
Genomic Alterations, Gene Expression Profiles and Functional Enrichment of Normal-Karyotype Acute Myeloid Leukaemia Based on Targeted Next-Generation Sequencing
 , , , and
, , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Samples and Ethics Statement
2.2. Targeted DNA Sequencing Using Archer Dx
2.3. Sanger Sequencing
2.4. RNA Sequencing
2.5. Functional and Pathway Enrichment Analysis of Genes with Somatic Variants Using WEB-Based Gene Set Analysis Toolkit (WebGestalt) for Overrepresentation Analysis (ORA)
2.6. Statistical Analysis
3. Results
3.1. Demographic and Clinical Summary of Patients in This Study
3.2. Variants Discovered in This Study
3.3. Size of FLT3 Internal Tandem Duplication (ITD)
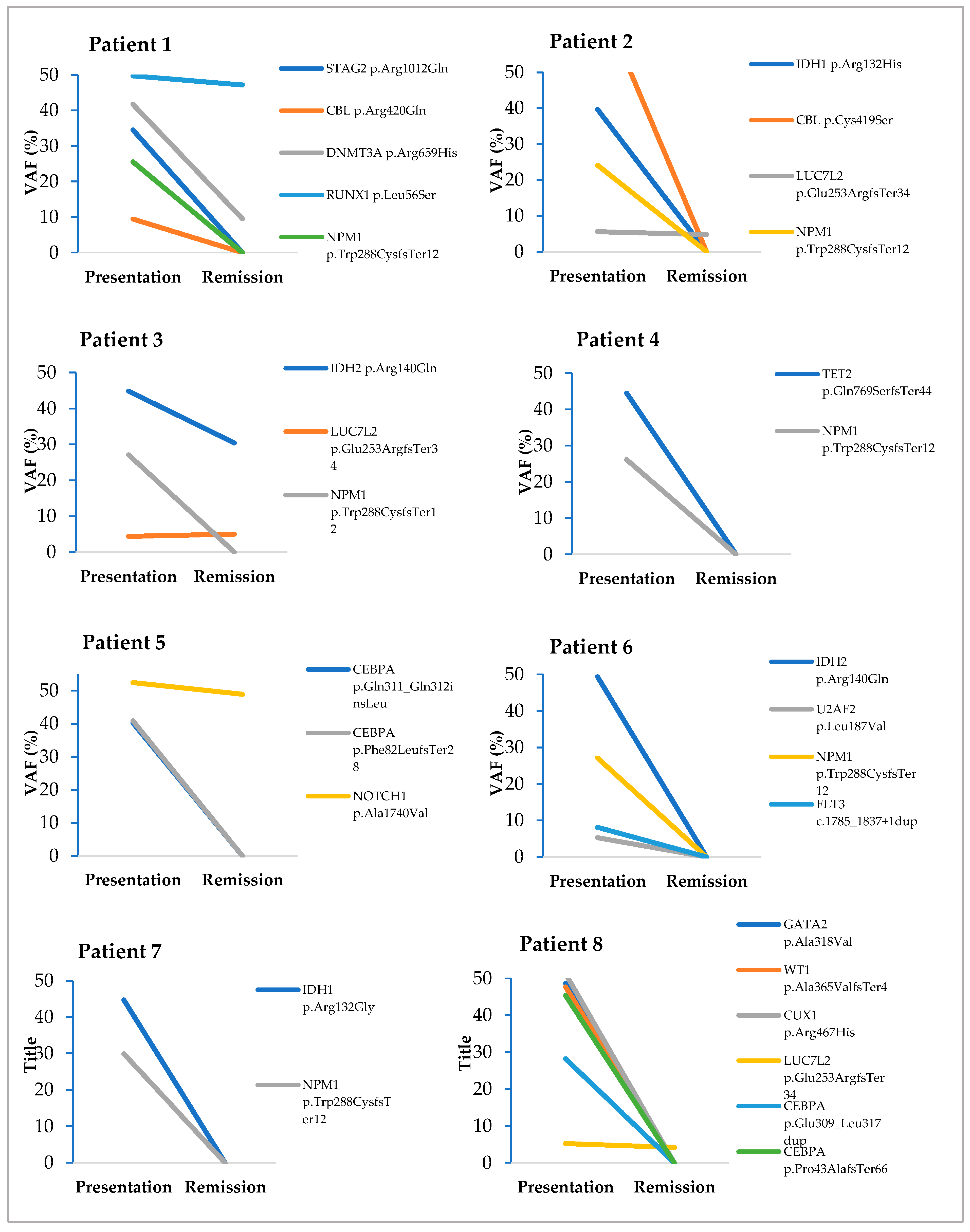
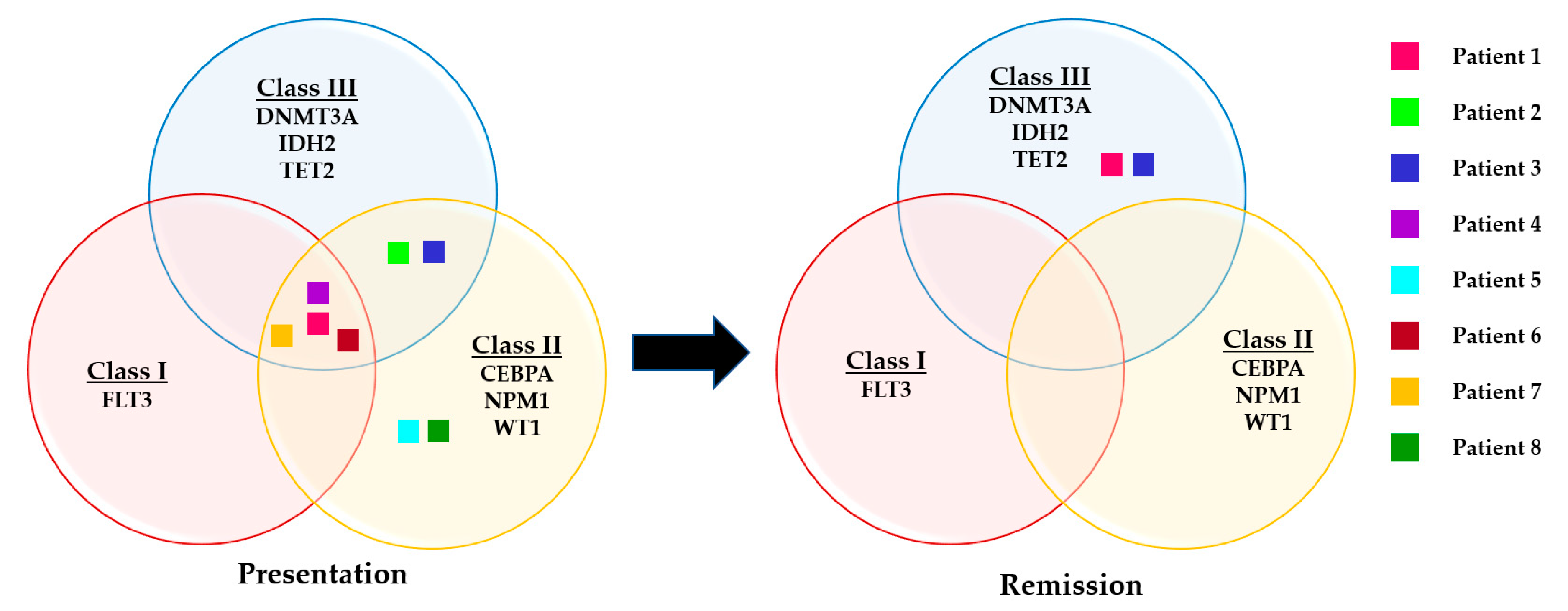
3.4. Evaluation of Somatic Variants Detected at Presentation and CR1/CR2
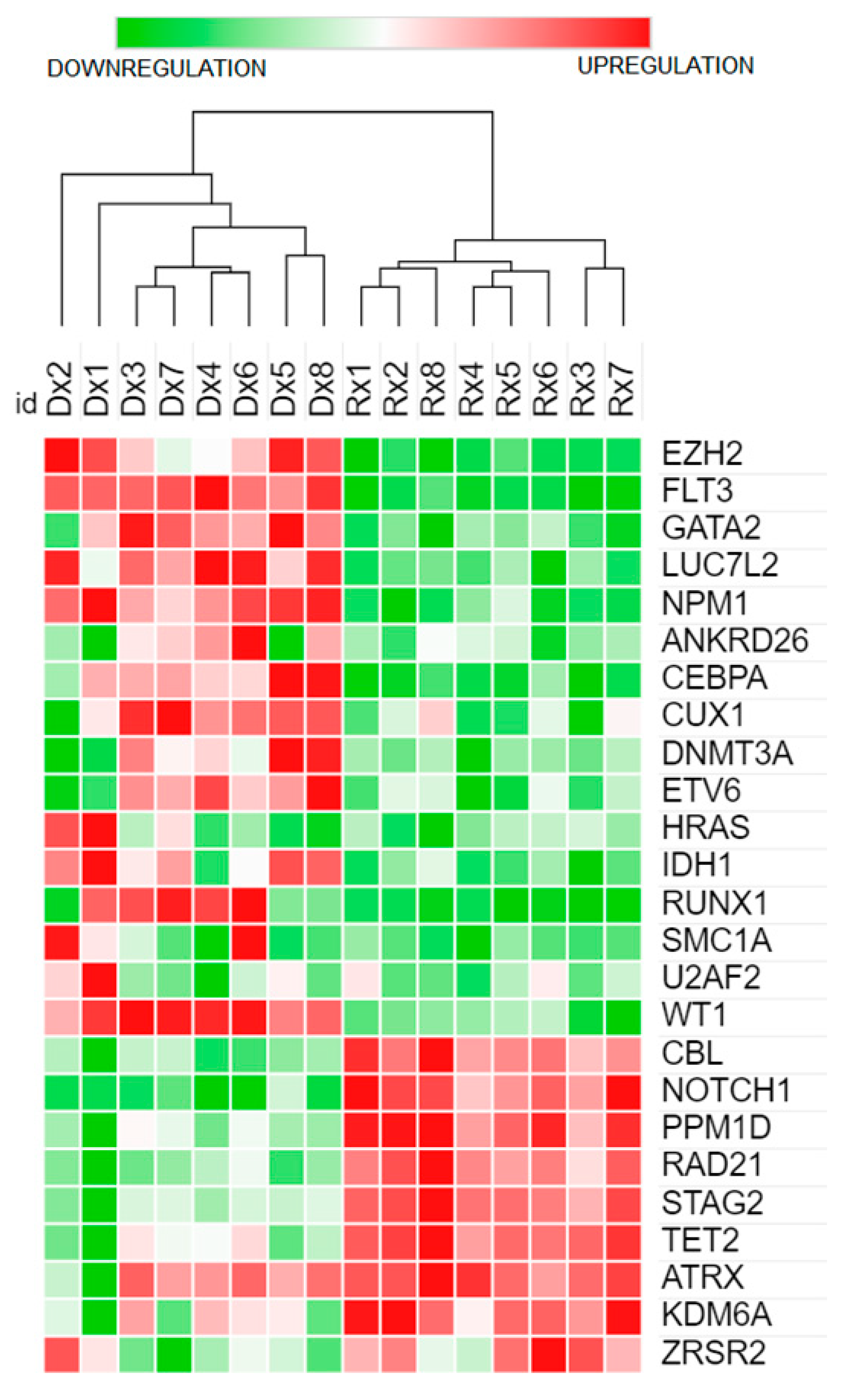
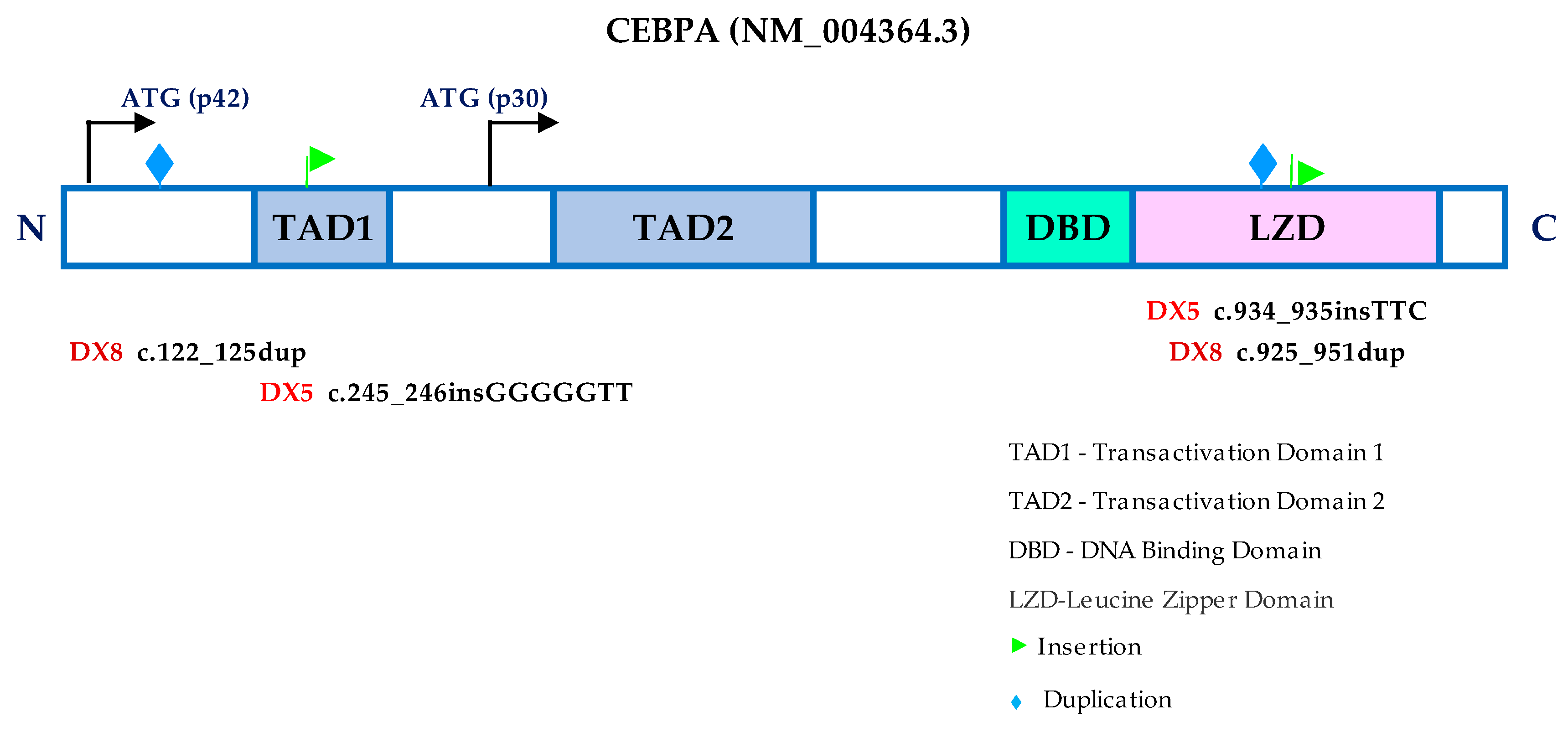
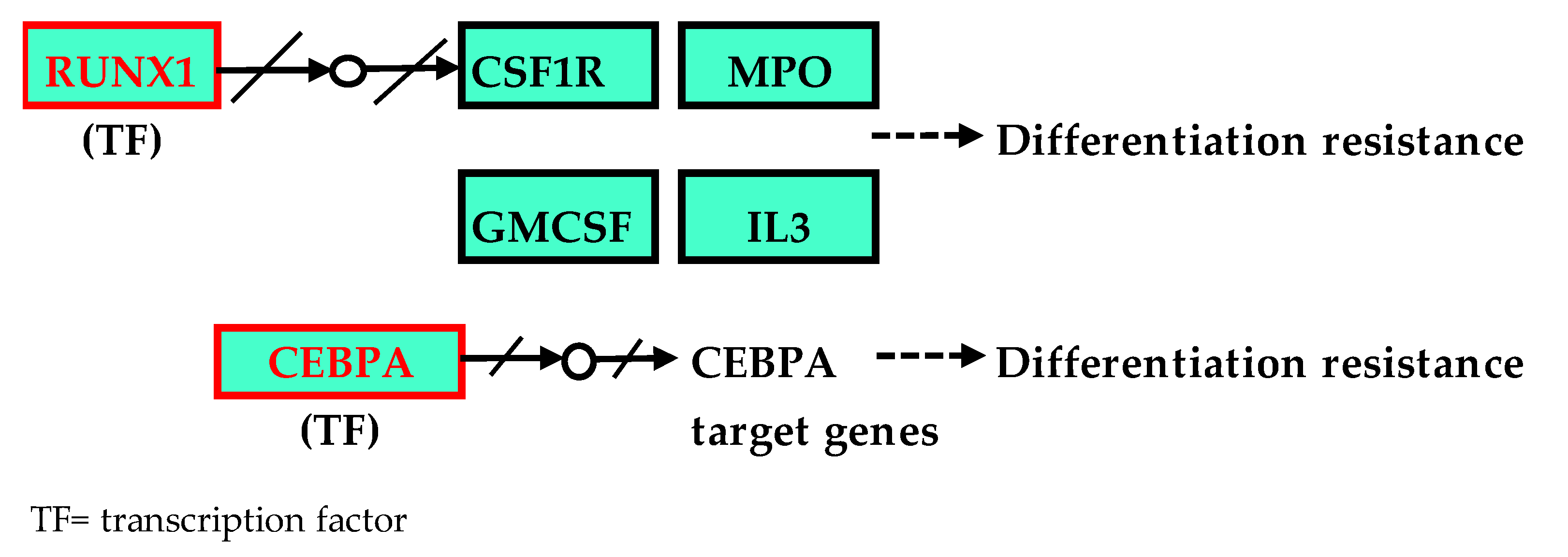
3.5. CEBPA Mutation and Gene Expression Profile
3.6. Functional and Pathway Enrichment Analysis of 26 Genes with Somatic Variants Using WebGestalt ORA Analysis
4. Discussion
Significance of the Study
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Betz, B.L.; Hess, J.L. Acute Myeloid Leukemia Diagnosis in the 21st Century. Arch. Pathol. Lab. Med. 2010, 134, 1427–1433. [Google Scholar] [CrossRef]
- Lazarevic, V.; Hörstedt, A.-S.; Johansson, B.; Antunovic, P.; Billström, R.; Derolf, Å.; Hulegårdh, E.; Lehmann, S.; Möllgård, L.; Nilsson, C.; et al. Incidence and prognostic significance of karyotypic subgroups in older patients with acute myeloid leukemia: The Swedish population-based experience. Blood Cancer J. 2014, 4, e188. [Google Scholar] [CrossRef] [Green Version]
- Meng, C.Y.; Noor, P.J.; Ismail, A.; Ahid, M.F.; Zakaria, Z. Cytogenetic Profile of de novo Acute Myeloid Leukemia Patients in Malaysia. Int. J. Biomed. Sci. IJBS 2013, 9, 26–32. [Google Scholar]
- Yang, L.; Zhang, H.; Yang, X.; Lu, T.; Ma, S.; Cheng, H.; Yen, K.; Cheng, T. Prognostic Prediction of Cytogenetically Normal Acute Myeloid Leukemia Based on a Gene Expression Model. Front. Oncol. 2021, 11, 659201. [Google Scholar] [CrossRef]
- Mrózek, K.; Marcucci, G.; Paschka, P.; Whitman, S.P.; Bloomfield, C.D. Clinical relevance of mutations and gene-expression changes in adult acute myeloid leukemia with normal cytogenetics: Are we ready for a prognostically prioritized molecular classification? Blood 2007, 109, 431–448. [Google Scholar] [CrossRef] [Green Version]
- Döhner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef] [Green Version]
- Döhner, H.; Wei, A.H.; Appelbaum, F.R.; Craddock, C.; DiNardo, C.D.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Godley, L.A.; Hasserjian, R.P.; et al. Diagnosis and management of AML in adults: 2022 recommendations from an international expert panel on behalf of the ELN. Blood 2022, 140, 1345–1377. [Google Scholar] [CrossRef]
- O’Donnell, M.R.; Tallman, M.S.; Abboud, C.N.; Altman, J.K.; Appelbaum, F.R.; Arber, D.A.; Bhatt, V.; Bixby, D.; Blum, W.; Coutre, S.E.; et al. Acute Myeloid Leukemia, Version 3.2017, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2017, 15, 926–957. [Google Scholar] [CrossRef]
- Ding, L.; Ley, T.J.; Larson, D.E.; Miller, C.A.; Koboldt, D.C.; Welch, J.S.; Ritchey, J.K.; Young, M.A.; Lamprecht, T.; McLellan, M.D.; et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature 2012, 481, 506–510. [Google Scholar] [CrossRef] [Green Version]
- Garg, M.; Nagata, Y.; Kanojia, D.; Mayakonda, A.; Yoshida, K.; Keloth, S.H.; Zang, Z.J.; Okuno, Y.; Shiraishi, Y.; Chiba, K.; et al. Profiling of somatic mutations in acute myeloid leukemia with FLT3-ITD at diagnosis and relapse. Blood 2015, 126, 2491–2501. [Google Scholar] [CrossRef] [Green Version]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Khoury, J.D.; Solary, E.; Abla, O.; Akkari, Y.; Alaggio, R.; Apperley, J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan, J.K.C.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 2022, 36, 1703–1719. [Google Scholar] [CrossRef]
- Dickson, G.J.; Bustraan, S.; Hills, R.K.; Ali, A.; Goldstone, A.H.; Burnett, A.K.; Linch, D.C.; Gale, R.E. The value of molecular stratification forCEBPADM and NPM1MUTFLT3WT genotypes in older patients with acute myeloid leukaemia. Br. J. Haematol. 2016, 172, 573–580. [Google Scholar] [CrossRef] [Green Version]
- van Waalwijk van Doorn-Khosrovani, S.B.; Erpelinck, C.; Meijer, J.; Van Oosterhoud, S.; Van Putten, W.L.J.; Valk, P.J.M.; Beverloo, H.B.; Tenen, D.; Löwenberg, B.; Delwel, R. Biallelic mutations in the CEBPA gene and low CEBPA expression levels as prognostic markers in intermediate-risk AML. Hematol. J. 2003, 4, 31–40. [Google Scholar] [CrossRef]
- Gaidzik, V.I.; Paschka, P.; Späth, D.; Habdank, M.; Köhne, C.-H.; Germing, U.; von Lilienfeld-Toal, M.; Held, G.; Horst, H.-A.; Haase, D.; et al. TET2 Mutations in Acute Myeloid Leukemia (AML): Results from a Comprehensive Genetic and Clinical Analysis of the AML Study Group. J. Clin. Oncol. 2012, 30, 1350–1357. [Google Scholar] [CrossRef]
- Wang, R.; Chen, C.; Jing, Y.; Qin, J.; Li, Y.; Chen, G.; Zhou, W.; Li, Y.; Wang, J.; Li, D.; et al. Characteristics and prognostic significance of genetic mutations in acute myeloid leukemia based on a targeted next-generation sequencing technique. Cancer Med. 2020, 9, 8457–8467. [Google Scholar] [CrossRef]
- Valk, P.J.; Verhaak, R.G.; Beijen, M.A.; Erpelinck, C.A.; Doorn-Khosrovani, S.B.V.W.V.; Boer, J.M.; Beverloo, H.B.; Moorhouse, M.J.; van der Spek, P.J.; Löwenberg, B.; et al. Prognostically Useful Gene-Expression Profiles in Acute Myeloid Leukemia. N. Engl. J. Med. 2004, 350, 1617–1628. [Google Scholar] [CrossRef] [Green Version]
- Bullinger, L.; Döhner, K.; Bair, E.; Fröhling, S.; Schlenk, R.F.; Tibshirani, R.; Döhner, H.; Pollack, J.R. Use of Gene-Expression Profiling to Identify Prognostic Subclasses in Adult Acute Myeloid Leukemia. N. Engl. J. Med. 2004, 350, 1605–1616. [Google Scholar] [CrossRef] [Green Version]
- Grimwade, D.; Haferlach, T. Gene-Expression Profiling in Acute Myeloid Leukemia. N. Engl. J. Med. 2004, 350, 1676–1678. [Google Scholar] [CrossRef] [Green Version]
- Kohlmann, A.; Bullinger, L.; Thiede, C.; Schaich, M.; Schnittger, S.; Döhner, K.; Dugas, M.; Klein, H.-U.; Döhner, H.; Ehninger, G.; et al. Gene expression profiling in AML with normal karyotype can predict mutations for molecular markers and allows novel insights into perturbed biological pathways. Leukemia 2010, 24, 1216–1220. [Google Scholar] [CrossRef] [Green Version]
- Pomeroy, E.J.; Eckfeldt, C.E. Targeting Ras signaling in AML: RALB is a small GTPase with big potential. Small GTPases 2017, 11, 39–44. [Google Scholar] [CrossRef]
- Voso, M.T.; Ottone, T.; Lavorgna, S.; Venditti, A.; Maurillo, L.; Lo-Coco, F.; Buccisano, F. MRD in AML: The Role of New Techniques. Front. Oncol. 2019, 9, 655. [Google Scholar] [CrossRef] [Green Version]
- The Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [CrossRef] [Green Version]
- Jongen-Lavrencic, M.; Grob, T.; Hanekamp, D.; Kavelaars, F.G.; al Hinai, A.; Zeilemaker, A.; Erpelinck-Verschueren, C.A.; Gradowska, P.L.; Meijer, R.; Cloos, J.; et al. Molecular Minimal Residual Disease in Acute Myeloid Leukemia. New Engl. J. Med. 2018, 378, 1189–1199. [Google Scholar] [CrossRef]
- Ravandi, F. Is it time to routinely incorporate MRD into practice? Best Pr. Res. Clin. Haematol. 2018, 31, 396–400. [Google Scholar] [CrossRef]
- Terwijn, M.; Van Putten, W.L.; Kelder, A.; Van Der Velden, V.H.; Brooimans, R.A.; Pabst, T.; Maertens, J.; Boeckx, N.; De Greef, G.E.; Valk, P.J.; et al. High Prognostic Impact of Flow Cytometric Minimal Residual Disease Detection in Acute Myeloid Leukemia: Data From the HOVON/SAKK AML 42A Study. J. Clin. Oncol. 2013, 31, 3889–3897. [Google Scholar] [CrossRef]
- Vonk, C.M.; Al Hinai, A.S.A.; Hanekamp, D.; Valk, P.J.M. Molecular Minimal Residual Disease Detection in Acute Myeloid Leukemia. Cancers 2021, 13, 5431. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef]
- De Greef, G.E.; Van Putten, W.L.J.; Boogaerts, M.; Huijgens, P.C.; Verdonck, L.F.; Vellenga, E.; Theobald, M.; Jacky, E.; Löwenberg, B.; The Dutch-Belgian Hemato-Oncology Co-operative Group HOVON; et al. Criteria for defining a complete remission in acute myeloid leukaemia revisited. An analysis of patients treated in HOVON-SAKK co-operative group studies. Br. J. Haematol. 2005, 128, 184–191. [Google Scholar] [CrossRef]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [Green Version]
- He, M.M.; Li, Q.; Yan, M.; Cao, H.; Hu, Y.; He, K.Y.; Cao, K.; Li, M.M.; Wang, K. Variant Interpretation for Cancer (VIC): A computational tool for assessing clinical impacts of somatic variants. Genome Med. 2019, 11, 53. [Google Scholar] [CrossRef] [Green Version]
- Eilbeck, K.; Lewis, S.E.; Mungall, C.J.; Yandell, M.; Stein, L.; Durbin, R.; Ashburner, M. The Sequence Ontology: A tool for the unification of genome annotations. Genome Biol. 2005, 6, R44. [Google Scholar] [CrossRef] [Green Version]
- Ensembl Variation—Calculated Variant Consequences. Available online: https://mart.ensembl.org/info/genome/variation/prediction/predicted_data.html (accessed on 16 March 2022).
- Yan, Y.H.; Chen, S.X.; Cheng, L.Y.; Rodriguez, A.Y.; Tang, R.; Cabrera, K.; Zhang, D.Y. Confirming putative variants at ≤ 5% allele frequency using allele enrichment and Sanger sequencing. Sci. Rep. 2021, 11, 11640. [Google Scholar] [CrossRef]
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Aguilera, M.A.; Meyer, R.; Massouras, A. VarSome: The human genomic variant search engine. Bioinformatics 2019, 35, 1978–1980. [Google Scholar] [CrossRef] [Green Version]
- Sherry, S.T.; Ward, M.-H.; Kholodov, M.; Baker, J.; Phan, L.; Smigielski, E.M.; Sirotkin, K. dbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 2001, 29, 308–311. [Google Scholar] [CrossRef] [Green Version]
- Landrum, M.J.; Lee, J.M.; Riley, G.R.; Jang, W.; Rubinstein, W.S.; Church, D.M.; Maglott, D.R. ClinVar: Public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014, 42, D980–D985. [Google Scholar] [CrossRef] [Green Version]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [Green Version]
- Sim, N.-L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef]
- Choi, Y.; Sims, G.E.; Murphy, S.; Miller, J.R.; Chan, A.P. Predicting the Functional Effect of Amino Acid Substitutions and Indels. PLoS ONE 2012, 7, e46688. [Google Scholar] [CrossRef] [Green Version]
- Reva, B.; Antipin, Y.; Sander, C. Predicting the functional impact of protein mutations: Application to cancer genomics. Nucleic Acids Res. 2011, 39, e118. [Google Scholar] [CrossRef] [Green Version]
- Shihab, H.A.; Gough, J.; Cooper, D.N.; Stenson, P.D.; Barker, G.L.A.; Edwards, K.J.; Day, I.N.M.; Gaunt, T.R. Predicting the Functional, Molecular, and Phenotypic Consequences of Amino Acid Substitutions using Hidden Markov Models. Hum. Mutat. 2013, 34, 57–65. [Google Scholar] [CrossRef] [Green Version]
- Qian, M.; Shao, Y. A Likelihood Ratio Test for Genome-Wide Association under Genetic Heterogeneity. Ann. Hum. Genet. 2013, 77, 174–182. [Google Scholar] [CrossRef] [Green Version]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef]
- Li, M.M.; Datto, M.; Duncavage, E.J.; Kulkarni, S.; Lindeman, N.I.; Roy, S.; Tsimberidou, A.M.; Vnencak-Jones, C.L.; Wolff, D.J.; Younes, A.; et al. Standards and Guidelines for the Interpretation and Reporting of Sequence Variants in Cancer: A Joint Consensus Recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J. Mol. Diagn. 2017, 19, 4–23. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Anesthesia Analg. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Vasaikar, S.; Shi, Z.; Greer, M.; Zhang, B. WebGestalt 2017: A more comprehensive, powerful, flexible and interactive gene set enrichment analysis toolkit. Nucleic Acids Res. 2017, 45, W130–W137. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Deeg, H.J. Not all patients with AML over 60 years of age should be offered early allogeneic stem cell transplantation. Blood Adv. 2022, 6, 1623–1627. [Google Scholar] [CrossRef]
- Rosenthal, S.H.; Gerasimova, A.; Ma, C.; Li, H.-R.; Grupe, A.; Chong, H.; Acab, A.; Smolgovsky, A.; Owen, R.; Elzinga, C.; et al. Analytical validation and performance characteristics of a 48-gene next-generation sequencing panel for detecting potentially actionable genomic alterations in myeloid neoplasms. PLoS ONE 2021, 16, e0243683. [Google Scholar] [CrossRef]
- Mills, R.E.; Pittard, W.S.; Mullaney, J.M.; Farooq, U.; Creasy, T.H.; Mahurkar, A.A.; Kemeza, D.M.; Strassler, D.S.; Ponting, C.P.; Webber, C.; et al. Natural genetic variation caused by small insertions and deletions in the human genome. Genome Res. 2011, 21, 830–839. [Google Scholar] [CrossRef] [Green Version]
- Yusoff, Y.M.; Ahid, F.; Abu Seman, Z.; Abdullah, J.; Kamaluddin, N.R.; Esa, E.; Zakaria, Z. Comprehensive analysis of mutations and clonal evolution patterns in a cohort of patients with cytogenetically normal acute myeloid leukemia. Mol. Cytogenet. 2021, 14, 45. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Cortes, J. Mutations in AML: Prognostic and therapeutic implications. Hematology 2016, 2016, 348–355. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, L.; Heyes, E.; Grebien, F. Gain-of-Function Effects of N-Terminal CEBPA Mutations in Acute Myeloid Leukemia. Bioessays 2020, 42, e1900178. [Google Scholar] [CrossRef] [Green Version]
- Fasan, A.; Haferlach, C.; Alpermann, T.; Jeromin, S.; Grossmann, V.; Eder, C.; Weissmann, S.; Dicker, F.; Kohlmann, A.; Schindela, S.; et al. The role of different genetic subtypes of CEBPA mutated AML. Leukemia 2014, 28, 794–803. [Google Scholar] [CrossRef]
- Koschmieder, S.; Halmos, B.; Levantini, E.; Tenen, D.G. Dysregulation of the C/EBPα Differentiation Pathway in Human Cancer. J. Clin. Oncol. 2009, 27, 619–628. [Google Scholar] [CrossRef] [Green Version]
- Wouters, B.J.; Löwenberg, B.; Erpelinck-Verschueren, C.A.J.; van Putten, W.L.J.; Valk, P.J.M.; Delwel, R. Double CEBPA mutations, but not single CEBPA mutations, define a subgroup of acute myeloid leukemia with a distinctive gene expression profile that is uniquely associated with a favorable outcome. Blood 2009, 113, 3088–3091. [Google Scholar] [CrossRef] [Green Version]
- Van Vliet, M.H.; Burgmer, P.; De Quartel, L.; Brand, J.P.; De Best, L.C.; Viëtor, H.; Löwenberg, B.; Valk, P.J.; Van Beers, E.H. Detection ofCEBPADouble Mutants in Acute Myeloid Leukemia Using a Custom Gene Expression Array. Genet. Test. Mol. Biomark. 2013, 17, 395–400. [Google Scholar] [CrossRef] [Green Version]
- Arindrarto, W.; Borràs, D.M.; de Groen, R.A.L.; van den Berg, R.R.; Locher, I.J.; van Diessen, S.A.M.E.; van der Holst, R.; van der Meijden, E.D.; Honders, M.W.; de Leeuw, R.H.; et al. Comprehensive diagnostics of acute myeloid leukemia by whole transcriptome RNA sequencing. Leukemia 2021, 35, 47–61. [Google Scholar] [CrossRef] [Green Version]
- Salarpour, F.; Goudarzipour, K.; Mohammadi, M.; Ahmadzadeh, A.; Faraahi, S.; Farsani, M.A. Evaluation of CCAAT/Enhancer Binding Protein (C/EBP) Alpha (CEBPA) and Runt-Related Transcription Factor 1 (RUNX1) Expression in Patients with De Novo Acute Myeloid Leukemia. Ann. Hum. Genet. 2017, 81, 276–283. [Google Scholar] [CrossRef]
- Corley, E.M.; Ali, M.K.M.; Alharthy, H.; Kline, K.A.F.; Sewell, D.; Law, J.Y.; Lee, S.T.; Niyongere, S.; Duong, V.H.; Baer, M.R.; et al. Impact of FLT3-ITD Insertion Length on Outcomes in Acute Myeloid Leukemia: A Propensity Score-Adjusted Cohort Study. Biology 2022, 11, 916. [Google Scholar] [CrossRef]
- Meshinchi, S.; Woods, W.G.; Stirewalt, D.L.; Sweetser, D.A.; Buckley, J.D.; Tjoa, T.K.; Bernstein, I.D.; Radich, J.P. Prevalence and prognostic significance of Flt3 internal tandem duplication in pediatric acute myeloid leukemia. Blood 2001, 97, 89–94. [Google Scholar] [CrossRef]
- Fröhling, S.; Schlenk, R.F.; Breitruck, J.; Benner, A.; Kreitmeier, S.; Tobis, K.; Döhner, H.; Döhner, K. Prognostic significance of activating FLT3 mutations in younger adults (16 to 60 years) with acute myeloid leukemia and normal cytogenetics: A study of the AML Study Group Ulm. Blood 2002, 100, 4372–4380. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Lee, G.D.; Park, J.; Yoon, J.-H.; Kim, H.-J.; Min, W.-S.; Kim, M. Quantitative fragment analysis of FLT3-ITD efficiently identifying poor prognostic group with high mutant allele burden or long ITD length. Blood Cancer J. 2015, 5, e336. [Google Scholar] [CrossRef] [Green Version]
- Meshinchi, S.; Stirewalt, D.L.; Alonzo, T.A.; Boggon, T.J.; Gerbing, R.B.; Rocnik, J.L.; Lange, B.J.; Gilliland, D.G.; Radich, J.P. Structural and numerical variation of FLT3/ITD in pediatric AML. Blood 2008, 111, 4930–4933. [Google Scholar] [CrossRef] [Green Version]
- Stirewalt, D.L.; Kopecky, K.J.; Meshinchi, S.; Engel, J.H.; Pogosova-Agadjanyan, E.L.; Linsley, J.; Slovak, M.L.; Willman, C.L.; Radich, J.P. Size of FLT3 internal tandem duplication has prognostic significance in patients with acute myeloid leukemia. Blood 2006, 107, 3724–3726. [Google Scholar] [CrossRef] [Green Version]
- Kusec, R.; Jaksic, O.; Ostojic, S.; Kardum-Skelin, I.; Vrhovac, R.; Jaksic, B. More on prognostic significance of FLT3/ITD size in acute myeloid leukemia (AML). Blood 2006, 108, 405–406. [Google Scholar] [CrossRef]
- Castaño-Bonilla, T.; Alonso-Dominguez, J.M.; Barragán, E.; Rodríguez-Veiga, R.; Sargas, C.; Gil, C.; Chillón, C.; Vidriales, M.B.; García, R.; Martínez-López, J.; et al. Prognostic significance of FLT3-ITD length in AML patients treated with intensive regimens. Sci. Rep. 2021, 11, 20745. [Google Scholar] [CrossRef]
- Schnittger, S.; Bacher, U.; Haferlach, C.; Alpermann, T.; Kern, W.; Haferlach, T. Diversity of the juxtamembrane and TKD1 mutations (Exons 13-15) in the FLT3 gene with regards to mutant load, sequence, length, localization, and correlation with biological data. Genes, Chromosom. Cancer 2012, 51, 910–924. [Google Scholar] [CrossRef]
- Gale, R.E.; Green, C.; Allen, C.; Mead, A.J.; Burnett, A.K.; Hills, R.K.; Linch, D.C. The impact of FLT3 internal tandem duplication mutant level, number, size, and interaction with NPM1 mutations in a large cohort of young adult patients with acute myeloid leukemia. Blood 2008, 111, 2776–2784. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.; Yun, W.; Lee, S.-T.; Choi, J.R.; Yoo, K.H.; Koo, H.H.; Jung, C.W.; Kim, S.H. Prevalence and clinical implications of germline predisposition gene mutations in patients with acute myeloid leukemia. Sci. Rep. 2020, 10, 14297. [Google Scholar] [CrossRef]
- Ernst, M.P.T.; Kavelaars, F.G.; Löwenberg, B.; Valk, P.J.M.; Raaijmakers, M.H.G.P. RUNX1 germline variants in RUNX1-mutant AML: How frequent? Blood 2021, 137, 1428–1431. [Google Scholar] [CrossRef]
- Mikkelsen, S.U.; Safavi, S.; Dimopoulos, K.; O’Rourke, C.J.; Andersen, M.K.; Holm, M.S.; Marcher, C.W.; Andersen, J.B.; Hansen, J.W.; Grønbæk, K. Structural aberrations are associated with poor survival in patients with clonal cytopenia of undetermined significance. Haematologica 2020, 106, 1762–1766. [Google Scholar] [CrossRef]
- Singh, H.; A Lane, A.; Correll, M.; Przychodzen, B.; Sykes, D.B.; Stone, R.M.; Ballen, K.K.; Amrein, P.C.; Maciejewski, J.; Attar, E.C. Putative RNA-splicing gene LUC7L2 on 7q34 represents a candidate gene in pathogenesis of myeloid malignancies. Blood Cancer J. 2013, 3, e117. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Yang, J.; Wei, Q.; Weng, L.; Wu, F.; Shi, Y.; Cheng, X.; Cai, X.; Hu, C.; Cao, P. Identification of a selective inhibitor of IDH2/R140Q enzyme that induces cellular differentiation in leukemia cells. Cell Commun. Signal. 2020, 18, 55. [Google Scholar] [CrossRef] [Green Version]
- Lambert, J.; Lambert, J.; Thomas, X.; Marceau-Renaut, A.; Micol, J.-B.; Renneville, A.; Clappier, E.; Hayette, S.; Récher, C.; Raffoux, E.; et al. Early detection of WT1 measurable residual disease identifies high-risk patients, independent of transplantation in AML. Blood Adv. 2021, 5, 5258–5268. [Google Scholar] [CrossRef]
- Dillon, R.; Hills, R.; Freeman, S.; Potter, N.; Jovanovic, J.; Ivey, A.; Kanda, A.S.; Runglall, M.; Foot, N.; Valganon, M.; et al. Molecular MRD status and outcome after transplantation in NPM1-mutated AML. Blood 2020, 135, 680–688. [Google Scholar] [CrossRef] [Green Version]
- Carbonell, D.; Chicano, M.; Cardero, A.J.; Gómez-Centurión, I.; Bailén, R.; Oarbeascoa, G.; Martínez-Señarís, D.; Franco, C.; Muñiz, P.; Anguita, J.; et al. FLT3-ITD Expression as a Potential Biomarker for the Assessment of Treatment Response in Patients with Acute Myeloid Leukemia. Cancers 2022, 14, 4006. [Google Scholar] [CrossRef]
- Lee, T.I.; Young, R.A. Transcriptional Regulation and Its Misregulation in Disease. Cell 2013, 152, 1237–1251. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient Details | ||||||||
|---|---|---|---|---|---|---|---|---|
| DX1 | DX2 | DX3 | DX4 | DX5 | DX6 | DX7 | DX8 | |
| Demographic data | ||||||||
| Age | 31 | 29 | 54 | 55 | 36 | 49 | 45 | 21 |
| Ethnicity | Malay | Malay | Indian | Malay | Chinese | Malay | Indian | Malay |
| Gender | Female | Female | Female | Female | Female | Female | Male | Male |
| No. of mutations | 3 | 3 | 1 | 1 | 1 | 3 | 1 | 5 |
| Karyotype | ||||||||
| G-banding | 46 (X,X) [19] | 46 (X,X) [19] | 46 (X,X) [19] | 46 (X,X) [19] | 46 (X,X) [19] | 46 (X,X) [19] | 46 (X,Y) [19] | 46 (X,Y) [19] |
| Cell counts | ||||||||
| White blood cell count (109/L) | 26.9 | 57.7 | 31.1 | 170 | 19.5 | 45.8 | 39.5 | 152.8 |
| Haemoglobin (g/dL) | 7.3 | 8.1 | 8.1 | 8.4 | 11.3 | 7.9 | 9.9 | 6.1 |
| Platelet (109/L) | 32 | 76 | 83 | 70 | 68 | 21 | 22 | 27 |
| % Blast (PB) | 20% | 55% | 89% | 90% | 40% | 90% | 95% | 94% |
| % Blast (BMA) | 90% | 80% | 90% | 92% | 90% | 96% | 90% | 95% |
| Flow cytometry immunophenotyping | ||||||||
| Aberrant antigen expression | CD2+ | CD2+ | - | CD56+ | CD2+ | - | - | - |
| Gene mutations | ||||||||
| Leukaemia Q-Fusion (30 fusion genes) | Negative | Negative | Negative | Negative | Negative | Negative | Negative | Negative |
| FLT3-ITD | Detected | wt | wt | Detected | wt | Detected | Detected | wt |
| NPM1 mutation | Detected | Detected | Detected | Detected | wt | Detected | Detected | wt |
| Archer HGC VariantPlex Myeloid Panel | ||||||||
| Genes with pathogenic/likely pathogenic somatic variants/ITD | CBL | CBL | IDH2 | TET2 | CEBPA | FLT3 | IDH1 | CEBPA |
| DNMT3A | IDH1 | NPM1 | NPM1 | IDH2 | NPM1 | CUX1 | ||
| RUNX1 | LUC7L2 | FLT3-ITD | U2AF2 | FLT3-ITD | GATA2 | |||
| STAG2 | NPM1 | NPM1 | IDH1 | |||||
| NPM1 | FLT3-ITD | LUC7L2 | ||||||
| FLT3-ITD | WT1 | |||||||
| FLT3-ITD insertion length (Archer HGC) | 42 | - | - | 45 | - | 54 | 144 | - |
| Number of mutations per patients | 4 | 3 | 1 | 1 | 1 | 3 | 1 | 5 |
| DEG | ||||||||
| Upregulated genes | EZH2 | EZH2 | CUX1 | CUX1 | EZH2 | ANKRD26 | CUX1 | EZH2 |
| FLT3 | FLT3 | FLT3 | ETV6 | CEBPA | CUX1 | FLT3 | CEBPA | |
| HRAS | HRAS | GATA2 | FLT3 | CUX1 | FLT3 | GATA2 | CUX1 | |
| IDH1 | LUC7L2 | LUC7L2 | GATA2 | DNMT3A | GATA2 | NPM1 | DNMT3A | |
| NPM1 | NPM1 | NPM1 | LUC7L2 | FLT3 | LUC7L2 | RUNX1 | ETV6 | |
| RUNX1 | SMC1A | RUNX1 | NPM1 | GATA2 | NPM1 | WT1 | FLT3 | |
| U2AF2 | WT1 | RUNX1 | IDH1 | RUNX1 | GATA2 | |||
| WT1 | NPM1 | SMC1A | IDH1 | |||||
| WT1 | LUC7L2 | |||||||
| NPM1 | ||||||||
| Downregulated genes | ATRX | ATRX | CBL | CBL | CBL | CBL | CBL | TET2 |
| CBL | CBL | NOTCH1 | NOTCH1 | NOTCH1 | NOTCH1 | KDM6A | STAG2 | |
| KDM6A | KDM6A | PPM1D | PPM1D | PPM1D | PPM1D | NOTCH1 | RAD21 | |
| NOTCH1 | NOTCH1 | RAD21 | RAD21 | RAD21 | RAD21 | PPM1D | PPM1D | |
| PPM1D | PPM1D | STAG2 | STAG2 | STAG2 | STAG2 | RAD21 | NOTCH1 | |
| RAD21 | RAD21 | TET2 | TET2 | TET2 | TET2 | STAG2 | KDM6A | |
| STAG2 | STAG2 | TET2 | CBL | |||||
| TET2 | TET2 | |||||||
| ELN 2017 Classification | ||||||||
| Prognosis | Intermediate | Good | Good | Intermediate | Good | Intermediate | Intermediate | Good |
| Treatment Protocol | ||||||||
| Induction | DA 3+7 | DA 3+7 | DA 3+7 | DA 3+7 | DA 3+7 | DA 3+7 | DA 3+7 | DA 3+7 |
| Consolidation | MIDAC/HIDAC | HIDAC/HIDAC/HIDAC | MIDAC/HIDAC/FLAG | MIDAC/FLAG/HIDAC | MIDAC/HIDAC/ARAC | MIDAC/FLAG/FLAG | MIDAC/HIDAC/ARAC | FLAG-IDA/HIDAC/ARAC |
| SCT | Nil | Nil | Nil | Allo-SCT after CR1 | Nil | Auto-SCT after CR1 | Allo-SCT after CR1 | Allo-SCT after CR2 |
| Response to treatment | ||||||||
| Remission status | CR1 | CR1 | CR1 | CR1 | CR1 | CR1 | CR1 | CR2 |
| OS | >5 | >5 | >5 | >5 | >5 | >5 | >5 | >5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ambayya, A.; Razali, R.; Sulong, S.; Zulkefli, E.S.; Yap, Y.Y.; Sathar, J.; Hassan, R. Genomic Alterations, Gene Expression Profiles and Functional Enrichment of Normal-Karyotype Acute Myeloid Leukaemia Based on Targeted Next-Generation Sequencing. Cancers 2023, 15, 1386. https://doi.org/10.3390/cancers15051386
Ambayya A, Razali R, Sulong S, Zulkefli ES, Yap YY, Sathar J, Hassan R. Genomic Alterations, Gene Expression Profiles and Functional Enrichment of Normal-Karyotype Acute Myeloid Leukaemia Based on Targeted Next-Generation Sequencing. Cancers. 2023; 15(5):1386. https://doi.org/10.3390/cancers15051386
Chicago/Turabian StyleAmbayya, Angeli, Rozaimi Razali, Sarina Sulong, Ezzanie Suffya Zulkefli, Yee Yee Yap, Jameela Sathar, and Rosline Hassan. 2023. "Genomic Alterations, Gene Expression Profiles and Functional Enrichment of Normal-Karyotype Acute Myeloid Leukaemia Based on Targeted Next-Generation Sequencing" Cancers 15, no. 5: 1386. https://doi.org/10.3390/cancers15051386