
SOCS1 Deficiency Promotes Hepatocellular Carcinoma via SOCS3-Dependent CDKN1A Induction and NRF2 Activation

 , , , ,
, , , ,
Abstract
:Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Mouse Strains
2.2. Experimental HCC
2.3. Induction of Genotoxic and Oxidative Stress
2.4. Immunofluorescence Staining of Liver Sections
2.5. Gene and Protein Expression Analysis
2.6. TCGA-LIHC Dataset and Analyses
2.7. Statistical Analysis
3. Results
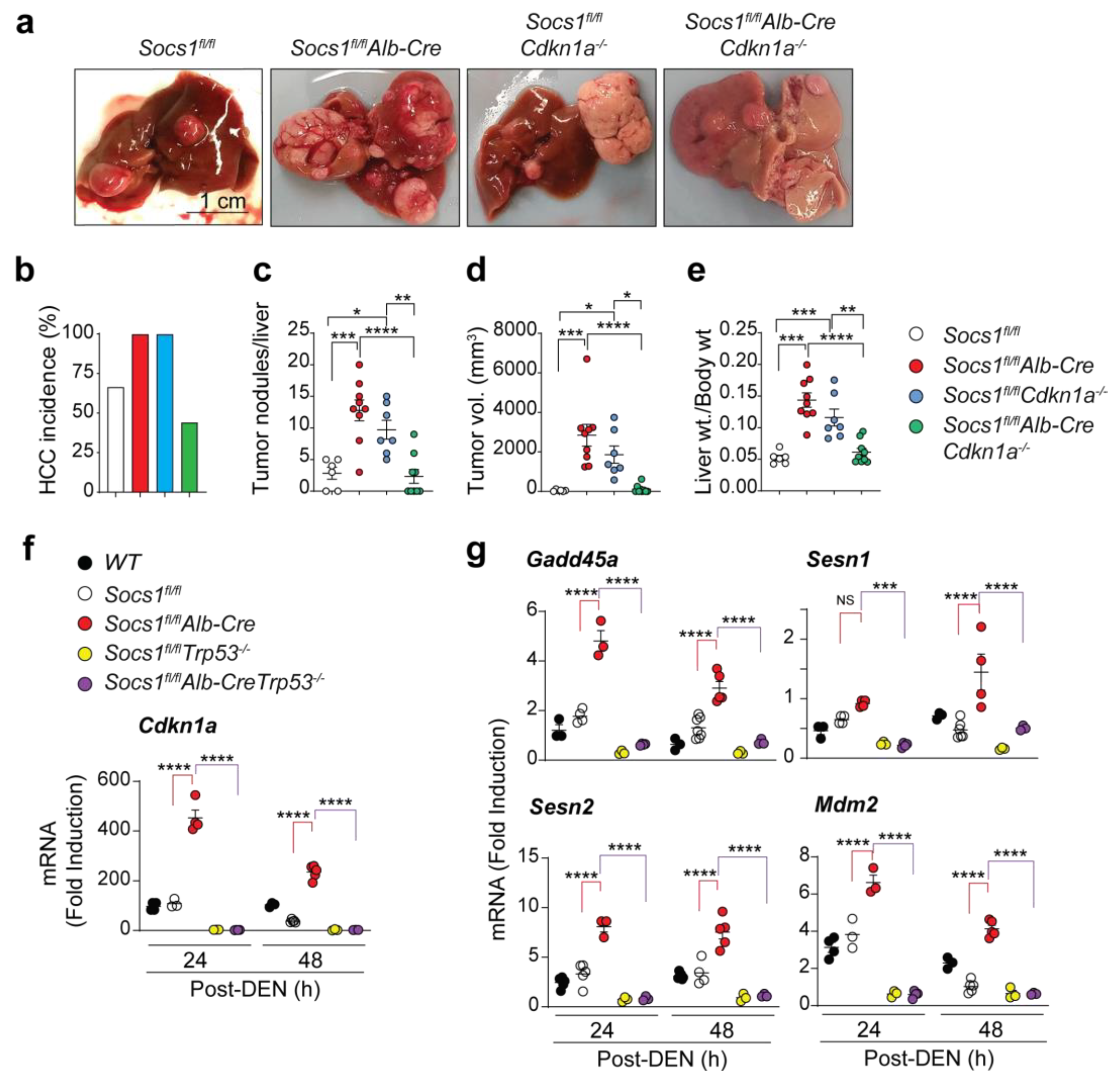
3.1. SOCS1-Mediated Tumor Suppression in the Liver Requires CDKN1A
3.2. Cdkn1a Induction in SOCS1-Deficient Livers Is Driven by p53
3.3. SOCS3 Activates p53 in SOCS1-Deficient Livers
3.4. SOCS3 Promotes HCC Progression in SOCS1-Deficient Livers
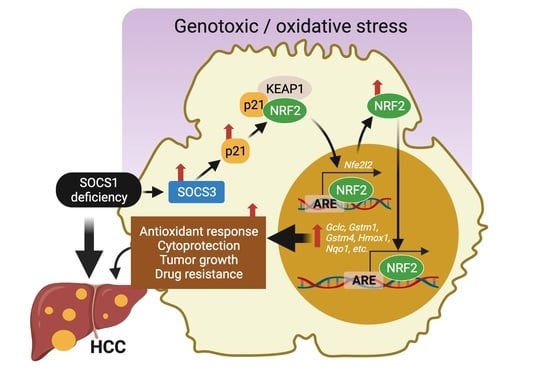
3.5. SOCS1 Deficiency Promotes NFR2 Activation in a SOCS3 and CDKN1A Dependent Manner
3.6. SOCS1-Low/SOCS3-High HCC Cases Display Enrichment of NRF2 Signature Genes and Predict Poor Progression-Free Survival
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yoshikawa, H.; Matsubara, K.; Qian, G.S.; Jackson, P.; Groopman, J.D.; Manning, J.E.; Harris, C.C.; Herman, J.G. SOCS-1, a negative regulator of the JAK/STAT pathway, is silenced by methylation in human hepatocellular carcinoma and shows growth-suppression activity. Nat. Genet. 2001, 28, 29–35. [Google Scholar] [CrossRef]
- Yang, B.; Guo, M.; Herman, J.G.; Clark, D.P. Aberrant promoter methylation profiles of tumor suppressor genes in hepatocellular carcinoma. Am. J. Pathol. 2003, 163, 1101–1107. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, T.; Ogata, H.; Kamio, M.; Joo, A.; Shiraishi, H.; Tokunaga, Y.; Sata, M.; Nagai, H.; Yoshimura, A. SOCS1 is a suppressor of liver fibrosis and hepatitis-induced carcinogenesis. J. Exp. Med. 2004, 199, 1701–1707. [Google Scholar] [CrossRef] [Green Version]
- Yeganeh, M.; Gui, Y.; Kandhi, R.; Bobbala, D.; Tobelaim, W.S.; Saucier, C.; Yoshimura, A.; Ferbeyre, G.; Ramanathan, S.; Ilangumaran, S. Suppressor of cytokine signaling 1-dependent regulation of the expression and oncogenic functions of p21(CIP1/WAF1) in the liver. Oncogene 2016, 35, 4200–4211. [Google Scholar] [CrossRef] [PubMed]
- Niwa, Y.; Kanda, H.; Shikauchi, Y.; Saiura, A.; Matsubara, K.; Kitagawa, T.; Yamamoto, J.; Kubo, T.; Yoshikawa, H. Methylation silencing of SOCS-3 promotes cell growth and migration by enhancing JAK/STAT and FAK signalings in human hepatocellular carcinoma. Oncogene 2005, 24, 6406–6417. [Google Scholar] [CrossRef] [Green Version]
- Riehle, K.J.; Campbell, J.S.; McMahan, R.S.; Johnson, M.M.; Beyer, R.P.; Bammler, T.K.; Fausto, N. Regulation of liver regeneration and hepatocarcinogenesis by suppressor of cytokine signaling 3. J. Exp. Med. 2008, 205, 91–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, M.G.M.; Ghosh, A.; Variya, B.; Santharam, M.A.; Kandhi, R.; Ramanathan, S.; Ilangumaran, S. Hepatocyte growth control by SOCS1 and SOCS3. Cytokine 2019, 121, 154733. [Google Scholar] [CrossRef]
- Gui, Y.; Yeganeh, M.; Ramanathan, S.; Leblanc, C.; Pomerleau, V.; Ferbeyre, G.; Saucier, C.; Ilangumaran, S. SOCS1 controls liver regeneration by regulating HGF signaling in hepatocytes. J. Hepatol. 2011, 55, 1300–1308. [Google Scholar] [CrossRef]
- Gui, Y.; Yeganeh, M.; Donates, Y.C.; Tobelaim, W.S.; Chababi, W.; Mayhue, M.; Yoshimura, A.; Ramanathan, S.; Saucier, C.; Ilangumaran, S. Regulation of MET receptor tyrosine kinase signaling by suppressor of cytokine signaling 1 in hepatocellular carcinoma. Oncogene 2015, 34, 5718–5728. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, V.; Mallette, F.A.; Deschenes-Simard, X.; Ramanathan, S.; Gagnon, J.; Moores, A.; Ilangumaran, S.; Ferbeyre, G. SOCS1 links cytokine signaling to p53 and senescence. Mol. Cell 2009, 36, 754–767. [Google Scholar] [CrossRef]
- Abbas, T.; Dutta, A. p21 in cancer: Intricate networks and multiple activities. Nat. Rev. Cancer 2009, 9, 400–414. [Google Scholar] [CrossRef] [PubMed]
- Gartel, A.L. p21(WAF1/CIP1) and cancer: A shifting paradigm? Biofactors 2009, 35, 161–164. [Google Scholar] [CrossRef] [PubMed]
- Koster, R.; di Pietro, A.; Timmer-Bosscha, H.; Gibcus, J.H.; van den Berg, A.; Suurmeijer, A.J.; Bischoff, R.; Gietema, J.A.; de Jong, S. Cytoplasmic p21 expression levels determine cisplatin resistance in human testicular cancer. J. Clin. Investig. 2010, 120, 3594–3605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kansanen, E.; Kuosmanen, S.M.; Leinonen, H.; Levonen, A.L. The Keap1-Nrf2 pathway: Mechanisms of activation and dysregulation in cancer. Redox Biol. 2013, 1, 45–49. [Google Scholar] [CrossRef] [Green Version]
- Menegon, S.; Columbano, A.; Giordano, S. The Dual Roles of NRF2 in Cancer. Trends Mol. Med. 2016, 22, 578–593. [Google Scholar] [CrossRef]
- Eggler, A.L.; Liu, G.; Pezzuto, J.M.; van Breemen, R.B.; Mesecar, A.D. Modifying specific cysteines of the electrophile-sensing human Keap1 protein is insufficient to disrupt binding to the Nrf2 domain Neh2. Proc. Natl. Acad. Sci. USA 2005, 102, 10070–10075. [Google Scholar] [CrossRef] [Green Version]
- Baird, L.; Lleres, D.; Swift, S.; Dinkova-Kostova, A.T. Regulatory flexibility in the Nrf2-mediated stress response is conferred by conformational cycling of the Keap1-Nrf2 protein complex. Proc. Natl. Acad. Sci. USA 2013, 110, 15259–15264. [Google Scholar] [CrossRef] [Green Version]
- Baird, L.; Dinkova-Kostova, A.T. Diffusion dynamics of the Keap1-Cullin3 interaction in single live cells. Biochem. Biophys. Res. Commun. 2013, 433, 58–65. [Google Scholar] [CrossRef]
- Chen, W.; Sun, Z.; Wang, X.J.; Jiang, T.; Huang, Z.; Fang, D.; Zhang, D.D. Direct interaction between Nrf2 and p21(Cip1/WAF1) upregulates the Nrf2-mediated antioxidant response. Mol. Cell 2009, 34, 663–673. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213–223. [Google Scholar] [CrossRef] [PubMed]
- DeNicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011, 475, 106–109. [Google Scholar] [CrossRef] [Green Version]
- Naugler, W.E.; Sakurai, T.; Kim, S.; Maeda, S.; Kim, K.; Elsharkawy, A.M.; Karin, M. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science 2007, 317, 121–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas Research Network. Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 2017, 169, 1327–1341.e23. [Google Scholar] [CrossRef] [Green Version]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.S.; Malta, T.M.; Pagnotta, S.M.; Castiglioni, I.; et al. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016, 44, e71. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Therneau, T.M. A Package for Survival Analysis in R. Available online: https://CRAN.R-project.org/package=survival (accessed on 3 March 2021).
- Kassambara, A. Drawing Survival Curves Using ‘ggplot2’. R Package Version 0.4.6. Available online: https://CRAN.r-project.org/web/packages/survminer (accessed on 3 March 2021).
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdottir, H.; Tamayo, P.; Mesirov, J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef] [PubMed]
- Dolgalev, I. msigdbr: MSigDB Gene Sets for Multiple Organisms in a Tidy Data Format. Available online: https://CRAN.R-project.org/package=msigdbr (accessed on 3 March 2021).
- Kohl, M. MKmisc: Miscellaneous Functions from M. Kohl_. Available online: https://www.stamats.de (accessed on 3 March 2021).
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS A J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Foroutan, M.; Bhuva, D.D.; Lyu, R.; Horan, K.; Cursons, J.; Davis, M.J. Single sample scoring of molecular phenotypes. BMC Bioinform. 2018, 19, 404. [Google Scholar] [CrossRef] [Green Version]
- Fausto, N.; Campbell, J.S. Mouse models of hepatocellular carcinoma. Semin. Liver Dis. 2010, 30, 87–98. [Google Scholar] [CrossRef]
- Yan, H.X.; Wu, H.P.; Zhang, H.L.; Ashton, C.; Tong, C.; Wu, J.; Qian, Q.J.; Wang, H.Y.; Ying, Q.L. DNA damage-induced sustained p53 activation contributes to inflammation-associated hepatocarcinogenesis in rats. Oncogene 2013, 32, 4565–4571. [Google Scholar] [CrossRef] [Green Version]
- Saint-Germain, E.; Mignacca, L.; Huot, G.; Acevedo, M.; Moineau-Vallee, K.; Calabrese, V.; Bourdeau, V.; Rowell, M.C.; Ilangumaran, S.; Lessard, F.; et al. Phosphorylation of SOCS1 Inhibits the SOCS1-p53 Tumor Suppressor Axis. Cancer Res. 2019, 79, 3306–3319. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Feng, D.; Wang, H.; Hong, F.; Bertola, A.; Wang, F.S.; Gao, B. Interleukin-22 induces hepatic stellate cell senescence and restricts liver fibrosis in mice. Hepatology 2012, 56, 1150–1159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwak, M.K.; Itoh, K.; Yamamoto, M.; Kensler, T.W. Enhanced expression of the transcription factor Nrf2 by cancer chemopreventive agents: Role of antioxidant response element-like sequences in the nrf2 promoter. Mol. Cell Biol. 2002, 22, 2883–2892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buitrago-Molina, L.E.; Marhenke, S.; Longerich, T.; Sharma, A.D.; Boukouris, A.E.; Geffers, R.; Guigas, B.; Manns, M.P.; Vogel, A. The degree of liver injury determines the role of p21 in liver regeneration and hepatocarcinogenesis in mice. Hepatology 2013, 58, 1143–1152. [Google Scholar] [CrossRef]
- Ngo, H.K.C.; Kim, D.H.; Cha, Y.N.; Na, H.K.; Surh, Y.J. Nrf2 Mutagewwnic Activation Drives Hepatocarcinogenesis. Cancer Res. 2017, 77, 4797–4808. [Google Scholar] [CrossRef] [Green Version]
- Sakurai, T.; He, G.; Matsuzawa, A.; Yu, G.Y.; Maeda, S.; Hardiman, G.; Karin, M. Hepatocyte necrosis induced by oxidative stress and IL-1 alpha release mediate carcinogen-induced compensatory proliferation and liver tumorigenesis. Cancer Cell 2008, 14, 156–165. [Google Scholar] [CrossRef] [Green Version]
- Polonen, P.; Jawahar Deen, A.; Leinonen, H.M.; Jyrkkanen, H.K.; Kuosmanen, S.; Mononen, M.; Jain, A.; Tuomainen, T.; Pasonen-Seppanen, S.; Hartikainen, J.M.; et al. Nrf2 and SQSTM1/p62 jointly contribute to mesenchymal transition and invasion in glioblastoma. Oncogene 2019, 38, 7473–7490. [Google Scholar] [CrossRef] [Green Version]
- Topley, G.I.; Okuyama, R.; Gonzales, J.G.; Conti, C.; Dotto, G.P. p21(WAF1/Cip1) functions as a suppressor of malignant skin tumor formation and a determinant of keratinocyte stem-cell potential. Proc. Natl. Acad. Sci. USA 1999, 96, 9089–9094. [Google Scholar] [CrossRef] [Green Version]
- Martin-Caballero, J.; Flores, J.M.; Garcia-Palencia, P.; Serrano, M. Tumor susceptibility of p21(Waf1/Cip1)-deficient mice. Cancer Res. 2001, 61, 6234–6238. [Google Scholar]
- Poole, A.J.; Heap, D.; Carroll, R.E.; Tyner, A.L. Tumor suppressor functions for the Cdk inhibitor p21 in the mouse colon. Oncogene 2004, 23, 8128–8134. [Google Scholar] [CrossRef] [Green Version]
- Ehedego, H.; Boekschoten, M.V.; Hu, W.; Doler, C.; Haybaeck, J.; Gabetaler, N.; Muller, M.; Liedtke, C.; Trautwein, C. p21 ablation in liver enhances DNA damage, cholestasis, and carcinogenesis. Cancer Res. 2015, 75, 1144–1155. [Google Scholar] [CrossRef] [Green Version]
- Wagayama, H.; Shiraki, K.; Sugimoto, K.; Ito, T.; Fujikawa, K.; Yamanaka, T.; Takase, K.; Nakano, T. High expression of p21WAF1/CIP1 is correlated with human hepatocellular carcinoma in patients with hepatitis C virus-associated chronic liver diseases. Hum. Pathol. 2002, 33, 429–434. [Google Scholar] [CrossRef]
- Shiraki, K.; Wagayama, H. Cytoplasmic p21(WAF1/CIP1) expression in human hepatocellular carcinomas. Liver Int. 2006, 26, 1018–1019. [Google Scholar] [CrossRef]
- Marhenke, S.; Buitrago-Molina, L.E.; Endig, J.; Orlik, J.; Schweitzer, N.; Klett, S.; Longerich, T.; Geffers, R.; Sanchez Munoz, A.; Dorrell, C.; et al. p21 promotes sustained liver regeneration and hepatocarcinogenesis in chronic cholestatic liver injury. Gut 2014, 63, 1501–1512. [Google Scholar] [CrossRef] [PubMed]
- LaBaer, J.; Garrett, M.D.; Stevenson, L.F.; Slingerland, J.M.; Sandhu, C.; Chou, H.S.; Fattaey, A.; Harlow, E. New functional activities for the p21 family of CDK inhibitors. Genes Dev. 1997, 11, 847–862. [Google Scholar] [CrossRef] [Green Version]
- Shiraha, H.; Yamamoto, K.; Namba, M. Human hepatocyte carcinogenesis (review). Int. J. Oncol. 2013, 42, 1133–1138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milkovic, L.; Zarkovic, N.; Saso, L. Controversy about pharmacological modulation of Nrf2 for cancer therapy. Redox Biol. 2017, 12, 727–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orru, C.; Giordano, S.; Columbano, A. Nrf2 in Neoplastic and Non-Neoplastic Liver Diseases. Cancers 2020, 12, 2932. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zhang, C.; Zhang, L.; Yang, Q.; Zhou, S.; Wen, Q.; Wang, J. Nrf2 is a potential prognostic marker and promotes proliferation and invasion in human hepatocellular carcinoma. BMC Cancer 2015, 15, 531. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.; Kim, S.; Lee, Y.; Lee, H.; Lee, Y.; Park, H.; Nahm, J.H.; Ahn, S.; Yu, S.J.; Lee, K.; et al. The Clinicopathological and Prognostic Significance of Nrf2 and Keap1 Expression in Hepatocellular Carcinoma. Cancers 2020, 12, 2128. [Google Scholar] [CrossRef]
- Guichard, C.; Amaddeo, G.; Imbeaud, S.; Ladeiro, Y.; Pelletier, L.; Maad, I.B.; Calderaro, J.; Bioulac-Sage, P.; Letexier, M.; Degos, F.; et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat. Genet. 2012, 44, 694–698. [Google Scholar] [CrossRef] [PubMed]
- Zavattari, P.; Perra, A.; Menegon, S.; Kowalik, M.A.; Petrelli, A.; Angioni, M.M.; Follenzi, A.; Quagliata, L.; Ledda-Columbano, G.M.; Terracciano, L.; et al. Nrf2, but not beta-catenin, mutation represents an early event in rat hepatocarcinogenesis. Hepatology 2015, 62, 851–862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umemura, A.; He, F.; Taniguchi, K.; Nakagawa, H.; Yamachika, S.; Font-Burgada, J.; Zhong, Z.; Subramaniam, S.; Raghunandan, S.; Duran, A.; et al. p62, Upregulated during Preneoplasia, Induces Hepatocellular Carcinogenesis by Maintaining Survival of Stressed HCC-Initiating Cells. Cancer Cell 2016, 29, 935–948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, F.; Antonucci, L.; Yamachika, S.; Zhang, Z.; Taniguchi, K.; Umemura, A.; Hatzivassiliou, G.; Roose-Girma, M.; Reina-Campos, M.; Duran, A.; et al. NRF2 activates growth factor genes and downstream AKT signaling to induce mouse and human hepatomegaly. J. Hepatol. 2020, 72, 1182–1195. [Google Scholar] [CrossRef] [Green Version]
- Parkinson, E.I.; Hergenrother, P.J. Deoxynyboquinones as NQO1-Activated Cancer Therapeutics. ACC Chem. Res. 2015, 48, 2715–2723. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khan, M.G.M.; Boufaied, N.; Yeganeh, M.; Kandhi, R.; Petkiewicz, S.; Sharma, A.; Yoshimura, A.; Ferbeyre, G.; Labbé, D.P.; Ramanathan, S.; et al. SOCS1 Deficiency Promotes Hepatocellular Carcinoma via SOCS3-Dependent CDKN1A Induction and NRF2 Activation. Cancers 2023, 15, 905. https://doi.org/10.3390/cancers15030905
Khan MGM, Boufaied N, Yeganeh M, Kandhi R, Petkiewicz S, Sharma A, Yoshimura A, Ferbeyre G, Labbé DP, Ramanathan S, et al. SOCS1 Deficiency Promotes Hepatocellular Carcinoma via SOCS3-Dependent CDKN1A Induction and NRF2 Activation. Cancers. 2023; 15(3):905. https://doi.org/10.3390/cancers15030905
Chicago/Turabian StyleKhan, Md Gulam Musawwir, Nadia Boufaied, Mehdi Yeganeh, Rajani Kandhi, Stephanie Petkiewicz, Ankur Sharma, Akihiko Yoshimura, Gerardo Ferbeyre, David P. Labbé, Sheela Ramanathan, and et al. 2023. "SOCS1 Deficiency Promotes Hepatocellular Carcinoma via SOCS3-Dependent CDKN1A Induction and NRF2 Activation" Cancers 15, no. 3: 905. https://doi.org/10.3390/cancers15030905