
Mechanisms of Resistance to Antibody–Drug Conjugates

Abstract
:Simple Summary
Abstract
1. Introduction
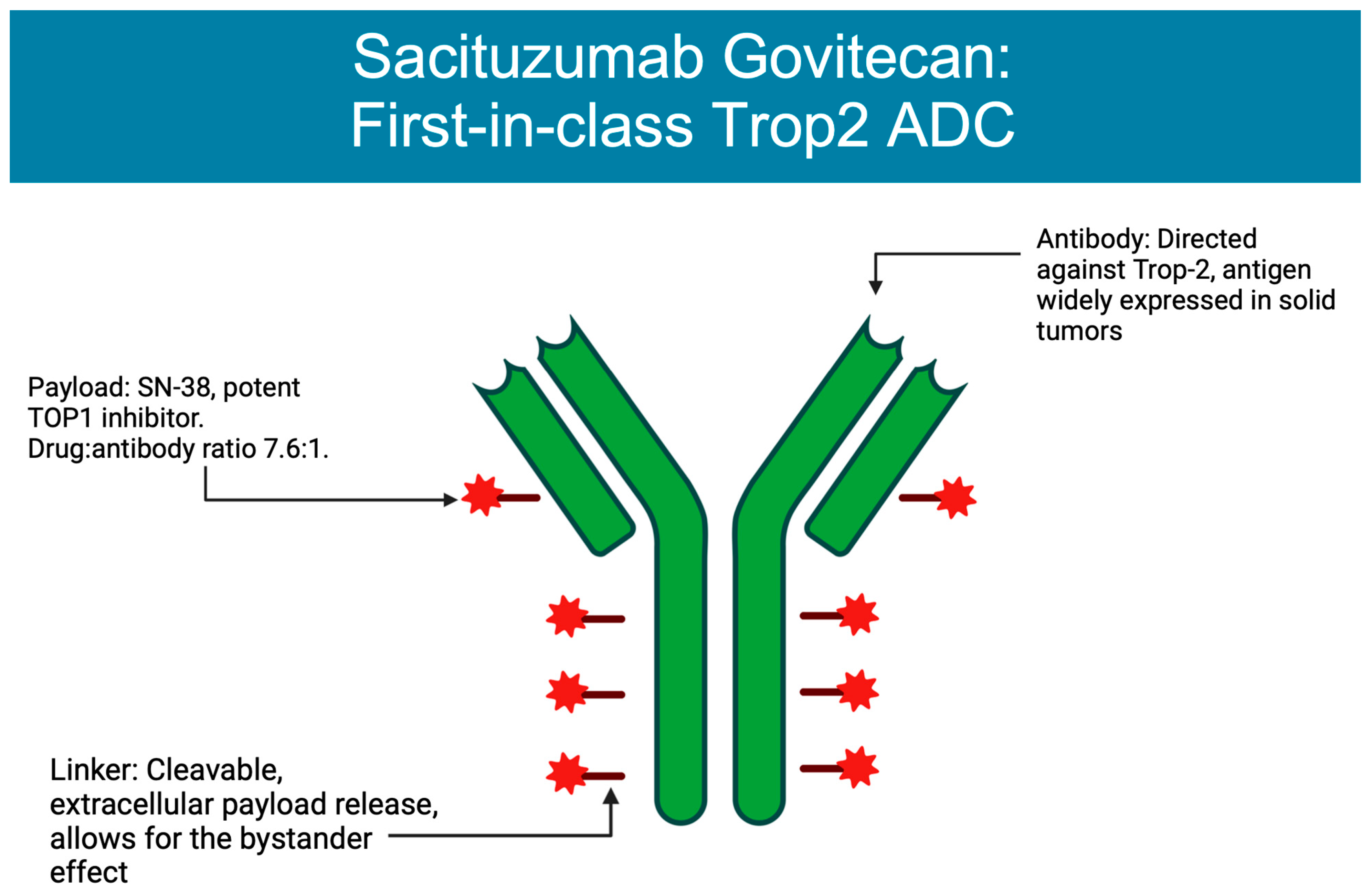
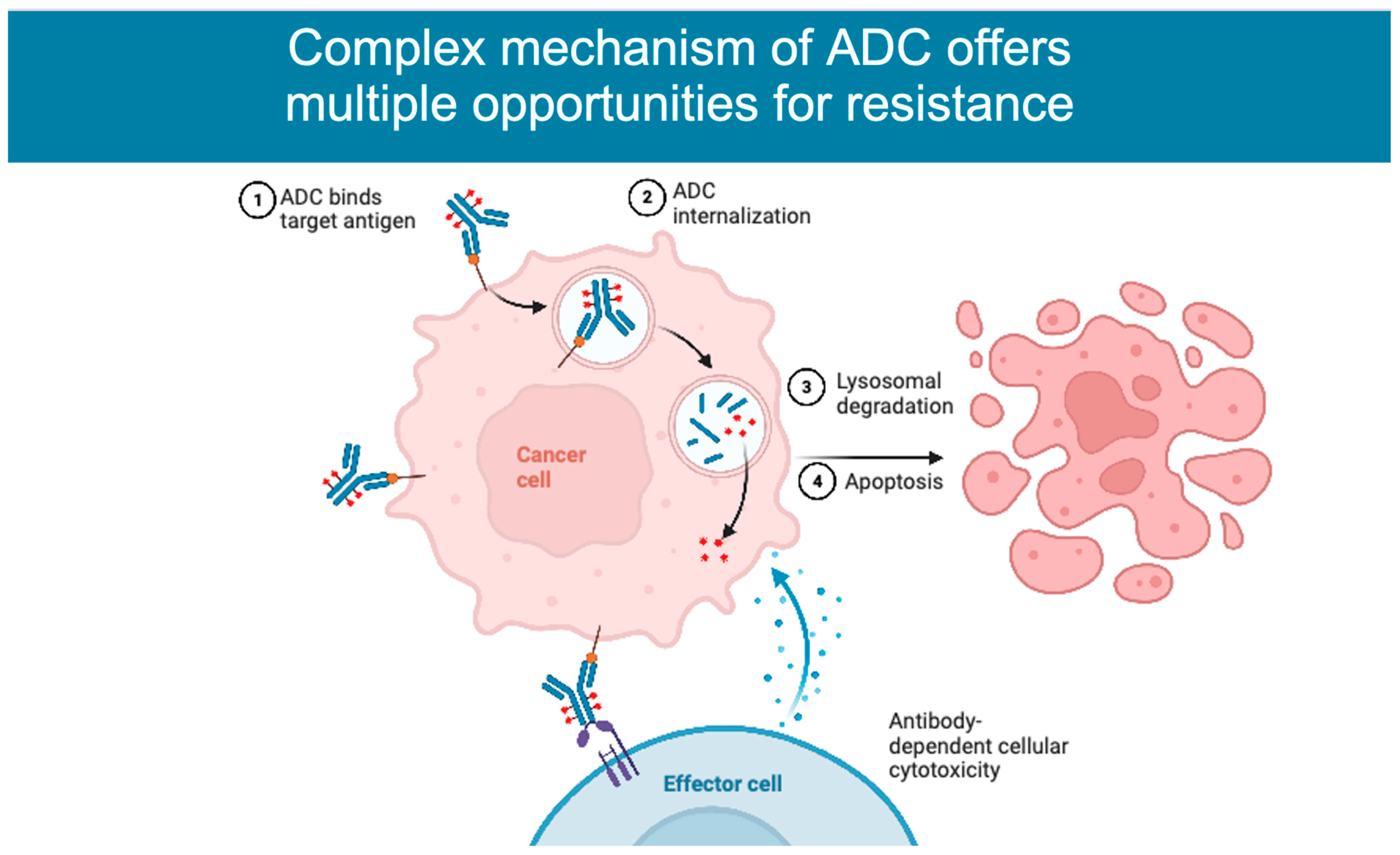
2. Mechanism of Action
3. ADCs Approved in Breast Cancer
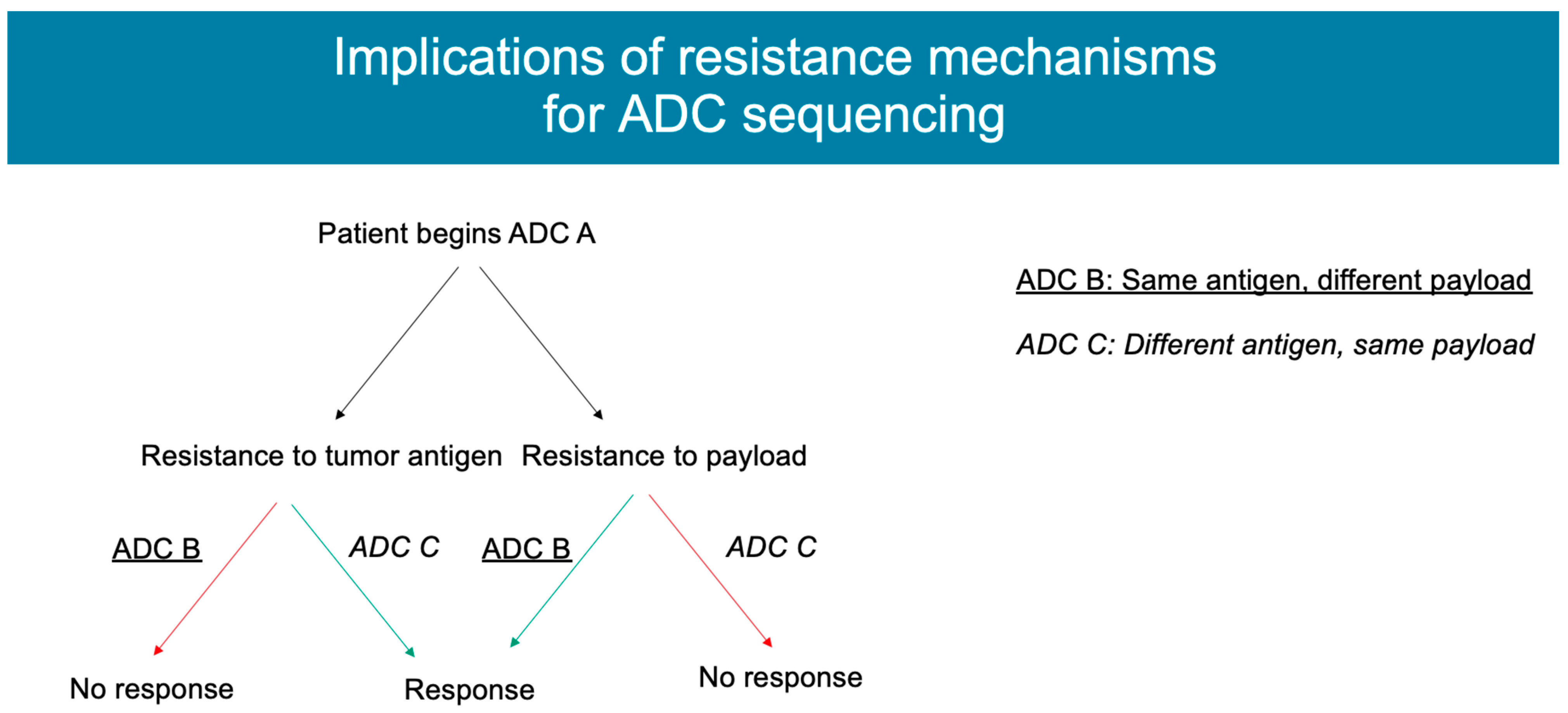
4. Mechanisms of Resistance
4.1. Antigen Expression
4.2. ADC Processing
4.3. Payload

{kind=link}
{kind=link}
{kind=link}
| Resistance Mechanism | ADC Where This Has Been Seen | Cancer Type | Examples |
|---|---|---|---|
| Antigen expression |
|
|
|
| ADC processing |
|
| |
| Payload |
|
|
|
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nagayama, A.; Ellisen, L.W.; Chabner, B.; Bardia, A. Antibody–Drug Conjugates for the Treatment of Solid Tumors: Clinical Experience and Latest Developments. Target. Oncol. 2017, 12, 719–739. [Google Scholar] [CrossRef] [PubMed]
- Drago, J.Z.; Modi, S.; Chandarlapaty, S. Unlocking the potential of antibody-drug conjugates for cancer therapy. Nat. Rev. Clin. Oncol. 2021, 18, 327–344. [Google Scholar] [CrossRef] [PubMed]
- Jain, N.; Smith, S.W.; Ghone, S.; Tomczuk, B. Current ADC Linker Chemistry. Pharm. Res. 2015, 32, 3526–3540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamblett, K.J.; Senter, P.D.; Chace, D.F.; Sun, M.M.C.; Lenox, J.; Cerveny, C.G.; Kissler, K.M.; Bernhardt, S.X.; Kopcha, A.K.; Zabinski, R.F.; et al. Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin. Cancer Res. 2004, 10, 7063–7070. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Ponte, J.F.; Yoder, N.C.; Laleau, R.; Coccia, J.; Lanieri, L.; Qiu, Q.; Wu, R.; Hong, E.; Bogalhas, M.; et al. Effects of Drug–Antibody Ratio on Pharmacokinetics, Biodistribution, Efficacy, and Tolerability of Antibody–Maytansinoid Conjugates. Bioconjug. Chem. 2017, 28, 1371–1381. [Google Scholar] [CrossRef]
- Ogitani, Y.; Hagihara, K.; Oitate, M.; Naito, H.; Agatsuma, T. Bystander killing effect of DS-8201a, a novel anti-human epidermal growth factor receptor 2 antibody-drug conjugate, in tumors with human epidermal growth factor receptor 2 heterogeneity. Cancer Sci. 2016, 107, 1039–1046. [Google Scholar] [CrossRef]
- Modi, S.; Park, H.; Murthy, R.K.; Iwata, H.; Tamura, K.; Tsurutani, J.; Moreno-Aspitia, A.; Doi, T.; Sagara, Y.; Redfern, C.; et al. Antitumor Activity and Safety of Trastuzumab Deruxtecan in Patients With HER2-Low-Expressing Advanced Breast Cancer: Results From a Phase Ib Study. J. Clin. Oncol. 2020, 38, 1887–1896. [Google Scholar] [CrossRef]
- Krop, I.E.; Kim, S.B.; González-Martín, A.; LoRusso, P.M.; Ferrero, J.M.; Smitt, M.; Yu, R.; Leung, A.C.; Wildiers, H. Trastuzumab emtansine versus treatment of physician’s choice for pretreated HER2-positive advanced breast cancer (TH3RESA): A randomised, open-label, phase 3 trial. Lancet Oncol. 2014, 15, 689–699. [Google Scholar] [CrossRef]
- Verma, S.; Miles, D.; Gianni, L.; Krop, I.E.; Welslau, M.; Baselga, J.; Pegram, M.; Oh, D.-Y.; Diéras, V.; Guardino, E.; et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. N. Engl. J. Med. 2012, 367, 1783–1791. [Google Scholar] [CrossRef] [Green Version]
- Perez, E.A.; Barrios, C.; Eiermann, W.; Toi, M.; Im, Y.H.; Conte, P.; Martin, M.; Pienkowski, T.; Pivot, X.; Burris, H., 3rd; et al. Trastuzumab Emtansine With or Without Pertuzumab Versus Trastuzumab Plus Taxane for Human Epidermal Growth Factor Receptor 2-Positive, Advanced Breast Cancer: Primary Results From the Phase III MARIANNE Study. J. Clin. Oncol. 2017, 35, 141–148. [Google Scholar] [CrossRef]
- Hurvitz, S.A.; Martin, M.; Symmans, W.F.; Jung, K.H.; Huang, C.-S.; Thompson, A.M.; Harbeck, N.; Valero, V.; Stroyakovskiy, D.; Wildiers, H.; et al. Neoadjuvant trastuzumab, pertuzumab, and chemotherapy versus trastuzumab emtansine plus pertuzumab in patients with HER2-positive breast cancer (KRISTINE): A randomised, open-label, multicentre, phase 3 trial. Lancet Oncol. 2018, 19, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Hunter, F.W.; Barker, H.R.; Lipert, B.; Rothé, F.; Gebhart, G.; Piccart-Gebhart, M.J.; Sotiriou, C.; Jamieson, S.M.F. Mechanisms of resistance to trastuzumab emtansine (T-DM1) in HER2-positive breast cancer. Br. J. Cancer 2020, 122, 603–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von Minckwitz, G.; Huang, C.S.; Mano, M.S.; Loibl, S.; Mamounas, E.P.; Untch, M.; Wolmark, N.; Rastogi, P.; Schneeweiss, A.; Redondo, A.; et al. Trastuzumab Emtansine for Residual Invasive HER2-Positive Breast Cancer. N. Engl. J. Med. 2019, 380, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Bardia, A.; Mayer, I.A.; Vahdat, L.T.; Tolaney, S.M.; Isakoff, S.J.; Diamond, J.R.; O’Shaughnessy, J.; Moroose, R.L.; Santin, A.D.; Abramson, V.G.; et al. Sacituzumab Govitecan-hziy in Refractory Metastatic Triple-Negative Breast Cancer. N. Engl. J. Med. 2019, 380, 741–751. [Google Scholar] [CrossRef]
- Bardia, A.; Tolaney, S.M.; Loirat, D.; Punie, K.; Oliveira, M.; Rugo, H.S.; Brufsky, A.; Kalinsky, K.; Cortes, J.; O’Shaughnessy, J.; et al. Sacituzumab govitecan (SG) versus treatment of physician’s choice (TPC) in patients (pts) with previously treated, metastatic triple-negative breast cancer (mTNBC): Final results from the phase 3 ASCENT study. J. Clin. Orthod. 2022, 40, 1071. [Google Scholar] [CrossRef]
- Bardia, A.; Hurvitz, S.A.; Tolaney, S.M.; Loirat, D.; Punie, K.; Oliveira, M.; Brufsky, A.; Sardesai, S.D.; Kalinsky, K.; Zelnak, A.B.; et al. Sacituzumab Govitecan in Metastatic Triple-Negative Breast Cancer. N. Engl. J. Med. 2021, 384, 1529–1541. [Google Scholar] [CrossRef]
- García-Alonso, S.; Ocaña, A.; Pandiella, A. Resistance to Antibody-Drug Conjugates. Cancer Res. 2018, 78, 2159–2165. [Google Scholar] [CrossRef] [Green Version]
- Ocaña, A.; Amir, E.; Pandiella, A. HER2 heterogeneity and resistance to anti-HER2 antibody-drug conjugates. Breast Cancer Res. 2020, 22, 15. [Google Scholar] [CrossRef]
- Ferraro, E.; Drago, J.Z.; Modi, S. Implementing antibody-drug conjugates (ADCs) in HER2-positive breast cancer: State of the art and future directions. Breast Cancer Res. 2021, 23, 84. [Google Scholar] [CrossRef]
- Modi, S.; Saura, C.; Yamashita, T.; Park, Y.H.; Kim, S.-B.; Tamura, K.; Andre, F.; Iwata, H.; Ito, Y.; Tsurutani, J.; et al. Trastuzumab Deruxtecan in Previously Treated HER2-Positive Breast Cancer. N. Engl. J. Med. 2020, 382, 610–621. [Google Scholar] [CrossRef]
- Cortés, J.; Kim, S.-B.; Chung, W.-P.; Im, S.-A.; Park, Y.H.; Hegg, R.; Kim, M.H.; Tseng, L.-M.; Petry, V.; Chung, C.-F.; et al. Trastuzumab Deruxtecan versus Trastuzumab Emtansine for Breast Cancer. N. Engl. J. Med. 2022, 386, 1143–1154. [Google Scholar] [CrossRef] [PubMed]
- Modi, S.; Jacot, W.; Yamashita, T.; Sohn, J.; Vidal, M.; Tokunaga, E.; Tsurutani, J.; Ueno, N.T.; Prat, A.; Chae, Y.S.; et al. Trastuzumab Deruxtecan in Previously Treated HER2-Low Advanced Breast Cancer. N. Engl. J. Med. 2022, 387, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, C.M.; Yamaguchi, A.; Anami, Y.; Xiong, W.; Otani, Y.; Lee, J.; Ueno, N.T.; Zhang, N.; An, Z.; Tsuchikama, K. Antibody-drug conjugates with dual payloads for combating breast tumor heterogeneity and drug resistance. Nat. Commun. 2021, 12, 3528. [Google Scholar] [CrossRef] [PubMed]
- Filho, O.M.; Viale, G.; Stein, S.; Trippa, L.; Yardley, D.A.; Mayer, I.A.; Abramson, V.G.; Arteaga, C.L.; Spring, L.M.; Waks, A.G.; et al. Impact of HER2 Heterogeneity on Treatment Response of Early-Stage HER2-Positive Breast Cancer: Phase II Neoadjuvant Clinical Trial of T-DM1 Combined with Pertuzumab. Cancer Discov. 2021, 11, 2474–2487. [Google Scholar] [CrossRef]
- Swain, S.M.; Shastry, M.; Hamilton, E. Targeting HER2-positive breast cancer: Advances and future directions. Nat. Rev. Drug Discov. 2023, 22, 101–126. [Google Scholar] [CrossRef]
- Sakai, H.; Tsurutani, J.; Iwasa, T.; Komoike, Y.; Sakai, K.; Nishio, K.; Nakagawa, K. HER2 genomic amplification in circulating tumor DNA and estrogen receptor positivity predict primary resistance to trastuzumab emtansine (T-DM1) in patients with HER2-positive metastatic breast cancer. Breast Cancer 2018, 25, 605–613. [Google Scholar] [CrossRef] [Green Version]
- Loganzo, F.; Sung, M.; Gerber, H.-P. Mechanisms of Resistance to Antibody-Drug Conjugates. Mol. Cancer Ther. 2016, 15, 2825–2834. [Google Scholar] [CrossRef] [Green Version]
- Loganzo, F.; Tan, X.; Sung, M.; Jin, G.; Myers, J.S.; Melamud, E.; Wang, F.; Diesl, V.; Follettie, M.T.; Musto, S.; et al. Tumor cells chronically treated with a trastuzumab-maytansinoid antibody-drug conjugate develop varied resistance mechanisms but respond to alternate treatments. Mol. Cancer Ther. 2015, 14, 952–963. [Google Scholar] [CrossRef] [Green Version]
- Sung, M.S.; Tan, X.; Hosselet, C.; Cinque, M.; Upeslacis, E.; Golas, J.; Wang, F.; Lu, B.; Tylaska, L.; King, L.; et al. Abstract 2113: Caveolae-mediated endocytosis as a novel mechanism of resistance to T-DM1 ADC. Cancer Res. 2016, 76 (Suppl. S14), 2113. [Google Scholar] [CrossRef]
- Phillips, G.D.L.; Fields, C.T.; Li, G.; Dowbenko, D.; Schaefer, G.; Miller, K.; Andre, F.; Burris, H.A.; Albain, K.S.; Harbeck, N.; et al. Dual targeting of HER2-positive cancer with trastuzumab emtansine and pertuzumab: Critical role for neuregulin blockade in antitumor response to combination therapy. Clin. Cancer Res. 2014, 20, 456–468. [Google Scholar] [CrossRef] [Green Version]
- Oh, S.Y.; Lee, Y.W.; Lee, E.J.; Kim, J.H.; Park, Y.J.; Heo, S.G.; Yu, M.R.; Hong, M.H.; DaSilva, J.; Daly, C.; et al. Preclinical Study of a Biparatopic METxMET Antibody-Drug Conjugate, REGN5093-M114, Overcomes MET-driven Acquired Resistance to EGFR TKIs in EGFR-mutant NSCLC. Clin. Cancer Res. 2023, 29, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Diéras, V.; Deluche, E.; Lusque, A.; Pistilli, B.; Bachelot, T.; Pierga, J.-Y.; Viret, F.; Levy, C.; Salabert, L.; Le Du, F.; et al. Abstract PD8-02: Trastuzumab deruxtecan (T-DXd) for advanced breast cancer patients (ABC), regardless HER2 status: A phase II study with biomarkers analysis (DAISY). Cancer Res. 2022, 82 (Suppl. S4), PD8-02. [Google Scholar] [CrossRef]
- Li, B.T.; Michelini, F.; Misale, S.; Cocco, E.; Baldino, L.; Cai, Y.; Shifman, S.; Tu, H.-Y.; Myers, M.L.; Xu, C.; et al. HER2-Mediated Internalization of Cytotoxic Agents in ERBB2 Amplified or Mutant Lung Cancers. Cancer Discov. 2020, 10, 674–687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sung, M.; Tan, X.; Lu, B.; Golas, J.; Hosselet, C.; Wang, F.; Tylaska, L.; King, L.; Zhou, D.; Dushin, R.; et al. Caveolae-Mediated Endocytosis as a Novel Mechanism of Resistance to Trastuzumab Emtansine (T-DM1). Mol. Cancer Ther. 2018, 17, 243–253. [Google Scholar] [CrossRef] [Green Version]
- Pereira, P.M.R.; Mandleywala, K.; Monette, S.; Lumish, M.; Tully, K.M.; Panikar, S.S.; Cornejo, M.; Mauguen, A.; Ragupathi, A.; Keltee, N.C.; et al. Caveolin-1 temporal modulation enhances antibody drug efficacy in heterogeneous gastric cancer. Nat. Commun. 2022, 13, 2526. [Google Scholar] [CrossRef]
- Christianson, H.C.; Menard, J.A.; Chandran, V.I.; Bourseau-Guilmain, E.; Shevela, D.; Lidfeldt, J.; Månsson, A.-S.; Pastorekova, S.; Messinger, J.; Belting, M. Tumor antigen glycosaminoglycan modification regulates antibody-drug conjugate delivery and cytotoxicity. Oncotarget 2017, 8, 66960–66974. [Google Scholar] [CrossRef] [Green Version]
- Schultz, M.L.; Tecedor, L.; Chang, M.; Davidson, B.L. Clarifying lysosomal storage diseases. Trends Neurosci. 2011, 34, 401–410. [Google Scholar] [CrossRef] [Green Version]
- Grigoriev, I.; Splinter, D.; Keijzer, N.; Wulf, P.S.; Demmers, J.; Ohtsuka, T.; Modesti, M.; Maly, I.V.; Grosveld, F.; Hoogenraad, C.C.; et al. Rab6 regulates transport and targeting of exocytotic carriers. Dev. Cell 2007, 13, 305–314. [Google Scholar] [CrossRef] [Green Version]
- Callow, M.G.; Zozulya, S.; Gishizky, M.L.; Jallal, B.; Smeal, T. PAK4 mediates morphological changes through the regulation of GEF-H1. J. Cell Sci. 2005, 118 Pt 9, 1861–1872. [Google Scholar] [CrossRef] [Green Version]
- Tsui, C.K.; Barfield, R.M.; Fischer, C.R.; Morgens, D.W.; Li, A.; Smith, B.A.H.; Gray, M.A.; Bertozzi, C.R.; Rabuka, D.; Bassik, M.C. CRISPR-Cas9 screens identify regulators of antibody-drug conjugate toxicity. Nat. Chem. Biol. 2019, 15, 949–958. [Google Scholar] [CrossRef]
- Yu, S.-F.; Zheng, B.; Go, M.; Lau, J.; Spencer, S.; Raab, H.; Soriano, R.; Jhunjhunwala, S.; Cohen, R.; Caruso, M.; et al. A Novel Anti-CD22 Anthracycline-Based Antibody-Drug Conjugate (ADC) That Overcomes Resistance to Auristatin-Based ADCs. Clin. Cancer Res. 2015, 21, 3298–3306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takegawa, N.; Nonagase, Y.; Yonesaka, K.; Sakai, K.; Maenishi, O.; Ogitani, Y.; Tamura, T.; Nishio, K.; Nakagawa, K.; Tsurutani, J. DS-8201a, a new HER2-targeting antibody-drug conjugate incorporating a novel DNA topoisomerase I inhibitor, overcomes HER2-positive gastric cancer T-DM1 resistance. Int. J. Cancer 2017, 141, 1682–1689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conilh, L.; Sadilkova, L.; Viricel, W.; Dumontet, C. Payload diversification: A key step in the development of antibody-drug conjugates. J. Hematol. Oncol. 2023, 16, 3. [Google Scholar] [CrossRef] [PubMed]
- Coates, J.T.; Sun, S.; Leshchiner, I.; Thimmiah, N.; Martin, E.E.; McLoughlin, D.; Danysh, B.P.; Slowik, K.; Jacobs, R.A.; Rhrissorrakrai, K.; et al. Parallel Genomic Alterations of Antigen and Payload Targets Mediate Polyclonal Acquired Clinical Resistance to Sacituzumab Govitecan in Triple-Negative Breast Cancer. Cancer Discov. 2021, 11, 2436–2445. [Google Scholar] [CrossRef]
- Aldonza, M.B.D.; Hong, J.-Y.; Alinsug, M.V.; Song, J.; Lee, S.K. Multiplicity of acquired cross-resistance in paclitaxel-resistant cancer cells is associated with feedback control of TUBB3 via FOXO3a-mediated ABCB1 regulation. Oncotarget 2016, 7, 34395–34419. [Google Scholar] [CrossRef] [Green Version]
- Coleman, N.; Yap, T.A.; Heymach, J.V.; Meric-Bernstam, F.; Le, X. Antibody-drug conjugates in lung cancer: Dawn of a new era? NPJ Precis. Oncol. 2023, 7, 5. [Google Scholar] [CrossRef]
- Cabaud, O.; Berger, L.G.M.; Crompot, E.; Adélaide, J.; Finetti, P.; Garnier, S.; Guille, A.; Carbuccia, N.; Farina, A.; Agavnian, E.; et al. Overcoming Resistance to Anti-Nectin-4 Antibody-Drug Conjugate. Mol. Cancer Ther. 2022, 21, 1227–1235. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abelman, R.O.; Wu, B.; Spring, L.M.; Ellisen, L.W.; Bardia, A. Mechanisms of Resistance to Antibody–Drug Conjugates. Cancers 2023, 15, 1278. https://doi.org/10.3390/cancers15041278
Abelman RO, Wu B, Spring LM, Ellisen LW, Bardia A. Mechanisms of Resistance to Antibody–Drug Conjugates. Cancers. 2023; 15(4):1278. https://doi.org/10.3390/cancers15041278
Chicago/Turabian StyleAbelman, Rachel Occhiogrosso, Bogang Wu, Laura M. Spring, Leif W. Ellisen, and Aditya Bardia. 2023. "Mechanisms of Resistance to Antibody–Drug Conjugates" Cancers 15, no. 4: 1278. https://doi.org/10.3390/cancers15041278