Pharmacokinetic Analysis of Omomyc Shows Lasting Structural Integrity and Long Terminal Half-Life in Tumor Tissue


Abstract
:Simple Summary
Abstract
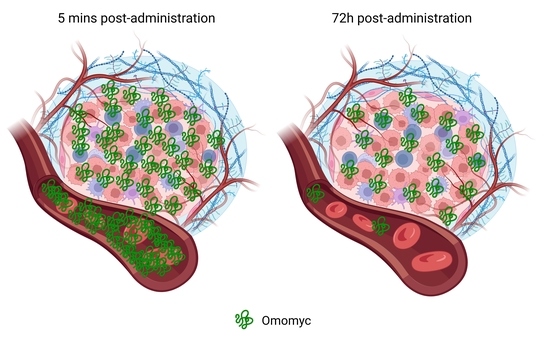
1. Introduction
2. Materials and Methods
2.1. Production of Serum and Tumor Samples
2.2. Sample Preparation for Mass Spectrometry
2.3. Shotgun LC–MS/MS for Spectral Library Generation
2.4. LC-PRM Mass Spectrometry Acquisition and Data Analysis
2.5. Statistical Analysis
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Dhanasekaran, R.; Deutzmann, A.; Mahauad-Fernandez, W.D.; Hansen, A.S.; Gouw, A.M.; Felsher, D.W. The MYC oncogene—the grand orchestrator of cancer growth and immune evasion. Nat. Rev. Clin. Oncol. 2022, 19, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Baluapuri, A.; Wolf, E.; Eilers, M. Target gene-independent functions of MYC oncoproteins. Nat. Rev. Mol. Cell Biol. 2020, 21, 255–267. [Google Scholar] [CrossRef]
- Schaub, F.X.; Dhankani, V.; Berger, A.C.; Trivedi, M.; Richardson, A.B.; Shaw, R.; Zhao, W.; Zhang, X.; Ventura, A.; Liu, Y.; et al. Pan-cancer Alterations of the MYC Oncogene and Its Proximal Network across the Cancer Genome Atlas. Cell Syst. 2018, 6, 282–300.e2. [Google Scholar] [CrossRef] [Green Version]
- Llombart, V.; Mansour, M.R. Therapeutic targeting of “undruggable” MYC. EBioMedicine 2022, 75, 103756. [Google Scholar] [CrossRef] [PubMed]
- Soucek, L.; Helmer-Citterich, M.; Sacco, A.; Jucker, R.; Cesareni, G.; Nasi, S. Design and Properties of a Myc Derivative That efficiently homodimerizes. Oncogene 1998, 17, 2463–2472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massó-Vallés, D.; Soucek, L. Blocking Myc to Treat Cancer: Reflecting on Two Decades of Omomyc. Cells 2020, 9, 883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savino, M.; Annibali, D.; Carucci, N.; Favuzzi, E.; Cole, M.D.; Evan, G.I.; Soucek, L.; Nasi, S. The Action Mechanism of the Myc Inhibitor Termed Omomyc May Give Clues on How to Target Myc for Cancer Therapy. PLoS ONE 2011, 6, e22284. [Google Scholar] [CrossRef] [Green Version]
- Beaulieu, M.-E.; Jauset, T.; Massó-Vallés, D.; Martínez-Martín, S.; Rahl, P.; Maltais, L.; Zacarias-Fluck, M.F.; Casacuberta-Serra, S.; del Pozo, E.S.; Fiore, C.; et al. Intrinsic cell-penetrating activity propels Omomyc from proof of concept to viable anti-MYC therapy. Sci. Transl. Med. 2019, 11, eaar5012. [Google Scholar] [CrossRef]
- Jung, L.A.; Gebhardt, A.; Koelmel, W.; Ade, C.P.; Walz, S.; Kuper, J.; von Eyss, B.; Letschert, S.; Redel, C.; D’Artista, L.; et al. OmoMYC blunts promoter invasion by oncogenic MYC to inhibit gene expression characteristic of MYC-dependent tumors. Oncogene 2017, 36, 1911–1924. [Google Scholar] [CrossRef] [Green Version]
- Soucek, L.; Whitfield, J.R.; Sodir, N.M.; Massó-Vallés, D.; Serrano, E.; Karnezis, A.N.; Swigart, L.B.; Evan, G.I. Inhibition of Myc family proteins eradicates KRas-driven lung cancer in mice. Genes Dev. 2013, 27, 504–513. [Google Scholar] [CrossRef]
- Soucek, L.; Whitfield, J.; Martins, C.P.; Finch, A.J.; Murphy, D.J.; Sodir, N.M.; Karnezis, A.N.; Swigart, L.B.; Nasi, S.; Evan, G.I. Modelling Myc inhibition as a cancer therapy. Nature 2008, 455, 679–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duffy, M.J.; O’Grady, S.; Tang, M.; Crown, J. MYC as a target for cancer treatment. Cancer Treat. Rev. 2021, 94, 102154. [Google Scholar] [CrossRef] [PubMed]
- Demma, M.J.; Mapelli, C.; Sun, A.; Bodea, S.; Ruprecht, B.; Javaid, S.; Wiswell, D.; Muise, E.; Chen, S.; Zelina, J.; et al. Omomyc Reveals New Mechanisms to Inhibit the MYC Oncogene. Mol. Cell. Biol. 2019, 39, e00248-19. [Google Scholar] [CrossRef] [Green Version]
- Massó-Vallés, D.; Beaulieu, M.-E.; Jauset, T.; Giuntini, F.; Zacarías-Fluck, M.F.; Foradada, L.; Martínez-Martín, S.; Serrano, E.; Martín-Fernández, G.; Casacuberta-Serra, S.; et al. MYC Inhibition Halts Metastatic Breast Cancer Progression by Blocking Growth, Invasion, and Seeding. Cancer Res. Commun. 2022, 2, 110–130. [Google Scholar] [CrossRef]
- Vizovisek, M.; Ristanovic, D.; Menghini, S.; Christiansen, M.; Schuerle, S. The Tumor Proteolytic Landscape: A Challenging Frontier in Cancer Diagnosis and Therapy. Int. J. Mol. Sci. 2021, 22, 2514. [Google Scholar] [CrossRef] [PubMed]
- Macklin, A.; Khan, S.; Kislinger, T. Recent advances in mass spectrometry based clinical proteomics: Applications to cancer research. Clin. Proteom. 2020, 17, 17. [Google Scholar] [CrossRef]
- Mullard, A. Climbing cancer’s MYC mountain. Nat. Rev. Drug Discov. 2022, 21, 865–867. [Google Scholar] [CrossRef]
- Kelstrup, C.D.; Young, C.; Lavallee, R.; Nielsen, M.L.; Olsen, J.V. Optimized Fast and Sensitive Acquisition Methods for Shotgun Proteomics on a Quadrupole Orbitrap Mass Spectrometer. J. Proteome Res. 2012, 11, 3487–3497. [Google Scholar] [CrossRef]
- Bruderer, R.; Bernhardt, O.M.; Gandhi, T.; Reiter, L. High-precision iRT prediction in the targeted analysis of data-independent acquisition and its impact on identification and quantitation. Proteomics 2016, 16, 2246–2256. [Google Scholar] [CrossRef] [Green Version]
- Reiter, L.; Rinner, O.; Picotti, P.; Hüttenhain, R.; Beck, M.; Brusniak, M.-Y.; Hengartner, M.O.; Aebersold, R. mProphet: Automated data processing and statistical validation for large-scale SRM experiments. Nat. Methods 2011, 8, 430–435. [Google Scholar] [CrossRef]
- Tully, B.; Balleine, R.L.; Hains, P.G.; Zhong, Q.; Reddel, R.R.; Robinson, P.J. Addressing the Challenges of High-Throughput Cancer Tissue Proteomics for Clinical Application: ProCan. Proteomics 2019, 19, 1900109. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
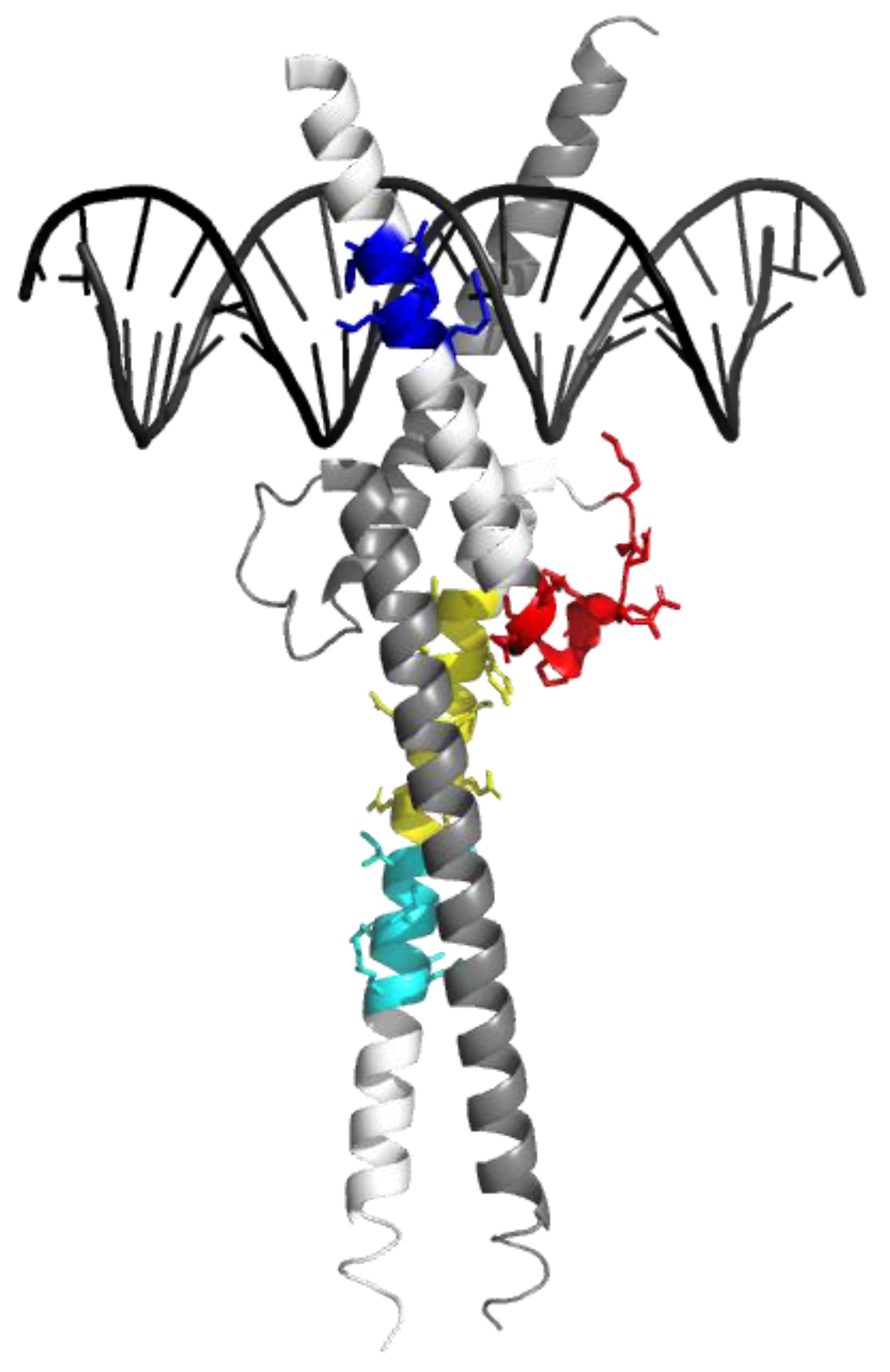{kind=link}
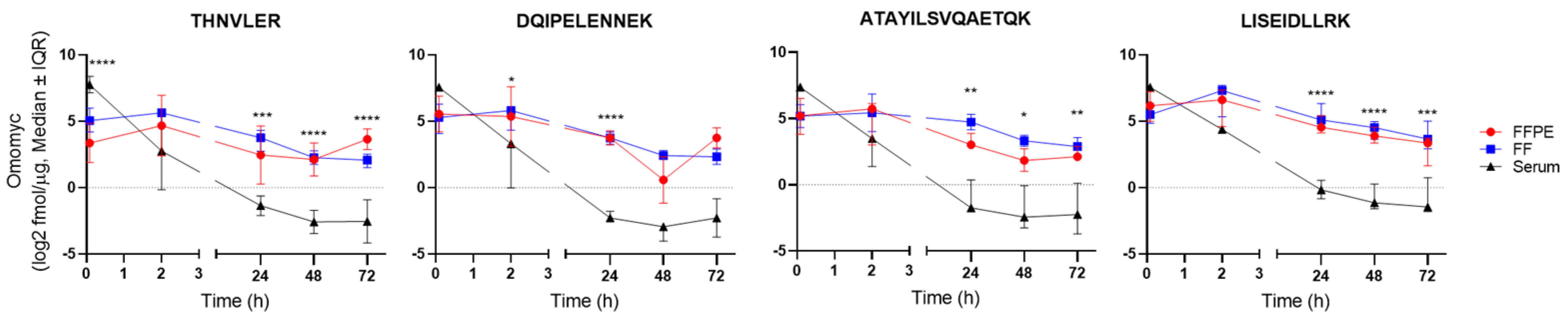{kind=link}
| FFPE Tissue | FF Tissue | Serum | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sample ID | THNVLER | DQIPELENNEK | ATAYILSVQAETQK | LISEIDLLR | THNVLER | DQIPELENNEK | ATAYILSVQAETQK | LISEIDLLR | THNVLER | DQIPELENNEK | ATAYILSVQAETQK | LISEIDLLR |
| DLD-1 O5m.1 | 43.01 | 137.46 | 194.26 | 181.37 | 109.67 | 129.47 | 150.84 | 103.42 | 95.68 | 85.56 | 91.99 | 79.75 |
| DLD-1 O5m.2 | 116.68 | 109.23 | 73.37 | 81.12 | 139.31 | 132.93 | 149.96 | 103.39 | 52.59 | 105.68 | 101.31 | 101.11 |
| DLD-1 O5m.3 | 30.54 | 167.08 | 171.01 | 159.15 | 41.77 | 35.26 | 41.97 | 57.92 | 114.21 | 97.62 | 91.65 | 134.14 |
| DLD-1 O5m.4 | 63.65 | 26.47 | 17.37 | 23.71 | 30.88 | 27.74 | 27.24 | 26.56 | 121.46 | 97.33 | 122.33 | 89.29 |
| DLD-1 O5m.5 | 246.12 | 59.76 | 44.00 | 54.65 | 178.37 | 174.60 | 129.99 | 208.71 | 116.05 | 113.81 | 92.73 | 95.70 |
| AVG | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 |
| DLD-1 O2h.1 | 358.05 | 142.10 | 105.59 | 110.06 | 109.30 | 114.67 | 264.48 | 258.69 | 7.07 | 6.57 | 8.70 | 10.34 |
| DLD-1 O2h.2 | 20.31 | 9.08 | 6.11 | 11.87 | 5.35 | 4.49 | 3.34 | 15.54 | 0.17 | 0.07 | 0.48 | 11.35 |
| DLD-1 O2h.3 | 270.88 | 56.98 | 41.57 | 61.96 | 89.15 | 77.55 | 195.52 | 276.63 | 5.31 | 3.74 | 4.42 | 12.58 |
| DLD-1 O2h.4 | 226.63 | 172.70 | 149.61 | 196.33 | 247.72 | 188.14 | 305.30 | 313.87 | 4.98 | 5.96 | 6.40 | 10.62 |
| DLD-1 O2h.5 | 504.71 | 161.50 | 129.65 | 174.81 | 64.60 | 84.80 | 77.49 | 85.35 | 7.77 | 5.12 | 6.23 | 9.04 |
| AVG | 276.12 | 108.47 | 86.50 | 111.00 | 103.22 | 93.93 | 169.23 | 190.01 | 5.06 | 4.29 | 5.25 | 10.79 |
| DLD-1 O24h.1 | 20.37 | 18.84 | 15.77 | 26.39 | 22.62 | 29.66 | 49.63 | 26.77 | 0.12 | 0.10 | 0.25 | 0.46 |
| DLD-1 O24h.2 | 6.42 | 38.54 | 47.11 | 80.96 | 52.40 | 58.23 | 140.77 | 142.67 | 0.31 | 0.16 | 2.09 | 1.04 |
| DLD-1 O24h.3 | 73.70 | 18.81 | 13.25 | 15.20 | 12.95 | 11.18 | 26.44 | 37.24 | 0.20 | 0.14 | 0.17 | 0.53 |
| DLD-1 O24h.4 | 108.16 | 21.63 | 16.06 | 25.38 | 30.97 | 23.78 | 66.73 | 106.08 | 0.19 | 0.11 | 0.15 | 0.36 |
| DLD-1 O24h.5 | 98.59 | 25.80 | 18.17 | 28.50 | 22.87 | 21.48 | 38.14 | 42.00 | 0.11 | 0.10 | 0.14 | 0.23 |
| AVG | 61.45 | 24.72 | 22.07 | 35.29 | 28.36 | 28.86 | 64.34 | 70.95 | 0.19 | 0.12 | 0.56 | 0.52 |
| DLD-1 O48h.1 | 18.60 | 1.20 | 4.35 | 15.87 | 6.94 | 10.33 | 13.43 | 17.56 | 0.04 | 0.03 | 0.05 | 0.15 |
| DLD-1 O48h.2 | 16.46 | 2.16 | 3.66 | 8.50 | 8.89 | 9.04 | 19.06 | 36.88 | 0.06 | ND | 0.08 | 0.19 |
| DLD-1 O48h.3 | 28.22 | 2.55 | 8.24 | 17.48 | 8.56 | 10.28 | 13.50 | 16.56 | 0.07 | ND | 0.21 | 0.34 |
| DLD-1 O48h.4 | 92.15 | ND | 20.66 | 39.83 | 12.32 | 10.49 | 31.25 | 59.31 | 0.10 | 0.07 | 0.10 | 0.23 |
| DLD-1 O48h.5 | 40.46 | 7.51 | 7.06 | 16.81 | 12.86 | 15.89 | 26.39 | 50.64 | 0.14 | 0.07 | 1.33 | 1.12 |
| AVG | 39.18 | 3.35 | 8.79 | 19.70 | 9.92 | 11.21 | 20.73 | 36.19 | 0.08 | 0.06 | 0.35 | 0.41 |
| DLD-1 O72h.1 | 169.93 | 42.40 | 15.67 | 15.90 | 10.46 | 12.72 | 35.94 | 50.85 | 0.05 | ND | 0.04 | 0.12 |
| DLD-1 O72h.2 | 100.76 | 26.85 | 8.59 | 8.72 | 7.72 | 9.07 | 9.42 | 11.79 | 0.04 | 0.04 | 0.12 | 0.18 |
| DLD-1 O72h.3 | 61.07 | 13.94 | 8.06 | 13.73 | 13.79 | 11.37 | 15.09 | 18.74 | 0.34 | 0.29 | 2.87 | 2.65 |
| DLD-1 O72h.4 | 104.04 | 27.32 | 7.23 | 1.41 | 5.29 | 7.52 | 12.60 | 17.89 | 0.09 | ND | 0.13 | 0.27 |
| DLD-1 O72h.5 | 59.59 | 17.95 | 13.78 | 11.53 | 10.31 | 12.29 | 20.13 | 27.98 | 0.04 | ND | 0.04 | 0.19 |
| AVG | 99.08 | 25.69 | 10.67 | 10.26 | 9.51 | 10.59 | 18.64 | 25.45 | 0.11 | 0.16 | 0.64 | 0.68 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beaulieu, M.-E.; Martínez-Martín, S.; Kaur, J.; Castillo Cano, V.; Massó-Vallés, D.; Foradada Felip, L.; López-Estévez, S.; Serrano del Pozo, E.; Thabussot, H.; Soucek, L. Pharmacokinetic Analysis of Omomyc Shows Lasting Structural Integrity and Long Terminal Half-Life in Tumor Tissue. Cancers 2023, 15, 826. https://doi.org/10.3390/cancers15030826
Beaulieu M-E, Martínez-Martín S, Kaur J, Castillo Cano V, Massó-Vallés D, Foradada Felip L, López-Estévez S, Serrano del Pozo E, Thabussot H, Soucek L. Pharmacokinetic Analysis of Omomyc Shows Lasting Structural Integrity and Long Terminal Half-Life in Tumor Tissue. Cancers. 2023; 15(3):826. https://doi.org/10.3390/cancers15030826
Chicago/Turabian StyleBeaulieu, Marie-Eve, Sandra Martínez-Martín, Jastrinjan Kaur, Virginia Castillo Cano, Daniel Massó-Vallés, Laia Foradada Felip, Sergio López-Estévez, Erika Serrano del Pozo, Hugo Thabussot, and Laura Soucek. 2023. "Pharmacokinetic Analysis of Omomyc Shows Lasting Structural Integrity and Long Terminal Half-Life in Tumor Tissue" Cancers 15, no. 3: 826. https://doi.org/10.3390/cancers15030826