and Neck Squamous Cell Carcinoma. Biomed. Res. Int. 2022, 2022, 3191474. [Google Scholar] [CrossRef]
- Wang, J.; Bian, Q.J.; Liu, J.L.; Moming, A. Identification and in vitro validation of prognostic lncRNA signature in head and neck squamous cell carcinoma. Bioengineered 2021, 12, 10049–10062. [Google Scholar] [CrossRef]
- Zhu, W.Y.; Ye, Z.P.; Chen, L.; Liang, H.F.; Cai, Q. A pyroptosis-related lncRNA signature predicts prognosis and immune microenvironment in head and neck squamous cell carcinoma. Int. Immunopharmacol. 2021, 101, 108268. [Google Scholar] [CrossRef]
- Yang, F.; Zhou, L.Q.; Yang, H.W.; Wang, Y.J. Nine-gene signature and nomogram for predicting survival in patients with head and neck squamous cell carcinoma. Front. Genet. 2022, 13, 927614. [Google Scholar] [CrossRef]
- Krijgsman, D.; Hokland, M.; Kuppen, P.J.K. The Role of Natural Killer T Cells in Cancer-A Phenotypical and Functional Approach. Front. Immunol. 2018, 9, 367. [Google Scholar] [CrossRef]
- Ingram, Z.; Madan, S.; Merchant, J.; Carter, Z.; Gordon, Z.; Carey, G.; Webb, T.J. Targeting Natural Killer T Cells in Solid Malignancies. Cells 2021, 10, 1329. [Google Scholar] [CrossRef] [PubMed]
- Terabe, M.; Berzofsky, J.A. Tissue-Specific Roles of NKT Cells in Tumor Immunity. Front. Immunol. 2018, 9, 1838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneiders, F.L.; de Bruin, R.C.G.; van den Eertwegh, A.J.M.; Scheper, R.J.; Leemans, C.R.; Brakenhoff, R.H.; Langendijk, J.A.; Verheul, H.M.W.; de Gruijl, T.D.; Molling, J.W.; et al. Circulating Invariant Natural Killer T-Cell Numbers Predict Outcome in Head and Neck Squamous Cell Carcinoma: Updated Analysis With 10-Year Follow-Up. J. Clin. Oncol. 2012, 30, 567–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, A.; Lukacs, J.D.; Johnston, B. The Current Landscape of NKT Cell Immunotherapy and the Hills Ahead. Cancers 2021, 13, 5174. [Google Scholar] [CrossRef]
- Denaro, N.; Merlano, M.; Garrone, O. In reply to immune desert in head and neck squamous cell carcinoma: A review of challenges and opportunities for modulating the tumor immune microenvironment. Oral Oncol. 2022, 124, 105640. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.; Chhatar, S.; Mishra, A.; Lal, G. Natural killer T cell activation increases iNOS(+)CD206(-) M1 macrophage and controls the growth of solid tumor. J. Immunother. Cancer 2019, 7, 208. [Google Scholar] [CrossRef] [Green Version]
- Hung, J.T.; Huang, J.R.; Yu, A.L. Tailored design of NKT-stimulatory glycolipids for polarization of immune responses. J. Biomed. Sci. 2017, 24, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kratzmeier, C.; Singh, S.; Asiedu, E.B.; Webb, T.J. Current Developments in the Preclinical and Clinical use of Natural Killer T cells. Biodrugs 2023, 37, 57–71. [Google Scholar] [CrossRef]
- Mele, D.; Pessino, G.; Trisolini, G.; Luchena, A.; Benazzo, M.; Morbini, P.; Mantovani, S.; Oliviero, B.; Mondelli, M.U.; Varchetta, S. Impaired intratumoral natural killer cell function in head and neck carcinoma. Front. Immunol. 2022, 13, 997806. [Google Scholar] [CrossRef]
- Groth, A.; Kloss, S.; von Strandmann, E.P.; Koehl, U.; Koch, J. Mechanisms of Tumor and Viral Immune Escape from Natural Killer Cell-Mediated Surveillance. J. Innate Immun. 2011, 3, 344–354. [Google Scholar] [CrossRef]
- Charap, A.J.; Enokida, T.; Brody, R.; Sfakianos, J.; Miles, B.; Bhardwaj, N.; Horowitz, A. Landscape of natural killer cell activity in head and neck squamous cell carcinoma. J. Immunother. Cancer 2020, 8, e001523. [Google Scholar] [CrossRef]
- Moreno-Nieves, U.Y.; Tay, J.K.; Saumyaa, S.; Horowitz, N.B.; Shin, J.H.; Mohammad, I.A.; Luca, B.; Mundy, D.C.; Gulati, G.S.; Bedi, N.; et al. Landscape of innate lymphoid cells in human head and neck cancer reveals divergent NK cell states in the tumor microenvironment. Proc. Natl. Acad. Sci. USA 2021, 118, e2101169118. [Google Scholar] [CrossRef] [PubMed]
- Bisheshar, S.K.; De Ruiter, E.J.; Devriese, L.A.; Willems, S.M. The prognostic role of NK cells and their ligands in squamous cell carcinoma of the head and neck: A systematic review and meta-analysis. Oncoimmunology 2020, 9, 1747345. [Google Scholar] [CrossRef] [Green Version]
- Buckle, I.; Guillerey, C. Inhibitory Receptors and Immune Checkpoints Regulating Natural Killer Cell Responses to Cancer. Cancers 2021, 13, 4263. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Arooj, S.; Wang, H. NK Cell-Based Immune Checkpoint Inhibition. Front. Immunol. 2020, 11, 167. [Google Scholar] [CrossRef] [PubMed]
- Morvan, M.G.; Lanier, L.L. NK cells and cancer: You can teach innate cells new tricks. Nat. Rev. Cancer 2016, 16, 7–19. [Google Scholar] [CrossRef]
- Muntasell, A.; Ochoa, M.C.; Cordeiro, L.; Berraondo, P.; de Cerio, A.L.D.; Cabo, M.; Lopez-Botet, M.; Melero, I. Targeting NK-cell checkpoints for cancer immunotherapy. Curr. Opin. Immunol. 2017, 45, 73–81. [Google Scholar] [CrossRef]
- Ferris, R.L. Immunology and Immunotherapy of Head and Neck Cancer. J. Clin. Oncol. 2015, 33, 3293–3304. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Bi, J.C.; Zheng, X.D.; Chen, Y.Y.; Wang, H.; Wu, W.Y.; Wang, Z.G.; Wu, Q.; Peng, H.; Wei, H.M.; et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat. Immunol. 2018, 19, 723–732. [Google Scholar] [CrossRef]
- Jung, E.K.; Chu, T.H.; Vo, M.C.; Nguyen, H.P.Q.; Lee, D.H.; Lee, J.K.; Lim, S.C.; Jung, S.H.; Yoon, T.M.; Yoon, M.S.; et al. Natural killer cells have a synergistic anti-tumor effect in combination with chemoradiotherapy against head and neck cancer. Cytotherapy 2022, 24, 905–915. [Google Scholar] [CrossRef]
- Wang, H.T.; Nan, S.J.; Wang, Y.; Xu, C.B. CDX2 enhances natural killer cell-mediated immunotherapy against head and neck squamous cell carcinoma through up-regulating CXCL14. J. Cell. Mol. Med. 2021, 25, 4596–4607. [Google Scholar] [CrossRef] [PubMed]
- Luz-Crawford, P.; Kurte, M.; Bravo-Alegria, J.; Contreras, R.; Nova-Lamperti, E.; Tejedor, G.; Noel, D.; Jorgensen, C.; Figueroa, F.; Djouad, F.; et al. Mesenchymal stem cells generate a CD4(+)CD25(+) Foxp3(+) regulatory T cell population during the differentiation process of Th1 and Th17 cells. Stem Cell Res. Ther. 2013, 4, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kesselring, R.; Thiel, A.; Pries, R.; Trenkle, T.; Wollenberg, B. Human Th17 cells can be induced through head and neck cancer and have a functional impact on HNSCC development. Br. J. Cancer 2010, 103, 1245–1254. [Google Scholar] [CrossRef] [Green Version]
- Partlova, S.; Boucek, J.; Kloudova, K.; Lukesova, E.; Zabrodsky, M.; Grega, M.; Fucikova, J.; Truxova, I.; Tachezy, R.; Spisek, R.; et al. Distinct patterns of intratumoral immune cell infiltrates in patients with HPV-associated compared to non-virally induced head and neck squamous cell carcinoma. Oncoimmunology 2015, 4, e965570. [Google Scholar] [CrossRef] [PubMed]
- Gameiro, S.F.; Ghasemi, F.; Barrett, J.W.; Koropatnick, J.; Nichols, A.C.; Mymryk, J.S.; Vareki, S.M. Treatment-naive HPV plus head and neck cancers display a T-cell-inflamed phenotype distinct from their HPV- counterparts that has implications for immunotherapy. Oncoimmunology 2018, 7, e1498439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.H.; Yan, B.Q.; Lou, H.H.; Shen, Z.J.; Tong, F.J.; Zhai, A.X.; Wei, L.L.; Zhang, F.M. Immunological network analysis in HPV associated head and neck squamous cancer and implications for disease prognosis. Mol. Immunol. 2018, 96, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Wagner, S.; Wittekindt, C.; Reuschenbach, M.; Hennig, B.; Thevarajah, M.; Wurdemann, N.; Prigge, E.S.; Doeberitz, M.V.; Dreyer, T.; Gattenlohner, S.; et al. CD56-positive lymphocyte infiltration in relation to human papillomavirus association and prognostic significance in oropharyngeal squamous cell carcinoma. Int. J. Cancer 2016, 138, 2263–2273. [Google Scholar] [CrossRef]
- Shamseddine, A.A.; Burman, B.; Lee, N.Y.; Zamarin, D.; Riaz, N. Tumor Immunity and Immunotherapy for HPV-Related Cancers. Cancer Discov. 2021, 11, 1896–1912. [Google Scholar] [CrossRef]
- Zhou, X.; Su, Y.X.; Lao, X.M.; Liang, Y.J.; Liao, G.Q. CD19(+)IL-10(+) regulatory B cells affect survival of tongue squamous cell carcinoma patients and induce resting CD4(+) T cells to CD4(+)Foxp3(+) regulatory T cells. Oral Oncol. 2016, 53, 27–35. [Google Scholar] [CrossRef]
- Kulasinghe, A.; Taheri, T.; O’Byrne, K.; Hughes, B.G.M.; Kenny, L.; Punyadeera, C. Highly Multiplexed Digital Spatial Profiling of the Tumor Microenvironment of Head and Neck Squamous Cell Carcinoma Patients. Front. Oncol. 2021, 10, 607349. [Google Scholar] [CrossRef]
- Solomon, B.; Young, R.J.; Bressel, M.; Urban, D.; Hendry, S.; Thai, A.; Angel, C.; Haddad, A.; Kowanetz, M.; Fua, T.; et al. Prognostic Significance of PD-L1(+) and CD8(+) Immune Cells in HPV+ Oropharyngeal Squamous Cell Carcinoma. Cancer Immunol. Res. 2018, 6, 295–304. [Google Scholar] [CrossRef] [Green Version]
- Hladikova, K.; Koucky, V.; Boucek, J.; Laco, J.; Grega, M.; Hodek, M.; Zabrodsky, M.; Vosmik, M.; Rozkosova, K.; Vosmikova, H.; et al. Tumor-infiltrating B cells affect the progression of oropharyngeal squamous cell carcinoma via cell-to-cell interactions with CD8(+) T cells. J. Immunother. Cancer 2019, 7, 261. [Google Scholar] [CrossRef]
- Santegoets, S.J.; Duurland, C.L.; Jordanova, E.S.; van Ham, J.J.; Ehsan, I.; van Egmond, S.L.; Welters, M.J.P.; van der Burg, S.H. Tbet-positive regulatory T cells accumulate in oropharyngeal cancers with ongoing tumor-specific type 1T cell responses. J. Immunother. Cancer 2019, 7, 14. [Google Scholar] [CrossRef]
- Succaria, F.; Kvistborg, P.; Stein, J.E.; Engle, E.L.; McMiller, T.L.; Rooper, L.M.; Thompson, E.; Berger, A.E.; van den Brekel, M.; Zuur, C.L.; et al. Characterization of the tumor immune microenvironment in human papillomavirus-positive and -negative head and neck squamous cell carcinomas. Cancer Immunol. Immunother. 2021, 70, 1227–1237. [Google Scholar] [CrossRef]
- Ruangritchankul, K.; Sandison, A.; Warburton, F.; Guerrero-Urbano, T.; Ferreira, M.R.; Lei, M.; Thavaraj, S. Clinical evaluation of tumour-infiltrating lymphocytes as a prognostic factor in patients with human papillomavirus-associated oropharyngeal squamous cell carcinoma. Histopathology 2019, 75, 146–150. [Google Scholar] [CrossRef]
- Corredor, G.; Toro, P.; Koyuncu, C.; Lu, C.; Buzzy, C.; Bera, K.; Fu, P.F.; Mehrad, M.; Ely, K.A.; Mokhtari, M.; et al. An Imaging Biomarker of Tumor-Infiltrating Lymphocytes to Risk-Stratify Patients With HPV-Associated Oropharyngeal Cancer. J. Natl. Cancer Inst. 2022, 114, 609–617. [Google Scholar] [CrossRef]
- Faraji, F.; Fung, N.; Zaidi, M.; Gourin, C.C.; Eisele, D.W.; Rooper, L.M.; Fakhry, C. Tumor-infiltrating lymphocyte quantification stratifies early-stage human papillomavirus oropharynx cancer prognosis. Laryngoscope 2020, 130, 930–938. [Google Scholar] [CrossRef]
- Al-Sahaf, S.; Hendawi, N.B.; Ollington, B.; Bolt, R.; Ottewell, P.D.; Hunter, K.D.; Murdoch, C. Increased Abundance of Tumour-Associated Neutrophils in HPV-Negative Compared to HPV-Positive Oropharyngeal Squamous Cell Carcinoma Is Mediated by IL-1R Signalling. Front. Oral Health 2021, 2, 604565. [Google Scholar] [CrossRef]
- Snietura, M.; Brewczynski, A.; Kopec, A.; Rutkowski, T. Infiltrates of M2-Like Tumour-Associated Macrophages Are Adverse Prognostic Factor in Patients with Human Papillomavirus-Negative but Not in Human Papillomavirus-Positive Oropharyngeal Squamous Cell Carcinoma. Pathobiology 2020, 87, 75–86. [Google Scholar] [CrossRef]
- Al-Sahaf, S.; Hunter, K.D.; Bolt, R.; Ottewell, P.D.; Murdoch, C. The IL-1/IL-1R axis induces greater fibroblast-derived chemokine release in human papillomavirus-negative compared to positive oropharyngeal cancer. Int. J. Cancer 2019, 144, 334–344. [Google Scholar] [CrossRef] [Green Version]
- Matlung, S.E.; Wilhelmina van Kempen, P.M.; Bovenschen, N.; van Baarle, D.; Willems, S.M. Differences in T-cell infiltrates and survival between HPV+ and HPV- oropharyngeal squamous cell carcinoma. Future Sci. OA 2016, 2, FSO88. [Google Scholar] [CrossRef] [Green Version]
- Ludwig, S.; Sharma, P.; Theodoraki, M.N.; Pietrowska, M.; Yerneni, S.S.; Lang, S.; Ferrone, S.; Whiteside, T.L. Molecular and Functional Profiles of Exosomes From HPV(+) and HPV(-) Head and Neck Cancer Cell Lines. Front. Oncol. 2018, 8, 445. [Google Scholar] [CrossRef]
- Wang, J.; Sun, H.; Zeng, Q.; Guo, X.J.; Wang, H.; Liu, H.H.; Dong, Z.Y. HPV-positive status associated with inflamed immune microenvironment and improved response to anti-PD-1 therapy in head and neck squamous cell carcinoma. Sci. Rep. 2019, 9, 13404. [Google Scholar] [CrossRef] [Green Version]
- Baruah, P.; Bullenkamp, J.; Wilson, P.O.G.; Lee, M.; Kaski, J.C.; Dumitriu, I.E. TLR9 Mediated Tumor-Stroma Interactions in Human Papilloma Virus (HPV)-Positive Head and Neck Squamous Cell Carcinoma Up-Regulate PD-L1 and PD-L2. Front. Immunol. 2019, 10, 1644. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.M.; Zhu, G.C.; Maroun, C.A.; Wu, I.X.Y.; Huang, D.H.; Seiwert, T.Y.; Liu, Y.; Mandal, R.; Zhang, X. Programmed Death-1/Programmed Death-Ligand 1-Axis Blockade in Recurrent or Metastatic Head and Neck Squamous Cell Carcinoma Stratified by Human Papillomavirus Status: A Systematic Review and Meta-Analysis. Front. Immunol. 2021, 12, 645170. [Google Scholar] [CrossRef]
- Botticelli, A.; Cirillo, A.; Strigari, L.; Valentini, F.; Cerbelli, B.; Scagnoli, S.; Cerbelli, E.; Zizzari, I.G.; Rocca, C.D.; D’Amati, G.; et al. Anti-PD-1 and Anti-PD-L1 in Head and Neck Cancer: A Network Meta-Analysis. Front. Immunol. 2021, 12, 705096. [Google Scholar] [CrossRef]
- Saada-Bouzid, E.; Peyrade, F.; Guigay, J. Immunotherapy in recurrent and or metastatic squamous cell carcinoma of the head and neck. Curr. Opin. Oncol. 2019, 31, 146–151. [Google Scholar] [CrossRef]
- Zhu, P.; Wang, Y.W.; Zhang, W.D.; Liu, X. Anti-PD1/PD-L1 monotherapy vs standard of care in patients with recurrent or metastatic head and neck squamous cell carcinoma A meta-analysis of randomized controlled trials. Medicine 2021, 100, e24339. [Google Scholar] [CrossRef]
- Clarke, E.; Eriksen, J.G.; Barrett, S. The effects of PD-1/PD-L1 checkpoint inhibitors on recurrent/metastatic head and neck squamous cell carcinoma: A critical review of the literature and meta-analysis. Acta Oncol. 2021, 60, 1534–1542. [Google Scholar] [CrossRef]
- Horton, J.D.; Knochelmann, H.M.; Day, T.A.; Paulos, C.M.; Neskey, D.M. Immune Evasion by Head and Neck Cancer: Foundations for Combination Therapy. Trends Cancer 2019, 5, 208–232. [Google Scholar] [CrossRef]
- Chakraborty, P.; Karmakar, T.; Arora, N.; Mukherjee, G. Immune and genomic signatures in oral (head and neck) cancer. Heliyon 2018, 4, e00880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galizia, D.; Minei, S.; Maldi, E.; Chila, G.; Polidori, A.; Merlano, M.C. How Risk Factors Affect Head and Neck Squamous Cell Carcinoma (HNSCC) Tumor Immune Microenvironment (TIME): Their Influence on Immune Escape Mechanisms and Immunotherapy Strategy. Biomedicines 2022, 10, 2498. [Google Scholar] [CrossRef] [PubMed]
- Boras, V.V.; Fucic, A.; Virag, M.; Gabric, D.; Blivajs, I.; Tomasovic-Loncaric, C.; Rakusic, Z.; Bisof, V.; Le Novere, N.; Vrdoljak, D.V. Significance of stroma in biology of oral squamous cell carcinoma. Tumori J. 2018, 104, 9–14. [Google Scholar] [CrossRef]
- Seliger, B.; Massa, C.; Yang, B.; Bethmann, D.; Kappler, M.; Eckert, A.W.; Wickenhauser, C. Immune Escape Mechanisms and Their Clinical Relevance in Head and Neck Squamous Cell Carcinoma. Int. J. Mol. Sci. 2020, 21, 7032. [Google Scholar] [CrossRef]
- Tang, S.; Ning, Q.; Yang, L.; Mo, Z.C.; Tang, S.S. Mechanisms of immune escape in the cancer immune cycle. Int. Immunopharmacol. 2020, 86, 106700. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Nathanson, T.; Rizvi, H.; Creelan, B.C.; Sanchez-Vega, F.; Ahuja, A.; Ni, A.; Novik, J.B.; Mangarin, L.M.B.; Abu-Akeel, M.; et al. Genomic Features of Response to Combination Immunotherapy in Patients with Advanced Non-Small-Cell Lung Cancer. Cancer Cell 2018, 33, 843–852.e4. [Google Scholar] [CrossRef] [Green Version]
- McGranahan, N.; Furness, A.J.S.; Rosenthal, R.; Ramskov, S.; Lyngaa, R.; Saini, S.K.; Jamal-Hanjani, M.; Wilson, G.A.; Birkbak, N.J.; Hiley, C.T.; et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016, 351, 1463–1469. [Google Scholar] [CrossRef] [Green Version]
- Ready, N.; Hellmann, M.D.; Awad, M.M.; Otterson, G.A.; Gutierrez, M.; Gainor, J.F.; Borghaei, H.; Jolivet, J.; Horn, L.; Mates, M.; et al. First-Line Nivolumab Plus Ipilimumab in Advanced Non-Small-Cell Lung Cancer (CheckMate 568): Outcomes by Programmed Death Ligand 1 and Tumor Mutational Burden as Biomarkers. J. Clin. Oncol. 2019, 37, 992–1000. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Callahan, M.K.; Awad, M.M.; Calvo, E.; Ascierto, P.A.; Atmaca, A.; Rizvi, N.A.; Hirsch, F.R.; Selvaggi, G.; Szustakowski, J.D.; et al. Tumor Mutational Burden and Efficacy of Nivolumab Monotherapy and in Combination with Ipilimumab in Small-Cell Lung Cancer. Cancer Cell 2018, 33, 853–861.e4. [Google Scholar] [CrossRef] [Green Version]
- Yarchoan, M.; Johnson, B.A.; Lutz, E.R.; Laheru, D.A.; Jaffee, E.M. Targeting neoantigens to augment antitumour immunity. Nat. Rev. Cancer 2017, 17, 209–222. [Google Scholar] [CrossRef]
- Anagnostou, V.; Smith, K.N.; Forde, P.M.; Niknafs, N.; Bhattacharya, R.; White, J.; Zhang, T.; Adleff, V.; Phallen, J.; Wali, N.; et al. Evolution of Neoantigen Landscape during Immune Checkpoint Blockade in Non-Small Cell Lung Cancer. Cancer Dis. 2017, 7, 264–276. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.P.; Vale, N.R.; Zacharakis, N.; Krishna, S.; Yu, Z.Y.; Gasmi, B.; Gartner, J.J.; Sindiri, S.; Malekzadeh, P.; Deniger, D.C.; et al. Adoptive Cellular Therapy with Autologous Tumor-Infiltrating Lymphocytes and T-cell Receptor- Engineered T Cells Targeting Common p53 Neoantigens in Human Solid Tumors. Cancer Immunol. Res. 2022, 10, 932–946. [Google Scholar] [CrossRef]
- Robbins, P.F. Tumor-Infiltrating Lymphocyte Therapy and Neoantigens. Cancer J. 2017, 23, 138–143. [Google Scholar] [CrossRef]
- Kitamura, H.; Ohno, Y.; Toyoshima, Y.; Ohtake, J.; Homma, S.; Kawamura, H.; Takahashi, N.; Taketomi, A. Interleukin-6/STAT3 signaling as a promising target to improve the efficacy of cancer immunotherapy. Cancer Sci. 2017, 108, 1947–1952. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Hao, S.N.; Zhao, Z.H.; Liu, J.; Shao, Q.Q.; Wang, F.; Sun, D.; He, Y.; Gao, W.J.; Mao, H.T. Interleukin 35: Inhibitory regulator in monocyte-derived dendritic cell maturation and activation. Cytokine 2018, 108, 43–52. [Google Scholar] [CrossRef]
- Veglia, F.; Tyurin, V.A.; Mohammadyani, D.; Blasi, M.; Duperret, E.K.; Donthireddy, L.; Hashimoto, A.; Kapralov, A.; Amoscato, A.; Angelini, R.; et al. Lipid bodies containing oxidatively truncated lipids block antigen cross-presentation by dendritic cells in cancer. Nat. Commun. 2017, 8, 2122. [Google Scholar] [CrossRef] [Green Version]
- Ning, Y.L.; Shen, K.; Wu, Q.Y.; Sun, X.; Bai, Y.; Xie, Y.W.; Pan, J.; Qi, C.J. Tumor exosomes block dendritic cells maturation to decrease the T cell immune response. Immunol. Lett. 2018, 199, 36–43. [Google Scholar] [CrossRef]
- Toyoshima, Y.; Kitamura, H.; Xiang, H.H.; Ohno, Y.; Homma, S.; Kawamura, H.; Takahashi, N.; Kamiyama, T.; Tanino, M.; Taketomi, A. IL6 Modulates the Immune Status of the Tumor Microenvironment to Facilitate Metastatic Colonization of Colorectal Cancer Cells. Cancer Immunol. Res. 2019, 7, 1944–1957. [Google Scholar] [CrossRef] [Green Version]
- Ohno, Y.; Toyoshima, Y.; Yurino, H.; Monma, N.; Xiang, H.H.; Sumida, K.; Kaneumi, S.; Terada, S.; Hashimoto, S.; Ikeo, K.; et al. Lack of interleukin-6 in the tumor microenvironment augments type-1 immunity and increases the efficacy of cancer immunotherapy. Cancer Sci. 2017, 108, 1959–1966. [Google Scholar] [CrossRef] [Green Version]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef] [Green Version]
- Boutros, C.; Tarhini, A.; Routier, E.; Lambotte, O.; Ladurie, F.L.; Carbonnel, F.; Izzeddine, H.; Marabelle, A.; Champiat, S.; Berdelou, A.; et al. Safety profiles of anti-CTLA-4 and anti-PD-1 antibodies alone and in combination. Nat. Rev. Clin. Oncol. 2016, 13, 473–486. [Google Scholar] [CrossRef]
- Dianat-Moghadam, H.; Sharifi, M.; Salehi, R.; Keshavarz, M.; Shahgolzari, M.; Amoozgar, Z. Engaging stemness improves cancer immunotherapy. Cancer Lett. 2023, 554, 216007. [Google Scholar] [CrossRef]
- Zhao, Y.H.; Yang, W.; Huang, Y.Y.; Cui, R.J.; Li, X.Y.; Li, B.J. Evolving Roles for Targeting CTLA-4 in Cancer Immunotherapy. Cell. Physiol. Biochem. 2018, 47, 721–734. [Google Scholar] [CrossRef]
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Disc. 2018, 8, 1069–1086. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Subudhi, S.K.; Blando, J.; Scutti, J.; Vence, L.; Wargo, J.; Allison, J.P.; Ribas, A.; Sharma, P. Anti-CTLA-4 Immunotherapy Does Not Deplete FOXP3(+) Regulatory T Cells (Tregs) in Human Cancers. Clin. Cancer Res. 2019, 25, 1233–1238. [Google Scholar] [CrossRef] [Green Version]
- Du, X.X.; Tang, F.; Liu, M.Y.; Su, J.J.; Zhang, Y.; Wu, W.; Devenport, M.; Lazarski, C.A.; Zhang, P.; Wang, X.; et al. A reappraisal of CTLA-4 checkpoint blockade in cancer immunotherapy. Cell Res. 2018, 28, 416–432. [Google Scholar] [CrossRef] [Green Version]
- Ribatti, D. Immunosuppressive effects of vascular endothelial growth factor. Oncol. Lett. 2022, 24, 369. [Google Scholar] [CrossRef]
- Pore, N. Vascular endothelial growth factor and tumor immune microenvironment—Chapter 10. In Endothelial Signaling in Vascular Dysfunction and Disease from Bench to Bedside; Elsevier: Amsterdam, The Netherlands, 2021; pp. 105–111. [Google Scholar] [CrossRef]
- Khan, K.A.; Kerbel, R.S. Improving immunotherapy outcomes with anti-angiogenic treatments and vice versa. Nat. Rev. Clin. Oncol. 2018, 15, 310–324. [Google Scholar] [CrossRef]
- Fukurnura, D.; Kloepper, J.; Amoozgar, Z.; Duda, D.G.; Jain, R.K. Enhancing cancer immunotherapy using antiangiogenics: Opportunities and challenges. Nat. Rev. Clin. Oncol. 2018, 15, 325–340. [Google Scholar] [CrossRef] [Green Version]
- Tada, Y.; Togashi, Y.; Kotani, D.; Kuwata, T.; Sato, E.; Kawazoe, A.; Doi, T.; Wada, H.; Nishikawa, H.; Shitara, K. Targeting VEGFR2 with Ramucirumab strongly impacts effector/ activated regulatory T cells and CD8(+) T cells in the tumor microenvironment. J. Immunother. Cancer 2018, 6, 106. [Google Scholar] [CrossRef]
- Kalles, V.; Papapanagiotou, I.; Matiatou, M.; Georgiou, G.; Theodoropoulos, C.; Triantafyllou, T.; Zografos, E.; Mitrousias, A.; Provatopoulou, X.; Michalopoulos, N.V. Evaluation of plasma and tissue expression levels of Endothelins (ET-1, Big ET-1) and VEGF in lobular neoplasia of the breast. J. Buon 2019, 24, 1913–1919. [Google Scholar]
- Kandalaft, L.E.; Facciabene, A.; Buckanovich, R.J.; Coukos, G. Endothelin B Receptor, a New Target in Cancer Immune Therapy. Clin. Cancer Res. 2009, 15, 4521–4528. [Google Scholar] [CrossRef] [Green Version]
- Buckanovich, R.J.; Facciabene, A.; Kim, S.; Benencia, F.; Sasaroli, D.; Balint, K.; Katsaros, D.; O’Brien-Jenkins, A.; Gimotty, P.A.; Coukos, G. Endothelin B receptor mediates the endothelial barrier to T cell homing to tumors and disables immune therapy. Nat. Med. 2008, 14, 28–36. [Google Scholar] [CrossRef]
- Gu, X.; Han, S.; Cui, M.; Xue, J.; Ai, L.; Sun, L.; Zhu, X.; Wang, Y.; Liu, C. Knockdown of endothelin receptor B inhibits the progression of triple-negative breast cancer. Ann. N. Y. Acad. Sci. 2019, 1448, 5–18. [Google Scholar] [CrossRef]
- Roh, W.; Chen, P.L.; Reuben, A.; Spencer, C.N.; Prieto, P.A.; Miller, J.P.; Gopalakrishnan, V.; Wang, F.; Cooper, Z.A.; Reddy, S.M.; et al. Integrated molecular analysis of tumor biopsies on sequential CTLA-4 and PD-1 blockade reveals markers of response and resistance. Sci. Transl. Med. 2017, 9, eaah3560. [Google Scholar] [CrossRef] [Green Version]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef]
- Gettinger, S.; Choi, J.M.; Hastings, K.; Truini, A.; Datar, I.; Sowell, R.; Wurtz, A.; Dong, W.L.; Cai, G.P.; Melnick, M.A.; et al. Impaired HLA Class I Antigen Processing and Presentation as a Mechanism of Acquired Resistance to Immune Checkpoint Inhibitors in Lung Cancer. Cancer Discov. 2017, 7, 1420–1435. [Google Scholar] [CrossRef] [Green Version]
- Sade-Feldman, M.; Jiao, Y.X.J.; Chen, J.H.; Rooney, M.S.; Barzily-Rokni, M.; Eliane, J.P.; Bjorgaard, S.L.; Hammond, M.R.; Vitzthum, H.; Blackmon, S.M.; et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat. Commun. 2017, 8, 1136. [Google Scholar] [CrossRef] [Green Version]
- Ling, A.; Lofgren-Burstrom, A.; Larsson, P.; Li, X.R.; Wikberg, M.L.; Oberg, A.; Stenling, R.; Edin, S.; Palmqvist, R. TAP1 down-regulation elicits immune escape and poor prognosis in colorectal cancer. Oncoimmunology 2017, 6, e1356143. [Google Scholar] [CrossRef] [Green Version]
- Shionoya, Y.; Kanaseki, T.; Miyamoto, S.; Tokita, S.; Hongo, A.; Kikuchi, Y.; Kochin, V.; Watanabe, K.; Horibe, R.; Saijo, H.; et al. Loss of tapasin in human lung and colon cancer cells and escape from tumor-associated antigen-specific CTL recognition. Oncoimmunology 2017, 6, e1274476. [Google Scholar] [CrossRef] [Green Version]
- Sultan, M.; Vidovic, D.; Paine, A.S.; Huynh, T.T.; Coyle, K.M.; Thomas, M.L.; Cruickshank, B.M.; Dean, C.A.; Clements, D.R.; Kim, Y.; et al. Epigenetic Silencing of TAP1 in Aldefluor(+) Breast Cancer Stem Cells Contributes to Their Enhanced Immune Evasion. Stem Cells 2018, 36, 641–654. [Google Scholar] [CrossRef] [Green Version]
- Sim, F.; Leidner, R.; Bell, R.B. Immunotherapy for Head and Neck Cancer. Oral Maxillofac. Surg. Clin. N. Am. 2019, 31, 85. [Google Scholar] [CrossRef]
- Peña-Romero, A.C.; Orenes-Piñero, E. Dual Effect of Immune Cells within Tumour Microenvironment: Pro- and Anti-Tumour Effects and Their Triggers. Cancers 2022, 14, 1681. [Google Scholar] [CrossRef]
- Damasio, M.P.S.; Nascimento, C.S.; Andrade, L.M.; de Oliveira, V.L.; Calzavara-Silva, C.E. The role of T-cells in head and neck squamous cell carcinoma: From immunity to immunotherapy. Front. Oncol. 2022, 12, 1021609. [Google Scholar] [CrossRef]
- Zhai, L.J.; Bell, A.; Ladomersky, E.; Lauing, K.L.; Bollu, L.; Sosman, J.A.; Zhang, B.; Wu, J.D.; Miller, S.D.; Meeks, J.J.; et al. Immunosuppressive IDO in Cancer: Mechanisms of Action, Animal Models, and Targeting Strategies. Front. Immunol. 2020, 11, 1185. [Google Scholar] [CrossRef]
- Platten, M.; Nollen, E.A.A.; Rohrig, U.F.; Fallarino, F.; Opitz, C.A. Tryptophan metabolism as a common therapeutic target in cancer, neurodegeneration and beyond. Nat. Rev. Drug Discov. 2019, 18, 379–401. [Google Scholar] [CrossRef]
- Prendergast, G.C.; Mondal, A.; Dey, S.; Laury-Kleintop, L.D.; Muller, A.J. Inflammatory Reprogramming with IDO1 Inhibitors: Turning Immunologically Unresponsive ‘Cold’ Tumors ‘Hot’. Trends Cancer 2018, 4, 38–58. [Google Scholar] [CrossRef]
- Cheong, J.E.; Sun, L.J. Targeting the IDO1/TDO2-KYN-AhR Pathway for Cancer Immunotherapy—Challenges and Opportunities. Trends Pharmacol. Sci. 2018, 39, 307–325. [Google Scholar] [CrossRef]
- Liu, Y.; Liang, X.; Dong, W.; Fang, Y.; Lv, J.; Zhang, T.; Fiskesund, R.; Xie, J.; Liu, J.; Yin, X.; et al. Tumor-Repopulating Cells Induce PD-1 Expression in CD8+ T Cells by Transferring Kynurenine and AhR Activation. Cancer Cell 2018, 33, 480–494.e7. [Google Scholar] [CrossRef] [Green Version]
- Bishnupuri, K.S.; Alvarado, D.M.; Khouri, A.N.; Shabsovich, M.; Chen, B.; Dieckgraefe, B.K.; Ciorba, M.A. IDO1 and Kynurenine Pathway Metabolites Activate PI3K-Akt Signaling in the Neoplastic Colon Epithelium to Promote Cancer Cell Proliferation and Inhibit Apoptosis. Cancer Res. 2019, 79, 1138–1150. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.P.; Fu, S.F.; Xuan, C.; Jing, Y.; Yuan, Y.; Kong, L.D.; Zhu, Z.M. The Clinicopathological and Prognostic Significance of IDO1 Expression in Human Solid Tumors: Evidence from a Systematic Review and Meta-Analysis. Cell. Physiol. Biochem. 2018, 49, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.Z.; Sun, Y.Y.; Yin, Z.C.; Feng, S.; Sun, L.P.; Li, Z.Y. Research progress of indoleamine 2,3-dioxygenase inhibitors. Future Med. Chem. 2015, 7, 185–201. [Google Scholar] [CrossRef] [PubMed]
- Gibney, G.T.; Hamid, O.; Lutzky, J.; Olszanski, A.J.; Mitchell, T.C.; Gajewski, T.F.; Chmielowski, B.; Hanks, B.A.; Zhao, Y.F.; Newton, R.C.; et al. Phase 1/2 study of epacadostat in combination with ipilimumab in patients with unresectable or metastatic melanoma. J. Immunother. Cancer 2019, 7, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchell, T.C.; Hamid, O.; Smith, D.C.; Bauer, T.M.; Wasser, J.S.; Olszanski, A.J.; Luke, J.J.; Balmanoukian, A.S.; Schmidt, E.V.; Zhao, Y.F.; et al. Epacadostat Plus Pembrolizumab in Patients With Advanced Solid Tumors: Phase I Results From a Multicenter, Open-Label Phase I/II Trial (ECHO-202/KEYNOTE-037). J. Clin. Oncol. 2018, 36, 3223–3230. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Li, X.; Zheng, S.; Du, G.; Ma, J.; Zhang, L.; Wang, H.; Tian, J. Comparison study of different indoleamine-2,3 dioxygenase inhibitors from the perspective of pharmacodynamic effects. Int. J. Immunopathol. Pharmacol. 2022, 34, 2058738420950584. [Google Scholar] [CrossRef]
- Marin-Acevedo, J.A.; Dholaria, B.; Soyano, A.E.; Knutson, K.L.; Chumsri, S.; Lou, Y. Next generation of immune checkpoint therapy in cancer: New developments and challenges. J. Hematol. Oncol. 2018, 11, 39. [Google Scholar] [CrossRef]
- Gato-Canas, M.; Zuazo, M.; Arasanz, H.; Ibanez-Vea, M.; Lorenzo, L.; Fernandez-Hinojal, G.; Vera, R.; Smerdou, C.; Martisova, E.; Arozarena, I.; et al. PDL1 Signals through Conserved Sequence Motifs to Overcome Interferon-Mediated Cytotoxicity. Cell Rep. 2017, 20, 1818–1829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ettl, T.; Grube, M.; Schulz, D.; Bauer, R.J. Checkpoint Inhibitors in Cancer Therapy: Clinical Benefits for Head and Neck Cancers. Cancers 2022, 14, 4985. [Google Scholar] [CrossRef]
- Iwai, Y.; Hamanishi, J.; Chamoto, K.; Honjo, T. Cancer immunotherapies targeting the PD-1 signaling pathway. J. Biomed. Sci. 2017, 24, 26. [Google Scholar] [CrossRef] [Green Version]
- Que, Y.; Fang, Z.X.; Guan, Y.X.; Xiao, W.; Xu, B.S.; Zhao, J.J.; Chen, H.Y.; Zhang, X.K.; Zeng, M.S.; Liang, Y.; et al. LAG-3 expression on tumor-infiltrating T cells in soft tissue sarcoma correlates with poor survival. Cancer Biol. Med. 2019, 16, 331–340. [Google Scholar] [CrossRef] [Green Version]
- Saleh, R.; Elkord, E. Treg-mediated acquired resistance to immune checkpoint inhibitors. Cancer Lett. 2019, 457, 168–179. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sanmamed, M.F.; Datar, I.; Su, T.T.; Ji, L.; Sun, J.W.; Chen, L.; Chen, Y.S.; Zhu, G.F.; Yin, W.W.; et al. Fibrinogen-like Protein 1 Is a Major Immune Inhibitory Ligand of LAG-3. Cell 2019, 176, 334–347.e12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrews, L.P.; Marciscano, A.E.; Drake, C.G.; Vignali, D.A.A. LAG3 (CD223) as a cancer immunotherapy target. Immunol. Rev. 2017, 276, 80–96. [Google Scholar] [CrossRef] [Green Version]
- Burugu, S.; Gao, D.; Leung, S.; Chia, S.K.; Nielsen, T.O. LAG-3+tumor infiltrating lymphocytes in breast cancer: Clinical correlates and association with PD-1/PD-L1+tumors. Ann. Oncol. 2017, 28, 2977–2984. [Google Scholar] [CrossRef] [PubMed]
- Burova, E.; Hermann, A.; Dai, J.; Ullman, E.; Halasz, G.; Potocky, T.; Hong, S.; Liu, M.; Allbritton, O.; Woodruff, A.; et al. Preclinical Development of the Anti-LAG-3 Antibody REGN3767: Characterization and Activity in Combination with the Anti-PD-1 Antibody Cemiplimab in Human PD-1xLAG-3-Knockin Mice. Mol. Cancer Ther. 2019, 18, 2051–2062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, S.; Sharma, G.; Travers, J.; Kumar, S.; Choi, J.; Jun, H.T.; Kehry, M.; Ramaswamy, S.; Jenkins, D. TSR-033, a Novel Therapeutic Antibody Targeting LAG-3, Enhances T-Cell Function and the Activity of PD-1 Blockade In Vitro and In Vivo. Mol. Cancer Ther. 2019, 18, 632–641. [Google Scholar] [CrossRef] [Green Version]
- Harris-Bookman, S.; Mathios, D.; Martin, A.M.; Xia, Y.X.; Kim, E.; Xu, H.Y.; Belcaid, Z.; Polanczyk, M.; Barberi, T.; Theodros, D.; et al. Expression of LAG-3 and efficacy of combination treatment with anti-LAG-3 and anti-PD-1 monoclonal antibodies in glioblastoma. Int. J. Cancer. 2018, 143, 3201–3208. [Google Scholar] [CrossRef] [Green Version]
- Wolf, Y.; Anderson, A.C.; Kuchroo, V.K. TIM3 comes of age as an inhibitory receptor. Nat. Rev. Immunol. 2020, 20, 173–185. [Google Scholar] [CrossRef]
- Zheng, Y.J.; Li, Y.; Lian, J.Y.; Yang, H.Y.; Li, F.; Zhao, S.; Qi, Y.; Zhang, Y.; Huang, L. TNF--induced Tim-3 expression marks the dysfunction of infiltrating natural killer cells in human esophageal cancer. J. Transl. Med. 2019, 17, 165. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.F.; Wu, L.; Yang, L.L.; Deng, W.W.; Mao, L.; Wu, H.; Zhang, W.F.; Sun, Z.J. Blockade of TIM3 relieves immunosuppression through reducing regulatory T cells in head and neck cancer. J. Exp. Clin. Cancer Res. 2018, 37, 44. [Google Scholar] [CrossRef] [Green Version]
- Tang, R.H.; Rangachari, M.; Kuchroo, V.K. Tim-3: A co-receptor with diverse roles in T cell exhaustion and tolerance. Semin. Immunol. 2019, 42, 101302. [Google Scholar] [CrossRef] [PubMed]
- Shayan, G.; Srivastava, R.; Li, J.; Schmitt, N.; Kane, L.P.; Ferris, R.L. Adaptive resistance to anti-PD1 therapy by Tim-3 upregulation is mediated by the PI3K-Akt pathway in head and neck cancer. Oncoimmunology 2017, 6, e1261779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Herter-Sprie, G.S.; Buczkowski, K.A.; Richards, W.G.; Gandhi, L.; Redig, A.J.; Rodig, S.J.; Asahina, H.; et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat. Commun. 2016, 7, 10501. [Google Scholar] [CrossRef] [Green Version]
- Yasinska, I.M.; Sakhnevych, S.S.; Pavlova, L.; Selno, A.T.H.; Abeleira, A.M.T.; Benlaouer, O.; Silva, I.G.; Mosimann, M.; Varani, L.; Bardelli, M.; et al. The Tim-3-Galectin-9 Pathway and Its Regulatory Mechanisms in Human Breast Cancer. Front. Immunol. 2019, 10, 1594. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.T.; Zhang, H.H.; Sun, S.B.; Sun, S.W.; Li, L.P. The effects of Tim-3 activation on T-cells in gastric cancer progression. Oncol. Lett. 2019, 17, 1461–1466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pu, F.F.; Chen, F.X.; Zhang, Z.C.; Qing, X.C.; Lin, H.; Zhao, L.; Xia, P.; Shao, Z.W. TIM-3 expression and its association with overall survival in primary osteosarcoma. Oncol. Lett. 2019, 18, 5294–5300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ClinicalTrials.gov Identifier: NCT03099109 for “A Study of LY3321367 Alone or with LY3300054 in Participants with Advanced Relapsed/Refractory Solid Tumours”. Available online: ClinicalTrials.gov (accessed on 31 January 2023).
- ClinicalTrials.gov Identifier: NCT02608268 for “Safety and Efficacy of MBG453 as Single Agent and in Combination with PDR001 in Patients with Advanced Malignancies”. Available online: ClinicalTrials.gov (accessed on 31 January 2023).
- Clinical Trial Number NCT02817633 for “Study of TSR-022, an Anti-TIM-3 Monoclonal Antibody, in Patients With Advanced Solid Tumours”. Available online: ClinicalTrials.gov (accessed on 31 January 2023).
- Abdeladhim, M.; Karnell, J.L.; Rieder, S.A. In or out of control: Modulating regulatory T cell homeostasis and function with immune checkpoint pathways. Front. Immunol. 2022, 13, 1033705. [Google Scholar] [CrossRef]
- Solomon, B.L.; Garrido-Laguna, I. TIGIT: A novel immunotherapy target moving from bench to bedside. Cancer Immunol. Immunother. 2018, 67, 1659–1667. [Google Scholar] [CrossRef]
- He, W.L.; Zhang, H.; Han, F.; Chen, X.L.; Lin, R.; Wang, W.; Qiu, H.B.; Zhuang, Z.H.; Liao, Q.; Zhang, W.J.; et al. CD155T/TIGIT Signaling Regulates CD8(+) T-cell Metabolism and Promotes Tumor Progression in Human Gastric Cancer. Cancer Res. 2017, 77, 6375–6388. [Google Scholar] [CrossRef] [Green Version]
- Annese, T.; Tamma, R.; Ribatti, D. Update in TIGIT Immune-Checkpoint Role in Cancer. Front. Oncol. 2022, 12, 871085. [Google Scholar] [CrossRef]
- Wu, L.; Mao, L.; Liu, J.F.; Chen, L.; Yu, G.T.; Yang, L.L.; Wu, H.; Bu, L.L.; Kulkarni, A.B.; Zhang, W.F.; et al. Blockade of TIGIT/CD155 Signaling Reverses T-cell Exhaustion and Enhances Antitumor Capability in Head and Neck Squamous Cell Carcinoma. Cancer Immuno. Res. 2019, 7, 1700–1713. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Li, L.J.; Zuo, C.; Chen, M.Y.; Zhang, X.L.; Myers, N.B.; Hogg, G.D.; DeNardo, D.G.; Goedegebuure, S.P.; Hawkins, W.G.; et al. Combination TIGIT/PD-1 blockade enhances the efficacy of neoantigen vaccines in a model of pancreatic cancer. Front. Immunol. 2022, 13, 1039226. [Google Scholar] [CrossRef] [PubMed]
- Pawlowska, A.; Skiba, W.; Suszczyk, D.; Kurylo, W.; Jakubowicz-Gil, J.; Paduch, R.; Wertel, I. The Dual Blockade of the TIGIT and PD-1/PD-L1 Pathway as a New Hope for Ovarian Cancer Patients. Cancers 2022, 14, 5757. [Google Scholar] [CrossRef] [PubMed]
- Ge, Z.H.; Peppelenbosch, M.P.; Sprengers, D.; Kwekkeboom, J. TIGIT, the Next Step Towards Successful Combination Immune Checkpoint Therapy in Cancer. Front. Immunol. 2021, 12, 699895. [Google Scholar] [CrossRef]
- Hung, A.L.; Maxwell, R.; Theodros, D.; Belcaid, Z.; Mathios, D.; Luksik, A.S.; Kim, E.; Wu, A.; Xia, Y.X.; Garzon-Muvdi, T.; et al. TIGIT and PD-1 dual checkpoint blockade enhances antitumor immunity and survival in GBM. Oncoimmunology 2018, 7, e1466769. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.H.; Zhang, Y.; Zhou, F.P.; Chen, X.C.; Sheng, J.P.; Zhang, J. TIGIT: A promising target to overcome the barrier of immunotherapy in hematological malignancies. Front. Oncol. 2022, 12, 1091782. [Google Scholar] [CrossRef]
- Wang, M.; Bu, J.; Zhou, M.; Sido, J.; Lin, Y.; Liu, G.; Lin, Q.; Xu, X.; Leavenworth, J.W.; Shen, E. CD8+T cells expressing both PD-1 and TIGIT but not CD226 are dysfunctional in acute myeloid leukemia (AML) patients. Clin. Immunol. 2018, 190, 64–73. [Google Scholar] [CrossRef]
- Zhang, X.L.; Zhang, H.F.; Chen, L.; Feng, Z.J.; Gao, L.; Li, Q. TIGIT expression is upregulated in T cells and causes T cell dysfunction independent of PD-1 and Tim-3 in adult B lineage acute lymphoblastic leukemia. Cell. Immunol. 2019, 344, 103958. [Google Scholar] [CrossRef]
- Cho, B.C.; Rodriguez-Abreu, D.; Hussein, M.; Cobo, M.; Patel, A.; Secen, N.; Gerstner, G.; Kim, D.W.; Lee, Y.G.; Su, W.C.; et al. Updated analysis and patient-reported outcomes (PROs) from CITYSCAPE: A randomised, double-blind, phase II study of the anti-TIGIT antibody tiragolumab plus atezolizumab (TA) versus placebo plus atezolizumab (PA) as first-line treatment for PD-L1+NSCLC. Ann. Oncolol. 2021, 32, S1428. [Google Scholar] [CrossRef]
- Murat, K.; Hamanishi, J.; Matsumura, N.; Chamoto, K.; Mise, N.; Abiko, K.; Baba, T.; Yamaguchi, K.; Murakami, R.; Hosoe, Y.; et al. VISTA expressed in tumor cells regulates T cell function. Cancer Sci. 2018, 109, 1323. [Google Scholar]
- Nowak, E.C.; Lines, J.L.; Varn, F.S.; Deng, J.; Sarde, A.; Mabaera, R.; Kuta, A.; Le Mercier, I.; Cheng, C.; Noelle, R.J. Immunoregulatory functions of VISTA. Immunol. Rev. 2017, 276, 66–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.H.; Wu, G.P.; Manick, B.; Hernandez, V.; Renelt, M.; Erickson, C.; Guan, J.N.; Singh, R.; Rollins, S.; Solorz, A.; et al. VSIG-3 as a ligand of VISTA inhibits human T-cell function. Immunology 2019, 156, 74–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ClinicalTrials.gov Identifier: NCT02812875 for “A Study of CA-170 Oral PD-L1, PD-L2 and VISTA Checkpoint Antagonist in Patients with Advanced Tumours and Lymphomas”. Available online: ClinicalTrials.gov (accessed on 31 January 2023).
- Argiris, A.; Harrington, K.J.; Tahara, M.; Schulten, J.; Chomette, P.; Ferreira Castro, A.; Licitra, L. Evidence-Based Treatment Options in Recurrent and/or Metastatic Squamous Cell Carcinoma of the Head and Neck. Front. Oncol. 2017, 7, 72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pontes, F.; Garcia, A.R.; Domingues, I.; João Sousa, M.; Felix, R.; Amorim, C.; Salgueiro, F.; Mariano, M.; Teixeira, M. Survival predictors and outcomes of patients with recurrent and/or metastatic head and neck cancer treated with chemotherapy plus cetuximab as first-line therapy: A real-world retrospective study. Cancer Treat. Res. Commun. 2021, 27, 100375. [Google Scholar] [CrossRef]
- Bhat, G.R.; Hyole, R.G.; Li, J. Head and neck cancer: Current challenges and future perspectives. Adv. Cancer Res. 2021, 152, 67–102. [Google Scholar] [CrossRef]
- Johnson, D.E.; Burtness, B.; Leemans, C.R.; Lui, V.W.Y.; Bauman, J.E.; Grandis, J.R. Head and neck squamous cell carcinoma. Nat. Rev. Dis. Primers 2020, 6, 92. [Google Scholar] [CrossRef]
- Mito, I.; Takahashi, H.; Kawabata-Iwakawa, R.; Ida, S.; Tada, H.; Chikamatsu, K. Comprehensive analysis of immune cell enrichment in the tumor microenvironment of head and neck squamous cell carcinoma. Sci. Rep. 2021, 11, 16134. [Google Scholar] [CrossRef]
- Outh-Gauer, S.; Alt, M.; Le Tourneau, C.; Augustin, J.; Broudin, C.; Gasne, C.; Denize, T.; Mirghani, H.; Fabre, E.; Menard, M.; et al. Immunotherapy in head and neck cancers: A new challenge for immunologists, pathologists and clinicians. Cancer Treat. Rev. 2018, 65, 54–64. [Google Scholar] [CrossRef]
- Peng, G.G.; Chi, H.; Gao, X.R.; Zhang, J.H.; Song, G.B.; Xie, X.X.; Su, K.; Song, B.Y.; Yang, J.Y.; Gu, T.; et al. Identification and validation of neurotrophic factor-related genes signature in HNSCC to predict survival and immune landscapes. Front. Genet. 2022, 13, 1010044. [Google Scholar] [CrossRef]
- Liu, C.; Wang, M.; Zhang, H.Y.; Li, C.Y.; Zhang, T.S.; Liu, H.; Zhu, S.; Chen, J. Tumor microenvironment and immunotherapy of oral cancer. Eur. J. Med. Res. 2022, 27, 198. [Google Scholar] [CrossRef] [PubMed]
- Sabado, R.L.; Balan, S.; Bhardwaj, N. Dendritic cell-based immunotherapy. Cell Res. 2017, 27, 74–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zandberg, D.P.; Strome, S.E. The role of the PD-L1: PD-1 pathway in squamous cell carcinoma of the head and neck. Oral Oncol. 2014, 50, 627–632. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, A.H.; Pauken, K.E. The diverse functions of the PD1 inhibitory pathway. Nat. Rev. Immunol. 2018, 18, 153–167. [Google Scholar] [CrossRef]
- Farkona, S.; Diamandis, E.P.; Blasutig, I.M. Cancer immunotherapy: The beginning of the end of cancer? BMC Med. 2016, 14, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golay, J.; Andrea, A.E. Combined Anti-Cancer Strategies Based on Anti-Checkpoint Inhibitor Antibodies. Antibodies 2020, 9, 17. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Chehrazi-Raffle, A.; Reddi, S.; Salgia, R. Development of PD-1 and PD-L1 inhibitors as a form of cancer immunotherapy: A comprehensive review of registration trials and future considerations. J. Immunother. Cancer 2018, 6, 8. [Google Scholar] [CrossRef]
- Ferris, R.L.; Blumenschein, G.; Fayette, J.; Guigay, J.; Colevas, A.D.; Licitra, L.; Harrington, K.; Kasper, S.; Vokes, E.E.; Even, C.; et al. Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. N. Engl. J. Med. 2016, 375, 1856–1867. [Google Scholar] [CrossRef] [Green Version]
- Haddad, R.; Concha-Benavente, F.; Blumenschein, G.; Fayette, J.; Guigay, J.; Colevas, A.D.; Licitra, L.; Kasper, S.; Vokes, E.E.; Worden, F.; et al. Nivolumab treatment beyond RECIST-defined progression in recurrent or metastatic squamous cell carcinoma of the head and neck in CheckMate 141: A subgroup analysis of a randomized phase 3 clinical trial. Cancer 2019, 125, 3208–3218. [Google Scholar] [CrossRef] [Green Version]
- Burtness, B.; Harrington, K.J.; Greil, R. Pembrolizumab alone or with chemotherapy versus cetuximab with chemotherapy for recurrent metastatic squamous cell carcinoma of the head and neck KEYNOTE-048): A randomised, open-label, phase 3 study (vol 394, pg 1915, 2019). Lancet 2021, 397, 2252. [Google Scholar] [CrossRef]
- Chow, L.Q.M.; Haddad, R.; Gupta, S.; Mahipal, A.; Mehra, R.; Tahara, M.; Berger, R.; Eder, J.P.; Burtness, B.; Lee, S.H.; et al. Antitumor Activity of Pembrolizumab in Biomarker-Unselected Patients With Recurrent and/or Metastatic Head and Neck Squamous Cell Carcinoma: Results From the Phase Ib KEYNOTE-012 Expansion Cohort. J. Clin. Oncol. 2016, 34, 3838. [Google Scholar] [CrossRef]
- Mehra, R.; Seiwert, T.Y.; Gupta, S.; Weiss, J.; Gluck, I.; Eder, J.P.; Burtness, B.; Tahara, M.; Keam, B.; Kang, H.; et al. Efficacy and safety of pembrolizumab in recurrent/metastatic head and neck squamous cell carcinoma: Pooled analyses after long-term follow-up in KEYNOTE-012. Br. J. Cancer 2018, 119, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Powell, S.F.; Liu, S.V.; Sukari, A.; Chung, C.H.; Bauml, J.; Haddad, R.I.; Gause, C.K.; Niewood, M.; Gammage, L.L.; Brown, H.; et al. KEYNOTE-055: A phase II trial of single agent pembrolizumab in patients (pts) with recurrent or metastatic head and neck squamous cell carcinoma (HNSCC) who have failed platinum and cetuximab. J. Clin. Oncol. 2015, 33, TPS3094. [Google Scholar] [CrossRef]
- Cohen, E.E.W.; Soulieres, D.; Le Tourneau, C.; Dinis, J.; Licitra, L.; Ahn, M.J.; Soria, A.; Machiels, J.P.; Mach, N.; Mehra, R.; et al. Pembrolizumab versus methotrexate, docetaxel, or cetuximab for recurrent or metastatic head-and-neck squamous cell carcinoma (KEYNOTE-040): A randomised, open-label, phase 3 study. Lancet 2019, 393, 156–167. [Google Scholar] [CrossRef]
- Wollenberg, B. PD-1 antibodies in head-and-neck cancer. Lancet 2019, 393, 108–109. [Google Scholar] [CrossRef] [PubMed]
- Marabelle, A.; Fakih, M.; Lopez, J.; Shah, M.; Shapira-Frommer, R.; Nakagawa, K.; Chung, H.C.; Kindler, H.L.; Lopez-Martin, J.A.; Miller, W.H.; et al. Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: Prospective biomarker analysis of the multicohort, open-label, phase 2 KEYNOTE-158 study. Lancet Oncol. 2020, 21, 1353–1365. [Google Scholar] [CrossRef] [PubMed]
- Hirata-Nozaki, Y.; Ohkuri, T.; Ohara, K.; Kumai, T.; Nagata, M.; Harabuchi, S.; Kosaka, A.; Nagato, T.; Ishibashi, K.; Oikawa, K.; et al. PD-L1-specific helper T-cells exhibit effective antitumor responses: New strategy of cancer immunotherapy targeting PD-L1 in head and neck squamous cell carcinoma. J. Transl. Med. 2019, 17, 207. [Google Scholar] [CrossRef] [Green Version]
- Meliante, P.G.; Barbato, C.; Zoccali, F.; Ralli, M.; Greco, A.; de Vincentiis, M.; Colizza, A.; Petrella, C.; Ferraguti, G.; Minni, A.; et al. Programmed Cell Death-Ligand 1 in Head and Neck Squamous Cell Carcinoma: Molecular Insights, Preclinical and Clinical Data, and Therapies. Int. J. Mol. Sci. 2022, 23, 15384. [Google Scholar] [CrossRef]
- Qiao, X.W.; Jiang, J.; Pang, X.; Huang, M.C.; Tang, Y.J.; Liang, X.H.; Tang, Y.L. The Evolving Landscape of PD-1/PD-L1 Pathway in Head and Neck Cancer. Front. Immunol. 2020, 11, 1721. [Google Scholar] [CrossRef]
- Colevas, A.D.; Bahleda, R.; Braiteh, F.; Balmanoukian, A.; Brana, I.; Chau, N.G.; Sarkar, I.; Molinero, L.; Grossman, W.; Kabbinavar, F.; et al. Safety and clinical activity of atezolizumab in head and neck cancer: Results from a phase I trial. Ann. Oncol. 2018, 29, 2247–2253. [Google Scholar] [CrossRef]
- Huang, Z.L.; Zheng, S.H.; Ding, S.R.; Wei, Y.H.; Chen, C.; Liu, X.; Li, H.; Xia, Y.F. Prognostic role of programmed cell death ligand-1 expression in head and neck cancer treated with programmed cell death protein-1/programmed cell death ligand-1 inhibitors: A meta-analysis based on clinical trials. J. Cancer Res. Ther. 2021, 17, 676–687. [Google Scholar] [CrossRef]
- El Idrissi, H.H.; Balar, I. Top 100 Most Cited Publications on CTLA-4 Molecule in Cancer Research: A Bibliometric Analysis. Middle East J. Cancer 2021, 14, 1–16. [Google Scholar] [CrossRef]
- Blank, C.U.; Enk, A. Therapeutic use of anti-CTLA-4 antibodies. Int. Immunol. 2015, 27, 3–10. [Google Scholar] [CrossRef] [Green Version]
- Ferris, R.L.; Haddad, R.; Even, C.; Tahara, M.; Dvorkin, M.; Ciuleanu, T.E.; Clement, P.M.; Mesia, R.; Kutukova, S.; Zholudeva, L.; et al. Durvalumab with or without tremelimumab in patients with recurrent or metastatic head and neck squamous cell carcinoma: EAGLE, a randomized, open -label phase III study. Ann. Oncol. 2020, 31, 942–950. [Google Scholar] [CrossRef]
- Guo, Y.; Luo, Y.; Zhang, Q.Y.; Huang, X.M.; Li, Z.D.; Shen, L.F.; Feng, J.F.; Sun, Y.; Yang, K.Y.; Ge, M.H.; et al. First-line treatment with chemotherapy plus cetuximab in Chinese patients with recurrent and/or metastatic squamous cell carcinoma of the head and neck: Efficacy and safety results of the randomised, phase III CHANGE-2 trial. Eur. J. Cancer 2021, 156, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Psyrri, A.; Fayette, J.; Harrington, K.; Gillison, M.; Ahn, M.J.; Takahashi, S.; Weiss, J.; Machiels, J.P.; Baxi, S.; Vasilyev, A.; et al. Durvalumab with or without tremelimumab versus the EXTREME regimen as first-line treatment for recurrent or metastatic squamous cell carcinoma of the head and neck: KESTREL, a randomized, open-label, phase III study. Ann. Oncol. 2022, 34, 262–274. [Google Scholar] [CrossRef] [PubMed]
- Guigay, J.; Lee, K.W.; Patel, M.R.; Daste, A.; Wong, D.J.; Goel, S.; Gordon, M.S.; Gutierrez, M.; Balmanoukian, A.; Le Tourneau, C.; et al. Avelumab for platinum-ineligible/refractory recurrent and/or metastatic squamous cell carcinoma of the head and neck: Phase Ib results from the JAVELIN Solid Tumor trial. J. Immunother. 2021, 9, e002998. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.Y.; Ferris, R.L.; Psyrri, A.; Haddad, R.I.; Tahara, M.; Bourhis, J.; Harrington, K.; Chang, P.M.H.; Lin, J.C.; Razaq, M.A.; et al. Avelumab plus standard-of-care chemoradiotherapy versus chemoradiotherapy alone in patients with locally advanced squamous cell carcinoma of the head and neck: A randomised, double-blind, placebo-controlled, multicentre, phase 3 trial. Lancet Oncol. 2021, 22, 450–462. [Google Scholar] [CrossRef]
- Siu, L.L.; Even, C.; Mesia, R.; Remenar, E.; Daste, A.; Delord, J.P.; Krauss, J.; Saba, N.F.; Nabell, L.; Ready, N.E.; et al. Safety and Efficacy of Durvalumab With or Without Tremelimumab in Patients With PD-L1-Low/Negative Recurrent or Metastatic HNSCC The Phase 2 CONDOR Randomized Clinical Trial. JAMA Oncol. 2019, 5, 195–203. [Google Scholar] [CrossRef]
- Argiris, A.; Harrington, K.; Tahara, M.; Ferris, R.L.; Gillison, M.; Fayette, J.; Daste, A.; Koralewski, P.; Nin, R.M.; Saba, N.F.; et al. Nivolumab (N) plus ipilimumab (I) vs EXTREME as first-line (1L) treatment (tx) for recurrent/metastatic squamous cell carcinoma of the head and neck (R/M SCCHN): Final results of CheckMate 651. Ann. Oncol. 2021, 32, S1310–S1311. [Google Scholar] [CrossRef]
- Julian, R.; Savani, M.; Bauman, J.E. Immunotherapy Approaches in HPV-Associated Head and Neck Cancer. Cancers 2021, 13, 5889. [Google Scholar] [CrossRef]
- Lee, J.B.; Ha, S.J.; Kim, H.R. Clinical Insights Into Novel Immune Checkpoint Inhibitors. Front. Pharmacol. 2021, 12, 681320. [Google Scholar] [CrossRef] [PubMed]
- Li, X.L.; Shao, C.S.; Shi, Y.F.; Han, W.D. Lessons learned from the blockade of immune checkpoints in cancer immunotherapy. J. Hematol. Oncol. 2018, 11, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baumeister, S.H.; Freeman, G.J.; Dranoff, G.; Sharpe, A.H. Coinhibitory Pathways in Immunotherapy for Cancer. Annu. Rev. Immunol. 2016, 34, 539–573. [Google Scholar] [CrossRef] [PubMed]
- Woo, S.R.; Turnis, M.E.; Goldberg, M.V.; Bankoti, J.; Selby, M.; Nirschl, C.J.; Bettini, M.L.; Gravano, D.M.; Vogel, P.; Liu, C.L.; et al. Immune Inhibitory Molecules LAG-3 and PD-1 Synergistically Regulate T-cell Function to Promote Tumoral Immune Escape. Cancer Res. 2012, 72, 917–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.D.; Hu, W.W.; Zheng, X.; Zhang, C.; Du, P.; Zheng, Z.J.; Yang, Y.; Wu, J.; Ji, M.; Jiang, J.T.; et al. Emerging immune checkpoints for cancer therapy. Acta Oncol. 2015, 54, 1706–1713. [Google Scholar] [CrossRef]
- Wang, H.; Mao, L.; Zhang, T.; Zhang, L.M.; Wu, Y.T.; Guo, W.; Hu, J.Z.; Ju, H.Y.; Ren, G.X. Altered expression of TIM-3, LAG-3, IDO, PD-L1, and CTLA-4 during nimotuzumab therapy correlates with responses and prognosis of oral squamous cell carcinoma patients. J. Oral Pathol. Med. 2019, 48, 669–676. [Google Scholar] [CrossRef]
- Peguero, J.A.; Bajaj, P.; Carcereny, E.; Clay, T.D.; Doger, B.; Felip, E.; Krebs, M.; Forster, M.; Aix, S.P.; Roxburgh, P.; et al. A multicenter, phase II study of soluble LAG-3 (Eftilagimod alpha) in combination with pembrolizumab (TACTI-002) in patients with advanced non-small cell lung cancer (NSCLC) or head and neck squamous cell carcinoma (HNSCC). J. Clin. Oncol. 2019, 37, TPS2667. [Google Scholar] [CrossRef]
- Yang, F.; Zeng, Z.Q.; Li, J.; Ren, X.B.; Wei, F. TIM-3 and CEACAM1 are Prognostic Factors in Head and Neck Squamous Cell Carcinoma. Front. Mol. Biosci. 2021, 8, 619765. [Google Scholar] [CrossRef]
- Hakemi, M.G.; Jafarinia, M.; Azizi, M.; Rezaeepoor, M.; Isayev, O.; Bazhin, A.V. The Role of TIM-3 in Hepatocellular Carcinoma: A Promising Target for Immunotherapy? Front. Oncol. 2020, 10, 601661. [Google Scholar] [CrossRef]
- Tundo, G.R.; Sbardella, D.; Lacal, P.M.; Graziani, G.; Marini, S. On the Horizon: Targeting Next-Generation Immune Checkpoints for Cancer Treatment. Chemotherapy 2019, 64, 62–80. [Google Scholar] [CrossRef] [PubMed]
- Hamid, O.; Chiappori, A.A.; Thompson, J.A.; Doi, T.; Hu-Lieskovan, S.; Eskens, F.; Ros, W.; Diab, A.; Spano, J.P.; Rizvi, N.A.; et al. First-in-human study of an OX40 (ivuxolimab) and 4-1BB (utomilumab) agonistic antibody combination in patients with advanced solid tumors. J. Immunother. Cancer 2022, 10, e005471. [Google Scholar] [CrossRef] [PubMed]
- Diab, A.; Hamid, O.; Thompson, J.A.; Ros, W.; Eskens, F.; Doi, T.; Hu-Lieskovan, S.; Klempner, S.J.; Ganguly, B.; Fleener, C.; et al. A Phase I, Open-Label, Dose-Escalation Study of the OX40 Agonist Ivuxolimab in Patients with Locally Advanced or Metastatic Cancers. Clin. Cancer Res. 2022, 28, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, R.J.; Hong, D.S.; Tolcher, A.W.; Patnaik, A.; Shapiro, G.; Chmielowski, B.; Ribas, A.; Brail, L.H.; Roberts, J.; Lee, L.; et al. Initial results from first-in-human study of IPI-549, a tumor macrophage-targeting agent, combined with nivolumab in advanced solid tumors. J. Clin. Oncol. 2018, 36, 3013. [Google Scholar] [CrossRef]
- Nywening, T.M.; Wang-Gillam, A.; Sanford, D.E.; Belt, B.A.; Panni, R.Z.; Cusworth, B.M.; Toriola, A.T.; Nieman, R.K.; Worley, L.A.; Yano, M.; et al. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: A single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol. 2016, 17, 651–662. [Google Scholar] [CrossRef] [Green Version]
- Brana, I.; Calles, A.; LoRusso, P.M.; Yee, L.K.; Puchalski, T.A.; Seetharam, S.; Zhong, B.; de Boer, C.J.; Tabernero, J.; Calvo, E. Carlumab, an anti-C-C chemokine ligand 2 monoclonal antibody, in combination with four chemotherapy regimens for the treatment of patients with solid tumors: An open-label, multicenter phase 1b study. Target. Oncol. 2015, 10, 111–123. [Google Scholar] [CrossRef]
- Ries, C.H.; Cannarile, M.A.; Hoves, S.; Benz, J.; Wartha, K.; Runza, V.; Rey-Giraud, F.; Pradel, L.P.; Feuerhake, F.; Klaman, I.; et al. Targeting Tumor-Associated Macrophages with Anti-CSF-1R Antibody Reveals a Strategy for Cancer Therapy. Cancer Cell 2014, 25, 846–859. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Roca, C.A.; Italiano, A.; Le Tourneau, C.; Cassier, P.A.; Toulmonde, M.; D’Angelo, S.P.; Campone, M.; Weber, K.L.; Loirat, D.; Cannarile, M.A.; et al. Phase I study of emactuzumab single agent or in combination with paclitaxel in patients with advanced/metastatic solid tumors reveals depletion of immunosuppressive M2-like macrophages. Ann. Oncol. 2019, 30, 1381–1392. [Google Scholar] [CrossRef]
- Pathria, P.; Louis, T.L.; Varner, J.A. Targeting Tumor-Associated Macrophages in Cancer. Trends Immunol. 2019, 40, 310–327. [Google Scholar] [CrossRef]
- Karapetyan, L.; Luke, J.J.; Davar, D. Toll-Like Receptor 9 Agonists in Cancer. OncoTargets Ther. 2020, 13, 10039–10060. [Google Scholar] [CrossRef]
- Cohen, E.E.W.; Nabell, L.; Wong, D.J.L.; Day, T.A.; Daniels, G.A.; Milhem, M.M.; Deva, S.; Jameson, M.B.; Guntinas-Lichius, O.; Almubarak, M.; et al. Phase 1b/2, open label, multicenter study of intratumoral SD-101 in combination with pembrolizumab in anti-PD-1 treatment naive patients with recurrent or metastatic head and neck squamous cell carcinoma (HNSCC). J. Clin. Oncol. 2019, 37, 6039. [Google Scholar] [CrossRef]
- Yu, J.P.; Du, W.J.; Yan, F.; Wang, Y.; Li, H.; Cao, S.; Yu, W.W.; Shen, C.; Liu, J.T.; Ren, X.B. Myeloid-Derived Suppressor Cells Suppress Antitumor Immune Responses through IDO Expression and Correlate with Lymph Node Metastasis in Patients with Breast Cancer. J. Immunol. 2013, 190, 3783–3797. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Clavijo, P.E.; Robbins, Y.; Patel, P.; Friedman, J.; Greene, S.; Das, R.; Silvin, C.; Van Waes, C.; Horn, L.A.; et al. Inhibiting myeloid-derived suppressor cell trafficking enhances T cell immunotherapy. JCI Insight 2019, 4, e126853. [Google Scholar] [CrossRef] [Green Version]
- Jung, K.H.; LoRusso, P.; Burris, H.; Gordon, M.; Bang, Y.J.; Hellmann, M.D.; Cervantes, A.; de Olza, M.O.; Marabelle, A.; Hodi, S.; et al. Phase I Study of the Indoleamine 2,3-Dioxygenase 1 (IDO1) Inhibitor Navoximod (GDC-0919) Administered with PD-L1 Inhibitor (Atezolizumab) in Advanced Solid Tumors. Clin. Cancer Res. 2019, 25, 3220–3228. [Google Scholar] [CrossRef] [Green Version]
- Greene, S.; Robbins, Y.; Mydlarz, W.K.; Huynh, A.P.; Schmitt, N.C.; Friedman, J.; Horn, L.A.; Palena, C.; Schlom, J.; Maeda, D.Y.; et al. Inhibition of MDSC Trafficking with SX-682, a CXCR1/2 Inhibitor, Enhances NK-Cell Immunotherapy in Head and Neck Cancer Models. Clin. Cancer Res. 2020, 26, 1420–1431. [Google Scholar] [CrossRef] [Green Version]
- Califano, J.A.; Khan, Z.; Noonan, K.A.; Rudraraju, L.; Zhang, Z.; Wang, H.; Goodman, S.; Gourin, C.G.; Ha, P.K.; Fakhry, C.; et al. Tadalafil Augments Tumor Specific Immunity in Patients with Head and Neck Squamous Cell Carcinoma. Clin. Cancer Res. 2015, 21, 30–38. [Google Scholar] [CrossRef] [Green Version]
- Guo, T.; Califano, J.A. Molecular Biology and Immunology of Head and Neck Cancer. Surg. Oncol. Clin. N. Am. 2015, 24, 397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weed, D.T.; Zilio, S.; Reis, I.M.; Sargi, Z.; Abouyared, M.; Gomez-Fernandez, C.R.; Civantos, F.J.; Rodriguez, C.P.; Serafini, P. The Reversal of Immune Exclusion Mediated by Tadalafil and an Anti-tumor Vaccine Also Induces PDL1 Upregulation in Recurrent Head and Neck Squamous Cell Carcinoma: Interim Analysis of a Phase I Clinical Trial. Front. Immunol. 2019, 10, 1206. [Google Scholar] [CrossRef] [PubMed]
- Oweida, A.; Hararah, M.K.; Phan, A.; Binder, D.; Bhatia, S.; Lennon, S.; Bukkapatnam, S.; Van Court, B.; Uyanga, N.; Darragh, L.; et al. Resistance to Radiotherapy and PD-L1 Blockade Is Mediated by TIM-3 Upregulation and Regulatory T-Cell Infiltration. Clin. Cancer Res. 2018, 24, 5368–5380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobs, J.F.M.; Punt, C.J.A.; Lesterhuis, W.J.; Sutmuller, R.P.M.; Brouwer, H.; Scharenborg, N.M.; Klasen, I.S.; Hilbrands, L.B.; Figdor, C.G.; de Vries, I.J.M.; et al. Dendritic Cell Vaccination in Combination with Anti-CD25 Monoclonal Antibody Treatment: A Phase I/II Study in Metastatic Melanoma Patients. Clin. Cancer Res. 2010, 16, 5067–5078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.Q.; Liu, Y.J.; Han, R.X.; Beier, U.H.; Thomas, R.M.; Wells, A.D.; Hancock, W.W. Mbd2 Promotes Foxp3 Demethylation and T-Regulatory-Cell Function. Mol. Cell. Biol. 2013, 33, 4106–4115. [Google Scholar] [CrossRef] [Green Version]
- Sinclair, C.; Revenko, A.S.; Johnson, R.B.; Peter, A.; Taylor, M.A.; Hettrick, L.A.; Klein, S.; Solanki, A.; Chapman, M.; Yates, J.; et al. Discovery and characterization of AZD8701, a high affinity antisense oligonucleotide targeting FOXP3 to relieve immunosuppression in cancer. Cancer Res. 2019, 79, 2713. [Google Scholar] [CrossRef]
- Kurose, K.; Ohue, Y.; Wada, H.; Iida, S.; Ishida, T.; Kojima, T.; Doi, T.; Suzuki, S.; Isobe, M.; Funakoshi, T.; et al. Phase Ia Study of FoxP3(+) CD4 Treg Depletion by Infusion of a Humanized Anti-CCR4 Antibody, KW-0761, in Cancer Patients. Clin. Cancer Res. 2015, 21, 4327–4336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, T.; Kurose, K.; Kojima, T.; Funakoshi, T.; Sato, E.; Nishikawa, H.; Nakajima, J.; Seto, Y.; Kakimi, K.; Iida, S.; et al. Phase Ib study on the humanized anti-CCR4 antibody, KW-0761, in advanced solid tumors. Nagoya J. Med. Sci. 2021, 83, 827–840. [Google Scholar] [CrossRef]
- Zamarin, D.; Hamid, O.; Nayak-Kapoor, A.; Sahebjam, S.; Sznol, M.; Collaku, A.; Fox, F.E.; Marshall, M.A.; Hong, D.S. y Mogamulizumab in Combination with Durvalumab or Tremelimumab in Patients with Advanced Solid Tumors: A Phase I Study. Clin. Cancer Res. 2020, 26, 4531–4541. [Google Scholar] [CrossRef] [PubMed]
- Doi, T.; Muro, K.; Ishii, H.; Kato, T.; Tsushima, T.; Takenoyama, M.; Oizumi, S.; Gemmoto, K.; Suna, H.; Enokitani, K.; et al. A Phase I Study of the Anti-CC Chemokine Receptor 4 Antibody, Mogamulizumab, in Combination with Nivolumab in Patients with Advanced or Metastatic Solid Tumors. Clin. Cancer Res. 2019, 25, 6614–6622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montler, R.; Bell, R.B.; Thalhofer, C.; Leidner, R.; Feng, Z.P.; Fox, B.A.; Cheng, A.C.; Bui, T.G.; Tucker, C.; Hoen, H.; et al. OX40, PD-1 and CTLA-4 are selectively expressed on tumor-infiltrating T cells in head and neck cancer. Clin. Transl. Immunol. 2016, 5, e70. [Google Scholar] [CrossRef] [PubMed]
- Duhen, R.; Ballesteros-Merino, C.; Frye, A.K.; Tran, E.; Rajamanickam, V.; Chang, S.C.; Koguchi, Y.; Bifulco, C.B.; Bernard, B.; Leidner, R.S.; et al. Neoadjuvant anti-OX40 (MEDI6469) therapy in patients with head and neck squamous cell carcinoma activates and expands antigen-specific tumor-infiltrating T cells. Nat. Commun. 2021, 12, 1047. [Google Scholar] [CrossRef]
- Bell, R.B.; Leidner, R.S.; Duhen, R.A.; Ballesteros-Merino, C.; Feng, Z.; Koguchi, Y.; Bifulco, C.B.; Curti, B.D.; Urba, W.J.; Fox, B.A.; et al. Abstract 37: Anti-OX40 (MEDI6469) prior to definitive surgical resection in patients with head and neck squamous cell carcinoma. Clin. Cancer Res. 2017, 23, 37. [Google Scholar] [CrossRef]
- Glisson, B.S.; Leidner, R.S.; Ferris, R.L.; Powderly, J.; Rizvi, N.A.; Keam, B.; Schneider, R.; Goel, S.; Ohr, J.P.; Burton, J.; et al. Safety and Clinical Activity of MEDI0562, a Humanized OX40 Agonist Monoclonal Antibody, in Adult Patients with Advanced Solid Tumors. Clin. Cancer Res. 2020, 26, 5358–5367. [Google Scholar] [CrossRef] [PubMed]
- Nishioka, T.; Nishida, E.; Iida, R.; Morita, A.; Shimizu, J. In vivo expansion of CD4(+)Foxp3(+) regulatory T cells mediated by GITR molecules. Immunol. Lett. 2008, 121, 97–104. [Google Scholar] [CrossRef]
- Chen, X.M.; Song, E.W. Turning foes to friends: Targeting cancer-associated fibroblasts. Nat. Rev. Drug Discov. 2019, 18, 99–115. [Google Scholar] [CrossRef] [PubMed]
- Saw, P.E.; Chen, J.N.; Song, E.W. Targeting CAFs to overcome anticancer therapeutic resistance. Trends Cancer 2022, 8, 527–555. [Google Scholar] [CrossRef] [PubMed]
- Pleshkan, V.V.; Alekseenko, I.V.; Tyulkina, D.V.; Kyzmich, A.I.; Zinovyeva, M.V.; Sverdlov, E.D. Fibroblast activation protein (FAP) as a possible target of the antitumor strategy. Mol. Gen. Mikrobiol. Virusol. 2016, 34, 90. [Google Scholar] [CrossRef] [PubMed]
- Ostermann, E.; Garin-Chesa, P.; Heider, K.H.; Kalat, M.; Lamche, H.; Puri, C.; Kerjaschki, D.; Rettig, W.J.; Adolf, G.R. Effective immunoconjugate therapy in cancer models targeting a serine protease of tumor fibroblasts. Clin. Cancer Res. 2008, 14, 4584–4592. [Google Scholar] [CrossRef] [Green Version]
- Soerensen, M.M.; Ros, W.; Rodriguez-Ruiz, M.E.; Robbrecht, D.; Rohrberg, K.S.; Martin-Liberal, J.; Lassen, U.N.; Bermejo, I.M.; Lolkema, M.P.; Tabernero, J.; et al. Safety, PK/PD, and anti-tumor activity of RO6874281, an engineered variant of interleukin-2 (IL-2v) targeted to tumor-associated fibroblasts via binding to fibroblast activation protein (FAP). J. Clin. Oncol. 2018, 36, e15155. [Google Scholar] [CrossRef]
- Angevin, E.; Tabernero, J.; Elez, E.; Cohen, S.J.; Bahleda, R.; van Laethem, J.L.; Ottensmeier, C.; Lopez-Martin, J.A.; Clive, S.; Joly, F.; et al. A Phase I/II, Multiple-Dose, Dose-Escalation Study of Siltuximab, an Anti-Interleukin-6 Monoclonal Antibody, in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2014, 20, 2192–2204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bharadwaj, U.; Eckols, T.K.; Xu, X.J.; Kasembeli, M.M.; Chen, Y.Y.; Adachi, M.; Song, Y.C.; Mo, Q.X.; Lai, S.Y.; Tweardy, D.J. Small-molecule inhibition of STAT3 in radioresistant head and neck squamous cell carcinoma. Oncotarget 2016, 7, 26307–26330. [Google Scholar] [CrossRef] [Green Version]
- Chiappori, A.A.; Eckhardt, S.G.; Bukowski, R.; Sullivan, D.M.; Ikeda, M.; Yano, Y.; Yamada-Sawada, T.; Kambayashi, Y.; Tanaka, K.; Javle, M.M.; et al. A phase I pharmacokinetic and pharmacodynamic study of S-3304, a novel matrix metalloproteinase inhibitor, in patients with advanced and refractory solid tumors. Clin. Cancer Res. 2007, 13, 2091–2099. [Google Scholar] [CrossRef] [Green Version]
- Wulff, S.; Pries, R.; Borngen, K.; Trenkle, T.; Wollenberg, B. Decreased Levels of Circulating Regulatory NK Cells in Patients with Head and Neck Cancer throughout all Tumor Stages. Anticancer Res. 2009, 29, 3053–3057. [Google Scholar]
- Schilling, B.; Halstead, E.S.; Schuler, P.; Harasymczuk, M.; Egan, J.E.; Whiteside, T.L. IRX-2, a novel immunotherapeutic, enhances and protects NK-cell functions in cancer patients. Cancer Immunol. Immunoth. 2012, 61, 1395–1405. [Google Scholar] [CrossRef] [Green Version]
- Concha-Benavente, F.; Kansy, B.; Moskovitz, J.; Moy, J.; Chandran, U.; Ferris, R.L. PD-L1 Mediates Dysfunction in Activated PD-1(+) NK Cells in Head and Neck Cancer Patients. Cancer Immunol. Res. 2018, 6, 1548–1560. [Google Scholar] [CrossRef] [Green Version]
- Lian, G.Y.; Mak, T.S.K.; Yu, X.Q.; Lan, H.Y. Challenges and Recent Advances in NK Cell-Targeted Immunotherapies in Solid Tumors. Int. J. Mol. Sci. 2022, 23, 164. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, B.K.; Yim, D.; Chow, I.T.; Gonzalez, S.; Dai, Z.; Mann, H.H.; Strong, R.K.; Groh, V.; Spies, T. Disulphide-isomerase-enabled shedding of tumour-associated NKG2D ligands. Nature 2007, 447, 482–485. [Google Scholar] [CrossRef] [PubMed]
- Xing, S.; Ferrari de Andrade, L. NKG2D and MICA/B shedding: A ‘tag game’ between NK cells and malignant cells. Clin. Trans. Immunol. 2020, 9, e1230. [Google Scholar] [CrossRef] [PubMed]
- Klöß, S.; Chambron, N.; Gardlowski, T.; Arseniev, L.; Koch, J.; Esser, R.; Glienke, W.; Seitz, O.; Köhl, U. Increased sMICA and TGFβ1 levels in HNSCC patients impair NKG2D-dependent functionality of activated NK cells. Oncoimmunology 2015, 4, e1055993. [Google Scholar] [CrossRef] [Green Version]
- Klöss, S.; Chambron, N.; Gardlowski, T.; Weil, S.; Koch, J.; Esser, R.; Pogge von Strandmann, E.; Morgan, M.A.; Arseniev, L.; Seitz, O.; et al. Cetuximab Reconstitutes Pro-Inflammatory Cytokine Secretions and Tumor-Infiltrating Capabilities of sMICA-Inhibited NK Cells in HNSCC Tumor Spheroids. Front. Immunol. 2015, 6, 543. [Google Scholar] [CrossRef] [Green Version]
- Weil, S.; Memmer, S.; Lechner, A.; Huppert, V.; Giannattasio, A.; Becker, T.; Muller-Runte, A.; Lampe, K.; Beutner, D.; Quaas, A.; et al. Natural Killer Group 2D Ligand Depletion Reconstitutes Natural Killer Cell Immunosurveillance of Head and Neck Squamous Cell Carcinoma. Front. Immunol. 2017, 8, 387. [Google Scholar] [CrossRef] [Green Version]
- Andre, P.; Denis, C.; Soulas, C.; Bourbon-Caillet, C.; Lopez, J.; Arnoux, T.; Blery, M.; Bonnafous, C.; Gauthier, L.; Morel, A.; et al. Anti-NKG2A mAb Is a Checkpoint Inhibitor that Promotes Anti-tumor Immunity by Unleashing Both T and NK Cells. Cell 2018, 175, 1731. [Google Scholar] [CrossRef] [Green Version]
- van Hall, T.; Andre, P.; Horowitz, A.; Ruan, D.F.; Borst, L.; Zerbib, R.; Narni-Mancinelli, E.; van der Burg, S.H.; Vivier, E. Monalizumab: Inhibiting the novel immune checkpoint NKG2A. J. Immunother. Cancer 2019, 7, 263. [Google Scholar] [CrossRef]
- Vermorken, J.B.; Remenar, E.; Hitt, R.; Kawecki, A.; Rottey, S.; Knierim, L.; Schulten, J.; Mesia, R. Platinum-based chemotherapy (CT) plus cetuximab in recurrent or metastatic squamous cell carcinoma of the head and neck cancer (R/M-SCCHN): 5-year follow-up data for the extreme trial. J. Clin. Oncol. 2014, 32, 6021. [Google Scholar] [CrossRef]
- Garcia-Foncillas, J.; Sunakawa, Y.; Aderka, D.; Wainberg, Z.; Ronga, P.; Witzler, P.; Stintzing, S. Distinguishing Features of Cetuximab and Panitumumab in Colorectal Cancer and Other Solid Tumors. Front. Immunol. 2019, 9, 849. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A.C.; Joller, N.; Kuchroo, V.K. Lag-3, Tim-3, and TIGIT: Co-inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity 2016, 44, 989–1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dougall, W.C.; Kurtulus, S.; Smyth, M.J.; Anderson, A.C. TIGIT and CD96: New checkpoint receptor targets for cancer immunotherapy. Immunol. Rev. 2017, 276, 112–120. [Google Scholar] [CrossRef]
- Xu, F.; Sunderland, A.; Zhou, Y.; Schulick, R.D.; Edil, B.H.; Zhu, Y.W. Blockade of CD112R and TIGIT signaling sensitizes human natural killer cell functions. Cancer Immunol. Immunother. 2017, 66, 1367–1375. [Google Scholar] [CrossRef]
- Puntigam, L.K.; Jeske, S.S.; Gotz, M.; Greiner, J.; Laban, S.; Theodoraki, M.N.; Doescher, J.; Weissinger, S.E.; Brunner, C.; Hoffmann, T.K.; et al. Immune Checkpoint Expression on Immune Cells of HNSCC Patients and Modulation by Chemo- and Immunotherapy. Int. J. Mol. Sci. 2020, 21, 5181. [Google Scholar] [CrossRef] [PubMed]
- Vence, L.; Bucktrout, S.L.; Curbelo, I.F.; Blando, J.; Smith, B.M.; Mahne, A.E.; Lin, J.C.; Park, T.; Pascua, E.; Sai, T.; et al. Characterization and Comparison of GITR Expression in Solid Tumors. Clin. Cancer Res. 2019, 25, 6501–6510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leyland, R.; Watkins, A.; Mulgrew, K.A.; Holoweckyj, N.; Bamber, L.; Tigue, N.J.; Offer, E.; Andrews, J.; Yan, L.; Mullins, S.; et al. A Novel Murine GITR Ligand Fusion Protein Induces Antitumor Activity as a Monotherapy that Is Further Enhanced in Combination with an OX40 Agonist. Clin. Cancer Res. 2017, 23, 3416–3427. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Hostager, B.S.; Arkee, T.; Bishop, G.A. Multiple mechanisms for TRAF3-mediated regulation of the T cell costimulatory receptor GITR. J. Biol. Chem. 2021, 297, 101097. [Google Scholar] [CrossRef]
- Zappasodi, R.; Sirard, C.; Li, Y.Y.; Budhu, S.; Abu-Akeel, M.; Liu, C.L.; Yang, X.; Zhong, H.; Newman, W.; Qi, J.J.; et al. Rational design of anti-GITR-based combination immunotherapy. Nat. Med. 2019, 25, 759. [Google Scholar] [CrossRef]
- Davar, D.; Zappasodi, R.; Wang, H.; Naik, G.S.; Sato, T.; Bauer, T.; Bajor, D.; Rixe, O.; Newman, W.; Qi, J.J.; et al. Phase IB Study of GITR Agonist Antibody TRX518 Singly and in Combination with Gemcitabine, Pembrolizumab, or Nivolumab in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2022, 28, 3990–4002. [Google Scholar] [CrossRef]
- Heinhuis, K.M.; Carlino, M.; Joerger, M.; Di Nicola, M.; Meniawy, T.; Rottey, S.; Moreno, V.; Gazzah, A.; Delord, J.P.; Paz-Ares, L.; et al. Safety, Tolerability, and Potential Clinical Activity of a Glucocorticoid-Induced TNF Receptor-Related Protein Agonist Alone or in Combination with Nivolumab for Patients with Advanced Solid Tumors A Phase 1/2a Dose-Escalation and Cohort-Expansion Clinical Trial. JAMA Oncol. 2020, 6, 100–107. [Google Scholar] [CrossRef] [Green Version]
- Balmanoukian, A.S.; Infante, J.R.; Aljumaily, R.; Naing, A.; Chintakuntlawar, A.V.; Rizvi, N.A.; Ross, H.J.; Gordon, M.; Mallinder, P.R.; Elgeioushi, N.; et al. Safety and Clinical Activity of MEDI1873, a Novel GITR Agonist, in Advanced Solid Tumors. Clin. Cancer Res. 2020, 26, 6196–6203. [Google Scholar] [CrossRef]
- Cazzetta, V.; Franzese, S.; Carenza, C.; Della Bella, S.; Mikulak, J.; Mavilio, D. Natural Killer-Dendritic Cell Interactions in Liver Cancer: Implications for Immunotherapy. Cancers 2021, 13, 2184. [Google Scholar] [CrossRef] [PubMed]
- Restifo, N.P.; Dudley, M.E.; Rosenberg, S.A. Adoptive immunotherapy for cancer: Harnessing the T cell response. Nat. Rev. Immunol. 2012, 12, 269–281. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.Y.; Fu, T.; Jiang, Y.Z.; Shao, Z.M. Natural killer cells in cancer biology and therapy. Mol. Cancer 2020, 19, 120. [Google Scholar] [CrossRef]
- Wang, W.X.; Jiang, J.T.; Wu, C.P. CAR-NK for tumor immunotherapy: Clinical transformation and future prospects. Cancer Lett. 2020, 472, 175–180. [Google Scholar] [CrossRef]
- Fesnak, A.D.; June, C.H.; Levine, B.L. Engineered T cells: The promise and challenges of cancer immunotherapy. Nat. Rev. Cancer 2016, 16, 566–581. [Google Scholar] [CrossRef]
- Jin, B.Y.; Campbell, T.E.; Draper, L.M.; Stevanovic, S.; Weissbrich, B.; Yu, Z.Y.; Restifo, N.P.; Rosenberg, S.A.; Trimble, C.L.; Hinrichs, C.S. Engineered T cells targeting E7 mediate regression of human papillomavirus cancers in a murine model. JCI Insight 2018, 3, e99488. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.Y.; Park, J.H. Engineered immune cells with nanomaterials to improve adoptive cell therapy. Biomed. Eng. Lett. 2021, 11, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Houot, R.; Schultz, L.M.; Marabelle, A.; Kohrt, H. T-cell-based Immunotherapy: Adoptive Cell Transfer and Checkpoint Inhibition. Cancer Immunol. Res. 2015, 3, 1115–1122. [Google Scholar] [CrossRef] [Green Version]
- Scarfo, I.; Maus, M.V. Current approaches to increase CAR T cell potency in solid tumors: Targeting the tumor microenvironment. J. Immunother. Cancer 2017, 5, 28. [Google Scholar] [CrossRef] [Green Version]
- Martinez, M.; Moon, E.K. CAR T Cells for Solid Tumors: New Strategies for Finding, Infiltrating, and Surviving in the Tumor Microenvironment. Front. Immunol. 2019, 10, 128. [Google Scholar] [CrossRef] [Green Version]
- Gustafson, M.P.; Ligon, J.A.; Bersenev, A.; McCann, C.D.; Shah, N.N.; Hanley, P.J. Emerging frontiers in immuno- and gene therapy for cancer. Cytotherapy 2023, 25, 20–32. [Google Scholar] [CrossRef] [PubMed]
- Mei, Z.; Zhang, K.; Lam, A.K.; Huang, J.; Qiu, F.; Qiao, B.; Zhang, Y. MUC1 as a target for CAR-T therapy in head and neck squamous cell carcinoma. Cancer Med. 2020, 9, 640–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selli, M.E.; Dalal, P.; Neelapu, S.; Singh, N. Mechanisms of Resistance and Relapse After CAR-T Cell Therapy. In Cancer Drug Discovery and Development; Humana Press Inc.: Totowa, NJ, USA, 2022; pp. 207–219. [Google Scholar] [CrossRef]
- Scarth, J.A.; Patterson, M.R.; Morgan, E.L.; Macdonald, A. The human papillomavirus oncoproteins: A review of the host pathways targeted on the road to transformation. J. Gen. Virol. 2021, 102, 001540. [Google Scholar] [CrossRef]
- Davis, Z.B.; Felices, M.; Verneris, M.R.; Miller, J.S. Natural Killer Cell Adoptive Transfer Therapy Exploiting the First Line of Defense Against Cancer. Cancer J. 2015, 21, 486–491. [Google Scholar] [CrossRef] [Green Version]
- Marofi, F.; Abdul-Rasheed, O.F.; Rahman, H.S.; Budi, H.S.; Jalil, A.T.; Yumashev, A.V.; Hassanzadeh, A.; Yazdanifar, M.; Motavalli, R.; Chartrand, M.S.; et al. CAR-NK cell in cancer immunotherapy; A promising frontier. Cancer Sci. 2021, 112, 3427–3436. [Google Scholar] [CrossRef]
- Biederstadt, A.; Rezvani, K. Engineering the next generation of CAR-NK immunotherapies. Int. J. Hematol. 2021, 114, 554–571. [Google Scholar] [CrossRef]
- Hu, Z.; Ott, P.A.; Wu, C.J. Towards personalized, tumour-specific, therapeutic vaccines for cancer. Nat. Rev. Immunol. 2018, 18, 168–182. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.W.; Ferrall, L.; Gaillard, S.; Wang, C.G.; Chi, W.Y.; Huang, C.H.; Roden, R.B.S.; Wu, T.C.; Chang, Y.N.; Hung, C.F. Development of DNA Vaccine Targeting E6 and E7 Proteins of Human Papillomavirus 16 (HPV16) and HPV18 for Immunotherapy in Combination with Recombinant Vaccinia Boost and PD-1 Antibody. Mbio 2021, 12, e03224-20. [Google Scholar] [CrossRef]
- Aggarwal, C.; Cohen, R.B.; Morrow, M.P.; Kraynyak, K.A.; Sylvester, A.J.; Knoblock, D.M.; Bauml, J.M.; Weinstein, G.S.; Lin, A.; Boyer, J.; et al. Immunotherapy Targeting HPV16/18 Generates Potent Immune Responses in HPV-Associated Head and Neck Cancer. Clin. Cancer Res. 2019, 25, 110–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aggarwal, C.; Saba, N.F.; Algazi, A.P.; Sukari, A.; Seiwert, T.; Haigentz, M.; Porosnicu, M.; Bonomi, M.; Boyer, J.; Durham, N.; et al. Safety and efficacy of MEDI0457 plus durvalumab in patients (pts) with human papillomavirus-associated recurrent/metastatic head and neck squamous cell carcinoma (HPV plus R/M HNSCC). Ann. Oncol. 2020, 31, S661–S662. [Google Scholar] [CrossRef]
- Massarelli, E.; William, W.; Johnson, F.; Kies, M.; Ferrarotto, R.; Guo, M.; Feng, L.; Lee, J.J.; Tran, H.; Kim, Y.U.; et al. Combining Immune Checkpoint Blockade and Tumor-Specific Vaccine for Patients with Incurable Human Papillomavirus 16-Related Cancer A Phase 2 Clinical Trial. JAMA Oncol. 2019, 5, 67–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chester, C.; Sanmamed, M.F.; Wang, J.; Melero, I. Immunotherapy targeting 4-1BB: Mechanistic rationale, clinical results, and future strategies. Blood 2018, 131, 49–57. [Google Scholar] [CrossRef]
- Srivastava, R.M.; Trivedi, S.; Concha-Benavente, F.; Gibson, S.P.; Reeder, C.; Ferrone, S.; Ferris, R.L. CD137 Stimulation Enhances Cetuximab-Induced Natural Killer: Dendritic Cell Priming of Antitumor T-Cell Immunity in Patients with Head and Neck Cancer. Clin. Cancer Res. 2017, 23, 707–716. [Google Scholar] [CrossRef] [Green Version]
- Le Tourneau, C.; Delord, J.P.; Cassier, P.; Loirat, D.; Tavernaro, A.; Bastien, B.; Bendjama, K. Phase Ib/II trial of TG4001 (Tipapkinogene sovacivec), a therapeutic HPV-vaccine, and Avelumab in patients with recurrent/metastatic (R/M) HPV-16+ cancers. Ann. Oncol. 2019, 30, v494–v495. [Google Scholar] [CrossRef]
- Chandra, J.; Woo, W.P.; Finlayson, N.; Liu, H.Y.; McGrath, M.; Ladwa, R.; Brauer, M.; Xu, Y.; Hanson, S.; Panizza, B.; et al. A phase 1, single centre, open label, escalating dose study to assess the safety, tolerability and immunogenicity of a therapeutic human papillomavirus (HPV) DNA vaccine (AMV002) for HPV-associated head and neck cancer (HNC). Cancer Immunol. Immunoth. 2021, 70, 743–753. [Google Scholar] [CrossRef]
- Byeon, H.K.; Ku, M.; Yang, J. Beyond EGFR inhibition: Multilateral combat strategies to stop the progression of head and neck cancer. Exp. Mol. Med. 2019, 51, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Ostrozka-Cieslik, A.; Sarecka-Hujar, B. The Use of Nanotechnology in Modern Pharmacotherapy. In Multifunctional Systems for Combined Delivery, Biosensing and Diagnostics; Grumezescu, A.M., Ed.; Elsevier Science BV: Amsterdam, The Netherlands, 2017; pp. 139–158. [Google Scholar] [CrossRef]
- Alasvand, N.; Urbanska, A.M.; Rahmati, M.; Saeidifar, M.; Gungor-Ozkerim, P.S.; Sefat, F.; Rajadas, J.; Mozafari, M. Therapeutic Nanoparticles for Targeted Delivery of Anticancer Drugs. In Multifunctional Systems for Combined Delivery, Biosensing and Diagnostics; Elsevier Science BV: Amsterdam, The Netherlands, 2017; pp. 245–259. [Google Scholar] [CrossRef]
- Zhao, H.; Li, Y.Q.; Wei, D.; Luo, H.R. The Application of Nanoparticle-Based Drug Delivery Systems in Checkpoint Blockade Cancer Immunotherapy. J. Immuno. Res. 2018, 2018, 3673295. [Google Scholar] [CrossRef]
- Andrade, L.M.; Martins, E.M.N.; Versiani, A.F.; Reis, D.S.; da Fonseca, F.G.; de Souza, I.P.; Paniago, R.M.; Pereira-Maia, E.; Ladeira, L.O. The physicochemical and biological characterization of a 24-month-stored nanocomplex based on gold nanoparticles conjugated with cetuximab demonstrated long-term stability, EGFR affinity and cancer cell death due to apoptosis. Mater. Sci. Eng. C. 2020, 107, 110203. [Google Scholar] [CrossRef]
- Zhou, J.J.; Li, X.H.; He, P.Y.; Qi, F.Y.; Ullah, M.W.; Li, S.J.; Liu, Y.T.; Bu, L.L.; Yang, G.; Sun, Z.J. Implantable versatile oxidized bacterial cellulose membrane for postoperative HNSCC treatment via photothermal-boosted immunotherapy. Nano Res. 2023, 16, 951–963. [Google Scholar] [CrossRef]
- Altintas, I.; Heukers, R.; van der Meel, R.; Lacombe, M.; Amidi, M.; Henegouwen, P.; Hennink, W.E.; Schiffelers, R.M.; Kok, R.J. Nanobody-albumin nanoparticles (NANAPs) for the delivery of a multikinase inhibitor 17864 to EGFR overexpressing tumor cells. J. Control. Release 2013, 165, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Ocadlikova, D.; Lecciso, M.; Broto, J.M.; Scotlandi, K.; Cavo, M.; Curti, A.; Palmerini, E. Sunitinib Exerts In Vitro Immunomodulatory Activity on Sarcomas via Dendritic Cells and Synergizes With PD-1 Blockade. Front. Immunol. 2021, 12, 577766. [Google Scholar] [CrossRef] [PubMed]
- Riestra-Ayora, J.; Sánchez-Rodríguez, C.; Palao-Suay, R.; Yanes-Díaz, J.; Martín-Hita, A.; Aguilar, M.R.; Sanz-Fernández, R. Paclitaxel-loaded polymeric nanoparticles based on α-tocopheryl succinate for the treatment of head and neck squamous cell carcinoma: In vivo murine model. Drug Deliv. 2021, 28, 1376–1388. [Google Scholar] [CrossRef] [PubMed]
- Thakur, N.; Thakur, S.; Chatterjee, S.; Das, J.; Sil, P.C. Nanoparticles as Smart Carriers for Enhanced Cancer Immunotherapy. Front. Chem. 2020, 8, 597806. [Google Scholar] [CrossRef]
- Yang, Z.G.; Ma, Y.F.; Zhao, H.; Yuan, Y.; Kim, B.Y.S. Nanotechnology platforms for cancer immunotherapy. Wiley Interdiscip. Rev. Nanomed. 2020, 12, e1590. [Google Scholar] [CrossRef]
- Buss, C.G.; Bhatia, S.N. Nanoparticle delivery of immunostimulatory oligonucleotides enhances response to checkpoint inhibitor therapeutics. Proc. Natl. Acad. Sci. USA 2020, 117, 13428–13436. [Google Scholar] [CrossRef]
- Espinosa-Cotton, M.; Rodman, S.N.; Ross, K.A.; Jensen, I.J.; Sangodeyi-Miller, K.; McLaren, A.J.; Dahl, R.A.; Gibson-Corley, K.N.; Koch, A.T.; Fu, Y.X.; et al. Interleukin-1 alpha increases anti-tumor efficacy of cetuximab in head and neck squamous cell carcinoma. J. Immunother. Cancer 2019, 7, 79. [Google Scholar] [CrossRef] [Green Version]
- Li, S.Y.; Liu, Y.; Xu, C.F.; Shen, S.; Sun, R.; Du, X.J.; Xia, J.X.; Zhu, Y.H.; Wang, J. Restoring anti-tumor functions of T cells via nanoparticle-mediated immune checkpoint modulation. J. Control. Release 2016, 231, 17–28. [Google Scholar] [CrossRef]
- Chen, H.W.; Luan, X.; Paholak, H.J.; Burnett, J.P.; Stevers, N.O.; Sansanaphongpricha, K.; He, M.; Chang, A.E.; Li, Q.; Sun, D.X. Depleting tumor-associated Tregs via nanoparticle-mediated hyperthermia to enhance anti-CTLA-4 immunotherapy. Nanomedicine 2020, 15, 77–92. [Google Scholar] [CrossRef]
- Nascimento, C.S.; Alves, E.A.R.; de Melo, C.P.; Correa-Oliveira, R.; Calzavara-Silva, C.E. Immunotherapy for cancer: Effects of iron oxide nanoparticles on polarization of tumor-associated macrophages. Nanomedicine 2021, 16, 2633–2650. [Google Scholar] [CrossRef]
- Tan, Y.S.; Sansanaphongpricha, K.; Xie, Y.Y.; Donnelly, C.R.; Luo, X.B.; Heath, B.R.; Zhao, X.Y.; Bellile, E.; Hu, H.X.; Chen, H.W.; et al. Mitigating SOX2-potentiated Immune Escape of Head and Neck Squamous Cell Carcinoma with a STING-inducing Nanosatellite Vaccine. Clin. Cancer Res. 2018, 24, 4242–4255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seiwert, T.Y.; Foster, C.C.; Le Tourneau, C.; Calugaru, V.; Bonvalot, S. Hafnium oxide nanoparticles activated by SABR in combination with PD-1 inhibitors for the treatment of patients with advanced HNSCC or NSCLC: A phase I/II trial. J. Clin. Oncol. 2019, 37, TPS23. [Google Scholar] [CrossRef]
- Bu, L.L.; Wang, H.Q.; Pan, Y.W.; Chen, L.; Wu, H.; Wu, X.J.; Zhao, C.C.; Rao, L.; Liu, B.; Sun, Z.J. Gelatinase-sensitive nanoparticles loaded with photosensitizer and STAT3 inhibitor for cancer photothermal therapy and immunotherapy. J. Nanobiotechnology 2021, 19, 379. [Google Scholar] [CrossRef] [PubMed]
- Wallington, D.G.; Contessa, J.N.; Hayman, T.J. STING Agonists in Head and Neck Squamous Cell Carcinoma. Cancer J. 2022, 28, 401–406. [Google Scholar] [CrossRef]
- Xu, Q.; Fang, M.Y.; Zhu, J.; Dong, H.R.; Cao, J.; Yan, L.; Leonard, F.; Oppel, F.; Sudhoff, H.; Kaufmann, A.M.; et al. Insights into Nanomedicine for Immunotherapeutics in Squamous Cell Carcinoma of the head and neck. Int. J. Biol. Sci. 2020, 16, 2506–2517. [Google Scholar] [CrossRef]
- Xu, Q.; Chen, Y.; Jin, Y.; Wang, Z.Y.; Dong, H.R.; Kaufmann, A.M.; Albers, A.E.; Qian, X. Advanced Nanomedicine for High-Risk HPV-Driven Head and Neck Cancer. Viruses 2022, 14, 2824. [Google Scholar] [CrossRef]
- Goldberg, M.S. Improving cancer immunotherapy through nanotechnology. Nat. Rev. Cancer 2019, 19, 587–602. [Google Scholar] [CrossRef]
- Grunwitz, C.; Salomon, N.; Vascotto, F.; Selmi, A.; Bukur, T.; Diken, M.; Kreiter, S.; Tureci, O.; Sahin, U. HPV16 RNA-LPX vaccine mediates complete regression of aggressively growing HPV-positive mouse tumors and establishes protective T cell memory. Oncoimmunology 2019, 8, e1629259. [Google Scholar] [CrossRef] [Green Version]
- Parmar, K.; Mohamed, A.; Vaish, E.; Thawani, R.; Cetnar, J.; Thein, K.Z. Immunotherapy in head and neck squamous cell carcinoma: An updated review. Cancer Treat. Res. Commun. 2022, 33, 100649. [Google Scholar] [CrossRef]
- Luo, M.; Wang, H.; Wang, Z.H.; Cai, H.C.; Lu, Z.G.; Li, Y.; Du, M.J.; Huang, G.; Wang, C.S.; Chen, X.; et al. A STING-activating nanovaccine for cancer immunotherapy. Nat. Nanotechnol. 2017, 12, 648. [Google Scholar] [CrossRef] [PubMed]
- Kohnepoushi, C.; Nejati, V.; Delirezh, N.; Biparva, P. Poly Lactic-co-Glycolic Acid Nanoparticles Containing Human Gastric Tumor Lysates as Antigen Delivery Vehicles for Dendritic Cell-Based Antitumor Immunotherapy. Immunol. Investig. 2019, 48, 794–808. [Google Scholar] [CrossRef]
- Iranpour, S.; Nejati, V.; Delirezh, N.; Biparva, P.; Shirian, S. Enhanced stimulation of anti-breast cancer T cells responses by dendritic cells loaded with poly lactic-co-glycolic acid (PLGA) nanoparticle encapsulated tumor antigens. J. Exp. Clin. Cancer Res. 2016, 35, 168. [Google Scholar] [CrossRef] [Green Version]
- Dharmaraj, N.; Piotrowski, S.L.; Huang, C.; Newton, J.M.; Golfman, L.S.; Hanoteau, A.; Koshy, S.T.; Li, A.W.; Pulikkathara, M.X.; Zhang, B.; et al. Anti-tumor immunity induced by ectopic expression of viral antigens is transient and limited by immune escape. Oncoimmunology 2019, 8, e1568809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, E.E.W.; Mell, L.K.; Bell, R.B.; Bifulco, C.B.; Leidner, R.; Burtness, B.; Gillison, M.L.; Harrington, K.J.; Le, Q.-T.; Lee, N.Y.; et al. The Society for Immunotherapy of Cancer consensus statement on immunotherapy for the treatment of squamous cell carcinoma of the head and neck (HNSCC). J. Immunother. Cancer. 2019, 7, 184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borcoman, E.; Marret, G.; Le Tourneau, C. Paradigm Change in First-Line Treatment of Recurrent and/or Metastatic Head and Neck Squamous Cell Carcinoma. Cancers 2021, 13, 2573. [Google Scholar] [CrossRef]
- Yilmaz, E.; Ismaila, N.; Bauman, J.E.; Dabney, R.; Gan, G.; Jordan, R.; Kaufman, M.; Kirtane, K.; McBride, S.M.; Old, M.O.; et al. Immunotherapy and Biomarker Testing in Recurrent and Metastatic Head and Neck Cancers: ASCO Guideline. Am. J. Clin. Oncol. 2023, 41, 1132–1146. [Google Scholar] [CrossRef]
- Fasano, M.; Corte, C.M.D.; Liello, R.D.; Viscardi, G.; Sparano, F.; Iacovino, M.L.; Paragliola, F.; Piccolo, A.; Napolitano, S.; Martini, G.; et al. Immunotherapy for head and neck cancer: Present and future. Crit. Rev. Oncol. Hematol. 2022, 174, 103679. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, Z. The history and advances in cancer immunotherapy: Understanding the characteristics of tumor-infiltrating immune cells and their therapeutic implications. Cell. Mol. Immunol 2020, 17, 807–821. [Google Scholar] [CrossRef]
- Atkinson, V.; Khattak, A.; Haydon, A.; Eastgate, M.; Roy, A.; Prithviraj, P.; Mueller, C.; Brignone, C.; Triebel, F. Eftilagimod alpha, a soluble lymphocyte activation gene-3 (LAG-3) protein plus pembrolizumab in patients with metastatic melanoma. J. Immunother. Cancer 2020, 8, e001681. [Google Scholar] [CrossRef]
- Majem, M.; Felip, E.; Doger, B.; Akay, M.; Carcereny, E.; Clay, T.; Krebs, M.G.; Peguero, J.; Triebel, F. 1266P Initial results from a phase II study (TACTI-002) of eftilagimod alpha (soluble LAG-3 protein) and pembrolizumab in patients with PD-L1 unselected first-line metastatic non-small cell lung carcinoma. Ann. Oncol. 2020, 31, S818. [Google Scholar] [CrossRef]
- Siu, L.L.; Burtness, B.; Cohen, E.E.W.; Harrington, K.J.; Licitra, L.F.; Rischin, D.; Zhu, Y.; Lee, C.P.; Pinheiro, C.; Swaby, R.F.; et al. Phase III LEAP-010 study: First-line pembrolizumab with or without lenvatinib in recurrent/metastatic (R/M) head and neck squamous cell carcinoma (HNSCC). J. Clin. Oncol. 2020, 38 (Suppl. 15), TPS6589. [Google Scholar] [CrossRef]
- Rischin, D.; Groenland, S.L.; Lim, A.M.L.; Martin-Liberal, J.; Moreno, V.; Trigo Perez, J.M.; Le Tourneau, C.; Mathew, M.; Cho, D.C.; Hansen, A.R.; et al. 119PD—Inducible T cell costimulatory (ICOS) receptor agonist, GSK3359609 (GSK609) alone and in combination with pembrolizumab (pembro): Preliminary results from INDUCE-1 expansion cohorts (EC) in head and neck squamous cell carcinoma (HNSCC). Ann. Oncol. 2019, 30, 454–455. [Google Scholar] [CrossRef]
- Massarelli, E.; Balmanoukian, A.S.; Vieito, M.; Le Tourneau, C.; Hernandez-Guerrero, T.; Trigo, J.M.; Aljumaily, R.; Chisamore, M.J.; Rogan, D.; Sung, R.; et al. INDUCE-1: Report on safety run-in cohorts combining Inducible T-cell co-stimulatory receptor (ICOS) agonist GSK3359609 (GSK609) with platinum+5-FU chemotherapy (5-FU/plat), with or without pembrolizumab (PE), for the treatment of advanced solid tumors. J. Clin. Oncol. 2020, 38 (Suppl. 15), 6544. [Google Scholar] [CrossRef]
- Villanueva, L.; Álvarez-Errico, D.; Esteller, M. The contribution of epigenetics to cancer immunotherapy. Trends Immunol. 2020, 41, 676–691. [Google Scholar] [CrossRef]






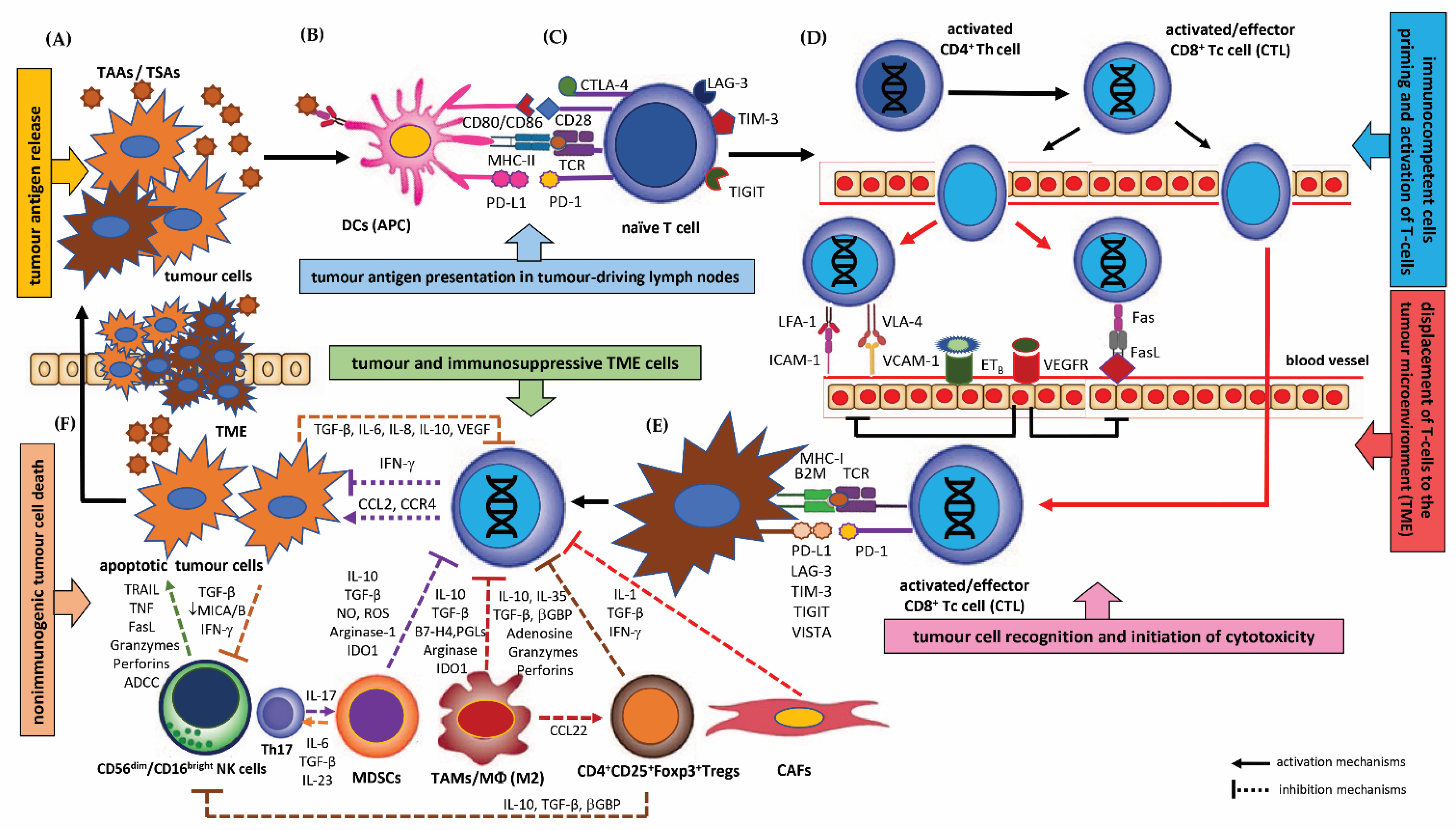{kind=link}
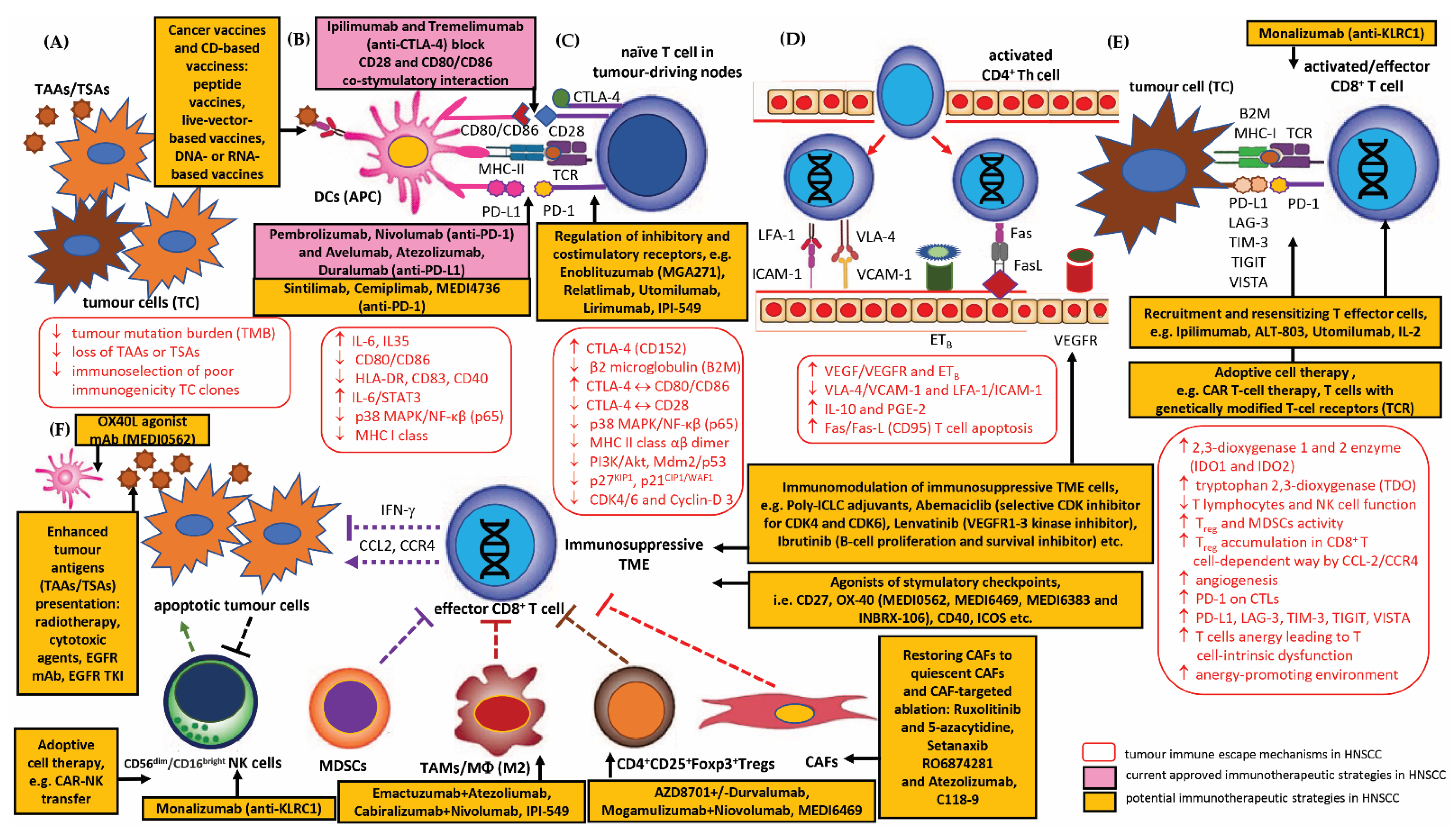{kind=link}
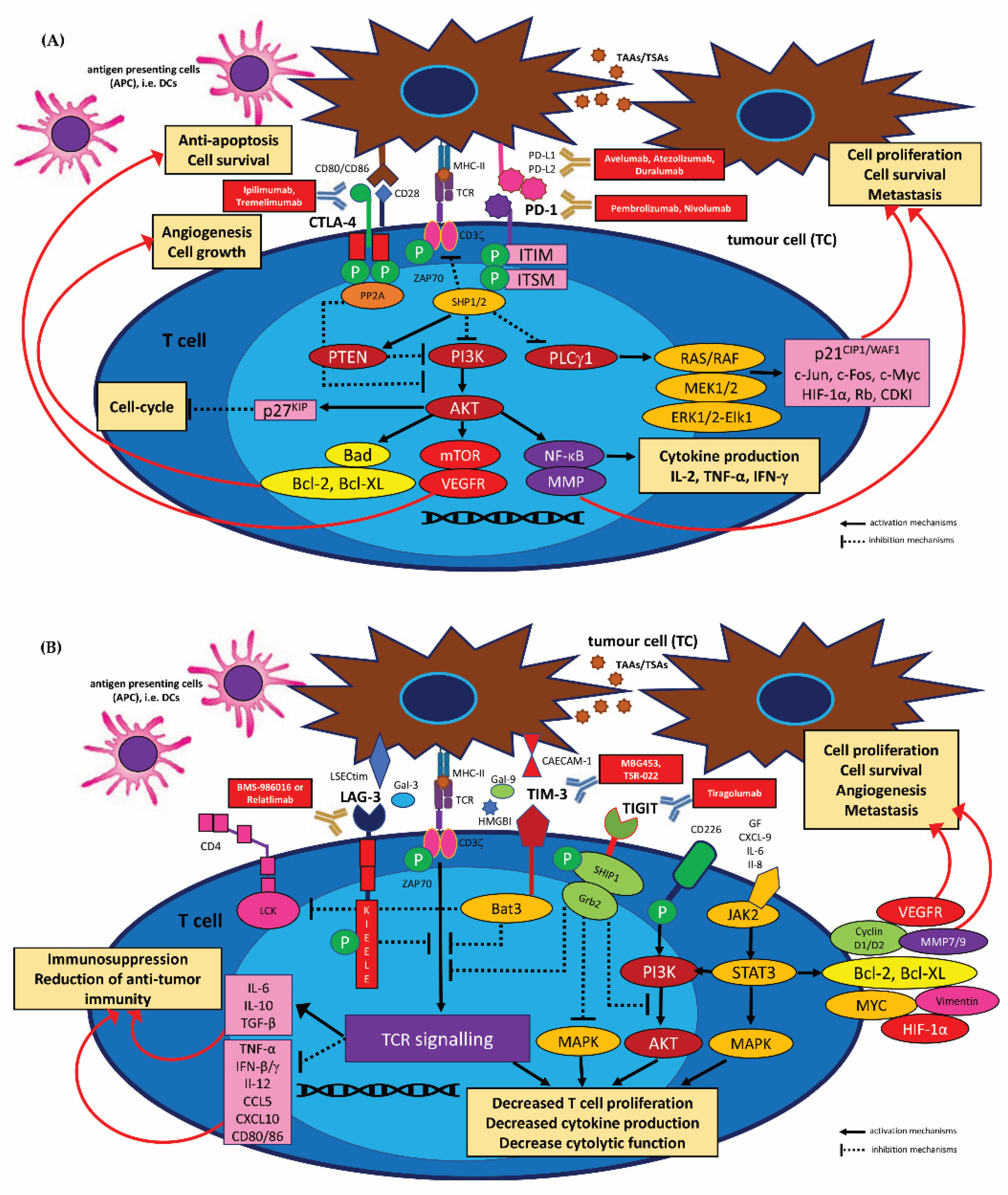{kind=link}
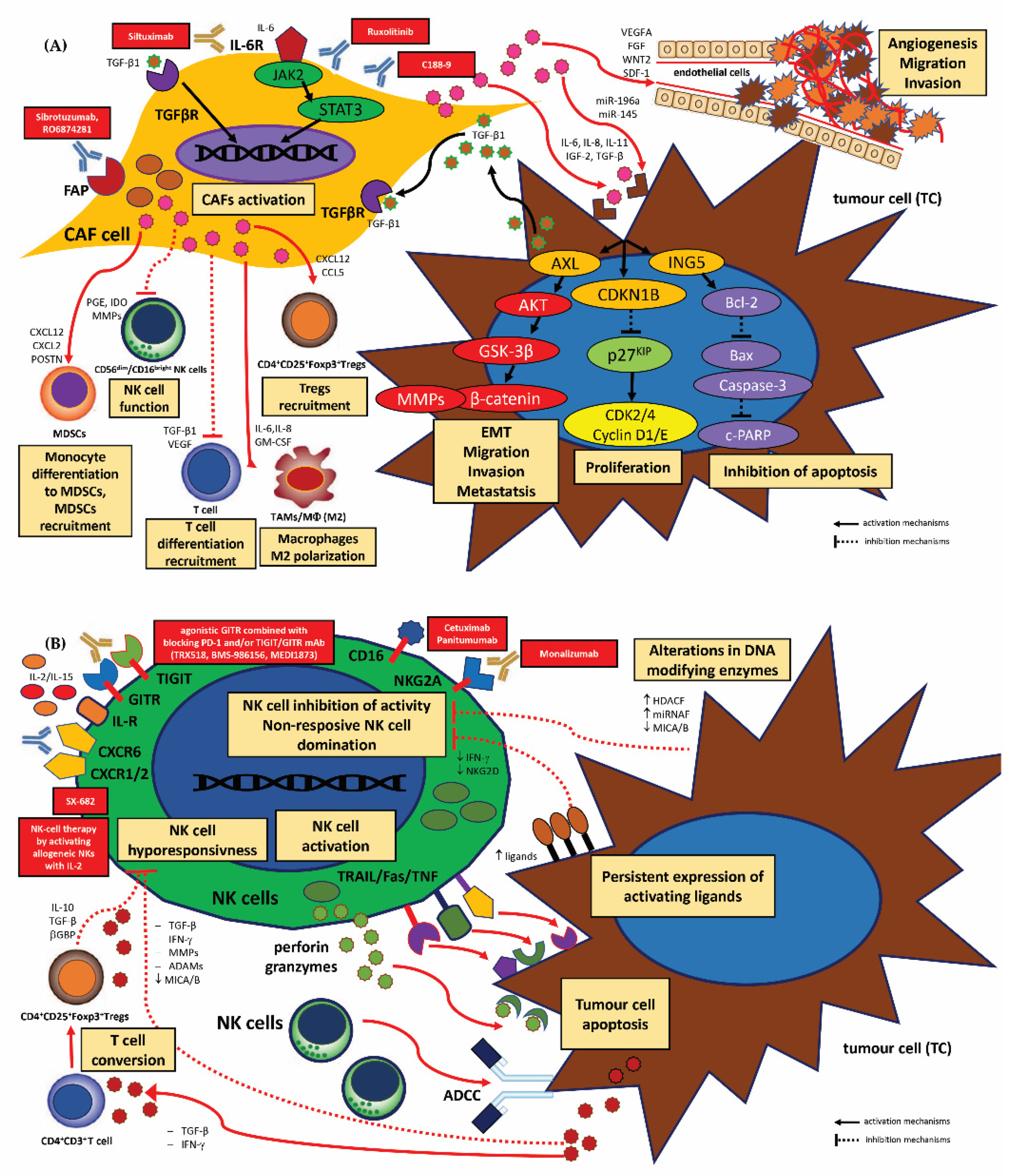{kind=link}
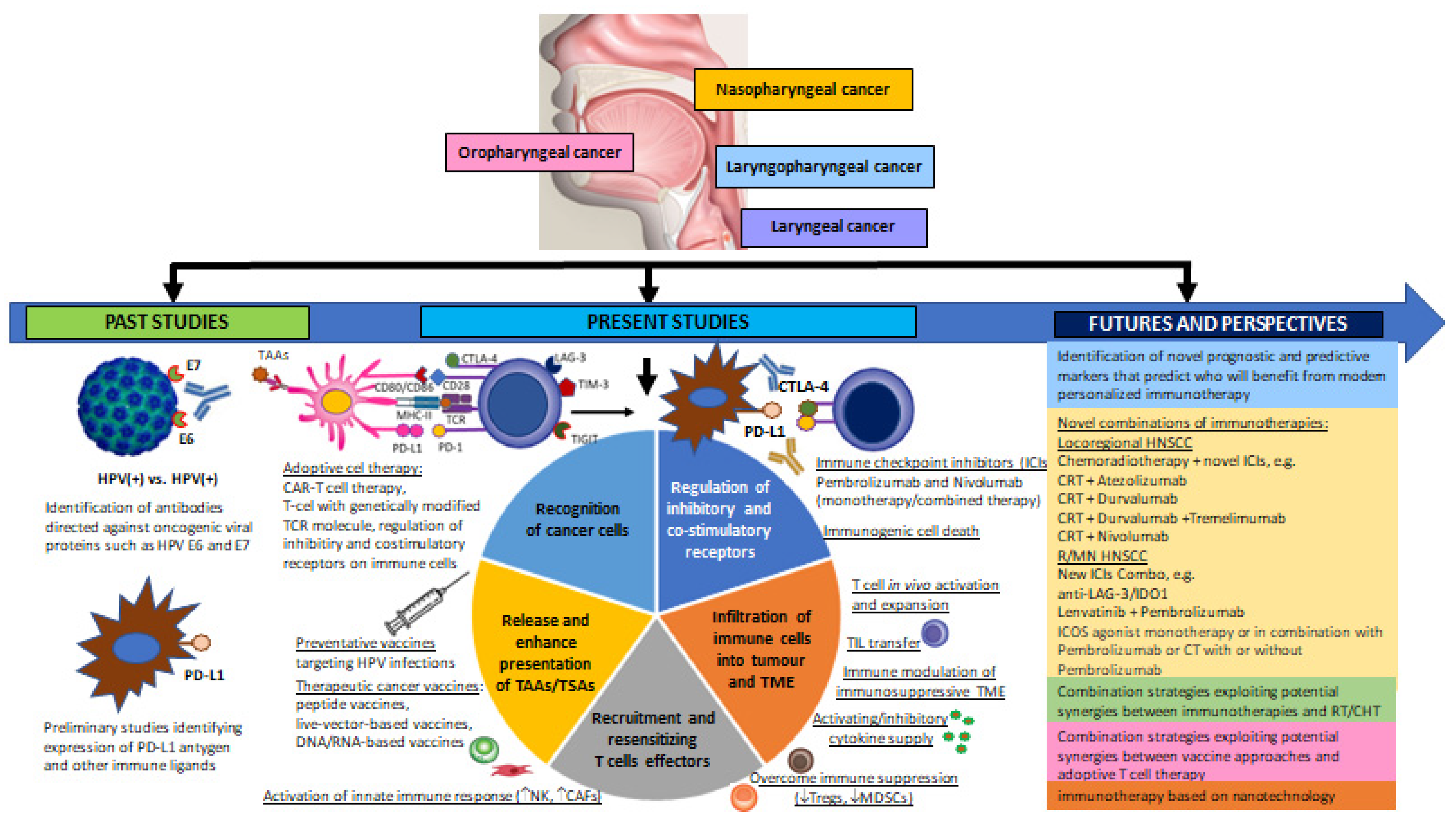{kind=link}
| Immune Cells | Markers of Immune Cells | Mechanisms | |
|---|---|---|---|
| Cytokine and Other Factors | Functions | ||
| Tumour-Antagonizing Immune Cell Activity | |||
| APCs (DCs) | CD11c+/CD11b+/CD8−/CD11a/CD15s/CD18/CD29/CD44/CD49d/CD50/CD54 | IRF7, NF-κB activation, IFN-α, IL-6, IL-8, TNF-α |
|
| Effector T cells | CD3+/CD4+ T cells Th1 phenotype lymphocytes | IFN-γ |
|
| CD3+/CD4+ T cells Th2 phenotype lymphocytes | IL-4, IL-5, IL-13 |
| |
| CD3+/CD4+ T cells Th17 phenotype lymphocytes | IL-17A, IL-17F |
| |
| CD3+/CD8+ T cytotoxic cells CTLs | perforin/granzyme B and granulysin release, IFN-γ, TNF-α |
| |
| NK cells | CD3−/CD16+/CD56+ | IFN-γ, TNF-α, GM-SCF, IL-5, IL-6, IL-8, IL-10, IL-13, CCL2, CCL3, CCL4, CCL5, CXCL10 |
|
| NKT cells | CD3+/CD16+/CD56+/CD161+/CD1a+ | IFN-γ, TNF-α, GM-SCF, TGF-β, IL-2, IL-4, IL-5, IL-6, IL-10, IL-13, IL-17A |
|
| M1 TAMs/MФ | CD68+ | IL-12, IL-23, TNF-α, CCL2, CCL5, CXCL5, CXCL9, CXCL10, CXCL16 PD-L1/PD-L2 expression MMP-9 release |
|
| N1 TANs | CD66b+/CD11b+/CD14−/ HLA-DR+/CD177+/CD15hig | ROS secretion ICAM-1, TNF-α cytokine secretion Fas signalling MMP-8 release |
|
| Tumour-Promoting Immune Cell Activity | |||
| M2 TAMs/MФ | CD68+ | IL-1ra, IL-4, IL-13 IL-10, TGF-β, PPARγ, VEGF, arginase-1 (Arg-1), IDO |
|
| N2 TANs | CD15+/HLA-DR+/CD11b+/CD14−/CD33+/Lox-1+ | ROS secretion chemokine/cytokine (CXCR4, MMP-9, VEGF, Lox1, FATP2) neutrophil elastase (NE) production NET formation cathepsin G, MMP-8/9 and VEGF secretion Arginase-1 (Arg-1) secretion |
|
| MDSCs | CD11b+/CD33+/CD14+/ CD15+/CD16+/HLA-DR−/ CD3−/CD19−/CD56− | NO, ROS, HIF-1α, iNOS, Arginase-1 (Arg-1) secretion IL-10, TGF-β1 secretion MIF, COX2, PGF2 (PGLs), SCF, M-CSF, IDO1, IL-6, IL-1β, IL-4, IL-13, TNF-α, GM-CSF, VEGF, PD-L1 expression MMP-9 release |
|
| Tregs | CD4+/CD25+/Foxp3+ | IL-10, TGF-β1, IL-35 secretion perforin/granzyme A/B release VEGF expression |
|
| CAFs | αSMA+/FAP+/FSP-1+/CD33−CAV-1/FSP-1/PDGFR-α/ PDGFR-β/Thy-1 absent cytokeratin (CK) | EGF, HGF, VEGF, MCP1, Ly6c, COX2, PGE2, BDNF, MFAP5, CXCL1, CXCL12, CXCL14, CCL2, CCL5, CCL7 IL-6, IL-17A, TGF-β1 and 2 secretion MMP2/3, vimentin release |
|
| Immune Settings | Immune Parameters | HPV(-)neg HNSCC | HPV(+)pos HNSCC |
|---|---|---|---|
| Immune cells | CD3+ zeta chain T cells (CD3ζ) | Lower (↓) | Higher (↑) |
| CD4+ T cells/ T cell proliferation and Th1,Th17 differentiation | Lower (↓)/Inhibition | Higher (↑)/Stimulation | |
| CD8+ IFN-γ T cells (CTLs) | Lower (↓) | Higher (↑) | |
| CD4+/CD8+ ratio | Increased (↑) | Decreased (↓) | |
| CD45RO+ cells | Lower (↓) | Higher (↑) | |
| CD3+ IL-17 T cells | Lower (↓) | Higher (↑) | |
| CD19+/CD20+ B cells | Lower (↓) | Higher (↑) | |
| CD4+CD25+Foxp3+Tregs | Higher (↑) | Lower (↓) | |
| CD56dim NK cells | Lower (↓) | Higher (↑) | |
| TI APCs/DCs | Lower (↓) | Higher (↑) | |
| TAMs CD68+ M1/M2 phenotype | M2 domination | M1 domination | |
| Myeloid dendritic cells (MDCs) | Lower (↓) | Higher (↑) | |
| Cytokines/ Chemokines | Secretion of chemokines via an IL-1/IL-1R-mediated axis | Higher (↑) | Lower (↓) |
| Neutrophil-specific chemokines (CXCL1, −5, −6, −8) | Higher (↑) | Lower (↓) | |
| Monocyte-specific chemokines (CCL7, −8) | Higher (↑) | Lower (↓) | |
| T-lymphocyte chemokines (CCL3, −4, −5) | Higher (↑) | Lower (↓) | |
| Expression of co-stimulatory CD80 and CD83 molecules on immature DCs | Lower (↓) | Higher (↑) | |
| Inhibitory cytokine IL-10, TGF-β, IL-6 production | Higher (↑) | Lower (↓) | |
| Cytokine IL-2 and/or IFN-γ production | Absence | Presence | |
| Response to mitogens or IL-2 | Decreased (↓) | Increased (↑) | |
| Level of cytotoxic mediators (granzyme A, granzyme B, perforins) | Lower (↓) | Higher (↑) | |
| Antigens/Transcription factors | Aberrant activation of the STAT3 and NF-ĸB related to TGF-β, IL-6 signalling | Increased (↑) | Decreased (↓) |
| HLA-DR expression | Decreased (↓) | Increased (↑) | |
| Immune inhibitory checkpoint ligand and receptors | T cell exhaustion markers: PD-1, CTLA-4, TIM-3, LAG-3, IDO1, KIR, TIGIT | Increased (↑) | Decreased (↓) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Starska-Kowarska, K. The Role of Different Immunocompetent Cell Populations in the Pathogenesis of Head and Neck Cancer—Regulatory Mechanisms of Pro- and Anti-Cancer Activity and Their Impact on Immunotherapy. Cancers 2023, 15, 1642. https://doi.org/10.3390/cancers15061642
Starska-Kowarska K. The Role of Different Immunocompetent Cell Populations in the Pathogenesis of Head and Neck Cancer—Regulatory Mechanisms of Pro- and Anti-Cancer Activity and Their Impact on Immunotherapy. Cancers. 2023; 15(6):1642. https://doi.org/10.3390/cancers15061642
Chicago/Turabian StyleStarska-Kowarska, Katarzyna. 2023. "The Role of Different Immunocompetent Cell Populations in the Pathogenesis of Head and Neck Cancer—Regulatory Mechanisms of Pro- and Anti-Cancer Activity and Their Impact on Immunotherapy" Cancers 15, no. 6: 1642. https://doi.org/10.3390/cancers15061642