
Molecular Insight into Drug Resistance Mechanism Conferred by Aberrant PIK3CD Splice Variant in African American Prostate Cancer

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Methods
2.1. Cell Culture
2.2. RT-PCR Assay
2.3. Immunohistochemistry (IHC) Assay
2.4. MTT (Cell Viability) Assay
2.5. Cell Apoptosis Assay
2.6. Invasion Assay
2.7. Western Blot Analysis
2.8. Molecular Docking
2.9. ATP-Competitive Binding Assay
2.10. Cell-Free Kinase Activity Assay
2.11. RNA Pulldown Assay
2.12. RNA Immunoprecipitation (RIP) Assay
3. Results
3.1. PIK3CD-S Splice Variant Is Overexpressed in AA PCa
3.2. AA PCa Cells Are More Resistant to PI3Kδ-Specific, Pan-PI3K, and PI3K/Mtor Dual Inhibitors
3.3. PCa Cells Expressing PIK3CD-S Splice Variant Exhibit Drug Resistance Phenotype When Treated with PI3Kδ-Specific, Pan-PI3K, and PI3K/Mtor Dual Inhibitors
3.4. Overexpression of PIK3CD-S Splice Variant Enhances Antiapoptotic and Invasive Capacities in PCa Cells
3.5. Overexpression of PI3Kδ-S Splice Isoform Causes Resistance to Drug Inhibitory Effect on PI3K/AKT/Mtor Signaling
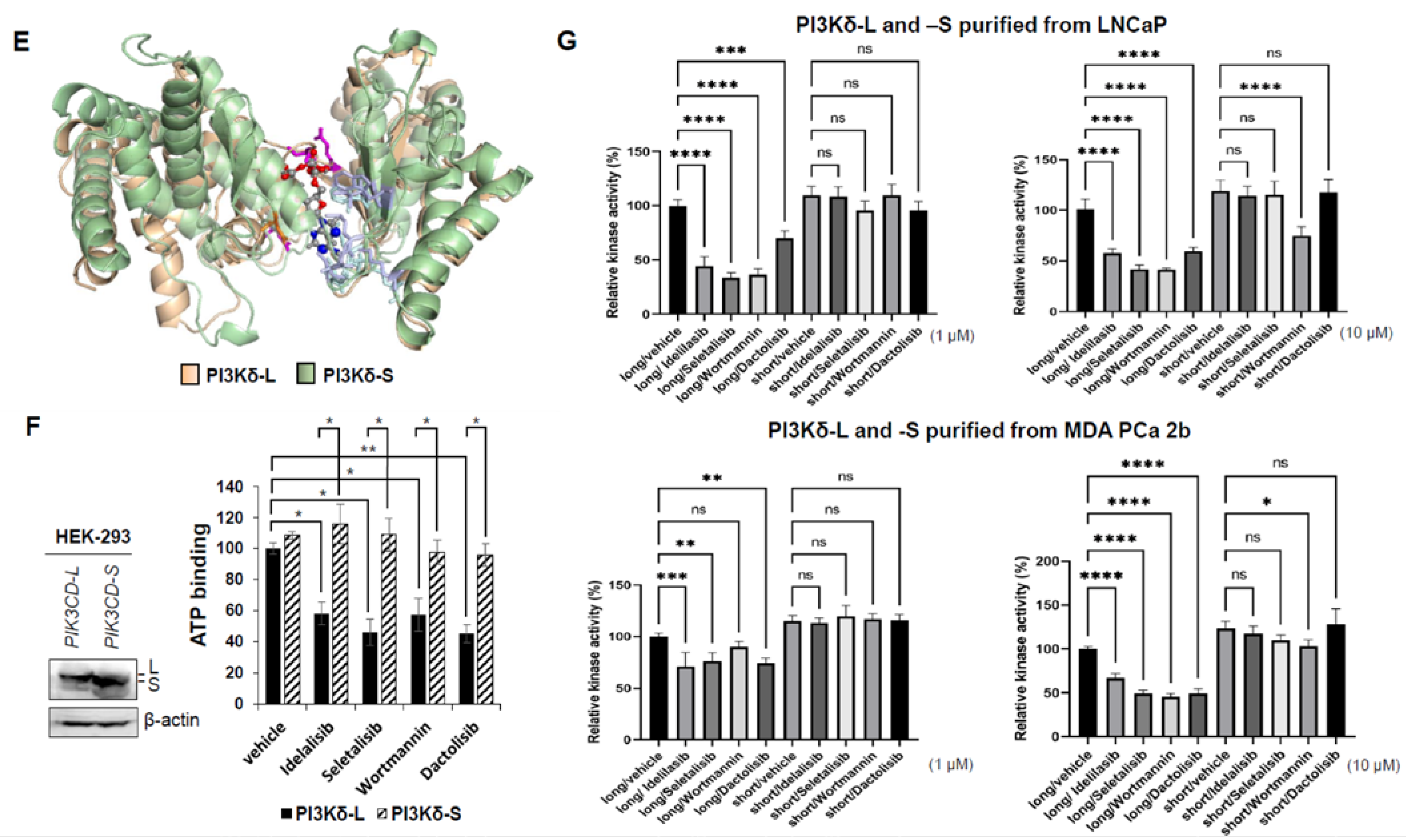
3.6. Molecular Docking Provides a Molecular Insight into Binding Affinities of PI3Kδ-L and PI3Kδ-S with Pan-PI3K and PI3Kδ-Specific Inhibitors
3.7. Competitive Drug/ATP-Binding Assays and Kinase Activity Assays Confirm a Reduced Binding Affinity between PI3Kδ-S and Drugs
3.8. Splicing Factor SRSF2 Modulates the Exon Skipping of PIK3CD
3.9. Inhibition of Splicing Kinase Sensitizes Pan-PI3Kδ and PI3Kδ-Selective Inhibitors against the PIK3CD-S-Expressing PCa
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Devaney, J.M.; Wang, S.; Furbert-Harris, P.; Apprey, V.; Ittmann, M.; Wang, B.D.; Olender, J.; Lee, N.H.; Kwabi-Addo, B. Genome-wide differentially methylated genes in prostate cancer tissues from African-American and Caucasian men. Epigenetics 2015, 10, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Rubicz, R.; Zhao, S.; Geybels, M.; Wright, J.L.; Kolb, S.; Klotzle, B.; Bibikova, M.; Troyer, D.; Lance, R.; Ostrander, E.A.; et al. DNA methylation profiles in African American prostate cancer patients in relation to disease progression. Genomics 2019, 111, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Tsai, C.W.; Chang, W.S.; Han, Y.; Huang, M.; Pettaway, C.A.; Bau, D.T.; Gu, J. Epigenome-Wide Association Study of Prostate Cancer in African Americans Identifies DNA Methylation Biomarkers for Aggressive Disease. Biomolecules 2021, 11, 1826. [Google Scholar] [CrossRef] [PubMed]
- Gujrati, H.; Ha, S.; Mohamed, A.; Wang, B.D. MicroRNA-mRNA Regulatory Network Mediates Activation of mTOR and VEGF Signaling in African American Prostate Cancer. Int. J. Mol. Sci. 2022, 23, 2926. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.D.; Ceniccola, K.; Yang, Q.; Andrawis, R.; Patel, V.; Ji, Y.; Rhim, J.; Olender, J.; Popratiloff, A.; Latham, P.; et al. Identification and Functional Validation of Reciprocal microRNA-mRNA Pairings in African American Prostate Cancer Disparities. Clin. Cancer Res. 2015, 21, 4970–4984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, A.; Goldberger, H.; Dimtchev, A.; Ramalinga, M.; Chijioke, J.; Marian, C.; Oermann, E.K.; Uhm, S.; Kim, J.S.; Chen, L.N.; et al. MicroRNA profiling in prostate cancer—The diagnostic potential of urinary miR-205 and miR-214. PLoS ONE 2013, 8, e76994. [Google Scholar] [CrossRef] [Green Version]
- Theodore, S.C.; Davis, M.; Zhao, F.; Wang, H.; Chen, D.; Rhim, J.; Dean-Colomb, W.; Turner, T.; Ji, W.; Zeng, G.; et al. MicroRNA profiling of novel African American and Caucasian Prostate Cancer cell lines reveals a reciprocal regulatory relationship of miR-152 and DNA methyltranferase 1. Oncotarget 2014, 5, 3512–3525. [Google Scholar] [CrossRef] [Green Version]
- Koga, Y.; Song, H.; Chalmers, Z.R.; Newberg, J.; Kim, E.; Carrot-Zhang, J.; Piou, D.; Polak, P.; Abdulkadir, S.A.; Ziv, E.; et al. Genomic Profiling of Prostate Cancers from Men with African and European Ancestry. Clin. Cancer Res. 2020, 26, 4651–4660. [Google Scholar] [CrossRef]
- Nagaya, N.; Rosenfeld, J.; Lee, G.T.; Kim, I.Y. RNA-seq profile of African American men with a clinically localized prostate cancer. Prostate Int. 2021, 9, 125–131. [Google Scholar] [CrossRef]
- Rayford, W.; Beksac, A.T.; Alger, J.; Alshalalfa, M.; Ahmed, M.; Khan, I.; Falagario, U.G.; Liu, Y.; Davicioni, E.; Spratt, D.E.; et al. Comparative analysis of 1152 African-American and European-American men with prostate cancer identifies distinct genomic and immunological differences. Commun. Biol. 2021, 4, 670. [Google Scholar] [CrossRef] [PubMed]
- Wallace, T.A.; Prueitt, R.L.; Yi, M.; Howe, T.M.; Gillespie, J.W.; Yfantis, H.G.; Stephens, R.M.; Caporaso, N.E.; Loffredo, C.A.; Ambs, S. Tumor immunobiological differences in prostate cancer between African-American and European-American men. Cancer Res. 2008, 68, 927–936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.D.; Yang, Q.; Ceniccola, K.; Bianco, F.; Andrawis, R.; Jarrett, T.; Frazier, H.; Patierno, S.R.; Lee, N.H. Androgen receptor-target genes in african american prostate cancer disparities. Prostate Cancer 2013, 2013, 763569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.D.; Ceniccola, K.; Hwang, S.; Andrawis, R.; Horvath, A.; Freedman, J.A.; Olender, J.; Knapp, S.; Ching, T.; Garmire, L.; et al. Alternative splicing promotes tumour aggressiveness and drug resistance in African American prostate cancer. Nat. Commun. 2017, 8, 15921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blencowe, B.J. Alternative splicing: New insights from global analyses. Cell 2006, 126, 37–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008, 40, 1413–1415. [Google Scholar] [CrossRef]
- Kahles, A.; Lehmann, K.V.; Toussaint, N.C.; Huser, M.; Stark, S.G.; Sachsenberg, T.; Stegle, O.; Kohlbacher, O.; Sander, C.; The Cancer Genome Atlas Research Network; et al. Comprehensive Analysis of Alternative Splicing Across Tumors from 8705 Patients. Cancer Cell 2018, 34, 211–224.e6. [Google Scholar] [CrossRef] [Green Version]
- Calderon-Aparicio, A.; Wang, B.D. Prostate cancer: Alternatively spliced mRNA transcripts in tumor progression and their uses as therapeutic targets. Int. J. Biochem. Cell Biol. 2021, 141, 106096. [Google Scholar] [CrossRef]
- Rajan, P.; Elliott, D.J.; Robson, C.N.; Leung, H.Y. Alternative splicing and biological heterogeneity in prostate cancer. Nat. Rev. Urol. 2009, 6, 454–460. [Google Scholar] [CrossRef]
- Wang, B.D.; Lee, N.H. Aberrant RNA Splicing in Cancer and Drug Resistance. Cancers 2018, 10, 458. [Google Scholar] [CrossRef] [Green Version]
- Olender, J.; Wang, B.D.; Ching, T.; Garmire, L.X.; Garofano, K.; Ji, Y.; Knox, T.; Latham, P.; Nguyen, K.; Rhim, J.; et al. A Novel FGFR3 Splice Variant Preferentially Expressed in African American Prostate Cancer Drives Aggressive Phenotypes and Docetaxel Resistance. Mol. Cancer Res. 2019, 17, 2115–2125. [Google Scholar] [CrossRef] [PubMed]
- Tzenaki, N.; Papakonstanti, E.A. p110delta PI3 kinase pathway: Emerging roles in cancer. Front. Oncol. 2013, 3, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tzenaki, N.; Andreou, M.; Stratigi, K.; Vergetaki, A.; Makrigiannakis, A.; Vanhaesebroeck, B.; Papakonstanti, E.A. High levels of p110delta PI3K expression in solid tumor cells suppress PTEN activity, generating cellular sensitivity to p110delta inhibitors through PTEN activation. FASEB J. 2012, 26, 2498–2508. [Google Scholar] [CrossRef] [PubMed]
- Xenou, L.; Papakonstanti, E.A. p110delta PI3K as a therapeutic target of solid tumours. Clin. Sci. 2020, 134, 1377–1397. [Google Scholar] [CrossRef]
- Yang, Q.; Modi, P.; Newcomb, T.; Queva, C.; Gandhi, V. Idelalisib: First-in-Class PI3K Delta Inhibitor for the Treatment of Chronic Lymphocytic Leukemia, Small Lymphocytic Leukemia, and Follicular Lymphoma. Clin. Cancer Res. 2015, 21, 1537–1542. [Google Scholar] [CrossRef] [Green Version]
- Yahiaoui, A.; Meadows, S.A.; Sorensen, R.A.; Cui, Z.H.; Keegan, K.S.; Brockett, R.; Chen, G.; Queva, C.; Li, L.; Tannheimer, S.L. PI3Kdelta inhibitor idelalisib in combination with BTK inhibitor ONO/GS-4059 in diffuse large B cell lymphoma with acquired resistance to PI3Kdelta and BTK inhibitors. PLoS ONE 2017, 12, e0171221. [Google Scholar] [CrossRef] [Green Version]
- Allen, R.A.; Brookings, D.C.; Powell, M.J.; Delgado, J.; Shuttleworth, L.K.; Merriman, M.; Fahy, I.J.; Tewari, R.; Silva, J.P.; Healy, L.J.; et al. Seletalisib: Characterization of a Novel, Potent, and Selective Inhibitor of PI3Kdelta. J. Pharmacol. Exp. Ther. 2017, 361, 429–440. [Google Scholar] [CrossRef] [Green Version]
- Arcaro, A.; Wymann, M.P. Wortmannin is a potent phosphatidylinositol 3-kinase inhibitor: The role of phosphatidylinositol 3,4,5-trisphosphate in neutrophil responses. Biochem. J. 1993, 296 Pt 2, 297–301. [Google Scholar] [CrossRef] [Green Version]
- Wise-Draper, T.M.; Moorthy, G.; Salkeni, M.A.; Karim, N.A.; Thomas, H.E.; Mercer, C.A.; Beg, M.S.; O’Gara, S.; Olowokure, O.; Fathallah, H.; et al. A Phase Ib Study of the Dual PI3K/mTOR Inhibitor Dactolisib (BEZ235) Combined with Everolimus in Patients with Advanced Solid Malignancies. Target. Oncol. 2017, 12, 323–332. [Google Scholar] [CrossRef] [Green Version]
- Gujrati, H.; Ha, S.; Waseem, M.; Wang, B.D. Downregulation of miR-99b-5p and Upregulation of Nuclear mTOR Cooperatively Promotes the Tumor Aggressiveness and Drug Resistance in African American Prostate Cancer. Int. J. Mol. Sci. 2022, 23, 9643. [Google Scholar] [CrossRef]
- Berndt, A.; Miller, S.; Williams, O.; Le, D.D.; Houseman, B.T.; Pacold, J.I.; Gorrec, F.; Hon, W.C.; Liu, Y.; Rommel, C.; et al. The p110 delta structure: Mechanisms for selectivity and potency of new PI(3)K inhibitors. Nat. Chem. Biol. 2010, 6, 117–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Somoza, J.R.; Koditek, D.; Villasenor, A.G.; Novikov, N.; Wong, M.H.; Liclican, A.; Xing, W.; Lagpacan, L.; Wang, R.; Schultz, B.E.; et al. Structural, biochemical, and biophysical characterization of idelalisib binding to phosphoinositide 3-kinase delta. J. Biol. Chem. 2015, 290, 8439–8446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albeck, S.; Unger, R.; Schreiber, G. Evaluation of direct and cooperative contributions towards the strength of buried hydrogen bonds and salt bridges. J. Mol. Biol. 2000, 298, 503–520. [Google Scholar] [CrossRef] [Green Version]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef] [PubMed]
- Biasini, M.; Bienert, S.; Waterhouse, A.; Arnold, K.; Studer, G.; Schmidt, T.; Kiefer, F.; Gallo Cassarino, T.; Bertoni, M.; Bordoli, L.; et al. SWISS-MODEL: Modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014, 42, W252–W258. [Google Scholar] [CrossRef] [PubMed]
- Maira, S.M.; Stauffer, F.; Brueggen, J.; Furet, P.; Schnell, C.; Fritsch, C.; Brachmann, S.; Chene, P.; De Pover, A.; Schoemaker, K.; et al. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol. Cancer Ther. 2008, 7, 1851–1863. [Google Scholar] [CrossRef] [Green Version]
- Vanhaesebroeck, B.; Perry, M.W.D.; Brown, J.R.; Andre, F.; Okkenhaug, K. PI3K inhibitors are finally coming of age. Nat. Rev. Drug Discov. 2021, 20, 741–769. [Google Scholar] [CrossRef]
- Walker, E.H.; Pacold, M.E.; Perisic, O.; Stephens, L.; Hawkins, P.T.; Wymann, M.P.; Williams, R.L. Structural determinants of phosphoinositide 3-kinase inhibition by wortmannin, LY294002, quercetin, myricetin, and staurosporine. Mol. Cell 2000, 6, 909–919. [Google Scholar] [CrossRef]
- Paz, I.; Akerman, M.; Dror, I.; Kosti, I.; Mandel-Gutfreund, Y. SFmap: A web server for motif analysis and prediction of splicing factor binding sites. Nucleic Acids Res. 2010, 38, W281–W285. [Google Scholar] [CrossRef] [Green Version]
- Fruman, D.A.; Rommel, C. PI3Kdelta inhibitors in cancer: Rationale and serendipity merge in the clinic. Cancer Discov. 2011, 1, 562–572. [Google Scholar] [CrossRef] [Green Version]
- Furman, R.R.; Sharman, J.P.; Coutre, S.E.; Cheson, B.D.; Pagel, J.M.; Hillmen, P.; Barrientos, J.C.; Zelenetz, A.D.; Kipps, T.J.; Flinn, I.; et al. Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. N. Engl. J. Med. 2014, 370, 997–1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gopal, A.K.; Kahl, B.S.; de Vos, S.; Wagner-Johnston, N.D.; Schuster, S.J.; Jurczak, W.J.; Flinn, I.W.; Flowers, C.R.; Martin, P.; Viardot, A.; et al. PI3Kdelta inhibition by idelalisib in patients with relapsed indolent lymphoma. N. Engl. J. Med. 2014, 370, 1008–1018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawyer, C.; Sturge, J.; Bennett, D.C.; O’Hare, M.J.; Allen, W.E.; Bain, J.; Jones, G.E.; Vanhaesebroeck, B. Regulation of breast cancer cell chemotaxis by the phosphoinositide 3-kinase p110delta. Cancer Res. 2003, 63, 1667–1675. [Google Scholar] [PubMed]
- Goulielmaki, E.; Bermudez-Brito, M.; Andreou, M.; Tzenaki, N.; Tzardi, M.; de Bree, E.; Tsentelierou, E.; Makrigiannakis, A.; Papakonstanti, E.A. Pharmacological inactivation of the PI3K p110delta prevents breast tumour progression by targeting cancer cells and macrophages. Cell Death Dis. 2018, 9, 678. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Chen, S.; Asara, J.M.; Balk, S.P. Phosphoinositide 3-kinase pathway activation in phosphate and tensin homolog (PTEN)-deficient prostate cancer cells is independent of receptor tyrosine kinases and mediated by the p110beta and p110delta catalytic subunits. J. Biol. Chem. 2010, 285, 14980–14989. [Google Scholar] [CrossRef] [Green Version]
- Ko, E.; Seo, H.W.; Jung, E.S.; Ju, S.; Kim, B.H.; Cho, H.; Kim, Y.J.; Park, Y.M.; Kim, J.S.; Jung, G. PI3Kdelta Is a Therapeutic Target in Hepatocellular Carcinoma. Hepatology 2018, 68, 2285–2300. [Google Scholar] [CrossRef] [Green Version]
- Lauder, S.N.; Vanhaesebroeck, B.; Gallimore, A. Sequential targeting of PI3Kdelta and LAG3 as an effective anti-cancer approach. Br. J. Cancer 2021, 125, 467–469. [Google Scholar] [CrossRef]
- Scheffold, A.; Jebaraj, B.M.C.; Tausch, E.; Bloehdorn, J.; Ghia, P.; Yahiaoui, A.; Dolnik, A.; Blatte, T.J.; Bullinger, L.; Dheenadayalan, R.P.; et al. IGF1R as druggable target mediating PI3K-delta inhibitor resistance in a murine model of chronic lymphocytic leukemia. Blood 2019, 134, 534–547. [Google Scholar] [CrossRef]
- Murali, I.; Kasar, S.; Naeem, A.; Tyekucheva, S.; Khalsa, J.K.; Thrash, E.M.; Itchaki, G.; Livitz, D.; Leshchiner, I.; Dong, S.; et al. Activation of the MAPK pathway mediates resistance to PI3K inhibitors in chronic lymphocytic leukemia. Blood 2021, 138, 44–56. [Google Scholar] [CrossRef]
- Wang, X.; Ding, J.; Meng, L.H. PI3K isoform-selective inhibitors: Next-generation targeted cancer therapies. Acta Pharmacol. Sin. 2015, 36, 1170–1176. [Google Scholar] [CrossRef] [Green Version]
- Siqueira, R.P.; Barbosa Ede, A.; Poleto, M.D.; Righetto, G.L.; Seraphim, T.V.; Salgado, R.L.; Ferreira, J.G.; Barros, M.V.; de Oliveira, L.L.; Laranjeira, A.B.; et al. Potential Antileukemia Effect and Structural Analyses of SRPK Inhibition by N-(2-(Piperidin-1-yl)-5-(Trifluoromethyl)Phenyl)Isonicotinamide (SRPIN340). PLoS ONE 2015, 10, e0134882. [Google Scholar] [CrossRef] [PubMed]
- Pandit, S.; Zhou, Y.; Shiue, L.; Coutinho-Mansfield, G.; Li, H.; Qiu, J.; Huang, J.; Yeo, G.W.; Ares, M., Jr.; Fu, X.D. Genome-wide analysis reveals SR protein cooperation and competition in regulated splicing. Mol. Cell 2013, 50, 223–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, C.; Cheng, Y.; Liu, Y.; Chen, L.; Liu, L.; Wei, N.; Xie, Z.; Wu, W.; Feng, Y. SRSF2 Regulates Alternative Splicing to Drive Hepatocellular Carcinoma Development. Cancer Res. 2017, 77, 1168–1178. [Google Scholar] [CrossRef] [PubMed] [Green Version]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ha, S.; Wang, B.-D. Molecular Insight into Drug Resistance Mechanism Conferred by Aberrant PIK3CD Splice Variant in African American Prostate Cancer. Cancers 2023, 15, 1337. https://doi.org/10.3390/cancers15041337
Ha S, Wang B-D. Molecular Insight into Drug Resistance Mechanism Conferred by Aberrant PIK3CD Splice Variant in African American Prostate Cancer. Cancers. 2023; 15(4):1337. https://doi.org/10.3390/cancers15041337
Chicago/Turabian StyleHa, Siyoung, and Bi-Dar Wang. 2023. "Molecular Insight into Drug Resistance Mechanism Conferred by Aberrant PIK3CD Splice Variant in African American Prostate Cancer" Cancers 15, no. 4: 1337. https://doi.org/10.3390/cancers15041337