
CAR-T Therapy in GBM: Current Challenges and Avenues for Improvement

Abstract
:Simple Summary
Abstract
1. Introduction
2. Completed Clinical Trials of CAR-T Cells in GBM Highlight Key Challenges
2.1. EGFRvIII
2.2. IL13Rα2
2.3. HER2
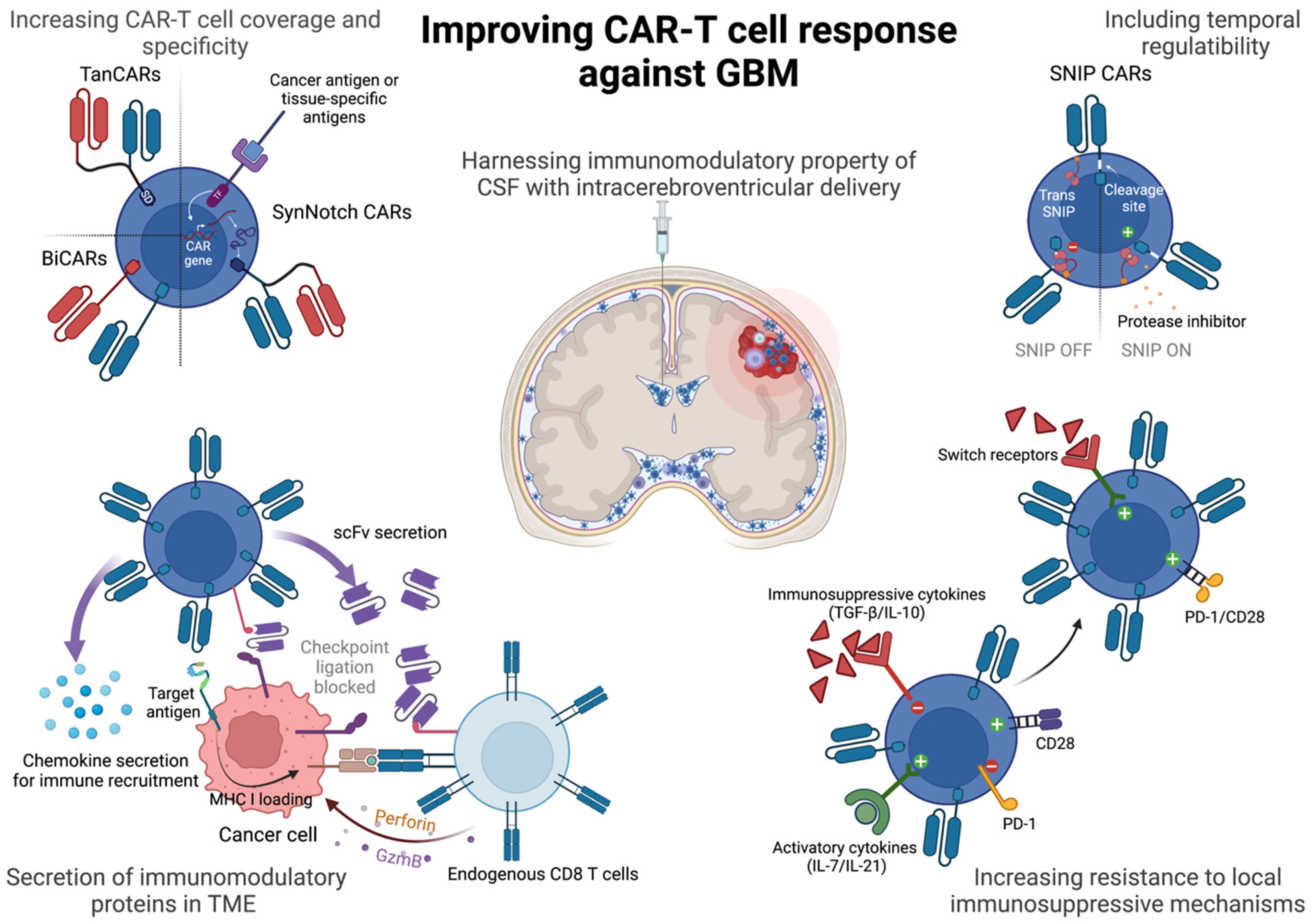
3. Strategies to Improve CAR-T Response
3.1. Bispecific CAR-T Cells
3.2. Engineering Temporal and Spatial Control over CAR-T Cell Activity
3.3. Engineering Resistance to Immunosuppressive Cytokines in the TME
3.4. Engineering Resistance to Exhaustion
3.5. Utilizing the Unique Anatomical and Immunological Niche of GBMs for CAR-T Delivery
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Guo, G.; Guan, H.; Yu, Y.; Lu, J.; Yu, J. Challenges and potential of PD-1/PD-L1 checkpoint blockade immunotherapy for glioblastoma. J. Exp. Clin. Cancer Res. 2019, 38, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T cell immunotherapy for human cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef] [Green Version]
- Porter, D.L.; Hwang, W.-T.; Frey, N.V.; Lacey, S.F.; Shaw, P.A.; Loren, A.W.; Bagg, A.; Marcucci, K.T.; Shen, A.; Gonzalez, V.; et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci. Transl. Med. 2015, 7, 303ra139. [Google Scholar] [CrossRef] [Green Version]
- Schuster, S.J.; Svoboda, J.; Chong, E.A.; Nasta, S.D.; Mato, A.R.; Anak, Ö.; Brogdon, J.L.; Pruteanu-Malinici, I.; Bhoj, V.; Landsburg, D.; et al. Chimeric Antigen Receptor T Cells in Refractory B-Cell Lymphomas. N. Engl. J. Med. 2017, 377, 2545–2554. [Google Scholar] [CrossRef]
- Piao, Y.; Liang, J.; Holmes, L.; Zurita, A.J.; Henry, V.; Heymach, J.V.; de Groot, J.F. Glioblastoma resistance to anti-VEGF therapy is associated with myeloid cell infiltration, stem cell accumulation, and a mesenchymal phenotype. Neuro-Oncol. 2012, 14, 1379–1392. [Google Scholar] [CrossRef] [Green Version]
- Sayour, E.J.; McLendon, P.; McLendon, R.; De Leon, G.; Reynolds, R.; Kresak, J.; Sampson, J.H.; Mitchell, D.A. Increased proportion of FoxP3+ regulatory T cells in tumor infiltrating lymphocytes is associated with tumor recurrence and reduced survival in patients with glioblastoma. Cancer Immunol. Immunother. CII 2015, 64, 419–427. [Google Scholar] [CrossRef]
- Seymour, T.; Nowak, A.; Kakulas, F. Targeting Aggressive Cancer Stem Cells in Glioblastoma. Front. Oncol. 2015, 5, 159. [Google Scholar] [CrossRef]
- Skaga, E.; Kulesskiy, E.; Fayzullin, A.; Sandberg, C.J.; Potdar, S.; Kyttälä, A.; Langmoen, I.A.; Laakso, A.; Gaál-Paavola, E.; Perola, M.; et al. Intertumoral heterogeneity in patient-specific drug sensitivities in treatment-naïve glioblastoma. BMC Cancer 2019, 19, 628. [Google Scholar] [CrossRef] [Green Version]
- Sottoriva, A.; Spiteri, I.; Piccirillo, S.G.M.; Touloumis, A.; Collins, V.P.; Marioni, J.C.; Curtis, C.; Watts, C.; Tavaré, S. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc. Natl. Acad. Sci. USA 2013, 110, 4009–4014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 2019, 178, 835–849.e21. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Chanoch-Myers, R.; Mathewson, N.D.; Myskiw, C.; Atta, L.; Bussema, L.; Eichhorn, S.W.; Greenwald, A.C.; Kinker, G.S.; Rodman, C.; et al. Interactions between cancer cells and immune cells drive transitions to mesenchymal-like states in glioblastoma. Cancer Cell 2021, 39, 779–792.e11. [Google Scholar] [CrossRef] [PubMed]
- Sotillo, E.; Barrett, D.M.; Black, K.L.; Bagashev, A.; Oldridge, D.; Wu, G.; Sussman, R.; Lanauze, C.; Ruella, M.; Gazzara, M.R.; et al. Convergence of Acquired Mutations and Alternative Splicing of CD19 Enables Resistance to CART-19 Immunotherapy. Cancer Discov. 2015, 5, 1282–1295. [Google Scholar] [CrossRef] [Green Version]
- An, Z.; Aksoy, O.; Zheng, T.; Fan, Q.-W.; Weiss, W.A. Epidermal growth factor receptor and EGFRvIII in glioblastoma: Signaling pathways and targeted therapies. Oncogene 2018, 37, 1561–1575. [Google Scholar] [CrossRef]
- Wikstrand, C.J.; McLendon, R.E.; Friedman, A.H.; Bigner, D.D. Cell Surface Localization and Density of the Tumor-associated Variant of the Epidermal Growth Factor Receptor, EGFRvIII1. Cancer Res. 1997, 57, 4130–4140. [Google Scholar]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017, 9, eaaa0984. [Google Scholar] [CrossRef] [Green Version]
- Goff, S.L.; Morgan, R.A.; Yang, J.C.; Sherry, R.M.; Robbins, P.F.; Restifo, N.P.; Feldman, S.A.; Lu, Y.-C.; Lu, L.; Zheng, Z.; et al. Pilot trial of adoptive transfer of chimeric antigen receptor transduced T cells targeting EGFRvIII in patients with glioblastoma. J. Immunother. 2019, 42, 126–135. [Google Scholar] [CrossRef]
- Brown, C.E.; Warden, C.D.; Starr, R.; Deng, X.; Badie, B.; Yuan, Y.-C.; Forman, S.J.; Barish, M.E. Glioma IL13Rα2 is associated with mesenchymal signature gene expression and poor patient prognosis. PLoS ONE 2013, 8, e77769. [Google Scholar] [CrossRef]
- Brown, C.E.; Badie, B.; Barish, M.E.; Weng, L.; Ostberg, J.R.; Chang, W.-C.; Naranjo, A.; Starr, R.; Wagner, J.; Wright, C.; et al. Bioactivity and Safety of IL13Rα2-Redirected Chimeric Antigen Receptor CD8+ T Cells in Patients with Recurrent Glioblastoma. Clin. Cancer Res. 2015, 21, 4062–4072. [Google Scholar] [CrossRef] [Green Version]
- Brown, C.E.; Alizadeh, D.; Starr, R.; Weng, L.; Wagner, J.R.; Naranjo, A.; Ostberg, J.R.; Blanchard, M.S.; Kilpatrick, J.; Simpson, J.; et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N. Engl. J. Med. 2016, 375, 2561–2569. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.E.; Rodriguez, A.; Palmer, J.; Ostberg, J.R.; Naranjo, A.; Wagner, J.R.; Aguilar, B.; Starr, R.; Weng, L.; Synold, T.W.; et al. Off-the-shelf, steroid-resistant, IL13Rα2-specific CAR T cells for treatment of glioblastoma. Neuro-Oncol. 2022, 24, 1318–1330. [Google Scholar] [CrossRef] [PubMed]
- Keu, K.V.; Witney, T.H.; Yaghoubi, S.; Rosenberg, J.; Kurien, A.; Magnusson, R.; Williams, J.; Habte, F.; Wagner, J.R.; Forman, S.; et al. Reporter Gene Imaging of Targeted T-Cell Immunotherapy in Recurrent Glioma. Sci. Transl. Med. 2017, 9, eaag2196. [Google Scholar] [CrossRef] [Green Version]
- Hsu, J.L.; Hung, M.-C. The role of HER2, EGFR, and other receptor tyrosine kinases in breast cancer. Cancer Metastasis Rev. 2016, 35, 575–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, L.; Li, H.; Bin, S.; Li, P.; Chen, J.; Gu, H.; Yuan, W. The efficacy of third generation anti-HER2 chimeric antigen receptor T cells in combination with PD1 blockade against malignant glioblastoma cells. Oncol. Rep. 2019, 42, 1549–1557. [Google Scholar] [CrossRef]
- Press, M.F.; Cordon-Cardo, C.; Slamon, D.J. Expression of the HER-2/neu proto-oncogene in normal human adult and fetal tissues. Oncogene 1990, 5, 953–962. [Google Scholar]
- Ahmed, N.; Brawley, V.; Hegde, M.; Bielamowicz, K.; Kalra, M.; Landi, D.; Robertson, C.; Gray, T.L.; Diouf, O.; Wakefield, A.; et al. HER2-Specific Chimeric Antigen Receptor–Modified Virus-Specific T Cells for Progressive Glioblastoma: A Phase 1 Dose-Escalation Trial. JAMA Oncol. 2017, 3, 1094–1101. [Google Scholar] [CrossRef]
- Hegde, M.; Corder, A.; Chow, K.K.; Mukherjee, M.; Ashoori, A.; Kew, Y.; Zhang, Y.J.; Baskin, D.S.; Merchant, F.A.; Brawley, V.S.; et al. Combinational Targeting Offsets Antigen Escape and Enhances Effector Functions of Adoptively Transferred T Cells in Glioblastoma. Mol. Ther. 2013, 21, 2087–2101. [Google Scholar] [CrossRef] [Green Version]
- Tian, M.; Cheuk, A.T.; Wei, J.S.; Abdelmaksoud, A.; Chou, H.-C.; Milewski, D.; Kelly, M.C.; Song, Y.K.; Dower, C.M.; Li, N.; et al. An optimized bicistronic chimeric antigen receptor against GPC2 or CD276 overcomes heterogeneous expression in neuroblastoma. J. Clin. Investig. 2022, 132, e155621. [Google Scholar] [CrossRef]
- Hegde, M.; Mukherjee, M.; Grada, Z.; Pignata, A.; Landi, D.; Navai, S.A.; Wakefield, A.; Fousek, K.; Bielamowicz, K.; Chow, K.K.H.; et al. Tandem CAR T cells targeting HER2 and IL13Rα2 mitigate tumor antigen escape. J. Clin. Investig. 2016, 126, 3036–3052. [Google Scholar] [CrossRef] [Green Version]
- Gust, J.; Taraseviciute, A.; Turtle, C.J. Neurotoxicity Associated with CD19-Targeted CAR-T Cell Therapies. CNS Drugs 2018, 32, 1091–1101. [Google Scholar] [CrossRef] [PubMed]
- Gust, J.; Hay, K.A.; Hanafi, L.-A.; Li, D.; Myerson, D.; Gonzalez-Cuyar, L.F.; Yeung, C.; Liles, W.C.; Wurfel, M.; Lopez, J.A.; et al. Endothelial Activation and Blood–Brain Barrier Disruption in Neurotoxicity after Adoptive Immunotherapy with CD19 CAR-T Cells. Cancer Discov. 2017, 7, 1404–1419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akhavan, D.; Alizadeh, D.; Wang, D.; Weist, M.R.; Shepphird, J.K.; Brown, C.E. CAR T cells for brain tumors: Lessons learned and road ahead. Immunol. Rev. 2019, 290, 60–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labanieh, L.; Majzner, R.G.; Klysz, D.; Sotillo, E.; Fisher, C.J.; Vilches-Moure, J.G.; Pacheco, K.Z.B.; Malipatlolla, M.; Xu, P.; Hui, J.H.; et al. Enhanced safety and efficacy of protease-regulated CAR-T cell receptors. Cell 2022, 185, 1745–1763.e22. [Google Scholar] [CrossRef]
- Sun, S.; Hao, H.; Yang, G.; Zhang, Y.; Fu, Y. Immunotherapy with CAR-Modified T Cells: Toxicities and Overcoming Strategies. J. Immunol. Res. 2018, 2018, 2386187. [Google Scholar] [CrossRef]
- Choe, J.H.; Watchmaker, P.B.; Simic, M.S.; Gilbert, R.D.; Li, A.W.; Krasnow, N.A.; Downey, K.M.; Yu, W.; Carrera, D.A.; Celli, A.; et al. SynNotch-CAR T cells overcome challenges of specificity, heterogeneity, and persistence in treating glioblastoma. Sci. Transl. Med. 2021, 13, eabe7378. [Google Scholar] [CrossRef]
- Morsut, L.; Roybal, K.T.; Xiong, X.; Gordley, R.M.; Coyle, S.M.; Thomson, M.; Lim, W.A. Engineering Customized Cell Sensing and Response Behaviors Using Synthetic Notch Receptors. Cell 2016, 164, 780–791. [Google Scholar] [CrossRef] [Green Version]
- Nduom, E.K.; Weller, M.; Heimberger, A.B. Immunosuppressive mechanisms in glioblastoma. Neuro-Oncol. 2015, 17, vii9–vii14. [Google Scholar] [CrossRef] [Green Version]
- Frederico, S.C.; Hancock, J.C.; Brettschneider, E.E.S.; Ratnam, N.M.; Gilbert, M.R.; Terabe, M. Making a Cold Tumor Hot: The Role of Vaccines in the Treatment of Glioblastoma. Front. Oncol. 2021, 11, 672508. [Google Scholar] [CrossRef]
- Adachi, K.; Kano, Y.; Nagai, T.; Okuyama, N.; Sakoda, Y.; Tamada, K. IL-7 and CCL19 expression in CAR-T cells improves immune cell infiltration and CAR-T cell survival in the tumor. Nat. Biotechnol. 2018, 36, 346–351. [Google Scholar] [CrossRef]
- Krenciute, G.; Prinzing, B.L.; Yi, Z.; Wu, M.-F.; Liu, H.; Dotti, G.; Balyasnikova, I.V.; Gottschalk, S. Transgenic Expression of IL15 Improves Antiglioma Activity of IL13Rα2-CAR T Cells but Results in Antigen Loss Variants. Cancer Immunol. Res. 2017, 5, 571–581. [Google Scholar] [CrossRef] [Green Version]
- Leen, A.M.; Sukumaran, S.; Watanabe, N.; Mohammed, S.; Keirnan, J.; Yanagisawa, R.; Anurathapan, U.; Rendon, D.; Heslop, H.E.; Rooney, C.M.; et al. Reversal of Tumor Immune Inhibition Using a Chimeric Cytokine Receptor. Mol. Ther. 2014, 22, 1211–1220. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Jiang, H.; Luo, H.; Sun, Y.; Shi, B.; Sun, R.; Li, Z. An IL-4/21 Inverted Cytokine Receptor Improving CAR-T Cell Potency in Immunosuppressive Solid-Tumor Microenvironment. Front. Immunol. 2019, 10, 1691. [Google Scholar] [CrossRef] [Green Version]
- Grabowski, M.M.; Sankey, E.W.; Ryan, K.J.; Chongsathidkiet, P.; Lorrey, S.J.; Wilkinson, D.S.; Fecci, P.E. Immune suppression in gliomas. J. Neurooncol. 2021, 151, 3–12. [Google Scholar] [CrossRef]
- Agliardi, G.; Liuzzi, A.R.; Hotblack, A.; De Feo, D.; Núñez, N.; Stowe, C.L.; Friebel, E.; Nannini, F.; Rindlisbacher, L.; Roberts, T.A.; et al. Intratumoral IL-12 delivery empowers CAR-T cell immunotherapy in a pre-clinical model of glioblastoma. Nat. Commun. 2021, 12, 444. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhang, Z.; Zhong, K.; Wang, Z.; Yang, N.; Tang, X.; Li, H.; Lu, Q.; Wu, Z.; Yuan, B.; et al. CXCL11-armed oncolytic adenoviruses enhance CAR-T cell therapeutic efficacy and reprogram tumor microenvironment in glioblastoma. Mol. Ther. 2023, 31, 134–153. [Google Scholar] [CrossRef] [PubMed]
- Cherkassky, L.; Morello, A.; Villena-Vargas, J.; Feng, Y.; Dimitrov, D.S.; Jones, D.R.; Sadelain, M.; Adusumilli, P.S. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J. Clin. Investig. 2016, 126, 3130–3144. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Ranganathan, R.; Jiang, S.; Fang, C.; Sun, J.; Kim, S.; Newick, K.; Lo, A.; June, C.H.; Zhao, Y.; et al. A Chimeric Switch-Receptor Targeting PD1 Augments the Efficacy of Second-Generation CAR T Cells in Advanced Solid Tumors. Cancer Res. 2016, 76, 1578–1590. [Google Scholar] [CrossRef] [Green Version]
- Woroniecka, K.; Chongsathidkiet, P.; Rhodin, K.; Kemeny, H.; Dechant, C.; Farber, S.H.; Elsamadicy, A.A.; Cui, X.; Koyama, S.; Jackson, C.; et al. T-Cell Exhaustion Signatures Vary with Tumor Type and Are Severe in Glioblastoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 4175–4186. [Google Scholar] [CrossRef] [Green Version]
- Rafiq, S.; Yeku, O.O.; Jackson, H.J.; Purdon, T.J.; van Leeuwen, D.G.; Drakes, D.J.; Song, M.; Miele, M.M.; Li, Z.; Wang, P.; et al. Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances anti-tumor efficacy in vivo. Nat. Biotechnol. 2018, 36, 847–856. [Google Scholar] [CrossRef]
- Suarez, E.R.; Chang, D.-K.; Sun, J.; Sui, J.; Freeman, G.J.; Signoretti, S.; Zhu, Q.; Marasco, W.A. Chimeric antigen receptor T cells secreting anti-PD-L1 antibodies more effectively regress renal cell carcinoma in a humanized mouse model. Oncotarget 2016, 7, 34341–34355. [Google Scholar] [CrossRef] [Green Version]
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Jia, A.; Bi, Y.; Wang, Y.; Liu, G. Metabolic Regulation of Myeloid-Derived Suppressor Cell Function in Cancer. Cells 2020, 9, 1011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, P.-C.; Bihuniak, J.D.; Macintyre, A.N.; Staron, M.; Liu, X.; Amezquita, R.; Tsui, Y.-C.; Cui, G.; Micevic, G.; Perales, J.C.; et al. Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-tumor T Cell Responses. Cell 2015, 162, 1217–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bengsch, B.; Johnson, A.L.; Kurachi, M.; Odorizzi, P.M.; Pauken, K.E.; Attanasio, J.; Stelekati, E.; McLane, L.M.; Paley, M.A.; Delgoffe, G.M.; et al. Bioenergetic insufficiencies due to metabolic alterations regulated by PD-1 are an early driver of CD8+ T cell exhaustion. Immunity 2016, 45, 358–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majzner, R.G.; Ramakrishna, S.; Yeom, K.W.; Patel, S.; Chinnasamy, H.; Schultz, L.M.; Richards, R.M.; Jiang, L.; Barsan, V.; Mancusi, R.; et al. GD2-CAR T cell therapy for H3K27M-mutated diffuse midline gliomas. Nature 2022, 603, 934–941. [Google Scholar] [CrossRef]
- Wang, X.; Huynh, C.; Urak, R.; Weng, L.; Walter, M.; Lim, L.; Vyas, V.; Chang, W.-C.; Aguilar, B.; Brito, A.; et al. The cerebroventricular environment modifies CAR T cells for potent activity against both central nervous system and systemic lymphoma. Cancer Immunol. Res. 2021, 9, 75–88. [Google Scholar] [CrossRef]
- Priceman, S.J.; Tilakawardane, D.; Jeang, B.; Aguilar, B.; Murad, J.P.; Park, A.K.; Chang, W.-C.; Ostberg, J.R.; Neman, J.; Jandial, R.; et al. Regional Delivery of Chimeric Antigen Receptor–Engineered T Cells Effectively Targets HER2+ Breast Cancer Metastasis to the Brain. Clin. Cancer Res. 2018, 24, 95–105. [Google Scholar] [CrossRef] [Green Version]
- Brown, C.E.; Aguilar, B.; Starr, R.; Yang, X.; Chang, W.-C.; Weng, L.; Chang, B.; Sarkissian, A.; Brito, A.; Sanchez, J.F.; et al. Optimization of IL13Rα2-Targeted Chimeric Antigen Receptor T Cells for Improved Anti-tumor Efficacy against Glioblastoma. Mol. Ther. 2018, 26, 31–44. [Google Scholar] [CrossRef] [Green Version]
- Ma, W.; Wang, Y.; Zhang, R.; Yang, F.; Zhang, D.; Huang, M.; Zhang, L.; Dorsey, J.F.; Binder, Z.A.; O’Rourke, D.M.; et al. Targeting PAK4 to reprogram the vascular microenvironment and improve CAR-T immunotherapy for glioblastoma. Nat. Cancer 2021, 2, 83–97. [Google Scholar] [CrossRef]
- Wang, D.; Prager, B.C.; Gimple, R.C.; Aguilar, B.; Alizadeh, D.; Tang, H.; Lv, D.; Starr, R.; Brito, A.; Wu, Q.; et al. CRISPR Screening of CAR T Cells and Cancer Stem Cells Reveals Critical Dependencies for Cell-Based Therapies. Cancer Discov. 2021, 11, 1192–1211. [Google Scholar] [CrossRef] [PubMed]
- Song, E.Z.; Wang, X.; Philipson, B.I.; Zhang, Q.; Thokala, R.; Zhang, L.; Assenmacher, C.-A.; Binder, Z.A.; Ming, G.; O’Rourke, D.M.; et al. The IAP antagonist birinapant enhances chimeric antigen receptor T cell therapy for glioblastoma by overcoming antigen heterogeneity. Mol. Ther.-Oncolytics 2022, 27, 288–304. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Target Antigen | Trial | Phase | No. of Patients | Current Status | Notes |
|---|---|---|---|---|---|
| EGFRvIII | NCT02209376 | I | 10 | Complete | Increased expression of inhibitory molecules and loss of EGFRvIII expression following therapy. |
| EGFRvIII | NCT01454596 | I/II | 18 | Complete | Preparative chemotherapy used before adoptive transfer of CAR-T cells. IL-2 infusion used to support CAR-T expansion. |
| IL-13Rα2 | NCT00730613 | I | 3 | Complete | Transient antitumor activity observed |
| IL-13Rα2 | NCT01082926 | I | 6 | Complete | Suitable for allogeneic off-the -shelf use. Glucocorticoid receptor inactivated in CAR-T cells. Modified to allow PET imaging. |
| IL-13Rα2 | NCT04003649 | I | 60 | Recruiting | Administering with and without nivolumab and ipilimumab. |
| IL-13Rα2 | NCT02208362 | I | 82 | Active, not recruiting | Using memory enriched T cells for CAR-expression. |
| IL-13Rα2 | NCT04661384 | I | 30 | Recruiting | Treating leptomeningeal diseases from GBM, ependymoma or medulloblastoma. |
| HER2 | NCT01109095 | I | 17 | Complete | Virus-specific T cells engineered to express CAR-T to increase persistence. |
| HER2 | NCT03389230 | I | 42 | Recruiting | Using memory enriched CAR-T cells in grade II-IV glioma patients. |
| MMP2 | NCT04214392 | I | 36 | Recruiting | Using chlorotoxin expressing CAR-T cells to recognize GBM cells expressing putative receptor metalloproteinase 2 (MMP2). |
| B7-H3 | NCT05366179 | I | 36 | Recruiting | Targeting B7-H3 expression in GBM. |
| B7-H3 | NCT05474378 | I | 39 | Recruiting | Targeting recurrent IDH-wild type GBM patients. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pant, A.; Lim, M. CAR-T Therapy in GBM: Current Challenges and Avenues for Improvement. Cancers 2023, 15, 1249. https://doi.org/10.3390/cancers15041249
Pant A, Lim M. CAR-T Therapy in GBM: Current Challenges and Avenues for Improvement. Cancers. 2023; 15(4):1249. https://doi.org/10.3390/cancers15041249
Chicago/Turabian StylePant, Ayush, and Michael Lim. 2023. "CAR-T Therapy in GBM: Current Challenges and Avenues for Improvement" Cancers 15, no. 4: 1249. https://doi.org/10.3390/cancers15041249