
Treatment Strategies for KRAS-Mutated Non-Small-Cell Lung Cancer
 ,
, 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Oncogenic KRAS Mutations
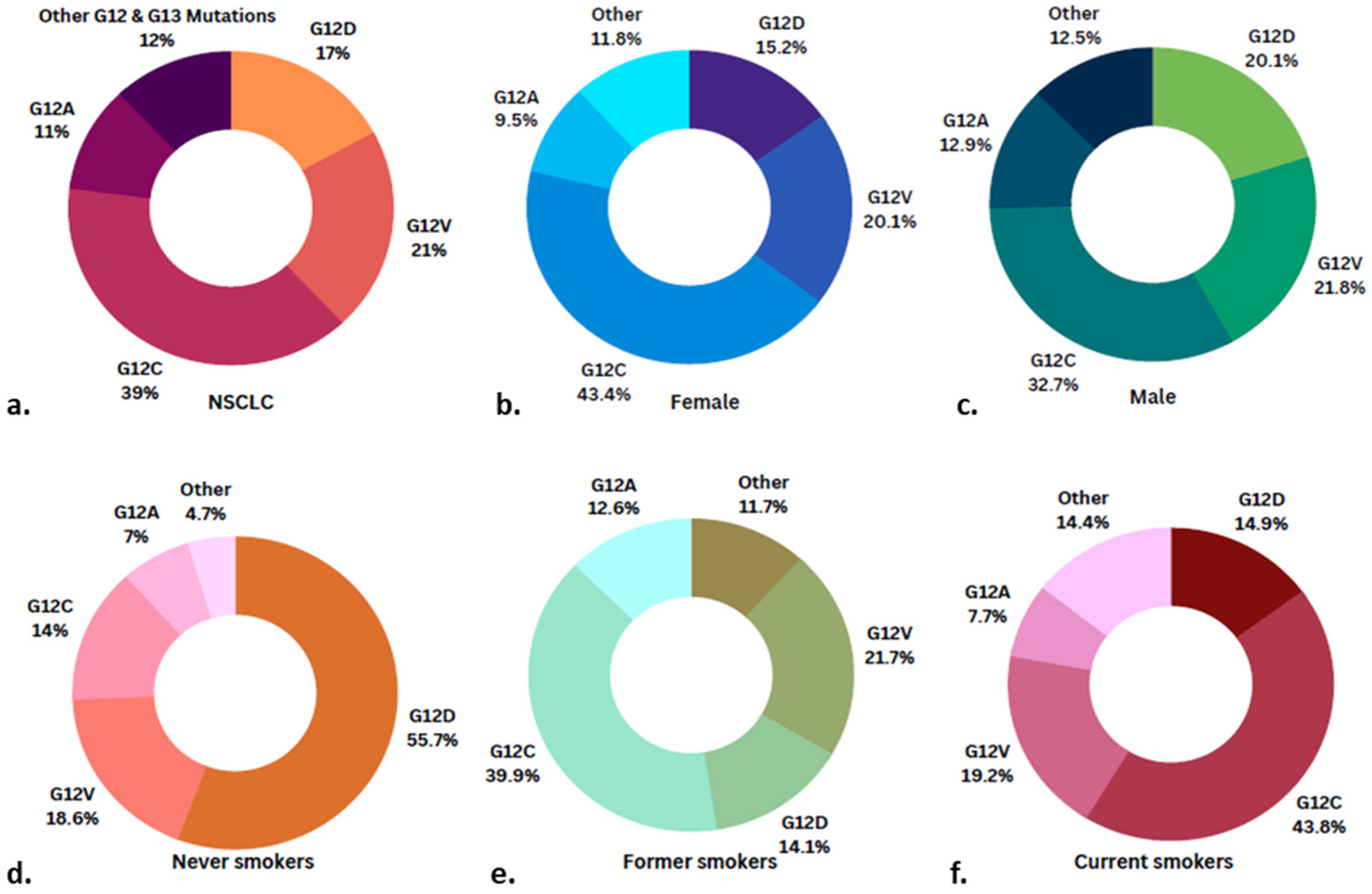
2.1. KRAS Mutations: Types and Prevalence
2.2. Genetic Alterations Co-Occurring with KRAS Mutations
3. KRAS a Therapeutic Target in Treatment Resistant Lung Adenocarcinomas (LUAD)
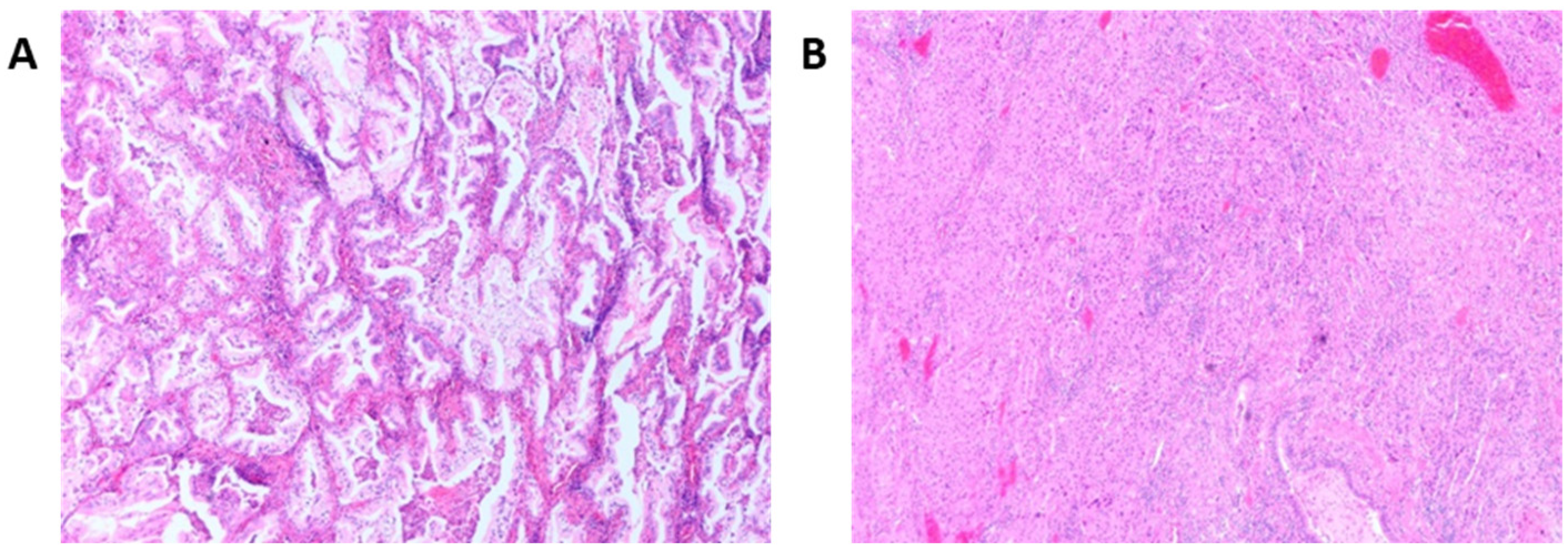
3.1. Histological Patterns Associated with Driver Mutations in Lung Cancer
3.2. Associations between KRAS Mutations and Invasive Mucinous Lung Adenocarcinomas
3.3. Novel Therapeutic Delivery Approaches to Target KRAS-Mutated NSCLC
4. Therapeutic Options for KRAS Mutated NSCLC
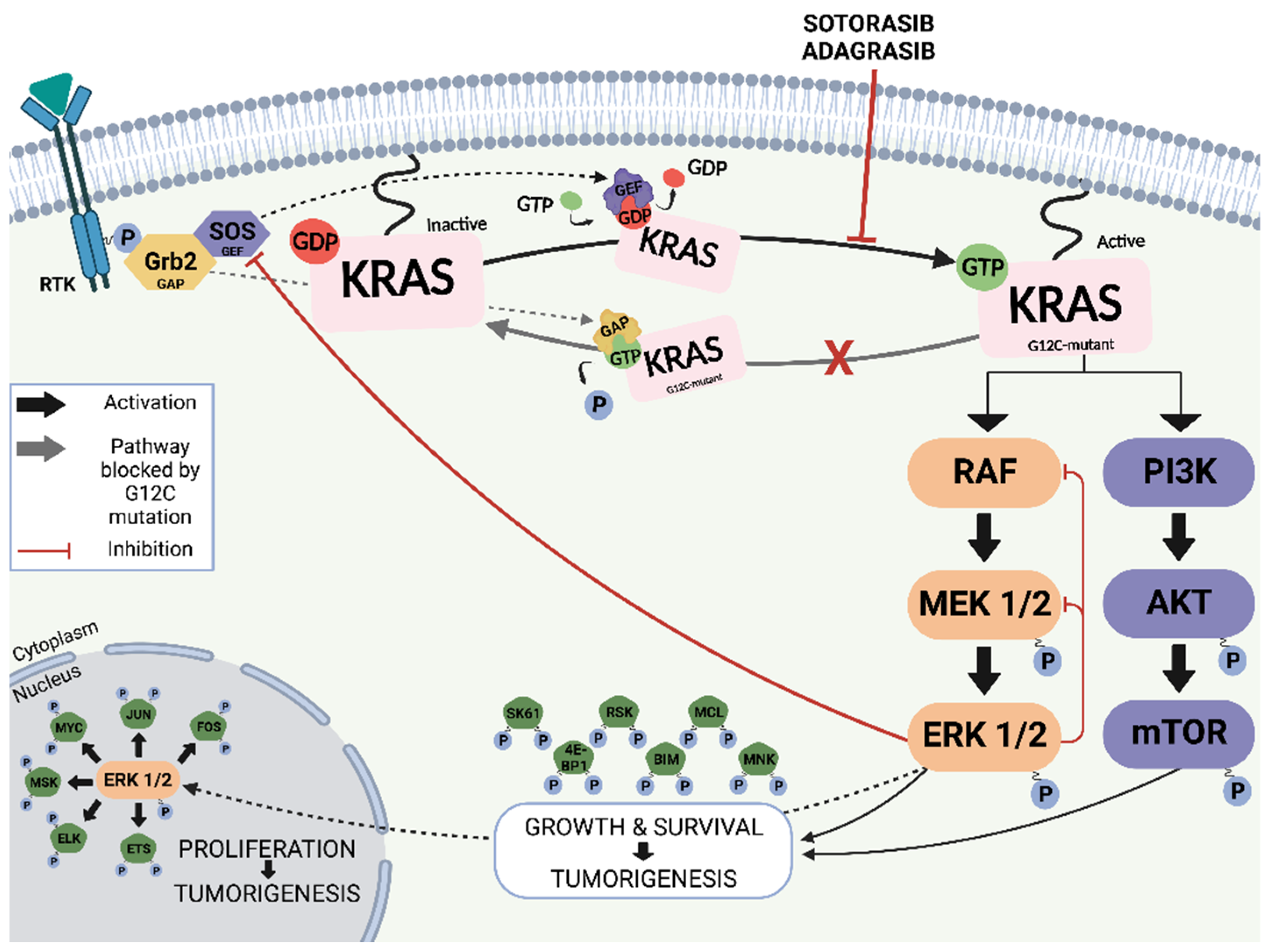
4.1. Targeting KRAS
4.2. Direct Inhibitors of KRAS G12C
4.2.1. Sotorasib (AMG510)
4.2.2. Adagrasib (MRTX849)
4.2.3. Other Direct KRAS G12C Inhibitors
4.2.4. Beyond KRAS G12C Inhibitors
5. Resisting KRAS Targeted Therapy
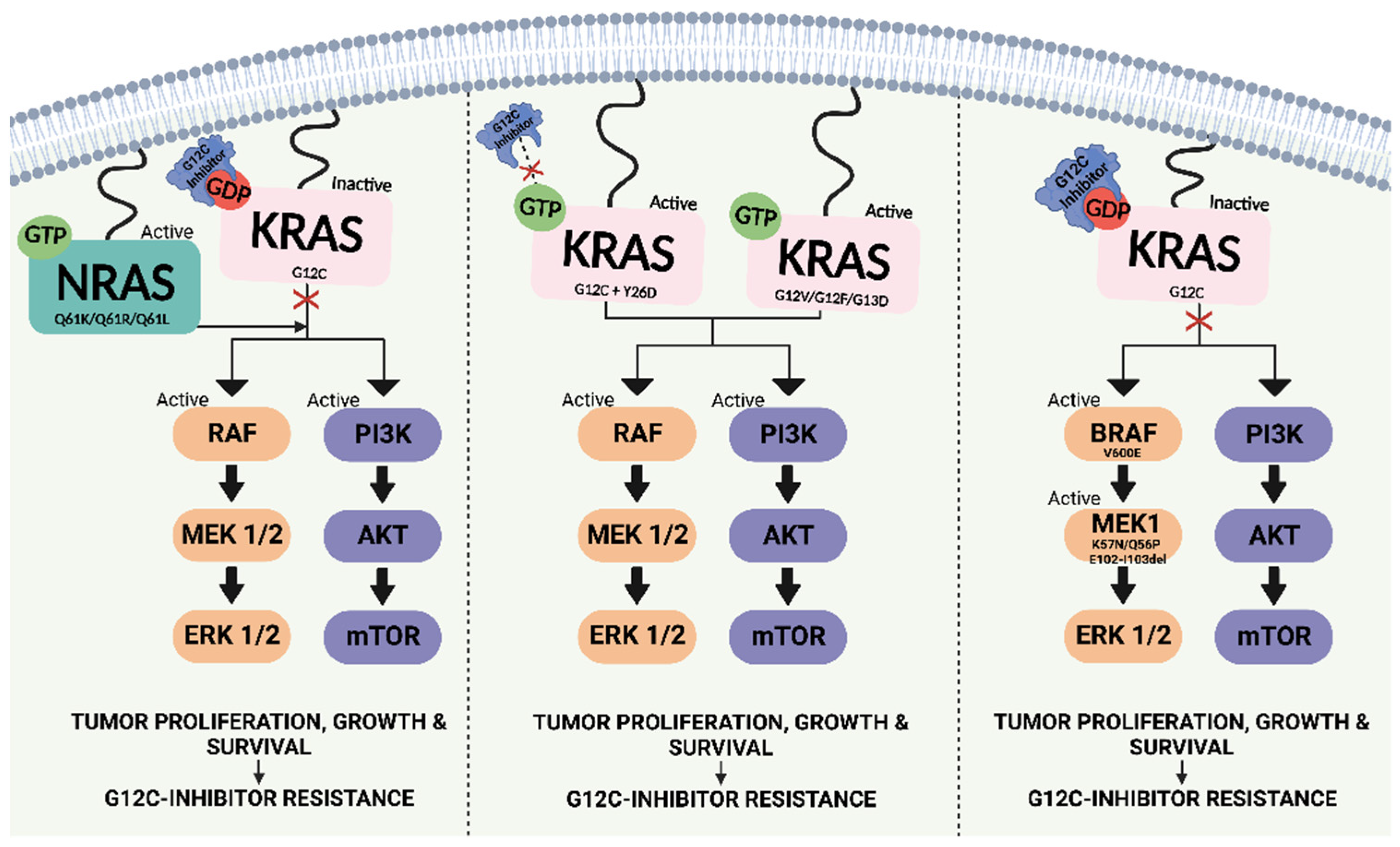
5.1. Resistance to KRAS G12C-Targeted Therapy
5.1.1. Intrinsic Resistance
5.1.2. Acquired Resistance
6. Overcoming Drug Resistance and the Future of KRAS Targeting
6.1. Synergistic Drug Combinations to Overcome Resistance to KRAS Targeted Therapy
6.2. Co-Targeting Metabolic Pathways in KRAS Mutant Lung Cancer
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Chang, E.H.; Gonda, M.A.; Ellis, R.W.; Scolnick, E.M.; Lowy, D.R. Human genome contains four genes homologous to transforming genes of Harvey and Kirsten murine sarcoma viruses. Proc. Natl. Acad. Sci. USA 1982, 79, 4848–4852. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Hancock, J.F. Ras nanoclusters: Versatile lipid-based signaling platforms. Biochim. Biophys. Acta 2015, 1853, 841–849. [Google Scholar] [CrossRef] [Green Version]
- Hall, B.E.; Bar-Sagi, D.; Nassar, N. The structural basis for the transition from Ras-GTP to Ras-GDP. Proc. Natl. Acad. Sci. USA 2002, 99, 12138–12142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. MAP kinase signaling pathways in cancer. Oncogene 2007, 26, 3279–3290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westra, W.H.; Baas, I.O.; Hruban, R.H.; Askin, F.B.; Wilson, K.; Offerhaus, G.J.; Slebos, R.J. K-ras oncogene activation in atypical alveolar hyperplasias of the human lung. Cancer Res. 1996, 56, 2224–2228. [Google Scholar]
- Riely, G.J.; Kris, M.G.; Rosenbaum, D.; Marks, J.; Li, A.; Chitale, D.A.; Nafa, K.; Riedel, E.R.; Hsu, M.; Pao, W.; et al. Frequency and distinctive spectrum of KRAS mutations in never smokers with lung adenocarcinoma. Clin. Cancer Res. 2008, 14, 5731–5734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calles, A.; Liao, X.; Sholl, L.M.; Rodig, S.J.; Freeman, G.J.; Butaney, M.; Lydon, C.; Dahlberg, S.E.; Hodi, F.; Oxnard, G.R.; et al. Expression of PD-1 and its ligands, PD-L1 and PD-L2, in smokers and never smokers with KRAS-mutant lung cancer. J. Thorac. Oncol. 2015, 10, 1726–1735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Racker, E.; Resnick, R.J.; Feldman, R. Glycolysis and methylaminoisobutyrate uptake in rat-1 cells transfected with ras or myc oncogenes. Proc. Natl. Acad. Sci. USA 1985, 82, 3535–3538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun, J.; Rago, C.; Cheong, I.; Pagliarini, R.; Angenendt, P.; Rajagopalan, H.; Schmidt, K.; Willson, J.K.; Markowitz, S.; Zhou, S.; et al. Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science 2009, 325, 1555–1559. [Google Scholar] [CrossRef] [Green Version]
- Ostrem, J.M.; Peters, U.; Sos, M.L.; Wells, J.A.; Shokat, K.M. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013, 503, 548–551. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.; Guo, Z.; Wang, F.; Fu, L. KRAS mutation: From undruggable to druggable in cancer. Signal Transduct. Target. Ther. 2021, 6, 386. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.S.; Wang, K.; Elkadi, O.R.; Tarasen, A.; Foulke, L.; Sheehan, C.E.; Otto, G.A.; Palmer, G.; Yelensky, R.; Lipson, D.; et al. Next-generation sequencing reveals frequent consistent genomic alterations in small cell undifferentiated lung cancer. J. Clin. Pathol. 2014, 67, 772–776. [Google Scholar] [CrossRef] [PubMed]
- Dogan, S.; Shen, R.; Ang, D.C.; Johnson, M.L.; D’Angelo, S.P.; Paik, P.K.; Brzostowski, E.B.; Riely, G.J.; Kris, M.G.; Zakowski, M.F.; et al. Molecular epidemiology of EGFR and KRAS mutations in 3026 lung adenocarcinomas: Higher susceptibility of women to smoking-related KRAS-mutant cancers. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 6169–6177. [Google Scholar] [CrossRef] [Green Version]
- Meng, M.; Zhong, K.; Jiang, T.; Liu, Z.; Kwan, H.Y.; Su, T. The current understanding on the impact of KRAS on colorectal cancer. Biomed. Pharmacother. 2021, 140, 111717. [Google Scholar] [CrossRef]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef]
- Amodio, V.; Yaeger, R.; Arcella, P.; Cancelliere, C.; Lamba, S.; Lorenzato, A.; Arena, S.; Montone, M.; Mussolin, B.; Bian, Y.; et al. EGFR Blockade Reverts Resistance to KRAS(G12C) Inhibition in Colorectal Cancer. Cancer Discov. 2020, 10, 1129–1139. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Lee, M.Y.; Ruan, A.; Chen, C.K.; Liu, H.P.; Wang, C.J.; Chao, W.R.; Han, C.P. Multipoint Kras oncogene mutations potentially indicate mucinous carcinoma on the entire spectrum of mucinous ovarian neoplasms. Oncotarget 2016, 7, 82097–82103. [Google Scholar] [CrossRef] [Green Version]
- Dearden, S.; Stevens, J.; Wu, Y.L.; Blowers, D. Mutation incidence and coincidence in non small-cell lung cancer: Meta-analyses by ethnicity and histology (mutMap). Ann. Oncol. 2013, 24, 2371–2376. [Google Scholar] [CrossRef]
- Skoulidis, F.; Byers, L.A.; Diao, L.; Papadimitrakopoulou, V.A.; Tong, P.; Izzo, J.; Behrens, C.; Kadara, H.; Parra, E.R.; Canales, J.R.; et al. Co-occurring genomic alterations define major subsets of KRAS-mutant lung adenocarcinoma with distinct biology, immune profiles, and therapeutic vulnerabilities. Cancer Discov. 2015, 5, 860–877. [Google Scholar] [CrossRef] [Green Version]
- Fang, C.; Zhang, C.; Zhao, W.Q.; Hu, W.W.; Wu, J.; Ji, M. Co-mutations of TP53 and KRAS serve as potential biomarkers for immune checkpoint blockade in squamous-cell non-small cell lung cancer: A case report. BMC Med. Genom. 2019, 12, 136. [Google Scholar] [CrossRef]
- Romero, R.; Sayin, V.I.; Davidson, S.M.; Bauer, M.R.; Singh, S.X.; LeBoeuf, S.E.; Karakousi, T.R.; Ellis, D.C.; Bhutkar, A.; Sánchez-Rivera, F.J.; et al. Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nat. Med. 2017, 23, 1362–1368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skoulidis, F.; Goldberg, M.E.; Greenawalt, D.M.; Hellmann, M.D.; Awad, M.M.; Gainor, J.F.; Schrock, A.B.; Hartmaier, R.J.; Trabucco, S.E.; Gay, L.; et al. STK11/LKB1 Mutations and PD-1 Inhibitor Resistance in KRAS-Mutant Lung Adenocarcinoma. Cancer Discov. 2018, 8, 822–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arbour, K.C.; Jordan, E.; Kim, H.R.; Dienstag, J.; Yu, H.A.; Sanchez-Vega, F.; Lito, P.; Berger, M.; Solit, D.B.; Hellmann, M.; et al. Effects of Co-occurring Genomic Alterations on Outcomes in Patients with KRAS-Mutant Non-Small Cell Lung Cancer. Clin Cancer Res. 2018, 24, 334–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuster, K.; Venkateswaran, N.; Rabellino, A.; Girard, L.; Peña-Llopis, S.; Scaglioni, P.P. Nullifying the CDKN2AB locus promotes mutant K-ras lung tumorigenesis. Mol. Cancer Res. 2014, 12, 912–923. [Google Scholar] [CrossRef] [Green Version]
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [Green Version]
- Russell, P.A.; Wainer, Z.; Wright, G.M.; Daniels, M.; Conron, M.; Williams, R.A. Does lung adenocarcinoma subtype predict patient survival?: A clinicopathologic study based on the new International Association for the Study of Lung Cancer/American Thoracic Society/European Respiratory Society international multidisciplinary lung adenocarcinoma classification. J. Thorac. Oncol. 2011, 6, 1496–1504. [Google Scholar] [CrossRef] [Green Version]
- Travis, W.D.; Brambilla, E.; Noguchi, M.; Nicholson, A.G.; Geisinger, K.; Yatabe, Y.; Powell, C.A.; Beer, D.; Riely, G.; Garg, K.; et al. International Association for the Study of Lung Cancer/American Thoracic Society/European Respiratory Society: International multidisciplinary classification of lung adenocarcinoma: Executive summary. Proc. Am. Thorac. Soc. 2011, 8, 381–385. [Google Scholar] [CrossRef]
- Yoshizawa, A.; Motoi, N.; Riely, G.J.; Sima, C.S.; Gerald, W.L.; Kris, M.G.; Park, B.J.; Rusch, V.W.; Travis, W.D. Impact of proposed IASLC/ATS/ERS classification of lung adenocarcinoma: Prognostic subgroups and implications for further revision of staging based on analysis of 514 stage I cases. Mod. Pathol. 2011, 24, 653–664. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Zhu, M.; Li, Y.; Li, Q. Association between EGFR exon 19 or exon 21 mutations and survival rates after first-line EGFR-TKI treatment in patients with non-small cell lung cancer. Mol. Clin. Oncol. 2019, 11, 301–308. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Zhao, J.; Zheng, J.; Kong, M.; Sun, K.; Wang, B.; Chen, X.; Ding, W.; Zhou, J. A Prediction Model for ROS1-Rearranged Lung Adenocarcinomas based on Histologic Features. PLoS ONE 2016, 11, e0161861. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, A.; Tsuta, K.; Watanabe, S.; Sekine, I.; Fukayama, M.; Tsuda, H.; Furuta, K.; Shibata, T. Frequent ALK rearrangement and TTF-1/p63 co-expression in lung adenocarcinoma with signet-ring cell component. Lung Cancer 2011, 72, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Shim, H.S.; Kenudson, M.; Zheng, Z.; Liebers, M.; Cha, Y.J.; Hoang Ho, Q.; Onozato, M.; Phi Le, L.; Heist, R.S.; Iafrate, A.J. Unique Genetic and Survival Characteristics of Invasive Mucinous Adenocarcinoma of the Lung. J. Thorac. Oncol. 2015, 10, 1156–1162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuta, K.; Kawago, M.; Inoue, E.; Yoshida, A.; Takahashi, F.; Sakurai, H.; Watanabe, S.I.; Takeuchi, M.; Furuta, K.; Asamura, H.; et al. The utility of the proposed IASLC/ATS/ERS lung adenocarcinoma subtypes for disease prognosis and correlation of driver gene alterations. Lung Cancer 2013, 81, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Cha, Y.J.; Kim, H.R.; Lee, H.J.; Cho, B.C.; Shim, H.S. Clinical course of stage IV invasive mucinous adenocarcinoma of the lung. Lung Cancer 2016, 102, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Pignata, S.; Ferrandina, G.; Scarfone, G.; Scollo, P.; Odicino, F.; Cormio, G.; Katsaros, D.; Villa, A.; Mereu, L.; Ghezzi, F.; et al. Activity of chemotherapy in mucinous ovarian cancer with a recurrence free interval of more than 6 months: Results from the SOCRATES retrospective study. BMC Cancer 2008, 8, 252. [Google Scholar] [CrossRef]
- Cu, Y.; Saltzman, W.M. Drug delivery: Stealth particles give mucus the slip. Nat. Mater. 2009, 8, 11–13. [Google Scholar] [CrossRef]
- Hugen, N.; Verhoeven, R.H.; Radema, S.A.; de Hingh, I.H.; Pruijt, J.F.; Nagtegaal, I.D.; Lemmens, V.E.; de Wilt, J.H. Prognosis and value of adjuvant chemotherapy in stage III mucinous colorectal carcinoma. Ann. Oncol. 2013, 24, 2819–2824. [Google Scholar] [CrossRef]
- Macha, M.A.; Krishn, S.R.; Jahan, R.; Banerjee, K.; Batra, S.K.; Jain, M. Emerging potential of natural products for targeting mucins for therapy against inflammation and cancer. Cancer Treat. Rev. 2015, 41, 277–288. [Google Scholar] [CrossRef] [Green Version]
- Perrais, M.; Pigny, P.; Copin, M.C.; Aubert, J.P.; Van Seuningen, I. Induction of MUC2 and MUC5AC mucins by factors of the epidermal growth factor (EGF) family is mediated by EGF receptor/Ras/Raf/extracellular signal-regulated kinase cascade and Sp1. J. Biol. Chem. 2002, 277, 32258–32267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dilly, A.K.; Song, X.; Zeh, H.J.; Guo, Z.S.; Lee, Y.J.; Bartlett, D.L.; Choudry, H.A. Mitogen-activated protein kinase inhibition reduces mucin 2 production and mucinous tumor growth. Transl. Res. 2015, 166, 344–354. [Google Scholar] [CrossRef] [PubMed]
- Lai, S.K.; Wang, Y.Y.; Hanes, J. Mucus-penetrating nanoparticles for drug and gene delivery to mucosal tissues. Adv. Drug Deliv. Rev. 2009, 61, 158–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira de Sousa, I.; Cattoz, B.; Wilcox, M.D.; Griffiths, P.C.; Dalgliesh, R.; Rogers, S.; Bernkop-Schnürch, A. Nanoparticles decorated with proteolytic enzymes, a promising strategy to overcome the mucus barrier. Eur. J. Pharm. Biopharm. 2015, 97, 257–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jesorka, A.; Orwar, O. Liposomes: Technologies and analytical applications. Annu. Rev. Anal. Chem. 2008, 1, 801–832. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Fourniols, T.; Labrak, Y.; Preat, V.; Beloqui, A.; des Rieux, A. Surface Modification of Lipid-Based Nanoparticles. ACS Nano 2022, 16, 7168–7196. [Google Scholar] [CrossRef]
- Huckaby, J.T.; Lai, S.K. PEGylation for enhancing nanoparticle diffusion in mucus. Adv. Drug Deliv. Rev. 2018, 124, 125–139. [Google Scholar] [CrossRef] [PubMed]
- Wagner, U.; Marth, C.; Largillier, R.; Kaern, J.; Brown, C.; Heywood, M.; Bonaventura, T.; Vergote, I.; Piccirillo, M.C.; Fossati, R.; et al. Final overall survival results of phase III GCIG CALYPSO trial of pegylated liposomal doxorubicin and carboplatin vs paclitaxel and carboplatin in platinum-sensitive ovarian cancer patients. Br. J. Cancer 2012, 107, 588–591. [Google Scholar] [CrossRef] [Green Version]
- Von Hoff, D.D.; Mita, M.M.; Ramanathan, R.K.; Weiss, G.J.; Mita, A.C.; Lorusso, P.M.; Burris, H.A., 3rd; Hart, L.L.; Low, S.C.; Parsons, D.M.; et al. Phase I Study of PSMA-Targeted Docetaxel-Containing Nanoparticle BIND-014 in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2016, 22, 3157–3163. [Google Scholar] [CrossRef] [Green Version]
- Genprex Receives Safety Review Committee Approval to Proceed to the Final Cohort in Acclaim-1 Phase 1 Dose Escalation Trial of REQORSA® in Combination with Tagrisso® in Advanced Non-Small Cell Lung Cancer. News Release. 15 December 2022. Available online: https://www.genprex.com/news/genprex-receives-safety-review-committee-approval-to-proceed-to-final-cohort-in-acclaim-1-phase-1-dose-escalation-trial-of-reqorsa-in-combination-with-tagrisso-in-advanced-non-small-cell/ (accessed on 10 January 2023).
- Carrasco-Esteban, E.; Domínguez-Rullán, J.A.; Barrionuevo-Castillo, P.; Pelari-Mici, L.; Leaman, O.; Sastre-Gallego, S.; López-Campos, F. Current role of nanoparticles in the treatment of lung cancer. J. Clin. Transl. Res. 2021, 7, 140–155. [Google Scholar]
- McCormick, F. KRAS as a Therapeutic Target. Clin. Cancer Res. 2015, 21, 1797–1801. [Google Scholar] [CrossRef] [Green Version]
- Pantsar, T. The current understanding of KRAS protein structure and dynamics. Comput. Struct. Biotechnol. J. 2020, 18, 189–198. [Google Scholar] [CrossRef]
- Hong, D.S.; Fakih, M.G.; Strickler, J.H.; Desai, J.; Durm, G.A.; Shapiro, G.I.; Falchook, G.S.; Price, T.J.; Sacher, A.; Denlinger, C.S.; et al. KRAS(G12C) Inhibition with Sotorasib in Advanced Solid Tumors. N. Engl. J. Med. 2020, 383, 1207–1217. [Google Scholar] [CrossRef] [PubMed]
- Skoulidis, F.; Li, B.T.; Dy, G.K.; Price, T.J.; Falchook, G.S.; Wolf, J.; Italiano, A.; Schuler, M.; Borghaei, H.; Barlesi, F.; et al. Sotorasib for Lung Cancers with KRAS p.G12C Mutation. N. Engl. J. Med. 2021, 384, 2371–2381. [Google Scholar] [CrossRef]
- FDA Approves First KRAS Inhibitor: Sotorasib. Cancer Discov. 2021, 11, OF4. [CrossRef] [PubMed]
- Molina-Arcas, M.; Samani, A.; Downward, J. Drugging the Undruggable: Advances on RAS Targeting in Cancer. Genes 2021, 12, 899. [Google Scholar] [CrossRef] [PubMed]
- Ou, S.I.; Jänne, P.A.; Leal, T.A.; Rybkin, I.I.; Sabari, J.K.; Barve, M.A.; Bazhenova, L.; Johnson, M.L.; Velastegui, K.L.; Cilliers, C.; et al. First-in-Human Phase I/IB Dose-Finding Study of Adagrasib (MRTX849) in Patients With Advanced KRAS(G12C) Solid Tumors (KRYSTAL-1). J. Clin. Oncol. 2022, 40, 2530–2538. [Google Scholar] [CrossRef]
- Goebel, L.; Müller, M.P.; Goody, R.S.; Rauh, D. KRasG12C inhibitors in clinical trials: A short historical perspective. RSC Med. Chem. 2020, 11, 760–770. [Google Scholar] [CrossRef]
- Hallin, J.; Engstrom, L.D.; Hargis, L.; Calinisan, A.; Aranda, R.; Briere, D.M.; Sudhakar, N.; Bowcut, V.; Baer, B.R.; Ballard, J.A.; et al. The KRAS(G12C) Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discov. 2020, 10, 54–71. [Google Scholar] [CrossRef] [Green Version]
- Riely, G.J.; Ou, S.H.I.; Rybkin, I.; Spira, A.; Papadopoulos, K.; Sabari, J.K.; Johnson, M.; Heist, R.S.; Bazhenova, L.; Barve, M.; et al. 99O_PR KRYSTAL-1: Activity and preliminary pharmacodynamic (PD) analysis of adagrasib (MRTX849) in patients (Pts) with advanced non–small cell lung cancer (NSCLC) harboring KRASG12C mutation. J. Thorac. Oncol. 2021, 16, S751–S752. [Google Scholar] [CrossRef]
- Wang, X.; Allen, S.; Blake, J.F.; Bowcut, V.; Briere, D.M.; Calinisan, A.; Dahlke, J.R.; Fell, J.B.; Fischer, J.P.; Gunn, R.J.; et al. Identification of MRTX1133, a Noncovalent, Potent, and Selective KRAS(G12D) Inhibitor. J. Med. Chem. 2022, 65, 3123–3133. [Google Scholar] [CrossRef]
- Békés, M.; Langley, D.R.; Crews, C.M. PROTAC targeted protein degraders: The past is prologue. Nat. Rev. Drug Discov. 2022, 21, 181–200. [Google Scholar] [CrossRef]
- Zeng, M.; Xiong, Y.; Safaee, N.; Nowak, R.P.; Donovan, K.A.; Yuan, C.J.; Nabet, B.; Gero, T.W.; Feru, F.; Li, L.; et al. Exploring targeted degradation strategy for oncogenic KRASG12C. Cell Chem. Biol. 2020, 27, 19–231. [Google Scholar] [CrossRef] [PubMed]
- Bond, M.J.; Chu, L.; Nalawansha, D.A.; Li, K.; Crews, C.M. Targeted degradation of oncogenic KRAS G12C by VHL-recruiting PROTACs. ACS Central Sci. 2020, 6, 1367–1375. [Google Scholar] [CrossRef]
- Kraut, N. Expanding the reach of precision oncology by drugging all KRAS mutants. In Proceedings of the 112th Annual Meeting of the American Association for Cancer Research, Virtual, 17–21 May 2021; AACR: Philadelphia, PA, USA, 2021. [Google Scholar]
- ClinicalTrials.gov. A Study to Test Different Doses of BI 1701963 Alone and Combined with Trametinib in Patients with Different Types of Advanced Cancer (Solid Tumors with KRAS Mutation). Full Text View. ClinicalTrials.gov. 2019. Available online: https://clinicaltrials.gov/ct2/show/NCT04111458 (accessed on 10 January 2023).
- Heavey, S.; O’Byrne, K.J.; Gately, K. Strategies for co-targeting the PI3K/AKT/mTOR pathway in NSCLC. Cancer Treat. Rev. 2014, 40, 445–456. [Google Scholar] [CrossRef] [PubMed]
- Heavey, S.; Cuffe, S.; Finn, S.; Young, V.; Ryan, R.; Nicholson, S.; Leonard, N.; McVeigh, N.; Barr, M.; O’Byrne, K.; et al. In pursuit of synergy: An investigation of the PI3K/mTOR/MEK co-targeted inhibition strategy in NSCLC. Oncotarget 2016, 7, 79526–79543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solanki, H.S.; Welsh, E.A.; Fang, B.; Izumi, V.; Darville, L.; Stone, B.; Franzese, R.; Chavan, S.; Kinose, F.; Imbody, D.; et al. Cell Type-specific Adaptive Signaling Responses to KRASG12C Inhibition. Clin. Cancer Res. 2021, 27, 2533–2548. [Google Scholar] [CrossRef]
- Tanaka, N.; Lin, J.J.; Li, C.; Ryan, M.B.; Zhang, J.; Kiedrowski, L.A.; Michel, A.G.; Syed, M.U.; Fella, K.A.; Sakhi, M.; et al. Clinical Acquired Resistance to KRAS(G12C) Inhibition through a Novel KRAS Switch-II Pocket Mutation and Polyclonal Alterations Converging on RAS-MAPK Reactivation. Cancer Discov. 2021, 11, 1913–1922. [Google Scholar] [CrossRef] [PubMed]
- Awad, M.M.; Liu, S.; Rybkin, I.I.; Arbour, K.C.; Dilly, J.; Zhu, V.W.; Johnson, M.L.; Heist, R.S.; Patil, T.; Riely, G.J.; et al. Acquired Resistance to KRAS(G12C) Inhibition in Cancer. N. Engl. J. Med. 2021, 384, 2382–2393. [Google Scholar] [CrossRef]
- Koga, T.; Suda, K.; Fujino, T.; Ohara, S.; Hamada, A.; Nishino, M.; Chiba, M.; Shimoji, M.; Takemoto, T.; Arita, T.; et al. KRAS Secondary Mutations That Confer Acquired Resistance to KRAS G12C Inhibitors, Sotorasib and Adagrasib, and Overcoming Strategies: Insights From In Vitro Experiments. J. Thorac. Oncol. 2021, 16, 1321–1332. [Google Scholar] [CrossRef]
- Dunnett-Kane, V.; Nicola, P.; Blackhall, F.; Lindsay, C. Mechanisms of Resistance to KRAS(G12C) Inhibitors. Cancers 2021, 13, 151. [Google Scholar] [CrossRef]
- Nagasaka, M.; Potugari, B.; Nguyen, A.; Sukari, A.; Azmi, A.S.; Ou, S.I. KRAS Inhibitors- yes but what next? Direct targeting of KRAS- vaccines, adoptive T cell therapy and beyond. Cancer Treat. Rev. 2021, 101, 102309. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Sotorasib Activity in Subjects With Advanced Solid Tumors With KRAS p.G12C Mutation (CodeBreak 101). Full Text View. ClinicalTrials.gov. 2019. Available online: https://clinicaltrials.gov/ct2/show/NCT04185883 (accessed on 10 January 2023).
- Goodwin, C.M.; Waters, A.M.; Klomp, J.E.; Javaid, S.; Bryant, K.L.; Stalnecker, C.A.; Drizyte-Miller, K.; Papke, B.; Yang, R.; Amparo, A.M.; et al. Combination Therapies with CDK4/6 Inhibitors to Treat KRAS-Mutant Pancreatic Cancer. Cancer Res. 2023, 83, 141–157. [Google Scholar] [CrossRef]
- Thummuri, D.; Budamagunta, V.; Hua, N.; Jin, L.; Allegra, C.J.; Kopetz, S.E.; Zajac-Kaye, M.; Kaye, F.J.; Zheng, G.; Zhou, D. BCL-XL PROTAC degrader DT2216 synergizes with sotorasib in preclinical models of KRASG12C-mutated cancers. J. Hematol. Oncol. 2022, 15, 23. [Google Scholar] [CrossRef]
- Briere, D.M.; Li, S.; Calinisan, A.; Sudhakar, N.; Aranda, R.; Hargis, L.; Peng, D.H.; Deng, J.; Engstrom, L.D.; Hallin, J.; et al. The KRAS(G12C) Inhibitor MRTX849 Reconditions the Tumor Immune Microenvironment and Sensitizes Tumors to Checkpoint Inhibitor Therapy. Mol. Cancer Ther. 2021, 20, 975–985. [Google Scholar] [CrossRef]
- Tang, R.; Shuldiner, E.G.; Kelly, M.; Murray, C.W.; Hebert, J.D.; Andrejka, L.; Tsai, M.K.; Hughes, N.W.; Parker, M.I.; Cai, H.; et al. Multiplexed screens identify RAS paralogues HRAS and NRAS as suppressors of KRAS-driven lung cancer growth. Nat. Cell Biol. 2023, 25, 159–169. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Vander Heiden, M.G.; McCormick, F. The Metabolic Landscape of RAS-Driven Cancers from biology to therapy. Nat. Cancer 2021, 2, 271–283. [Google Scholar] [CrossRef] [PubMed]
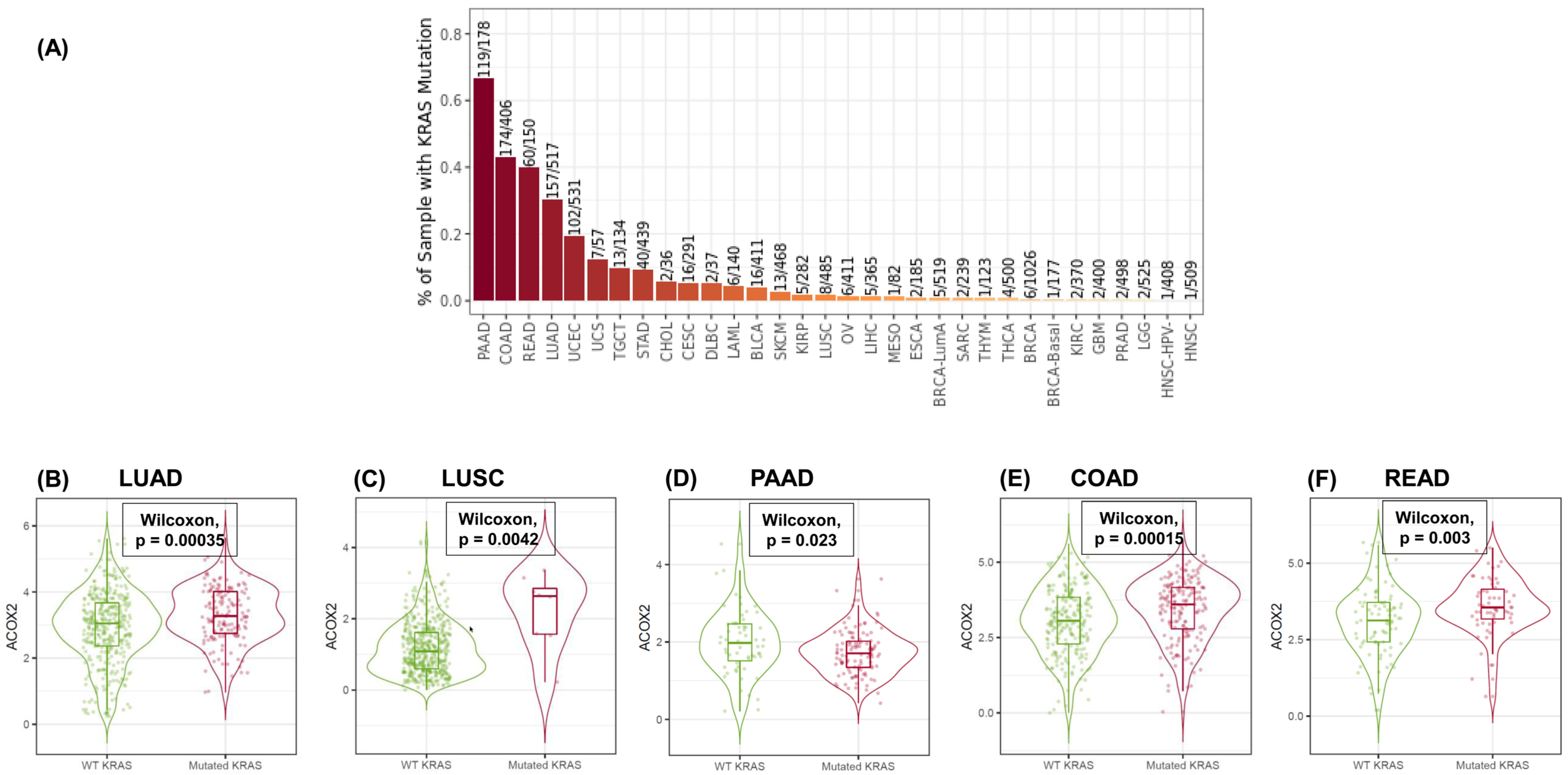
- Sui, J.S.Y.; Martin, P.; Keogh, A.; Murchan, P.; Ryan, L.; Nicholson, S.; Cuffe, S.; Broin, P.; Finn, S.P.; Fitzmaurice, G.J.; et al. Altered expression of ACOX2 in non-small cell lung cancer. BMC Pulm. Med. 2022, 22, 321. [Google Scholar] [CrossRef] [PubMed]
- Ferdinandusse, S.; Denis, S.; van Roermund, C.W.T.; Preece, M.A.; Koster, J.; Ebberink, M.S.; Waterham, H.R.; Wanders, R.J.A. A novel case of ACOX2 deficiency leads to recognition of a third human peroxisomal acyl-CoA oxidase. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 952–958. [Google Scholar] [CrossRef]
- Braverman, N.E.; Raymond, G.V.; Rizzo, W.B.; Moser, A.B.; Wilkinson, M.E.; Stone, E.M.; Steinberg, S.J.; Wangler, M.F.; Rush, E.T.; Hacia, J.G.; et al. Peroxisome biogenesis disorders in the Zellweger spectrum: An overview of current diagnosis, clinical manifestations, and treatment guidelines. Mol. Genet. Metab. 2016, 117, 313–321. [Google Scholar] [CrossRef] [Green Version]
- Narayanan, D.; Ma, S.; Özcelik, D. Targeting the Redox Landscape in Cancer Therapy. Cancers 2020, 12, 1706. [Google Scholar] [CrossRef]
- Nordgren, M.; Fransen, M. Peroxisomal metabolism and oxidative stress. Biochimie 2014, 98, 56–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerr, E.M.; Martins, C.P. Metabolic rewiring in mutant Kras lung cancer. FEBS J. 2018, 285, 28–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yousuf, U.; Sofi, S.; Makhdoomi, A.; Mir, M.A. Identification and analysis of dysregulated fatty acid metabolism genes in breast cancer subtypes. Med. Oncol. 2022, 39, 256. [Google Scholar] [CrossRef] [PubMed]
- Bjørklund, S.S.; Kristensen, V.N.; Seiler, M.; Kumar, S.; Alnæs, G.I.; Ming, Y.; Kerrigan, J.; Naume, B.; Sachidanandam, R.; Bhanot, G.; et al. Expression of an estrogen-regulated variant transcript of the peroxisomal branched chain fatty acid oxidase ACOX2 in breast carcinomas. BMC Cancer 2015, 15, 524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Yuan, D.; Jiang, K.; Li, R.; Qu, H.; Jiang, F.N.; Zhong, W.D.; Sun, F.; Jia, Z.; Zhu, J. Genome-wide CRISPR-Cas9 screening and identification of potential genes promoting prostate cancer growth and metastasis. Curr. Cancer Drug Targets 2023, 23, 71–86. [Google Scholar] [CrossRef]
- Yu, G.; Cheng, C.J.; Lin, S.C.; Lee, Y.C.; Frigo, D.E.; Yu-Lee, L.Y.; Gallick, G.E.; Titus, M.A.; Nutt, L.K.; Lin, S.H. Organelle-Derived Acetyl-CoA Promotes Prostate Cancer Cell Survival, Migration, and Metastasis via Activation of Calmodulin Kinase II. Cancer Res. 2018, 78, 2490–2502. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Zhang, Y.; Sun, S.; Wang, K.; Qian, J.; Cui, Z.; Tao, T.; Zhou, J. ACOX2 is a prognostic marker and impedes the progression of hepatocellular carcinoma via PPARα pathway. Cell Death Dis. 2021, 12, 15. [Google Scholar] [CrossRef]
- Dahabieh, M.S.; Di Pietro, E.; Jangal, M.; Goncalves, C.; Witcher, M.; Braverman, N.E.; Del Rincón, S.V. Peroxisomes and cancer: The role of a metabolic specialist in a disease of aberrant metabolism. Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 103–121. [Google Scholar] [CrossRef]
- Kim, J.A. Peroxisome Metabolism in Cancer. Cells 2020, 9, 1692. [Google Scholar] [CrossRef]
- Svensson, R.U.; Shaw, R.J. Lipid Synthesis Is a Metabolic Liability of Non-Small Cell Lung Cancer. Cold Spring Harb. Symp. Quant. Biol. 2016, 81, 93–103. [Google Scholar] [CrossRef] [Green Version]
- Dreyer, C.; Krey, G.; Keller, H.; Givel, F.; Helftenbein, G.; Wahli, W. Control of the peroxisomal beta-oxidation pathway by a novel family of nuclear hormone receptors. Cell 1992, 68, 879–887. [Google Scholar] [CrossRef]
- Liu, Y.; Deguchi, Y.; Wei, D.; Liu, F.; Moussalli, M.J.; Deguchi, E.; Li, D.; Wang, H.; Valentin, L.A.; Colby, J.K.; et al. Rapid acceleration of KRAS-mutant pancreatic carcinogenesis via remodeling of tumor immune microenvironment by PPARδ. Nat. Commun. 2022, 13, 2665. [Google Scholar] [CrossRef] [PubMed]
- Luo, F.; Brooks, D.G.; Ye, H.; Hamoudi, R.; Poulogiannis, G.; Patek, C.E.; Winton, D.J.; Arends, M.J. Mutated K-ras(Asp12) promotes tumourigenesis in Apc(Min) mice more in the large than the small intestines, with synergistic effects between K-ras and Wnt pathways. Int. J. Exp. Pathol. 2009, 90, 558–574. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Fu, J.; Zeng, Z.; Cohen, D.; Li, J.; Chen, Q.; Li, B.; Liu, X.S. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 2020, 48, W509–W514. [Google Scholar] [CrossRef] [PubMed]
- Baldelli, E.; El Gazzah, E.; Moran, J.C.; Hodge, K.A.; Manojlovic, Z.; Bassiouni, R.; Carpten, J.D.; Ludovini, V.; Baglivo, S.; Crinò, L.; et al. Wild-Type KRAS Allele Effects on Druggable Targets in KRAS Mutant Lung Adenocarcinomas. Genes 2021, 12, 1402. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.H.; Lin, H.Y.; Verma, V.; Xu-Welliver, M.; Thall, P.F.; Yao, L.; Kim, P.Y.; Gombos, D.S.; Kawedia, J.D.; Komaki, R.; et al. Phase I Trial of Definitive Concurrent Chemoradiotherapy and Trametinib for KRAS-Mutated Non-Small Cell Lung Cancer. Cancer Treat. Res. Commun. 2022, 30, 100514. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Goswami, D.; Adiseshaiah, P.P.; Burgan, W.; Yi, M.; Guerin, T.M.; Kozlov, S.V.; Nissley, D.V.; McCormick, F. Undermining Glutaminolysis Bolsters Chemotherapy While NRF2 Promotes Chemoresistance in KRAS-Driven Pancreatic Cancers. Cancer Res. 2020, 80, 1630–1643. [Google Scholar] [CrossRef] [Green Version]
- Lawless, M.W.; O’Byrne, K.J.; Gray, S.G. Oxidative stress induced lung cancer and COPD: Opportunities for epigenetic therapy. J. Cell. Mol. Med. 2009, 13, 2800–2821. [Google Scholar] [CrossRef] [Green Version]
- Xia, M.; Li, X.; Diao, Y.; Du, B.; Li, Y. Targeted inhibition of glutamine metabolism enhances the antitumor effect of selumetinib in KRAS-mutant NSCLC. Transl. Oncol. 2021, 14, 100920. [Google Scholar] [CrossRef]
- Riess, J.W.; Frankel, P.; Shackelford, D.; Dunphy, M.; Badawi, R.D.; Nardo, L.; Cherry, S.R.; Lanza, I.; Reid, J.; Gonsalves, W.I.; et al. Phase 1 Trial of MLN0128 (Sapanisertib) and CB-839 HCl (Telaglenastat) in Patients With Advanced NSCLC (NCI 10327): Rationale and Study Design. Clin. Lung Cancer 2021, 22, 67–70. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound(s) | Company | Cancer Type Tested | Combinations | NCT Number |
|---|---|---|---|---|
| AMG 510/sotorasib * CodeBreak 100 Ph1/2 CodeBreak 101 Ph1b CodeBreak 200 Ph3 A lung- MAP treatment Ph2 | Amgen | NSCLC CRC Solid tumours incl. PAC Only KRAS G12C mutation | Monotherapy PD-1/PD-L1 EGFR TKI Chemotherapy EGFR Ab ± chemo or + MEKi VEGF Ab ± Chemo SHP-2 mTOR CDK inhibitor | NCT03600883 NCT04185883 NCT04303780 NCT04625647 |
| MRTX849/adagrasib * KRYSTAL-1 Ph1/2 KRYSTAL-2 Ph1/2 KRYSTAL-7 Ph2 KRYSTAL-10 Ph3 KRYSTAL-12 Ph3 | Mirati | NSCLC CRC Solid tumours incl. PaC Only KRAS G12C mutation | Monotherapy PD-1 EGFR Ab EGFR TKI SHP-2 CDK inhibitor SOS1 | NCT03785249 NCT04330664 NCT04613596 NCT04793958 NCT04685135 |
| ARS-3248/ JNJ-74699157 Ph1 | Wellspring Biosciences J&J | Solid tumours KRAS G12C mut | Monotherapy | NCT04006301 Terminated after 10 pts enrolled |
| LY3499446 Ph1/2 | Eli Lily | NSCLC CRC Solid tumours KRAS G12C mutation | Monotherapy CDK inhibitor EGFR Ab EGFR TKI | NCT04165031 Terminated due to toxicity |
| GDC-6036 Ph1 | Genentech/Roche | NSCLC, CRC solid tumours incl. PaC Only KRAS G12C mutation | Monotherapy PD-L1 EGFR Ab VEGF Ab EGFR TKI | NCT04449874 |
| D-1553 Ph1/2 D-1553 Ph1/2 | InventisBio | Solid tumours KRAS G12C mut | Monotherapy | NCT04585035 NCT05383898 |
| JDQ443 Ph1/2 | Novartis | Solid tumours KRAS G12 mut | SHP-2 PD-1 | NCT04699188 |
| BI 1,823,911 Ph1a/1b | Boehringer Ingelheim | Solid tumours KRAS G12 mut | Monotherapy + BI 1701963 | NCT04973163 |
| LY3537982 Ph1a/1b | Eli Lilly | Solid tumours KRAS G12 mut | Monotherapy CDK inhibitor EGFR TKI PD1 ERK1/2 inhibitor Aurora A EGFR Ab SHP-2 | NCT04956640 |
| JAB-21822 Ph1/2 JAB-21822 Ph 1b/2 JAB-21822 Ph 1/2 JAB-21822 Ph 1/2a | Jacobio | Solid tumours KRAS G12 mut | Monotherapy EGFR Ab SHP-2 | NCT05009329 NCT05194995 NCT05002270 NCT05288205 |
| YL-15293 Ph 1/2 | Shanghai YinLi | Solid tumours KRAS G12 mut | Monotherapy | NCT05119933 |
| RMC-6291 Ph1/1b | Revolution Medicine | Solid tumours KRAS G12 mut | Monotherapy | NCT05462717 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
O’Sullivan, É.; Keogh, A.; Henderson, B.; Finn, S.P.; Gray, S.G.; Gately, K. Treatment Strategies for KRAS-Mutated Non-Small-Cell Lung Cancer. Cancers 2023, 15, 1635. https://doi.org/10.3390/cancers15061635
O’Sullivan É, Keogh A, Henderson B, Finn SP, Gray SG, Gately K. Treatment Strategies for KRAS-Mutated Non-Small-Cell Lung Cancer. Cancers. 2023; 15(6):1635. https://doi.org/10.3390/cancers15061635
Chicago/Turabian StyleO’Sullivan, Éabha, Anna Keogh, Brian Henderson, Stephen P. Finn, Steven G. Gray, and Kathy Gately. 2023. "Treatment Strategies for KRAS-Mutated Non-Small-Cell Lung Cancer" Cancers 15, no. 6: 1635. https://doi.org/10.3390/cancers15061635