
HDAC8 Deacetylates HIF-1α and Enhances Its Protein Stability to Promote Tumor Growth and Migration in Melanoma

Abstract
:Simple Summary
Abstract
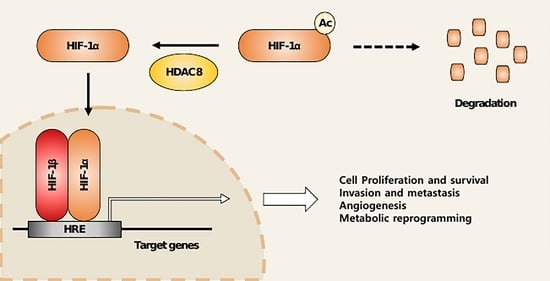
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Line and Culture
2.3. CRISPR/Cas9-Mediated Knockout (KO) of HDAC8 Gene
2.4. Cell Growth and Viability Assay
2.5. Colony Formation Assay
2.6. Luciferase Reporter Assay
2.7. Wound Healing Assay
2.8. Transwell Migration Assay
2.9. Western Blot Analysis
2.10. Co-Immunoprecipitation Assay (Co-IP)
2.11. RNA Extraction and qRT-PCR
2.12. Analysis of Online Databases
2.13. Statistical Analysis Figures, Tables, and Schemes
3. Results
3.1. HDAC8 Upregulates HIF-1α Expression in Melanoma
3.2. HDAC8 Deacetylates HIF-1α and Enhances Its Protein Stability
3.3. HDAC8 Regulates HIF-1α Transcriptional Activity
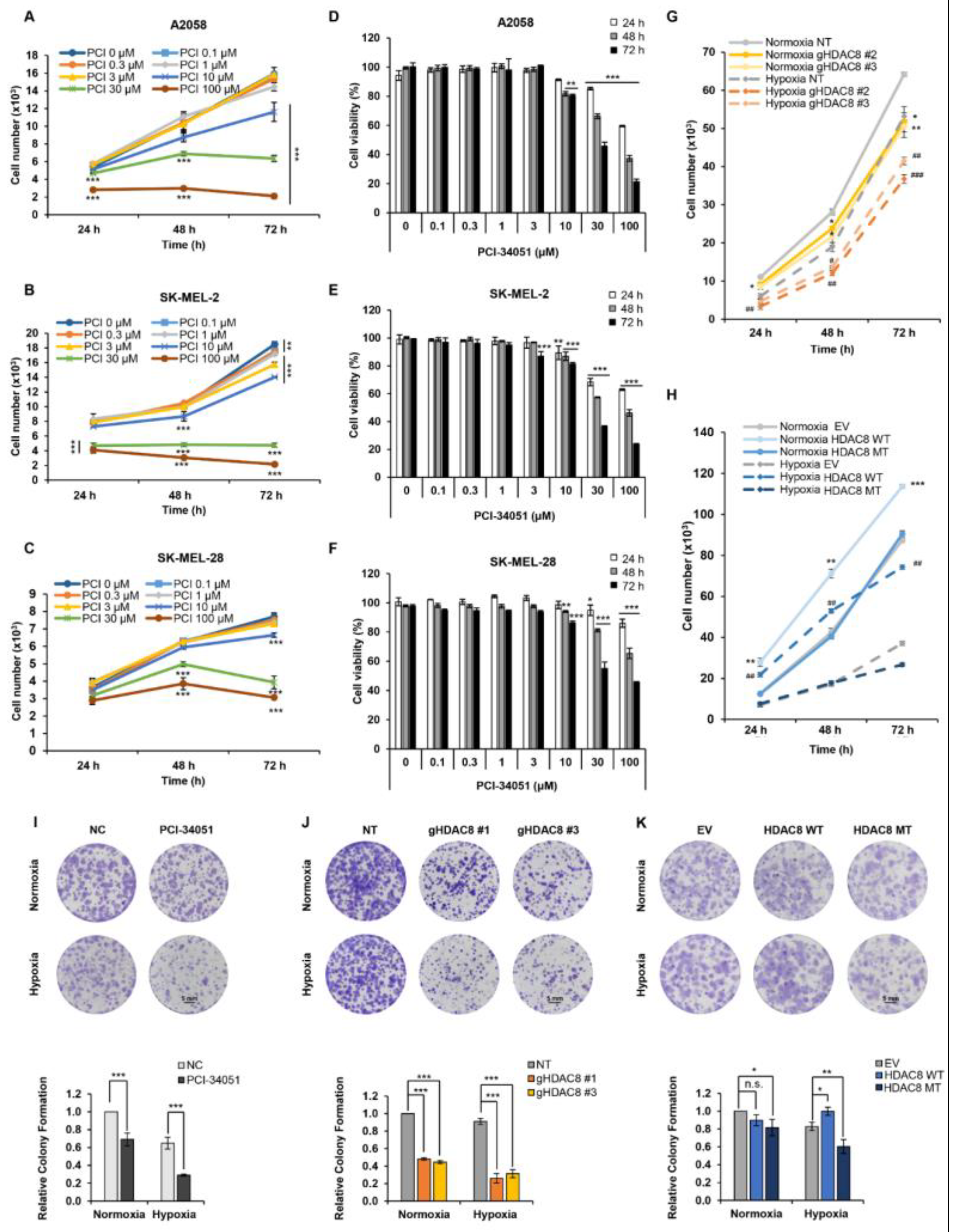
3.4. HDAC8 Promotes Cell Proliferation and Enhances Migration in Melanoma
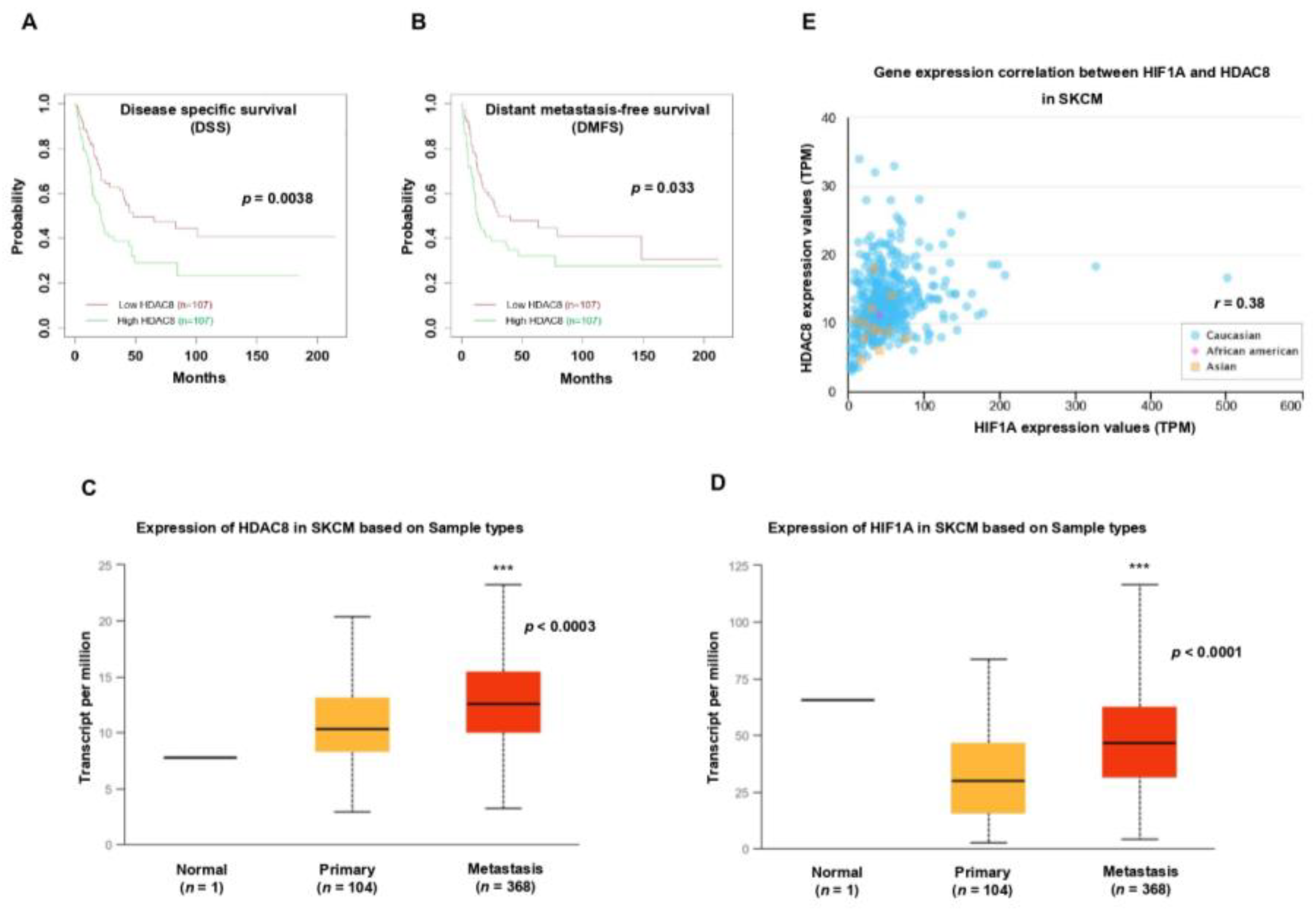
3.5. HDAC8 and HIF-1α Expression Is Correlated with Poor Prognosis in Melanoma
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Schadendorf, D.; van Akkooi, A.C.; Berking, C.; Griewank, K.G.; Gutzmer, R.; Hauschild, A.; Stang, A.; Roesch, A.; Ugurel, S. Melanoma. Lancet 2018, 392, 971–984. [Google Scholar] [CrossRef]
- Leonardi, G.C.; Falzone, L.; Salemi, R.; Zanghì, A.; Spandidos, D.A.; Mccubrey, J.A.; Candido, S.; Libra, M. Cutaneous melanoma: From pathogenesis to therapy. Int. J. Oncol. 2018, 52, 1071–1080. [Google Scholar] [CrossRef]
- Schadendorf, D.; Fisher, D.E.; Garbe, C.; Gershenwald, J.E.; Grob, J.-J.; Halpern, A.; Herlyn, M.; Marchetti, M.A.; McArthur, G.; Ribas, A. Melanoma. Nat. Rev. Dis. Primers 2015, 1, 15003. [Google Scholar] [CrossRef]
- National Cancer Institute. Melanoma of the Skin-Cancer Stat Facts. Available online: https://seer.cancer.gov/statfacts/html/melan.html (accessed on 27 December 2022).
- Domingues, B.; Lopes, J.M.; Soares, P.; Pópulo, H. Melanoma treatment in review. Immunotargets Ther. 2018, 7, 35–49. [Google Scholar] [CrossRef]
- Soni, S.; Padwad, Y.S. HIF-1 in cancer therapy: Two decade long story of a transcription factor. Acta Oncol. 2017, 56, 503–515. [Google Scholar] [CrossRef]
- Prabhakar, N.R.; Semenza, G.L. Oxygen sensing and homeostasis. Physiology 2015, 30, 340–348. [Google Scholar] [CrossRef]
- Wilson, W.R.; Hay, M.P. Targeting hypoxia in cancer therapy. Nat. Rev. Cancer 2011, 11, 393–410. [Google Scholar] [CrossRef]
- Weidemann, A.; Johnson, R. Biology of HIF-1α. Cell Death Differ. 2008, 15, 621–627. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-inducible factor 1 and cancer pathogenesis. IUBMB Life 2008, 60, 591–597. [Google Scholar] [CrossRef]
- Zhong, H.; De Marzo, A.M.; Laughner, E.; Lim, M.; Hilton, D.A.; Zagzag, D.; Buechler, P.; Isaacs, W.B.; Semenza, G.L.; Simons, J.W. Overexpression of hypoxia-inducible factor 1α in common human cancers and their metastases. Cancer Res. 1999, 59, 5830–5835. [Google Scholar]
- Berger, S.L.; Kouzarides, T.; Shiekhattar, R.; Shilatifard, A. An operational definition of epigenetics. Genes Dev. 2009, 23, 781–783. [Google Scholar] [CrossRef]
- Ahuja, N.; Sharma, A.R.; Baylin, S.B. Epigenetic therapeutics: A new weapon in the war against cancer. Ann. Rev. Med. 2016, 67, 73–89. [Google Scholar] [CrossRef]
- Gong, F.; Miller, K.M. Mammalian DNA repair: HATs and HDACs make their mark through histone acetylation. Mutat. Res. 2013, 750, 23–30. [Google Scholar] [CrossRef]
- Gallinari, P.; Marco, S.D.; Jones, P.; Pallaoro, M.; Steinkühler, C. HDACs, histone deacetylation and gene transcription: From molecular biology to cancer therapeutics. Cell Res. 2007, 17, 195–211. [Google Scholar] [CrossRef]
- Song, S.; Wang, Y.; Xu, P.; Yang, R.; Ma, Z.; Liang, S.; Zhang, G. The inhibition of histone deacetylase 8 suppresses proliferation and inhibits apoptosis in gastric adenocarcinoma. Int. J. Oncol. 2015, 47, 1819–1828. [Google Scholar] [CrossRef]
- Zhang, R.; Shen, M.; Wu, C.; Chen, Y.; Lu, J.; Li, J.; Zhao, L.; Meng, H.; Zhou, X.; Huang, G. HDAC8-dependent deacetylation of PKM2 directs nuclear localization and glycolysis to promote proliferation in hepatocellular carcinoma. Cell Death Dis. 2020, 11, 1036. [Google Scholar] [CrossRef]
- Ahn, M.-Y.; Yoon, J.-H. Histone deacetylase 8 as a novel therapeutic target in oral squamous cell carcinoma. Oncol. Rep. 2017, 37, 540–546. [Google Scholar] [CrossRef]
- Moreno, D.A.; Scrideli, C.A.; Cortez, M.A.A.; De Paula Queiroz, R.; Valera, E.T.; Da Silva Silveira, V.; Yunes, J.A.; Brandalise, S.R.; Tone, L.G. Differential expression of HDAC3, HDAC7 and HDAC9 is associated with prognosis and survival in childhood acute lymphoblastic leukaemia. Br. J. Haematol. 2010, 150, 665–673. [Google Scholar] [CrossRef]
- Oehme, I.; Deubzer, H.E.; Lodrini, M.; Milde, T.; Witt, O. Targeting of HDAC8 and investigational inhibitors in neuroblastoma. Expert Opin. Investig. Drugs 2009, 18, 1605–1617. [Google Scholar] [CrossRef]
- An, P.; Chen, F.; Li, Z.; Ling, Y.; Peng, Y.; Zhang, H.; Li, J.; Chen, Z.; Wang, H. HDAC8 promotes the dissemination of breast cancer cells via AKT/GSK-3β/Snail signals. Oncogene 2020, 39, 4956–4969. [Google Scholar] [CrossRef]
- Kim, J.Y.; Cho, H.; Yoo, J.; Kim, G.W.; Jeon, Y.H.; Lee, S.W.; Kwon, S.H. Pathological Role of HDAC8: Cancer and Beyond. Cells 2022, 11, 3161. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, N.; Amin, S.A.; Jha, T. Selective and nonselective HDAC8 inhibitors: A therapeutic patent review. Pharm. Pat. Anal. 2018, 7, 259–276. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, S.; Ramos, J.; Luo, W.; Sirisawad, M.; Verner, E.; Buggy, J. A novel histone deacetylase 8 (HDAC8)-specific inhibitor PCI-34051 induces apoptosis in T-cell lymphomas. Leukemia 2008, 22, 1026–1034. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, T.; Antony, J.; Braithwaite, A.W.; Horsfield, J.A. HDAC8 inhibition blocks SMC3 deacetylation and delays cell cycle progression without affecting cohesin-dependent transcription in MCF7 cancer cells. J. Biol. Chem. 2016, 291, 12761–12770. [Google Scholar] [CrossRef]
- Zbytek, B.; Peacock, D.L.; Seagroves, T.N.; Slominski, A. Putative role of HIF transcriptional activity in melanocytes and melanoma biology. Dermatoendocrinol. 2013, 5, 239–251. [Google Scholar] [CrossRef]
- Nsengimana, J.; Laye, J.; Filia, A.; Walker, C.; Jewell, R.; Van den Oord, J.J.; Wolter, P.; Patel, P.; Sucker, A.; Schadendorf, D. Independent replication of a melanoma subtype gene signature and evaluation of its prognostic value and biological correlates in a population cohort. Oncotarget 2015, 6, 11683–11693. [Google Scholar] [CrossRef]
- Chandrashekar, D.S.; Bashel, B.; Balasubramanya, S.A.H.; Creighton, C.J.; Ponce-Rodriguez, I.; Chakravarthi, B.V.; Varambally, S. UALCAN: A portal for facilitating tumor subgroup gene expression and survival analyses. Neoplasia 2017, 19, 649–658. [Google Scholar] [CrossRef]
- Kim, S.-H.; Jeong, J.-W.; Park, J.; Lee, J.-W.; Seo, J.H.; Jung, B.-K.; Bae, M.-K.; Kim, K.-W. Regulation of the HIF-1α stability by histone deacetylases. Oncol. Rep. 2007, 17, 647–651. [Google Scholar] [CrossRef]
- Chang, C.C.; Lin, B.R.; Chen, S.T.; Hsieh, T.H.; Li, Y.J.; Kuo, M.Y.P. HDAC2 promotes cell migration/invasion abilities through HIF-1α stabilization in human oral squamous cell carcinoma. J. Oral Pathol. Med. 2011, 40, 567–575. [Google Scholar] [CrossRef]
- Decroos, C.; Christianson, N.H.; Gullett, L.E.; Bowman, C.M.; Christianson, K.E.; Deardorff, M.A.; Christianson, D.W. Biochemical and structural characterization of HDAC8 mutants associated with Cornelia de Lange syndrome spectrum disorders. Biochemistry 2015, 54, 6501–6513. [Google Scholar] [CrossRef]
- Luo, W.; Wang, Y. Epigenetic regulators: Multifunctional proteins modulating hypoxia-inducible factor-α protein stability and activity. Cell. Mol. Life Sci. 2018, 75, 1043–1056. [Google Scholar] [CrossRef] [PubMed]
- Harris, A.L. Hypoxia--a key regulatory factor in tumour growth. Nat. Rev. Cancer 2002, 2, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Albadari, N.; Deng, S.; Li, W. The transcriptional factors HIF-1 and HIF-2 and their novel inhibitors in cancer therapy. Expert Opin. Drug Discov. 2019, 14, 667–682. [Google Scholar] [CrossRef] [PubMed]
- Wilmott, J.S.; Colebatch, A.J.; Kakavand, H.; Shang, P.; Carlino, M.S.; Thompson, J.F.; Long, G.V.; Scolyer, R.A.; Hersey, P. Expression of the class 1 histone deacetylases HDAC8 and 3 are associated with improved survival of patients with metastatic melanoma. Mod. Pathol. 2015, 28, 884–894. [Google Scholar] [CrossRef] [PubMed]
- Cirenajwis, H.; Ekedahl, H.; Lauss, M.; Harbst, K.; Carneiro, A.; Enoksson, J.; Rosengren, F.; Werner-Hartman, L.; Törngren, T.; Kvist, A. Molecular stratification of metastatic melanoma using gene expression profiling: Prediction of survival outcome and benefit from molecular targeted therapy. Oncotarget 2015, 6, 12297–12309. [Google Scholar] [CrossRef]
- Zbytek, B.; Carlson, J.A.; Granese, J.; Ross, J.; Mihm, M.C., Jr.; Slominski, A. Current concepts of metastasis in melanoma. Expert Rev. Dermatol. 2008, 3, 569–585. [Google Scholar] [CrossRef]
- Geng, H.; Liu, Q.; Xue, C.; David, L.L.; Beer, T.M.; Thomas, G.V.; Dai, M.-S.; Qian, D.Z. HIF1α protein stability is increased by acetylation at lysine 709. J. Biol. Chem. 2012, 287, 35496–35505. [Google Scholar] [CrossRef]
- Lim, J.-H.; Lee, Y.-M.; Chun, Y.-S.; Chen, J.; Kim, J.-E.; Park, J.-W. Sirtuin 1 modulates cellular responses to hypoxia by deacetylating hypoxia-inducible factor 1α. Mol. Cell 2010, 38, 864–878. [Google Scholar] [CrossRef]
- Yoo, Y.G.; Kong, G.; Lee, M.O. Metastasis-associated protein 1 enhances stability of hypoxia-inducible factor-1α protein by recruiting histone deacetylase 1. EMBO J. 2006, 25, 1231–1241. [Google Scholar] [CrossRef]
- Geng, H.; Harvey, C.T.; Pittsenbarger, J.; Liu, Q.; Beer, T.M.; Xue, C.; Qian, D.Z. HDAC4 protein regulates HIF1α protein lysine acetylation and cancer cell response to hypoxia. J. Biol. Chem. 2011, 286, 38095–38102. [Google Scholar] [CrossRef]
- Semenza, G.L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Bedogni, B.; Welford, S.M.; Cassarino, D.S.; Nickoloff, B.J.; Giaccia, A.J.; Powell, M.B. The hypoxic microenvironment of the skin contributes to Akt-mediated melanocyte transformation. Cancer Cell 2005, 8, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.-H.; Worley, L.A.; Onken, M.D.; Harbour, J.W. Prognostic biomarkers in uveal melanoma: Evidence for a stem cell-like phenotype associated with metastasis. Melanoma Res. 2008, 18, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Emmons, M.F.; Faião-Flores, F.; Sharma, R.; Thapa, R.; Messina, J.L.; Becker, J.C.; Schadendorf, D.; Seto, E.; Sondak, V.K.; Koomen, J.M.; et al. HDAC8 Regulates a Stress Response Pathway in Melanoma to Mediate Escape from BRAF Inhibitor Therapy. Cancer Res. 2019, 79, 2947–2961. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inhibitors | Selectivity | Concentration | Inhibitors | Selectivity | Concentration |
|---|---|---|---|---|---|
| Trichostatin A (TSA) | Pan-HDAC | 2 µM | PCI-34051 | HDAC8 | 20 µM |
| SAHA (Vorinostat) | Pan-HDAC | 2 µM | TMP195 | Class IIa HDAC | 2 µM |
| LBH589 (Panobinostat) | Pan-HDAC | 2 µM | TMP269 | Class IIa HDAC | 2 µM |
| Sodium Butyrate (NaB) | Pan-HDAC | 2 µM | LMK-235 | HDAC4/5 | 2 µM |
| FK228 (Romidepsin) | HDAC1/2 | 0.2 µM | ACY-241 (Citarinostat) | HDAC6 | 2 µM |
| T247 | HDAC3 | 2 uM | Tenovin-6 | SIRT1/2 | 10 µM |
| Target | Oligonucleotide Sequences |
|---|---|
| gHDAC8 #1 | F: 5′ CAC CGG CTG CCC AAT GCC TGA TTG A 3′ R: 5′ AAA CTC AAT CAG GCA TTG GGC AGC C 3′ |
| gHDAC8 #2 | F: 5′ CAC CGT AGG ATA GTT AAG CCT AAA G 3′ R: 5′ AAA CCT TTA GGC TTA ACT ATC CTA C 3′ |
| gHDAC8 #3 | F: 5′ CAC CGA TGC ACT GCA TAA GCA GAT G 3′ R: 5′ AAA CCA TCT GCT TAT GCA GTG CAT C 3′ |
| Target Gene | Oligonucleotide Sequences |
|---|---|
| HIF1A | F: 5′ GAT TCG CCA TGG AGG GC 3′ R: 5′ TTC GAC GTT CAG AAC TCA TCT TTT 3′ |
| VEGFA | F: 5′ TGA AGC CCT GGA GTG CGT 3′ R: 5′ AGG TTT GAT CCG CAT GAT CTG 3′ |
| SLC2A1 | F: 5′ TCT CTG TCG GCC TCT TTG TT 3′ R: 5′ GCA GAA GGG CAA CAG GAT AC 3′ |
| HK2 | F: 5′ AAG GCT TCA AGG CAT CTG 3′ R: 5′ CCA CAG GTC ATC ATA GTT CC 3′ |
| GAPDH | F: 5′ CAT GAG AAG TAT GAC AAC AGC CT 3′ R: 5′ AGT CCT TCC ACG ATA CCA AAG T 3′ |
| Time | 72 h | ||
|---|---|---|---|
| A2058 | SK-MEL-2 | SK-MEL-28 | |
| 1 GI50 (µM) | 15.38 | 14.39 | 17.62 |
| 2 IC50 (µM) | 21.03 | 15.72 | 21.64 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.Y.; Cho, H.; Yoo, J.; Kim, G.W.; Jeon, Y.H.; Lee, S.W.; Kwon, S.H. HDAC8 Deacetylates HIF-1α and Enhances Its Protein Stability to Promote Tumor Growth and Migration in Melanoma. Cancers 2023, 15, 1123. https://doi.org/10.3390/cancers15041123
Kim JY, Cho H, Yoo J, Kim GW, Jeon YH, Lee SW, Kwon SH. HDAC8 Deacetylates HIF-1α and Enhances Its Protein Stability to Promote Tumor Growth and Migration in Melanoma. Cancers. 2023; 15(4):1123. https://doi.org/10.3390/cancers15041123
Chicago/Turabian StyleKim, Ji Yoon, Hayoung Cho, Jung Yoo, Go Woon Kim, Yu Hyun Jeon, Sang Wu Lee, and So Hee Kwon. 2023. "HDAC8 Deacetylates HIF-1α and Enhances Its Protein Stability to Promote Tumor Growth and Migration in Melanoma" Cancers 15, no. 4: 1123. https://doi.org/10.3390/cancers15041123