
Osteosarcoma in Pediatric and Adult Populations: Are Adults Just Big Kids?
 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction

{kind=link}
{kind=link}
| Regimen | Age Range | Survival | Reference | |
|---|---|---|---|---|
| MAP vs. MAP/IE | 40 years or younger (does not report by age group) | -Mean time to first event 43.3–44.1 months -3 year EFS 53–55% | Marina et al., 2016 [3] | |
| Multiple regimens on COG trials CCG-7943, POG-9754, INT-0133, and AOST0121 | <10 10 to 17 ≥18 | 10-year EFS 55% 55 37 | 10-year OS 68% 60 41 | Janeway et al., 2012 [4] |
| MAP based chemo | Child (0–11/12) * Adolescent (12–17) Adult (≥17) | HR (EFS primary endpoint) 1.00 1.25 1.32 | Smeland et al., 2019 [5] | |
| High-dose methotrexate, Adriamycin, and BCD | <20 ≥21 | DSS, % at 5, 10, 15 years 34, 31, 31 40, 27, 19 | Bernthal et al., 2012 [6] | |
| Multiple regimens included | <40 ≥40 | 10-year OS 60.2% 41.6% | Bielack et al., 2002 [7] | |
| Multiple regimens included | <15 ≥15 | 10-year OS 76% 68% | Fuchs et al., 1998 [8] | |
| MAP vs. MAP/IE | <40 | 5-year OS 74% | Smeland et al., 2003 [12] | |
| Multiple regimens included | Child (0–11/12) * Adolescent (12–17) and Adult (≥17) | HR (OS from study entry) 1.0 1.23 | Collins et al., 2013 [16] | |
| Doxorubicin, cisplatin, ifosfamide, and methotrexate suggested | 41–65 | 5-year OS 66% | Ferrari et al., 2018 [20] | |
2. Differences in Chemotherapy Tolerance
3. Differences in Underlying Tumor Presentation
3.1. Tumor Location
3.2. Primary versus Secondary Osteosarcoma
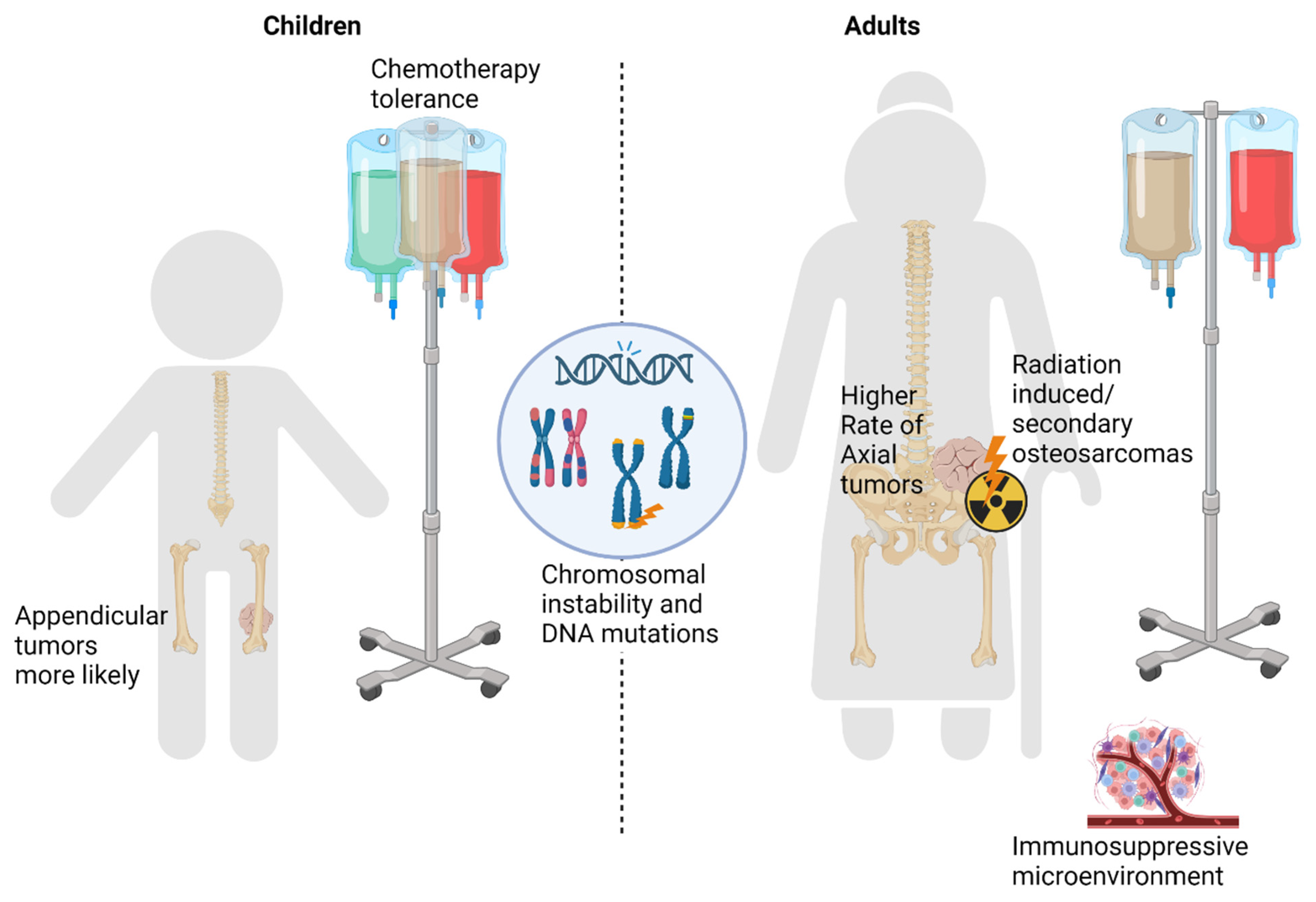
4. Differences in Underlying Tumor Biology (Summarized in Figure 1)
4.1. Chromosomal Instability

4.2. Mitogenic Signaling and Cell Cycle Checkpoints
4.3. Telomere Dysregulation
4.4. Chromosomal Phenomena
4.5. miRNA Expression
5. Immune Microenvironment
6. Other Heritable Cancer Predisposition Syndromes
7. Treatment Implications
8. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ottaviani, G.; Jaffe, N. The epidemiology of osteosarcoma. Cancer Treat. Res. 2009, 152, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Mirabello, L.; Troisi, R.J.; Savage, S.A. Osteosarcoma incidence and survival rates from 1973 to 2004: Data from the Surveillance, Epidemiology, and End Results Program. Cancer 2009, 115, 1531–1543. [Google Scholar] [CrossRef]
- Marina, N.M.; Smeland, S.; Bielack, S.S.; Bernstein, M.; Jovic, G.; Krailo, M.D.; Hook, J.M.; Arndt, C.; van den Berg, H.; Brennan, B.; et al. Comparison of MAPIE versus MAP in patients with a poor response to preoperative chemotherapy for newly diagnosed high-grade osteosarcoma (EURAMOS-1): An open-label, international, randomised controlled trial. Lancet Oncol. 2016, 17, 1396–1408. [Google Scholar] [CrossRef]
- Janeway, K.A.; Barkauskas, D.A.; Krailo, M.D.; Meyers, P.A.; Schwartz, C.L.; Ebb, D.H.; Seibel, N.L.; Grier, H.E.; Gorlick, R.; Marina, N. Outcome for adolescent and young adult patients with osteosarcoma: A report from the Children’s Oncology Group. Cancer 2012, 118, 4597–4605. [Google Scholar] [CrossRef]
- Smeland, S.; Bielack, S.S.; Whelan, J.; Bernstein, M.; Hogendoorn, P.; Krailo, M.D.; Gorlick, R.; Janeway, K.A.; Ingleby, F.C.; Anninga, J.; et al. Survival and prognosis with osteosarcoma: Outcomes in more than 2000 patients in the EURAMOS-1 (European and American Osteosarcoma Study) cohort. Eur. J. Cancer 2019, 109, 36–50. [Google Scholar] [CrossRef] [PubMed]
- Bernthal, N.M.; Federman, N.; Eilber, F.R.; Nelson, S.D.; Eckardt, J.J.; Eilber, F.C.; Tap, W.D. Long-term results (>25 years) of a randomized, prospective clinical trial evaluating chemotherapy in patients with high-grade, operable osteosarcoma. Cancer 2012, 118, 5888–5893. [Google Scholar] [CrossRef]
- Bielack, S.S.; Kempf-Bielack, B.; Delling, G.; Exner, G.U.; Flege, S.; Helmke, K.; Kotz, R.; Salzer-Kuntschik, M.; Werner, M.; Winkelmann, W.; et al. Prognostic factors in high-grade osteosarcoma of the extremities or trunk: An analysis of 1,702 patients treated on neoadjuvant cooperative osteosarcoma study group protocols. J. Clin. Oncol. 2002, 20, 776–790. [Google Scholar] [CrossRef]
- Fuchs, N.; Bielack, S.S.; Epler, D.; Bieling, P.; Delling, G.; Körholz, D.; Graf, N.; Heise, U.; Jürgens, H.; Kotz, R.; et al. Long-term results of the co-operative German-Austrian-Swiss osteosarcoma study group’s protocol COSS-86 of intensive multidrug chemotherapy and surgery for osteosarcoma of the limbs. Ann. Oncol. 1998, 9, 893–899. [Google Scholar] [CrossRef] [PubMed]
- Wagner, M.J.; Livingston, J.A.; Patel, S.R.; Benjamin, R.S. Chemotherapy for Bone Sarcoma in Adults. J. Oncol. Pract. 2016, 12, 208–216. [Google Scholar] [CrossRef]
- Whelan, J.S.; Bielack, S.S.; Marina, N.; Smeland, S.; Jovic, G.; Hook, J.M.; Krailo, M.; Anninga, J.; Butterfass-Bahloul, T.; Bohling, T.; et al. EURAMOS-1, an international randomised study for osteosarcoma: Results from pre-randomisation treatment. Ann. Oncol. 2015, 26, 407–414. [Google Scholar] [CrossRef]
- Winkler, K.; Bielack, S.S.; Delling, G.; Jürgens, H.; Kotz, R.; Salzer-Kuntschik, M. Treatment of osteosarcoma: Experience of the Cooperative Osteosarcoma Study Group (COSS). Cancer Treat. Res. 1993, 62, 269–277. [Google Scholar] [CrossRef]
- Smeland, S.; Müller, C.; Alvegard, T.A.; Wiklund, T.; Wiebe, T.; Björk, O.; Stenwig, A.E.; Willén, H.; Holmström, T.; Follerås, G.; et al. Scandinavian Sarcoma Group Osteosarcoma Study SSG VIII: Prognostic factors for outcome and the role of replacement salvage chemotherapy for poor histological responders. Eur. J. Cancer 2003, 39, 488–494. [Google Scholar] [CrossRef]
- Damron, T.A.; Ward, W.G.; Stewart, A. Osteosarcoma, chondrosarcoma, and Ewing’s sarcoma: National Cancer Data Base Report. Clin. Orthop. Relat. Res. 2007, 459, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Aksnes, L.H.; Hall, K.S.; Folleraas, G.; Stenwig, A.E.; Bjerkehagen, B.; Taksdal, I.; Winderen, M.; Bruland, O.S.; Saeter, G. Management of high-grade bone sarcomas over two decades: The Norwegian Radium Hospital experience. Acta Oncol. 2006, 45, 38–46. [Google Scholar] [CrossRef]
- Jeon, D.G.; Lee, S.Y.; Cho, W.H.; Song, W.S.; Park, J.H. Primary osteosarcoma in patients older than 40 years of age. J. Korean Med. Sci. 2006, 21, 715–718. [Google Scholar] [CrossRef] [PubMed]
- Collins, M.; Wilhelm, M.; Conyers, R.; Herschtal, A.; Whelan, J.; Bielack, S.; Kager, L.; Kühne, T.; Sydes, M.; Gelderblom, H.; et al. Benefits and adverse events in younger versus older patients receiving neoadjuvant chemotherapy for osteosarcoma: Findings from a meta-analysis. J. Clin. Oncol. 2013, 31, 2303–2312. [Google Scholar] [CrossRef] [PubMed]
- Harting, M.T.; Lally, K.P.; Andrassy, R.J.; Vaporciyan, A.A.; Cox, C.S., Jr.; Hayes-Jordan, A.; Blakely, M.L. Age as a prognostic factor for patients with osteosarcoma: An analysis of 438 patients. J. Cancer Res. Clin. Oncol. 2010, 136, 561–570. [Google Scholar] [CrossRef] [PubMed]
- Bacci, G.; Ferrari, S.; Bertoni, F.; Ruggieri, P.; Picci, P.; Longhi, A.; Casadei, R.; Fabbri, N.; Forni, C.; Versari, M.; et al. Long-term outcome for patients with nonmetastatic osteosarcoma of the extremity treated at the istituto ortopedico rizzoli according to the istituto ortopedico rizzoli/osteosarcoma-2 protocol: An updated report. J. Clin. Oncol. 2000, 18, 4016–4027. [Google Scholar] [CrossRef]
- Bacci, G.; Longhi, A.; Bertoni, F.; Bacchini, P.; Ruggeri, P.; Versari, M.; Picci, P. Primary high-grade osteosarcoma: Comparison between preadolescent and older patients. J. Pediatr. Hematol. Oncol. 2005, 27, 129–134. [Google Scholar] [CrossRef]
- Ferrari, S.; Bielack, S.S.; Smeland, S.; Longhi, A.; Egerer, G.; Sundby Hall, K.; Donati, D.; Kevric, M.; Brosjo, O.; Comandone, A.; et al. EURO-B.O.S.S.: A European study on chemotherapy in bone-sarcoma patients aged over 40: Outcome in primary high-grade osteosarcoma. Tumori J. 2018, 104, 30–36. [Google Scholar] [CrossRef]
- Carsi, B.; Rock, M.G. Primary osteosarcoma in adults older than 40 years. Clin. Orthop. Relat. Res. 2002, 397, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Huvos, A.G. Osteogenic sarcoma of bones and soft tissues in older persons. A clinicopathologic analysis of 117 patients older than 60 years. Cancer 1986, 57, 1442–1449. [Google Scholar] [CrossRef]
- Miller, R.P.; Tadagavadi, R.K.; Ramesh, G.; Reeves, W.B. Mechanisms of Cisplatin nephrotoxicity. Toxins 2010, 2, 2490–2518. [Google Scholar] [CrossRef] [PubMed]
- De Jongh, F.E.; van Veen, R.N.; Veltman, S.J.; de Wit, R.; van der Burg, M.E.; van den Bent, M.J.; Planting, A.S.; Graveland, W.J.; Stoter, G.; Verweij, J. Weekly high-dose cisplatin is a feasible treatment option: Analysis on prognostic factors for toxicity in 400 patients. Br. J. Cancer 2003, 88, 1199–1206. [Google Scholar] [CrossRef] [PubMed]
- Widemann, B.C.; Balis, F.M.; Kempf-Bielack, B.; Bielack, S.; Pratt, C.B.; Ferrari, S.; Bacci, G.; Craft, A.W.; Adamson, P.C. High-dose methotrexate-induced nephrotoxicity in patients with osteosarcoma. Cancer 2004, 100, 2222–2232. [Google Scholar] [CrossRef]
- Tett, S.E.; Triggs, E.J. Use of methotrexate in older patients. A risk-benefit assessment. Drugs Aging 1996, 9, 458–471. [Google Scholar] [CrossRef]
- Chatterjee, K.; Zhang, J.; Honbo, N.; Karliner, J.S. Doxorubicin cardiomyopathy. Cardiology 2010, 115, 155–162. [Google Scholar] [CrossRef]
- Swain, S.M.; Whaley, F.S.; Ewer, M.S. Congestive heart failure in patients treated with doxorubicin: A retrospective analysis of three trials. Cancer 2003, 97, 2869–2879. [Google Scholar] [CrossRef] [PubMed]
- Balducci, L. Myelosuppression and its consequences in elderly patients with cancer. Oncology 2003, 17, 27–32. [Google Scholar]
- Dhotre, K.; Adams, S.A.; Hebert, J.R.; Bottai, M.; Heiney, S.P. Oncology Nurses’ Experiences with Patients Who Choose to Discontinue Cancer Chemotherapy. Oncol. Nurs. Forum 2016, 43, 617–623. [Google Scholar] [CrossRef]
- Longhi, A.; Errani, C.; Gonzales-Arabio, D.; Ferrari, C.; Mercuri, M. Osteosarcoma in patients older than 65 years. J. Clin. Oncol. 2008, 26, 5368–5373. [Google Scholar] [CrossRef] [PubMed]
- Ottesen, T.D.; Shultz, B.N.; Munger, A.M.; Sibindi, C.; Yurter, A.; Varthi, A.G.; Grauer, J.N. Characteristics, Management, and Outcomes of Patients with Osteosarcoma: An Analysis of Outcomes from the National Cancer Database. J. Am. Acad. Orthop. Surg. Glob. Res. Rev. 2022, 6, e22-00009. [Google Scholar] [CrossRef] [PubMed]
- Cotterill, S.J.; Wright, C.M.; Pearce, M.S.; Craft, A.W. Stature of young people with malignant bone tumors. Pediatr. Blood Cancer 2004, 42, 59–63. [Google Scholar] [CrossRef]
- Hainaut, P.; Pfeifer, G.P. Somatic TP53 Mutations in the Era of Genome Sequencing. Cold Spring Harb. Perspect. Med. 2016, 6, a026179. [Google Scholar] [CrossRef] [PubMed]
- Sabharwal, R.; Gupta, S.; Sepolia, S.; Panigrahi, R.; Mohanty, S.; Subudhi, S.K.; Kumar, M. An Insight in to Paget’s Disease of Bone. Niger. J. Surg. 2014, 20, 9–15. [Google Scholar] [CrossRef]
- Paul Tuck, S.; Layfield, R.; Walker, J.; Mekkayil, B.; Francis, R. Adult Paget’s disease of bone: A review. Rheumatology 2017, 56, 2050–2059. [Google Scholar] [CrossRef]
- Hansen, M.F.; Nellissery, M.J.; Bhatia, P. Common mechanisms of osteosarcoma and Paget’s disease. J. Bone Miner. Res. 1999, 14 (Suppl. 2), 39–44. [Google Scholar] [CrossRef]
- Deyrup, A.T.; Montag, A.G.; Inwards, C.Y.; Xu, Z.; Swee, R.G.; Krishnan Unni, K. Sarcomas arising in Paget disease of bone: A clinicopathologic analysis of 70 cases. Arch. Pathol. Lab. Med. 2007, 131, 942–946. [Google Scholar] [CrossRef]
- Lyles, K.W.; Siris, E.S.; Singer, F.R.; Meunier, P.J. A clinical approach to diagnosis and management of Paget’s disease of bone. J. Bone Miner. Res. 2001, 16, 1379–1387. [Google Scholar] [CrossRef]
- Tucker, M.A.; D’Angio, G.J.; Boice, J.D., Jr.; Strong, L.C.; Li, F.P.; Stovall, M.; Stone, B.J.; Green, D.M.; Lombardi, F.; Newton, W.; et al. Bone sarcomas linked to radiotherapy and chemotherapy in children. N. Engl. J. Med. 1987, 317, 588–593. [Google Scholar] [CrossRef]
- Kristenson, S.; Mann, R.; Leafblad, K.; Cook, B.; Chang, J. Radiation-induced osteosarcoma following treatment of Ewing’s sarcoma. Radiol. Case Rep. 2020, 15, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Kalra, S.; Grimer, R.J.; Spooner, D.; Carter, S.R.; Tillman, R.M.; Abudu, A. Radiation-induced sarcomas of bone: Factors that affect outcome. J. Bone Joint Surg. Br. 2007, 89, 808–813. [Google Scholar] [CrossRef]
- Giannini, L.; Incandela, F.; Fiore, M.; Gronchi, A.; Stacchiotti, S.; Sangalli, C.; Piazza, C. Radiation-Induced Sarcoma of the Head and Neck: A Review of the Literature. Front. Oncol. 2018, 8, 449. [Google Scholar] [CrossRef]
- Hamre, M.R.; Severson, R.K.; Chuba, P.; Lucas, D.R.; Thomas, R.L.; Mott, M.P. Osteosarcoma as a second malignant neoplasm. Radiother. Oncol. 2002, 65, 153–157. [Google Scholar] [CrossRef] [PubMed]
- Bjerkehagen, B.; Smastuen, M.C.; Hall, K.S.; Skjeldal, S.; Smeland, S.; Fossa, S.D. Why do patients with radiation-induced sarcomas have a poor sarcoma-related survival? Br. J. Cancer 2012, 106, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Spałek, M.J.; Poleszczuk, J.; Czarnecka, A.M.; Dudzisz-Śledź, M.; Napieralska, A.; Matysiakiewicz, J.; Chojnacka, M.; Raciborska, A.; Sztuder, A.; Maciejczyk, A.; et al. Radiotherapy in the Management of Pediatric and Adult Osteosarcomas: A Multi-Institutional Cohort Analysis. Cells 2021, 10, 366. [Google Scholar] [CrossRef]
- Gonin-Laurent, N.; Gibaud, A.; Huygue, M.; Lefevre, S.H.; Le Bras, M.; Chauveinc, L.; Sastre-Garau, X.; Doz, F.; Lumbroso, L.; Chevillard, S.; et al. Specific TP53 mutation pattern in radiation-induced sarcomas. Carcinogenesis 2006, 27, 1266–1272. [Google Scholar] [CrossRef]
- Poos, K.; Smida, J.; Maugg, D.; Eckstein, G.; Baumhoer, D.; Nathrath, M.; Korsching, E. Genomic heterogeneity of osteosarcoma—Shift from single candidates to functional modules. PLoS ONE 2015, 10, e0123082. [Google Scholar] [CrossRef]
- Seeber, A.; Elliott, A.; Modiano, J.; Untergasser, G.; von Mehren, M.; Rosenberg, A.; Khushman, M.d.; Dizon, D.S.; Riedel, R.F.; Trent, J.C.; et al. Age as a factor in the molecular landscape and the tumor-microenvironmental signature of osteosarcoma. J. Clin. Oncol. 2022, 40, 11525. [Google Scholar] [CrossRef]
- Martin, J.W.; Squire, J.A.; Zielenska, M. The genetics of osteosarcoma. Sarcoma 2012, 2012, 627254. [Google Scholar] [CrossRef]
- Olivos, D.J., III; Mayo, L.D. Emerging Non-Canonical Functions and Regulation by p53: p53 and Stemness. Int. J. Mol. Sci. 2016, 17, 1982. [Google Scholar] [CrossRef]
- Wu, C.C.; Beird, H.C.; Andrew Livingston, J.; Advani, S.; Mitra, A.; Cao, S.; Reuben, A.; Ingram, D.; Wang, W.L.; Ju, Z.; et al. Immuno-genomic landscape of osteosarcoma. Nat. Commun. 2020, 11, 1008. [Google Scholar] [CrossRef]
- Mirabello, L.; Yeager, M.; Mai, P.L.; Gastier-Foster, J.M.; Gorlick, R.; Khanna, C.; Patino-Garcia, A.; Sierrasesumaga, L.; Lecanda, F.; Andrulis, I.L.; et al. Germline TP53 variants and susceptibility to osteosarcoma. J. Natl. Cancer Inst. 2015, 107, djv101. [Google Scholar] [CrossRef]
- Chen, X.; Bahrami, A.; Pappo, A.; Easton, J.; Dalton, J.; Hedlund, E.; Ellison, D.; Shurtleff, S.; Wu, G.; Wei, L.; et al. Recurrent somatic structural variations contribute to tumorigenesis in pediatric osteosarcoma. Cell Rep. 2014, 7, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, S.; Barøy, T.; Sun, J.; Nome, T.; Vodák, D.; Bryne, J.C.; Håkelien, A.M.; Fernandez-Cuesta, L.; Möhlendick, B.; Rieder, H.; et al. Unscrambling the genomic chaos of osteosarcoma reveals extensive transcript fusion, recurrent rearrangements and frequent novel TP53 aberrations. Oncotarget 2016, 7, 5273–5288. [Google Scholar] [CrossRef] [PubMed]
- Perry, J.A.; Kiezun, A.; Tonzi, P.; Van Allen, E.M.; Carter, S.L.; Baca, S.C.; Cowley, G.S.; Bhatt, A.S.; Rheinbay, E.; Pedamallu, C.S.; et al. Complementary genomic approaches highlight the PI3K/mTOR pathway as a common vulnerability in osteosarcoma. Proc. Natl. Acad. Sci. USA 2014, 111, E5564–E5573. [Google Scholar] [CrossRef] [PubMed]
- Patino-Garcia, A.; Pineiro, E.S.; Diez, M.Z.; Iturriagagoitia, L.G.; Klussmann, F.A.; Ariznabarreta, L.S. Genetic and epigenetic alterations of the cell cycle regulators and tumor suppressor genes in pediatric osteosarcomas. J. Pediatr. Hematol. Oncol. 2003, 25, 362–367. [Google Scholar] [CrossRef]
- Sdek, P.; Ying, H.; Chang, D.L.; Qiu, W.; Zheng, H.; Touitou, R.; Allday, M.J.; Xiao, Z.X. MDM2 promotes proteasome-dependent ubiquitin-independent degradation of retinoblastoma protein. Mol. Cell 2005, 20, 699–708. [Google Scholar] [CrossRef]
- Momand, J.; Jung, D.; Wilczynski, S.; Niland, J. The MDM2 gene amplification database. Nucleic Acids Res. 1998, 26, 3453–3459. [Google Scholar] [CrossRef]
- Oh, J.H.; Kim, H.S.; Kim, H.H.; Kim, W.H.; Lee, S.H. Aberrant methylation of p14ARF gene correlates with poor survival in osteosarcoma. Clin. Orthop. Relat. Res. 2006, 442, 216–222. [Google Scholar] [CrossRef]
- Yan, T.; Wunder, J.S.; Gokgoz, N.; Gill, M.; Eskandarian, S.; Parkes, R.K.; Bull, S.B.; Bell, R.S.; Andrulis, I.L. COPS3 amplification and clinical outcome in osteosarcoma. Cancer 2007, 109, 1870–1876. [Google Scholar] [CrossRef] [PubMed]
- Czarnecka, A.M.; Synoradzki, K.; Firlej, W.; Bartnik, E.; Sobczuk, P.; Fiedorowicz, M.; Grieb, P.; Rutkowski, P. Molecular Biology of Osteosarcoma. Cancers 2020, 12, 2130. [Google Scholar] [CrossRef]
- Manning, A.L.; Dyson, N.J. pRB, a tumor suppressor with a stabilizing presence. Trends Cell Biol. 2011, 21, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Berman, S.D.; Calo, E.; Landman, A.S.; Danielian, P.S.; Miller, E.S.; West, J.C.; Fonhoue, B.D.; Caron, A.; Bronson, R.; Bouxsein, M.L.; et al. Metastatic osteosarcoma induced by inactivation of Rb and p53 in the osteoblast lineage. Proc. Natl. Acad. Sci. USA 2008, 105, 11851–11856. [Google Scholar] [CrossRef]
- Wong, F.L.; Boice, J.D., Jr.; Abramson, D.H.; Tarone, R.E.; Kleinerman, R.A.; Stovall, M.; Goldman, M.B.; Seddon, J.M.; Tarbell, N.; Fraumeni, J.F., Jr.; et al. Cancer Incidence After Retinoblastoma: Radiation Dose and Sarcoma Risk. JAMA 1997, 278, 1262–1267. [Google Scholar] [CrossRef] [PubMed]
- Toguchida, J.; Ishizaki, K.; Sasaki, M.S.; Ikenaga, M.; Sugimoto, M.; Kotoura, Y.; Yamamuro, T. Chromosomal Reorganization for the Expression of Recessive Mutation of Retinoblastoma Susceptibility Gene in the Development of Osteosarcoma. Cancer Res. 1988, 48, 3939–3943. [Google Scholar] [PubMed]
- Smida, J.; Baumhoer, D.; Rosemann, M.; Walch, A.; Bielack, S.; Poremba, C.; Remberger, K.; Korsching, E.; Scheurlen, W.; Dierkes, C.; et al. Genomic alterations and allelic imbalances are strong prognostic predictors in osteosarcoma. Clin. Cancer Res. 2010, 16, 4256–4267. [Google Scholar] [CrossRef]
- López-Guerrero, J.A.; López-Ginés, C.; Pellín, A.; Carda, C.; Llombart-Bosch, A. Deregulation of the G1 to S-phase cell cycle checkpoint is involved in the pathogenesis of human osteosarcoma. Diagn. Mol. Pathol. 2004, 13, 81–91. [Google Scholar] [CrossRef]
- Fujiwara, T.; Fujiwara, M.; Numoto, K.; Ogura, K.; Yoshida, A.; Yonemoto, T.; Suzuki, S.; Kawai, A. Second primary osteosarcomas in patients with retinoblastoma. Jpn. J. Clin. Oncol. 2015, 45, 1139–1145. [Google Scholar] [CrossRef]
- Woo, K.I.; Harbour, J.W. Review of 676 second primary tumors in patients with retinoblastoma: Association between age at onset and tumor type. Arch. Ophthalmol. 2010, 128, 865–870. [Google Scholar] [CrossRef]
- Jiang, Y.; Wang, J.; Sun, M.; Zuo, D.; Wang, H.; Shen, J.; Jiang, W.; Mu, H.; Ma, X.; Yin, F.; et al. Multi-omics analysis identifies osteosarcoma subtypes with distinct prognosis indicating stratified treatment. Nat. Commun. 2022, 13, 7207. [Google Scholar] [CrossRef]
- Chen, D.; Zhao, Z.; Huang, Z.; Chen, D.C.; Zhu, X.X.; Wang, Y.Z.; Yan, Y.W.; Tang, S.; Madhavan, S.; Ni, W.; et al. Super enhancer inhibitors suppress MYC driven transcriptional amplification and tumor progression in osteosarcoma. Bone Res. 2018, 6, 11. [Google Scholar] [CrossRef]
- Felsher, D.W. MYC Inactivation Elicits Oncogene Addiction through Both Tumor Cell-Intrinsic and Host-Dependent Mechanisms. Genes. Cancer 2010, 1, 597–604. [Google Scholar] [CrossRef]
- Wu, X.; Cai, Z.D.; Lou, L.M.; Zhu, Y.B. Expressions of p53, c-MYC, BCL-2 and apoptotic index in human osteosarcoma and their correlations with prognosis of patients. Cancer Epidemiol. 2012, 36, 212–216. [Google Scholar] [CrossRef]
- Ladanyi, M.; Park, C.K.; Lewis, R.; Jhanwar, S.C.; Healey, J.H.; Huvos, A.G. Sporadic amplification of the MYC gene in human osteosarcomas. Diagn. Mol. Pathol. 1993, 2, 163–167. [Google Scholar] [CrossRef]
- De Noon, S.; Ijaz, J.; Coorens, T.H.; Amary, F.; Ye, H.; Strobl, A.; Lyskjaer, I.; Flanagan, A.M.; Behjati, S. MYC amplifications are common events in childhood osteosarcoma. J. Pathology. Clin. Res. 2021, 7, 425–431. [Google Scholar] [CrossRef]
- Suehara, Y.; Alex, D.; Bowman, A.; Middha, S.; Zehir, A.; Chakravarty, D.; Wang, L.; Jour, G.; Nafa, K.; Hayashi, T.; et al. Clinical Genomic Sequencing of Pediatric and Adult Osteosarcoma Reveals Distinct Molecular Subsets with Potentially Targetable Alterations. Clin. Cancer Res. 2019, 25, 6346–6356. [Google Scholar] [CrossRef] [PubMed]
- Ballinger, M.L.; Pattnaik, S.; Mundra, P.A.; Zaheed, M.; Rath, E.; Priestley, P.; Baber, J.; Ray-Coquard, I.; Isambert, N.; Causeret, S.; et al. Heritable defects in telomere and mitotic function selectively predispose to sarcomas. Science 2023, 379, 253–260. [Google Scholar] [CrossRef]
- Scheel, C.; Schaefer, K.L.; Jauch, A.; Keller, M.; Wai, D.; Brinkschmidt, C.; van Valen, F.; Boecker, W.; Dockhorn-Dworniczak, B.; Poremba, C. Alternative lengthening of telomeres is associated with chromosomal instability in osteosarcomas. Oncogene 2001, 20, 3835–3844. [Google Scholar] [CrossRef] [PubMed]
- Bryan, T.M.; Englezou, A.; Dalla-Pozza, L.; Dunham, M.A.; Reddel, R.R. Evidence for an alternative mechanism for maintaining telomere length in human tumors and tumor-derived cell lines. Nat. Med. 1997, 3, 1271–1274. [Google Scholar] [CrossRef] [PubMed]
- Heaphy, C.M.; Subhawong, A.P.; Hong, S.M.; Goggins, M.G.; Montgomery, E.A.; Gabrielson, E.; Netto, G.J.; Epstein, J.I.; Lotan, T.L.; Westra, W.H.; et al. Prevalence of the alternative lengthening of telomeres telomere maintenance mechanism in human cancer subtypes. Am. J. Pathol. 2011, 179, 1608–1615. [Google Scholar] [CrossRef]
- Diotti, R.; Loayza, D. Shelterin complex and associated factors at human telomeres. Nucleus 2011, 2, 119–135. [Google Scholar] [CrossRef]
- Mirabello, L.; Richards, E.G.; Duong, L.M.; Yu, K.; Wang, Z.; Cawthon, R.; Berndt, S.I.; Burdett, L.; Chowdhury, S.; Teshome, K.; et al. Telomere length and variation in telomere biology genes in individuals with osteosarcoma. Int. J. Mol. Epidemiol. Genet. 2011, 2, 19–29. [Google Scholar] [PubMed]
- Sanders, R.P.; Drissi, R.; Billups, C.A.; Daw, N.C.; Valentine, M.B.; Dome, J.S. Telomerase expression predicts unfavorable outcome in osteosarcoma. J. Clin. Oncol. 2004, 22, 3790–3797. [Google Scholar] [CrossRef] [PubMed]
- Walsh, K.M.; Whitehead, T.P.; de Smith, A.J.; Smirnov, I.V.; Park, M.; Endicott, A.A.; Francis, S.S.; Codd, V.; Samani, N.J.; Metayer, C.; et al. Common genetic variants associated with telomere length confer risk for neuroblastoma and other childhood cancers. Carcinogenesis 2016, 37, 576–582. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Hansen, H.M.; Semmes, E.C.; Gonzalez-Maya, J.; Morimoto, L.; Wei, Q.; Eward, W.C.; DeWitt, S.B.; Hurst, J.H.; Metayer, C.; et al. Common genetic variation and risk of osteosarcoma in a multi-ethnic pediatric and adolescent population. Bone 2020, 130, 115070. [Google Scholar] [CrossRef]
- Forment, J.V.; Kaidi, A.; Jackson, S.P. Chromothripsis and cancer: Causes and consequences of chromosome shattering. Nat. Rev. Cancer 2012, 12, 663–670. [Google Scholar] [CrossRef] [PubMed]
- Cortes-Ciriano, I.; Lee, J.J.; Xi, R.; Jain, D.; Jung, Y.L.; Yang, L.; Gordenin, D.; Klimczak, L.J.; Zhang, C.Z.; Pellman, D.S.; et al. Comprehensive analysis of chromothripsis in 2,658 human cancers using whole-genome sequencing. Nat. Genet. 2020, 52, 331–341. [Google Scholar] [CrossRef]
- Behjati, S.; Tarpey, P.S.; Haase, K.; Ye, H.; Young, M.D.; Alexandrov, L.B.; Farndon, S.J.; Collord, G.; Wedge, D.C.; Martincorena, I.; et al. Recurrent mutation of IGF signalling genes and distinct patterns of genomic rearrangement in osteosarcoma. Nat. Commun. 2017, 8, 15936. [Google Scholar] [CrossRef]
- Nik-Zainal, S.; Alexandrov, L.B.; Wedge, D.C.; Van Loo, P.; Greenman, C.D.; Raine, K.; Jones, D.; Hinton, J.; Marshall, J.; Stebbings, L.A.; et al. Mutational processes molding the genomes of 21 breast cancers. Cell 2012, 149, 979–993. [Google Scholar] [CrossRef]
- D’Antonio, M.; Tamayo, P.; Mesirov, J.P.; Frazer, K.A. Kataegis Expression Signature in Breast Cancer Is Associated with Late Onset, Better Prognosis, and Higher HER2 Levels. Cell Rep. 2016, 16, 672–683. [Google Scholar] [CrossRef]
- Sasaki, R.; Osaki, M.; Okada, F. MicroRNA-Based Diagnosis and Treatment of Metastatic Human Osteosarcoma. Cancers 2019, 11, 553. [Google Scholar] [CrossRef]
- Kobayashi, E.; Hornicek, F.J.; Duan, Z. MicroRNA Involvement in Osteosarcoma. Sarcoma 2012, 2012, 359739. [Google Scholar] [CrossRef]
- Maire, G.; Martin, J.W.; Yoshimoto, M.; Chilton-MacNeill, S.; Zielenska, M.; Squire, J.A. Analysis of miRNA-gene expression-genomic profiles reveals complex mechanisms of microRNA deregulation in osteosarcoma. Cancer Genet. 2011, 204, 138–146. [Google Scholar] [CrossRef]
- Wang, J.S.; Duan, M.Y.; Zhong, Y.S.; Li, X.D.; Du, S.X.; Xie, P.; Zheng, G.Z.; Han, J.M. Investigating age-induced differentially expressed genes and potential molecular mechanisms in osteosarcoma based on integrated bioinformatics analysis. Mol. Med. Rep. 2019, 19, 2729–2739. [Google Scholar] [CrossRef]
- Rankin, E.B.; Giaccia, A.J. The role of hypoxia-inducible factors in tumorigenesis. Cell Death Differ. 2008, 15, 678–685. [Google Scholar] [CrossRef] [PubMed]
- Tawbi, H.A.; Burgess, M.; Bolejack, V.; Van Tine, B.A.; Schuetze, S.M.; Hu, J.; D’Angelo, S.; Attia, S.; Riedel, R.F.; Priebat, D.A.; et al. Pembrolizumab in advanced soft-tissue sarcoma and bone sarcoma (SARC028): A multicentre, two-cohort, single-arm, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 1493–1501. [Google Scholar] [CrossRef] [PubMed]
- Petitprez, F.; de Reyniès, A.; Keung, E.Z.; Chen, T.W.; Sun, C.M.; Calderaro, J.; Jeng, Y.M.; Hsiao, L.P.; Lacroix, L.; Bougoüin, A.; et al. B cells are associated with survival and immunotherapy response in sarcoma. Nature 2020, 577, 556–560. [Google Scholar] [CrossRef]
- Wang, L.L.; Gannavarapu, A.; Kozinetz, C.A.; Levy, M.L.; Lewis, R.A.; Chintagumpala, M.M.; Ruiz-Maldanado, R.; Contreras-Ruiz, J.; Cunniff, C.; Erickson, R.P.; et al. Association between osteosarcoma and deleterious mutations in the RECQL4 gene in Rothmund-Thomson syndrome. J. Natl. Cancer Inst. 2003, 95, 669–674. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.L.; Levy, M.L.; Lewis, R.A.; Chintagumpala, M.M.; Lev, D.; Rogers, M.; Plon, S.E. Clinical manifestations in a cohort of 41 Rothmund-Thomson syndrome patients. Am. J. Med. Genet. 2001, 102, 11–17. [Google Scholar] [CrossRef]
- Siitonen, H.A.; Sotkasiira, J.; Biervliet, M.; Benmansour, A.; Capri, Y.; Cormier-Daire, V.; Crandall, B.; Hannula-Jouppi, K.; Hennekam, R.; Herzog, D.; et al. The mutation spectrum in RECQL4 diseases. Eur. J. Hum. Genet. 2009, 17, 151–158. [Google Scholar] [CrossRef]
- Larizza, L.; Roversi, G.; Volpi, L. Rothmund-Thomson syndrome. Orphanet J. Rare Dis. 2010, 5, 2. [Google Scholar] [CrossRef]
- Siitonen, H.A.; Kopra, O.; Kaariainen, H.; Haravuori, H.; Winter, R.M.; Saamanen, A.M.; Peltonen, L.; Kestila, M. Molecular defect of RAPADILINO syndrome expands the phenotype spectrum of RECQL diseases. Hum. Mol. Genet. 2003, 12, 2837–2844. [Google Scholar] [CrossRef] [PubMed]
- Hameed, M.; Mandelker, D. Tumor Syndromes Predisposing to Osteosarcoma. Adv. Anat. Pathol. 2018, 25, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Cunniff, C.; Bassetti, J.A.; Ellis, N.A. Bloom’s Syndrome: Clinical Spectrum, Molecular Pathogenesis, and Cancer Predisposition. Mol. Syndromol. 2017, 8, 4–23. [Google Scholar] [CrossRef]
- Goto, M. Hierarchical deterioration of body systems in Werner’s syndrome: Implications for normal ageing. Mech. Ageing Dev. 1997, 98, 239–254. [Google Scholar] [CrossRef]
- De Renty, C.; Ellis, N.A. Bloom’s syndrome: Why not premature aging?: A comparison of the BLM and WRN helicases. Ageing Res. Rev. 2017, 33, 36–51. [Google Scholar] [CrossRef]
- Ishikawa, Y.; Miller, R.W.; Machinami, R.; Sugano, H.; Goto, M. Atypical osteosarcomas in Werner Syndrome (adult progeria). Jpn. J. Cancer Res. 2000, 91, 1345–1349. [Google Scholar] [CrossRef]
- Lipton, J.M.; Federman, N.; Khabbaze, Y.; Schwartz, C.L.; Hilliard, L.M.; Clark, J.I.; Vlachos, A.; Diamond-Black Anemia, R. Osteogenic sarcoma associated with Diamond-Blackfan anemia: A report from the Diamond-Blackfan Anemia Registry. J. Pediatr. Hematol. Oncol. 2001, 23, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Masliah-Planchon, J.; Levy, D.; Heron, D.; Giuliano, F.; Badens, C.; Freneaux, P.; Galmiche, L.; Guinebretierre, J.M.; Cellier, C.; Waterfall, J.J.; et al. Does ATRX germline variation predispose to osteosarcoma? Three additional cases of osteosarcoma in two ATR-X syndrome patients. Eur. J. Hum. Genet. 2018, 26, 1217–1221. [Google Scholar] [CrossRef]
- Cheung, N.K.; Zhang, J.; Lu, C.; Parker, M.; Bahrami, A.; Tickoo, S.K.; Heguy, A.; Pappo, A.S.; Federico, S.; Dalton, J.; et al. Association of age at diagnosis and genetic mutations in patients with neuroblastoma. JAMA 2012, 307, 1062–1071. [Google Scholar] [CrossRef]
- Skalniak, L.; Twarda-Clapa, A.; Neochoritis, C.G.; Surmiak, E.; Machula, M.; Wisniewska, A.; Labuzek, B.; Ali, A.M.; Krzanik, S.; Dubin, G.; et al. A fluorinated indole-based MDM2 antagonist selectively inhibits the growth of p53(wt) osteosarcoma cells. FEBS J. 2019, 286, 1360–1374. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Bao, H. Abemaciclib is synergistic with doxorubicin in osteosarcoma pre-clinical models via inhibition of CDK4/6–Cyclin D–Rb pathway. Cancer Chemother. Pharmacol. 2022, 89, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Avutu, V.; Slotkin, E.K.; Livingston, J.A.A.; Chawla, S.P.; Pressey, J.G.; Nandkumar, P.; Zheng, C.; Misir, S.; Pultar, P.; Voliotis, D.; et al. A phase 1/2 dose-escalation and dose-expansion study of ZN-c3 in combination with gemcitabine in adult and pediatric subjects with relapsed or refractory osteosarcoma. J. Clin. Oncol. 2022, 40, TPS11584. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, C.; Davis, L.E.; Albert, C.M.; Samuels, B.; Roberts, J.L.; Wagner, M.J. Osteosarcoma in Pediatric and Adult Populations: Are Adults Just Big Kids? Cancers 2023, 15, 5044. https://doi.org/10.3390/cancers15205044
Kim C, Davis LE, Albert CM, Samuels B, Roberts JL, Wagner MJ. Osteosarcoma in Pediatric and Adult Populations: Are Adults Just Big Kids? Cancers. 2023; 15(20):5044. https://doi.org/10.3390/cancers15205044
Chicago/Turabian StyleKim, Caleb, Lara E. Davis, Catherine M. Albert, Brian Samuels, Jesse L. Roberts, and Michael J. Wagner. 2023. "Osteosarcoma in Pediatric and Adult Populations: Are Adults Just Big Kids?" Cancers 15, no. 20: 5044. https://doi.org/10.3390/cancers15205044