
Inactivation of Tumor Suppressor CYLD Inhibits Fibroblast Reprogramming to Pluripotency
 , , , , and
, , , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Generation of Lentiviral Particles for Somatic Cell Reprogramming
2.2. Isolation of Mouse Embryonic Fibroblasts (MEFs)
2.3. Genotyping
2.4. Production of Pinducer20 C601S and Pinducer21 FCYLD
2.5. Western Blot
2.6. Cell Culture
2.7. Feeder Cell Preparation
2.8. Somatic Cell Reprogramming
2.9. ALP Staining
2.10. Colony Harvesting
2.11. RNA Extraction, cDNA Synthesis and Quantitative Real-Time PCR (qPCR)
2.12. Immunofluorescence
2.13. In Vitro Spontaneous Differentiation Assay
2.14. Proteomics
2.15. Statistical Analysis
3. Results
3.1. CYLD DUB Deficiency Affects Reprogramming Efficiency
3.2. Δ9 CYLD Does Not Affect iPSC Pluripotency
3.3. CYLD Is Dispensable for In Vitro Spontaneous Differentiation
3.4. CYLD Regulates Early Reprogramming
3.5. CYLD DUB Deficiency Delays Early Reprogramming Progression
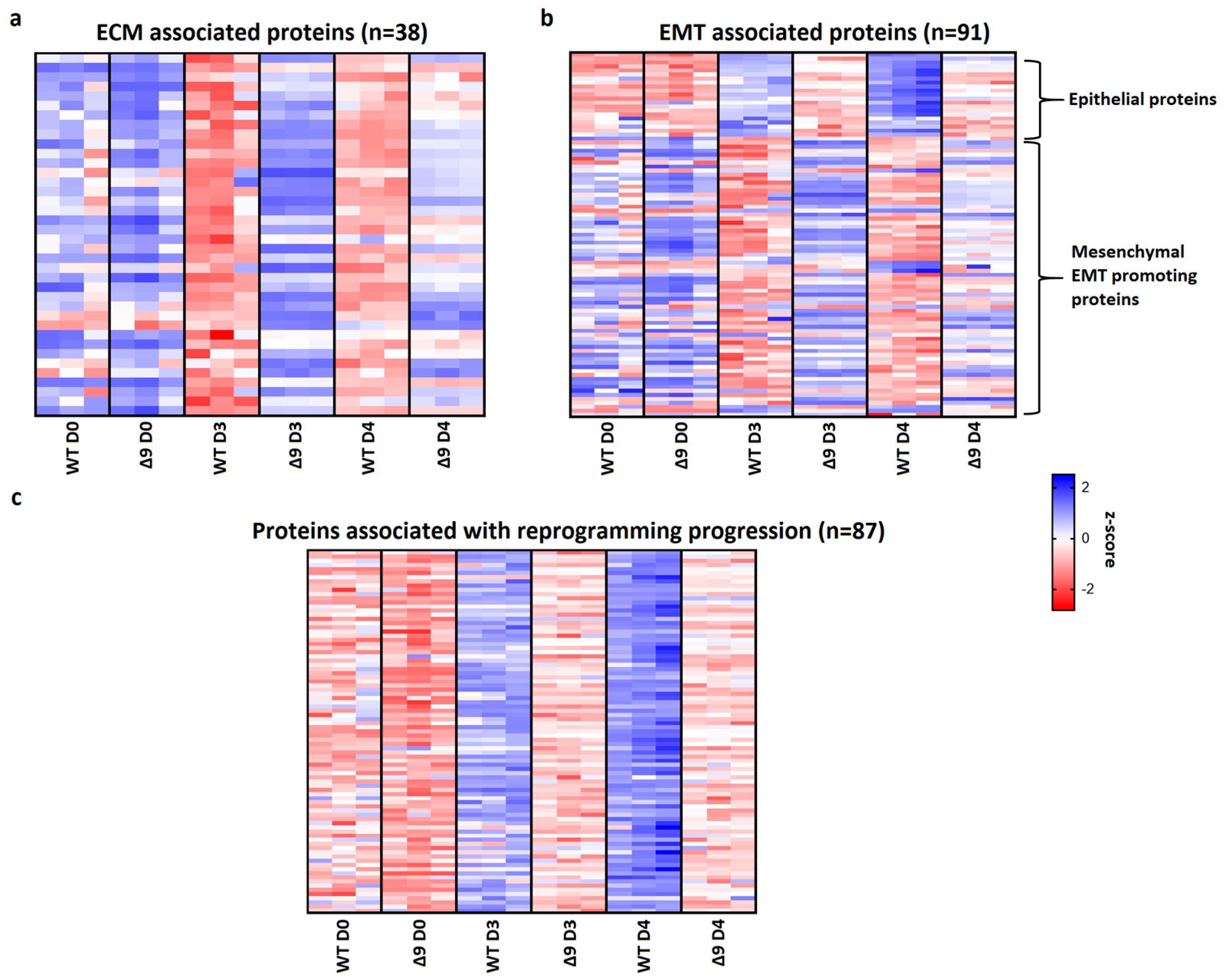
3.6. CYLD Regulates MET and ECM Organization
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef]
- Guan, J.; Wang, G.; Wang, J.; Zhang, Z.; Fu, Y.; Cheng, L.; Meng, G.; Lyu, Y.; Zhu, J.; Li, Y.; et al. Chemical Reprogramming of Human Somatic Cells to Pluripotent Stem Cells. Nature 2022, 605, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Fu, Y.; Zhu, J.; Liu, Y.; Zhang, Q.; Yi, Z.; Chen, S.; Jiao, Z.; Xu, X.; Xu, J.; et al. Single-Cell RNA-Seq Reveals Dynamic Early Embryonic-like Programs during Chemical Reprogramming. Cell Stem Cell 2018, 23, 31–45. [Google Scholar] [CrossRef] [PubMed]
- Hou, P.; Li, Y.; Zhang, X.; Liu, C.; Guan, J.; Li, H.; Zhao, T.; Ye, J.; Yang, W.; Liu, K.; et al. Pluripotent Stem Cells Induced from Mouse Somatic Cells by Small-Molecule Compounds. Science 2013, 341, 651–654. [Google Scholar] [CrossRef]
- Wang, A.Y.L.; Loh, C.Y.Y. Episomal Induced Pluripotent Stem Cells: Functional and Potential Therapeutic Applications. Cell Transplant. 2019, 28, 112S–131S. [Google Scholar] [CrossRef]
- Wang, A.Y.L. Application of Modified mRNA in Somatic Reprogramming to Pluripotency and Directed Conversion of Cell Fate. Int. J. Mol. Sci. 2021, 22, 8148. [Google Scholar] [CrossRef]
- Kim, D.; Kim, C.-H.; Moon, J.-I.; Chung, Y.-G.; Chang, M.-Y.; Han, B.-S.; Ko, S.; Yang, E.; Cha, K.Y.; Lanza, R.; et al. Generation of Human Induced Pluripotent Stem Cells by Direct Delivery of Reprogramming Proteins. Cell Stem Cell 2009, 4, 472–476. [Google Scholar] [CrossRef]
- Woltjen, K.; Michael, I.P.; Mohseni, P.; Desai, R.; Mileikovsky, M.; Hämäläinen, R.; Cowling, R.; Wang, W.; Liu, P.; Gertsenstein, M.; et al. piggyBac Transposition Reprograms Fibroblasts to Induced Pluripotent Stem Cells. Nature 2009, 458, 766–770. [Google Scholar] [CrossRef]
- Aboul-Soud, M.A.M.; Alzahrani, A.J.; Mahmoud, A. Induced Pluripotent Stem Cells (iPSCs)—Roles in Regenerative Therapies, Disease Modelling and Drug Screening. Cells 2021, 10, 2319. [Google Scholar] [CrossRef] [PubMed]
- Kouroupi, G.; Taoufik, E.; Vlachos, I.S.; Tsioras, K.; Antoniou, N.; Papastefanaki, F.; Chroni-Tzartou, D.; Wrasidlo, W.; Bohl, D.; Stellas, D.; et al. Defective Synaptic Connectivity and Axonal Neuropathology in a Human iPSC-Based Model of Familial Parkinson’s Disease. Proc. Natl. Acad. Sci. USA 2017, 114, E3679–E3688. [Google Scholar] [CrossRef]
- Rashid, S.T.; Corbineau, S.; Hannan, N.; Marciniak, S.J.; Miranda, E.; Alexander, G.; Huang-Doran, I.; Griffin, J.; Ahrlund-Richter, L.; Skepper, J.; et al. Modeling Inherited Metabolic Disorders of the Liver Using Human Induced Pluripotent Stem Cells. J. Clin. Investig. 2010, 120, 3127–3136. [Google Scholar] [CrossRef] [PubMed]
- Israel, M.A.; Yuan, S.H.; Bardy, C.; Reyna, S.M.; Mu, Y.; Herrera, C.; Hefferan, M.P.; Van Gorp, S.; Nazor, K.L.; Boscolo, F.S.; et al. Probing Sporadic and Familial Alzheimer’s Disease Using Induced Pluripotent Stem Cells. Nature 2012, 482, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Browder, K.C.; Reddy, P.; Yamamoto, M.; Haghani, A.; Guillen, I.G.; Sahu, S.; Wang, C.; Luque, Y.; Prieto, J.; Shi, L.; et al. In Vivo Partial Reprogramming Alters Age-Associated Molecular Changes during Physiological Aging in Mice. Nat. Aging 2022, 2, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Hishida, T.; Yamamoto, M.; Hishida-Nozaki, Y.; Shao, C.; Huang, L.; Wang, C.; Shojima, K.; Xue, Y.; Hang, Y.; Shokhirev, M.; et al. In Vivo Partial Cellular Reprogramming Enhances Liver Plasticity and Regeneration. Cell Rep. 2022, 39, 110730. [Google Scholar] [CrossRef]
- Wang, C.; Rabadan Ros, R.; Martinez-Redondo, P.; Ma, Z.; Shi, L.; Xue, Y.; Guillen-Guillen, I.; Huang, L.; Hishida, T.; Liao, H.-K.; et al. In Vivo Partial Reprogramming of Myofibers Promotes Muscle Regeneration by Remodeling the Stem Cell Niche. Nat. Commun. 2021, 12, 3094. [Google Scholar] [CrossRef]
- Chen, Y.; Lüttmann, F.F.; Schoger, E.; Schöler, H.R.; Zelarayán, L.C.; Kim, K.-P.; Haigh, J.J.; Kim, J.; Braun, T. Reversible Reprogramming of Cardiomyocytes to a Fetal State Drives Heart Regeneration in Mice. Science 2021, 373, 1537–1540. [Google Scholar] [CrossRef]
- Buganim, Y.; Faddah, D.A.; Jaenisch, R. Mechanisms and Models of Somatic Cell Reprogramming. Nat. Rev. Genet. 2013, 14, 427–439. [Google Scholar] [CrossRef]
- González, F.; Huangfu, D. Mechanisms Underlying the Formation of Induced Pluripotent Stem Cells. WIREs Dev. Biol. 2016, 5, 39–65. [Google Scholar] [CrossRef]
- Liu, K.; Song, Y.; Yu, H.; Zhao, T. Understanding the Roadmaps to Induced Pluripotency. Cell Death Dis. 2014, 5, e1232. [Google Scholar] [CrossRef]
- Meir, Y.-J.J.; Li, G. Somatic Reprogramming—Above and Beyond Pluripotency. Cells 2021, 10, 2888. [Google Scholar] [CrossRef]
- Fidalgo, M.; Faiola, F.; Pereira, C.-F.; Ding, J.; Saunders, A.; Gingold, J.; Schaniel, C.; Lemischka, I.R.; Silva, J.C.R.; Wang, J. Zfp281 Mediates Nanog Autorepression through Recruitment of the NuRD Complex and Inhibits Somatic Cell Reprogramming. Proc. Natl. Acad. Sci. USA 2012, 109, 16202–16207. [Google Scholar] [CrossRef]
- Lluis, F.; Ombrato, L.; Pedone, E.; Pepe, S.; Merrill, B.J.; Cosma, M.P. T-Cell Factor 3 (Tcf3) Deletion Increases Somatic Cell Reprogramming by Inducing Epigenome Modifications. Proc. Natl. Acad. Sci. USA 2011, 108, 11912–11917. [Google Scholar] [CrossRef]
- Popowski, M.; Templeton, T.D.; Lee, B.-K.; Rhee, C.; Li, H.; Miner, C.; Dekker, J.D.; Orlanski, S.; Bergman, Y.; Iyer, V.R.; et al. Bright/Arid3A Acts as a Barrier to Somatic Cell Reprogramming through Direct Regulation of Oct4, Sox2, and Nanog. Stem Cell Rep. 2014, 2, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Serrano, F.; Calatayud, C.F.; Blazquez, M.; Torres, J.; Castell, J.V.; Bort, R. Gata4 Blocks Somatic Cell Reprogramming By Directly Repressing Nanog. Stem Cells 2013, 31, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Han, Q.; Peng, T.; Peng, M.; Wei, B.; Li, D.; Wang, X.; Yu, S.; Yang, J.; Cao, S.; et al. The Oncogene C-Jun Impedes Somatic Cell Reprogramming. Nat. Cell Biol. 2015, 17, 856–867. [Google Scholar] [CrossRef]
- Yang, C.-S.; Lopez, C.G.; Rana, T.M. Discovery of Nonsteroidal Anti-Inflammatory Drug and Anticancer Drug Enhancing Reprogramming and Induced Pluripotent Stem Cell Generation. Stem Cells 2011, 29, 1528–1536. [Google Scholar] [CrossRef]
- Mali, P.; Chou, B.-K.; Yen, J.; Ye, Z.; Zou, J.; Dowey, S.; Brodsky, R.A.; Ohm, J.E.; Yu, W.; Baylin, S.B.; et al. Butyrate Greatly Enhances Derivation of Human Induced Pluripotent Stem Cells by Promoting Epigenetic Remodeling and the Expression of Pluripotency-Associated Genes. Stem Cells 2010, 28, 713–720. [Google Scholar] [CrossRef]
- Esteban, M.A.; Wang, T.; Qin, B.; Yang, J.; Qin, D.; Cai, J.; Li, W.; Weng, Z.; Chen, J.; Ni, S.; et al. Vitamin C Enhances the Generation of Mouse and Human Induced Pluripotent Stem Cells. Cell Stem Cell 2010, 6, 71–79. [Google Scholar] [CrossRef]
- Huangfu, D.; Maehr, R.; Guo, W.; Eijkelenboom, A.; Snitow, M.; Chen, A.E.; Melton, D.A. Induction of Pluripotent Stem Cells by Defined Factors Is Greatly Improved by Small-Molecule Compounds. Nat. Biotechnol. 2008, 26, 795–797. [Google Scholar] [CrossRef]
- Neganova, I.; Shmeleva, E.; Munkley, J.; Chichagova, V.; Anyfantis, G.; Anderson, R.; Passos, J.; Elliott, D.J.; Armstrong, L.; Lako, M. JNK/SAPK Signaling Is Essential for Efficient Reprogramming of Human Fibroblasts to Induced Pluripotent Stem Cells. Stem Cells 2016, 34, 1198–1212. [Google Scholar] [CrossRef] [PubMed]
- Hartman, A.A.; Scalf, S.M.; Zhang, J.; Hu, X.; Chen, X.; Eastman, A.E.; Yang, C.; Guo, S. YAP Non-Cell-Autonomously Promotes Pluripotency Induction in Mouse Cells. Stem Cell Rep. 2020, 14, 730–743. [Google Scholar] [CrossRef]
- Marson, A.; Foreman, R.; Chevalier, B.; Bilodeau, S.; Kahn, M.; Young, R.A.; Jaenisch, R. Wnt Signaling Promotes Reprogramming of Somatic Cells to Pluripotency. Cell Stem Cell 2008, 3, 132–135. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Feng, G.; Wang, L.; Teng, F.; Wang, L.; Li, W.; Zhang, Y.; Zhou, Q. MeCP2 Deficiency Promotes Cell Reprogramming by Stimulating IGF1/AKT/mTOR Signaling and Activating Ribosomal Protein-Mediated Cell Cycle Gene Translation. J. Mol. Cell Biol. 2018, 10, 515–526. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.; Barrandon, O.; Nichols, J.; Kawaguchi, J.; Theunissen, T.W.; Smith, A. Promotion of Reprogramming to Ground State Pluripotency by Signal Inhibition. PLoS Biol. 2008, 6, e253. [Google Scholar] [CrossRef]
- Maherali, N.; Hochedlinger, K. Tgfβ Signal Inhibition Cooperates in the Induction of iPSCs and Replaces Sox2 and cMyc. Curr. Biol. 2009, 19, 1718–1723. [Google Scholar] [CrossRef]
- Ichida, J.K.; Blanchard, J.; Lam, K.; Son, E.Y.; Chung, J.E.; Egli, D.; Loh, K.M.; Carter, A.C.; Di Giorgio, F.P.; Koszka, K.; et al. A Small-Molecule Inhibitor of Tgf-β Signaling Replaces Sox2 in Reprogramming by Inducing Nanog. Cell Stem Cell 2009, 5, 491–503. [Google Scholar] [CrossRef]
- Ho, R.; Papp, B.; Hoffman, J.A.; Merrill, B.J.; Plath, K. Stage-Specific Regulation of Reprogramming to Induced Pluripotent Stem Cells by Wnt Signaling and T Cell Factor Proteins. Cell Rep. 2013, 3, 2113–2126. [Google Scholar] [CrossRef]
- Tan, F.; Qian, C.; Tang, K.; Abd-Allah, S.M.; Jing, N. Inhibition of Transforming Growth Factor β (TGF-β) Signaling Can Substitute for Oct4 Protein in Reprogramming and Maintain Pluripotency. J. Biol. Chem. 2015, 290, 4500–4511. [Google Scholar] [CrossRef]
- Doege, C.A.; Inoue, K.; Yamashita, T.; Rhee, D.B.; Travis, S.; Fujita, R.; Guarnieri, P.; Bhagat, G.; Vanti, W.B.; Shih, A.; et al. Early-Stage Epigenetic Modification during Somatic Cell Reprogramming by Parp1 and Tet2. Nature 2012, 488, 652–655. [Google Scholar] [CrossRef]
- Chen, J.; Liu, H.; Liu, J.; Qi, J.; Wei, B.; Yang, J.; Liang, H.; Chen, Y.; Chen, J.; Wu, Y.; et al. H3K9 Methylation Is a Barrier during Somatic Cell Reprogramming into iPSCs. Nat. Genet. 2013, 45, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Bhutani, N.; Brady, J.J.; Damian, M.; Sacco, A.; Corbel, S.Y.; Blau, H.M. Reprogramming towards Pluripotency Requires AID-Dependent DNA Demethylation. Nature 2010, 463, 1042–1047. [Google Scholar] [CrossRef]
- Hu, X.; Zhang, L.; Mao, S.-Q.; Li, Z.; Chen, J.; Zhang, R.-R.; Wu, H.-P.; Gao, J.; Guo, F.; Liu, W.; et al. Tet and TDG Mediate DNA Demethylation Essential for Mesenchymal-to-Epithelial Transition in Somatic Cell Reprogramming. Cell Stem Cell 2014, 14, 512–522. [Google Scholar] [CrossRef]
- Gao, Y.; Chen, J.; Li, K.; Wu, T.; Huang, B.; Liu, W.; Kou, X.; Zhang, Y.; Huang, H.; Jiang, Y.; et al. Replacement of Oct4 by Tet1 during iPSC Induction Reveals an Important Role of DNA Methylation and Hydroxymethylation in Reprogramming. Cell Stem Cell 2013, 12, 453–469. [Google Scholar] [CrossRef] [PubMed]
- Kretsovali, A.; Hadjimichael, C.; Charmpilas, N. Histone Deacetylase Inhibitors in Cell Pluripotency, Differentiation, and Reprogramming. Stem Cells Int. 2012, 2012, 184154. [Google Scholar] [CrossRef] [PubMed]
- Liao, B.; Bao, X.; Liu, L.; Feng, S.; Zovoilis, A.; Liu, W.; Xue, Y.; Cai, J.; Guo, X.; Qin, B.; et al. MicroRNA Cluster 302–367 Enhances Somatic Cell Reprogramming by Accelerating a Mesenchymal-to-Epithelial Transition. J. Biol. Chem. 2011, 286, 17359–17364. [Google Scholar] [CrossRef]
- Xu, N.; Papagiannakopoulos, T.; Pan, G.; Thomson, J.A.; Kosik, K.S. MicroRNA-145 Regulates OCT4, SOX2, and KLF4 and Represses Pluripotency in Human Embryonic Stem Cells. Cell 2009, 137, 647–658. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.-S.; Li, Z.; Rana, T.M. microRNAs Modulate iPS Cell Generation. RNA 2011, 17, 1451–1460. [Google Scholar] [CrossRef]
- Li, Z.; Dang, J.; Chang, K.-Y.; Rana, T.M. MicroRNA-Mediated Regulation of Extracellular Matrix Formation Modulates Somatic Cell Reprogramming. RNA 2014, 20, 1900–1915. [Google Scholar] [CrossRef]
- Zhao, F.-Q.; Misra, Y.; Li, D.-B.; Wadsworth, M.; Krag, D.; Weaver, D.; Tessitore, J.; Li, D.-W.; Zhang, G.; Tian, Q.; et al. Differential Expression of Oct3/4 in Human Breast Cancer and Normal Tissues. Int. J. Oncol. 2018, 52, 2069–2078. [Google Scholar] [CrossRef]
- Clemente-Periván, S.I.; Gómez-Gómez, Y.; Leyva-Vázquez, M.A.; Lagunas-Martínez, A.; Organista-Nava, J.; Illades-Aguiar, B. Role of Oct3/4 in Cervical Cancer Tumorigenesis. Front. Oncol. 2020, 10, 247. [Google Scholar] [CrossRef]
- Yu, S.S.; Cirillo, N. The Molecular Markers of Cancer Stem Cells in Head and Neck Tumors. J. Cell. Physiol. 2020, 235, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Novak, D.; Hüser, L.; Elton, J.J.; Umansky, V.; Altevogt, P.; Utikal, J. SOX2 in Development and Cancer Biology. Semin. Cancer Biol. 2020, 67, 74–82. [Google Scholar] [CrossRef]
- Chaudhary, S.; Islam, Z.; Mishra, V.; Rawat, S.; Ashraf, G.M.; Kolatkar, P.R. Sox2: A Regulatory Factor in Tumorigenesis and Metastasis. Curr. Protein Pept. Sci. 2019, 20, 495–504. [Google Scholar] [CrossRef]
- Deng, X.; Kong, F.; Li, S.; Jiang, H.; Dong, L.; Xu, X.; Zhang, X.; Yuan, H.; Xu, Y.; Chu, Y.; et al. A KLF4/PiHL/EZH2/HMGA2 Regulatory Axis and Its Function in Promoting Oxaliplatin-Resistance of Colorectal Cancer. Cell Death Dis. 2021, 12, 485. [Google Scholar] [CrossRef]
- Chen, J.; Li, H.; Zhang, B.; Xiong, Z.; Jin, Z.; Chen, J.; Zheng, Y.; Zhu, X.; Zhang, S. ABI2 -mediated MEOX2/KLF4-NANOG Axis Promotes Liver Cancer Stem Cell and Drives Tumour Recurrence. Liver Int. 2022, 42, 2562–2576. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Wang, L.; Shi, G.; Sun, S. The Deubiquitinase OTUD1 Inhibits Non-small Cell Lung Cancer Progression by Deubiquitinating and Stabilizing KLF4. Thorac. Cancer 2022, 13, 761–770. [Google Scholar] [CrossRef]
- Dhanasekaran, R.; Deutzmann, A.; Mahauad-Fernandez, W.D.; Hansen, A.S.; Gouw, A.M.; Felsher, D.W. The MYC Oncogene—The Grand Orchestrator of Cancer Growth and Immune Evasion. Nat. Rev. Clin. Oncol. 2022, 19, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Fatma, H.; Maurya, S.K.; Siddique, H.R. Epigenetic Modifications of C-MYC: Role in Cancer Cell Reprogramming, Progression and Chemoresistance. Semin. Cancer Biol. 2022, 83, 166–176. [Google Scholar] [CrossRef]
- Dang, C.V. MYC on the Path to Cancer. Cell 2012, 149, 22–35. [Google Scholar] [CrossRef]
- Hong, H.; Takahashi, K.; Ichisaka, T.; Aoi, T.; Kanagawa, O.; Nakagawa, M.; Okita, K.; Yamanaka, S. Suppression of Induced Pluripotent Stem Cell Generation by the P53–P21 Pathway. Nature 2009, 460, 1132–1135. [Google Scholar] [CrossRef]
- Li, H.; Collado, M.; Villasante, A.; Strati, K.; Ortega, S.; Cañamero, M.; Blasco, M.A.; Serrano, M. The Ink4/Arf Locus Is a Barrier for iPS Cell Reprogramming. Nature 2009, 460, 1136–1139. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, S.; Panopoulos, A.D.; Herrerías, A.; Bissig, K.-D.; Lutz, M.; Berggren, W.T.; Verma, I.M.; Izpisua Belmonte, J.C. A High Proliferation Rate Is Required for Cell Reprogramming and Maintenance of Human Embryonic Stem Cell Identity. Curr. Biol. 2011, 21, 45–52. [Google Scholar] [CrossRef] [PubMed]
- David, L.; Polo, J.M. Phases of Reprogramming. Stem Cell Res. 2014, 12, 754–761. [Google Scholar] [CrossRef]
- Lai, X.; Li, Q.; Wu, F.; Lin, J.; Chen, J.; Zheng, H.; Guo, L. Epithelial-Mesenchymal Transition and Metabolic Switching in Cancer: Lessons From Somatic Cell Reprogramming. Front. Cell Dev. Biol. 2020, 8, 760. [Google Scholar] [CrossRef]
- Smith, Z.D.; Nachman, I.; Regev, A.; Meissner, A. Dynamic Single-Cell Imaging of Direct Reprogramming Reveals an Early Specifying Event. Nat. Biotechnol. 2010, 28, 521–526. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Han, Q.; Pei, D. EMT and MET as Paradigms for Cell Fate Switching. J. Mol. Cell Biol. 2012, 4, 66–69. [Google Scholar] [CrossRef]
- Chen, T.; Yuan, D.; Wei, B.; Jiang, J.; Kang, J.; Ling, K.; Gu, Y.; Li, J.; Xiao, L.; Pei, G. E-Cadherin-Mediated Cell–Cell Contact Is Critical for Induced Pluripotent Stem Cell Generation. Stem Cells 2010, 28, 1315–1325. [Google Scholar] [CrossRef]
- Schwager, S.C.; Taufalele, P.V.; Reinhart-King, C.A. Cell–Cell Mechanical Communication in Cancer. Cel. Mol. Bioeng. 2019, 12, 1–14. [Google Scholar] [CrossRef]
- Roy, N.; Hebrok, M. Regulation of Cellular Identity in Cancer. Dev. Cell 2015, 35, 674–684. [Google Scholar] [CrossRef]
- Rasmussen, T.P. Parallels between Artificial Reprogramming and the Biogenesis of Cancer Stem Cells: Involvement of lncRNAs. Semin. Cancer Biol. 2019, 57, 36–44. [Google Scholar] [CrossRef]
- Friedmann-Morvinski, D.; Verma, I.M. Dedifferentiation and Reprogramming: Origins of Cancer Stem Cells. EMBO Rep. 2014, 15, 244–253. [Google Scholar] [CrossRef]
- Saito, S.; Lin, Y.-C.; Nakamura, Y.; Eckner, R.; Wuputra, K.; Kuo, K.-K.; Lin, C.-S.; Yokoyama, K.K. Potential Application of Cell Reprogramming Techniques for Cancer Research. Cell. Mol. Life Sci. 2019, 76, 45–65. [Google Scholar] [CrossRef] [PubMed]
- Granados, K.; Poelchen, J.; Novak, D.; Utikal, J. Cellular Reprogramming—A Model for Melanoma Cellular Plasticity. Int. J. Mol. Sci. 2020, 21, 8274. [Google Scholar] [CrossRef] [PubMed]
- Ohnishi, K.; Semi, K.; Yamamoto, T.; Shimizu, M.; Tanaka, A.; Mitsunaga, K.; Okita, K.; Osafune, K.; Arioka, Y.; Maeda, T.; et al. Premature Termination of Reprogramming In Vivo Leads to Cancer Development through Altered Epigenetic Regulation. Cell 2014, 156, 663–677. [Google Scholar] [CrossRef]
- Semi, K.; Matsuda, Y.; Ohnishi, K.; Yamada, Y. Cellular Reprogramming and Cancer Development. Int. J. Cancer 2013, 132, 1240–1248. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Zaret, K.S. Reprogramming of Human Cancer Cells to Pluripotency for Models of Cancer Progression. EMBO J. 2015, 34, 739–747. [Google Scholar] [CrossRef]
- Huyghe, A.; Furlan, G.; Schroeder, J.; Cascales, E.; Trajkova, A.; Ruel, M.; Stüder, F.; Larcombe, M.; Yang Sun, Y.B.; Mugnier, F.; et al. Comparative Roadmaps of Reprogramming and Oncogenic Transformation Identify Bcl11b and Atoh8 as Broad Regulators of Cellular Plasticity. Nat. Cell Biol. 2022, 24, 1350–1363. [Google Scholar] [CrossRef]
- Xiong, S.; Feng, Y.; Cheng, L. Cellular Reprogramming as a Therapeutic Target in Cancer. Trends Cell Biol. 2019, 29, 623–634. [Google Scholar] [CrossRef]
- Gong, L.; Yan, Q.; Zhang, Y.; Fang, X.; Liu, B.; Guan, X. Cancer Cell Reprogramming: A Promising Therapy Converting Malignancy to Benignity. Cancer Commun. 2019, 39, 48. [Google Scholar] [CrossRef]
- Lork, M.; Verhelst, K.; Beyaert, R. CYLD, A20 and OTULIN Deubiquitinases in NF-κB Signaling and Cell Death: So Similar, yet so Different. Cell Death Differ. 2017, 24, 1172–1183. [Google Scholar] [CrossRef] [PubMed]
- Harhaj, E.W.; Dixit, V.M. Regulation of NF-κB by Deubiquitinases: Deubiquitinases Regulating NF-κB Signaling. Immunol. Rev. 2012, 246, 107–124. [Google Scholar] [CrossRef]
- Trompouki, E.; Hatzivassiliou, E.; Tsichritzis, T.; Farmer, H.; Ashworth, A.; Mosialos, G. CYLD Is a Deubiquitinating Enzyme That Negatively Regulates NF-κB Activation by TNFR Family Members. Nature 2003, 424, 793–796. [Google Scholar] [CrossRef] [PubMed]
- Schlicher, L.; Wissler, M.; Preiss, F.; Brauns-Schubert, P.; Jakob, C.; Dumit, V.; Borner, C.; Dengjel, J.; Maurer, U. SPATA 2 Promotes CYLD Activity and Regulates TNF -induced NF -κB Signaling and Cell Death. EMBO Rep. 2016, 17, 1485–1497. [Google Scholar] [CrossRef]
- Kupka, S.; De Miguel, D.; Draber, P.; Martino, L.; Surinova, S.; Rittinger, K.; Walczak, H. SPATA2-Mediated Binding of CYLD to HOIP Enables CYLD Recruitment to Signaling Complexes. Cell Rep. 2016, 16, 2271–2280. [Google Scholar] [CrossRef]
- Pseftogas, A.; Xanthopoulos, K.; Poutahidis, T.; Ainali, C.; Dafou, D.; Panteris, E.; Kern, J.G.; Varelas, X.; Hardas, A.; Gonidas, C.; et al. The Tumor Suppressor CYLD Inhibits Mammary Epithelial to Mesenchymal Transition by the Coordinated Inhibition of YAP/TAZ and TGFβ Signaling. Cancers 2020, 12, 2047. [Google Scholar] [CrossRef]
- Zhao, Y.; Thornton, A.M.; Kinney, M.C.; Ma, C.A.; Spinner, J.J.; Fuss, I.J.; Shevach, E.M.; Jain, A. The Deubiquitinase CYLD Targets Smad7 Protein to Regulate Transforming Growth Factor β (TGF-β) Signaling and the Development of Regulatory T Cells. J. Biol. Chem. 2011, 286, 40520–40530. [Google Scholar] [CrossRef]
- Shinriki, S.; Jono, H.; Maeshiro, M.; Nakamura, T.; Guo, J.; Li, J.-D.; Ueda, M.; Yoshida, R.; Shinohara, M.; Nakayama, H.; et al. Loss of CYLD Promotes Cell Invasion via ALK5 Stabilization in Oral Squamous Cell Carcinoma: Association of CYLD with OSCC-Related Invasion. J. Pathol. 2018, 244, 367–379. [Google Scholar] [CrossRef]
- Van Andel, H.; Kocemba, K.A.; De Haan-Kramer, A.; Mellink, C.H.; Piwowar, M.; Broijl, A.; Van Duin, M.; Sonneveld, P.; Maurice, M.M.; Kersten, M.J.; et al. Loss of CYLD Expression Unleashes Wnt Signaling in Multiple Myeloma and Is Associated with Aggressive Disease. Oncogene 2017, 36, 2105–2115. [Google Scholar] [CrossRef] [PubMed]
- Tauriello, D.V.F.; Haegebarth, A.; Kuper, I.; Edelmann, M.J.; Henraat, M.; Canninga-van Dijk, M.R.; Kessler, B.M.; Clevers, H.; Maurice, M.M. Loss of the Tumor Suppressor CYLD Enhances Wnt/β-Catenin Signaling through K63-Linked Ubiquitination of Dvl. Mol. Cell 2010, 37, 607–619. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, Z.; Wang, P.; Li, D.; Zhou, J.; Wu, S. CYLD Negatively Regulates Hippo Signaling by Limiting Hpo Phosphorylation in Drosophila. Biochem. Biophys. Res. Commun. 2014, 452, 808–812. [Google Scholar] [CrossRef]
- Nikolaou, K.; Tsagaratou, A.; Eftychi, C.; Kollias, G.; Mosialos, G.; Talianidis, I. Inactivation of the Deubiquitinase CYLD in Hepatocytes Causes Apoptosis, Inflammation, Fibrosis, and Cancer. Cancer Cell 2012, 21, 738–750. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Patai, Á.V.; Hogenson, T.L.; Fernandez-Zapico, M.E.; Qin, B.; Sinicrope, F.A. Irreversible JNK Blockade Overcomes PD-L1-Mediated Resistance to Chemotherapy in Colorectal Cancer. Oncogene 2021, 40, 5105–5115. [Google Scholar] [CrossRef]
- Pannem, R.R.; Dorn, C.; Ahlqvist, K.; Bosserhoff, A.K.; Hellerbrand, C.; Massoumi, R. CYLD Controls C-MYC Expression through the JNK-Dependent Signaling Pathway in Hepatocellular Carcinoma. Carcinogenesis 2014, 35, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Herlyn, M.; Yang, X. TRIM15 and CYLD Regulate ERK Activation via Lysine-63-Linked Polyubiquitination. Nat. Cell Biol. 2021, 23, 978–991. [Google Scholar] [CrossRef] [PubMed]
- Qiu, C.; Liu, K.; Zhang, S.; Gao, S.; Chen, W.; Li, D.; Huang, Y. Bisdemethoxycurcumin Inhibits Hepatocellular Carcinoma Proliferation Through Akt Inactivation via CYLD-Mediated Deubiquitination. Drug Des. Dev. Ther. 2020, 14, 993–1001. [Google Scholar] [CrossRef]
- Yang, W.-L.; Jin, G.; Li, C.-F.; Jeong, Y.S.; Moten, A.; Xu, D.; Feng, Z.; Chen, W.; Cai, Z.; Darnay, B.; et al. Cycles of Ubiquitination and Deubiquitination Critically Regulate Growth Factor–Mediated Activation of Akt Signaling. Sci. Signal. 2013, 6, ra3. [Google Scholar] [CrossRef]
- Lim, J.H.; Jono, H.; Komatsu, K.; Woo, C.-H.; Lee, J.; Miyata, M.; Matsuno, T.; Xu, X.; Huang, Y.; Zhang, W.; et al. CYLD Negatively Regulates Transforming Growth Factor-β-Signalling via Deubiquitinating Akt. Nat. Commun. 2012, 3, 771. [Google Scholar] [CrossRef]
- Haq, S.; Sarodaya, N.; Karapurkar, J.K.; Suresh, B.; Jo, J.K.; Singh, V.; Bae, Y.S.; Kim, K.-S.; Ramakrishna, S. CYLD Destabilizes NoxO1 Protein by Promoting Ubiquitination and Regulates Prostate Cancer Progression. Cancer Lett. 2022, 525, 146–157. [Google Scholar] [CrossRef]
- Wang, L.; Lin, Y.; Zhou, X.; Chen, Y.; Li, X.; Luo, W.; Zhou, Y.; Cai, L. CYLD Deficiency Enhances Metabolic Reprogramming and Tumor Progression in Nasopharyngeal Carcinoma via PFKFB3. Cancer Lett. 2022, 532, 215586. [Google Scholar] [CrossRef]
- Cui, Z.; Kang, H.; Grandis, J.R.; Johnson, D.E. CYLD Alterations in the Tumorigenesis and Progression of Human Papillomavirus–Associated Head and Neck Cancers. Mol. Cancer Res. 2021, 19, 14–24. [Google Scholar] [CrossRef]
- Zhao, J.; Wang, X.; Mi, Z.; Jiang, X.; Sun, L.; Zheng, B.; Wang, J.; Meng, M.; Zhang, L.; Wang, Z.; et al. STAT3/miR-135b/NF-κB Axis Confers Aggressiveness and Unfavorable Prognosis in Non-Small-Cell Lung Cancer. Cell Death Dis. 2021, 12, 493. [Google Scholar] [CrossRef]
- Wang, J.; Tan, L.; Jia, B.; Yu, X.; Yao, R.; OUYang, N.; Yu, X.; Cao, X.; Tong, J.; Chen, T.; et al. Downregulation of m6A Reader YTHDC2 Promotes the Proliferation and Migration of Malignant Lung Cells via CYLD/NF-κB Pathway. Int. J. Biol. Sci. 2021, 17, 2633–2651. [Google Scholar] [CrossRef] [PubMed]
- Minderman, M.; Lantermans, H.C.; Grüneberg, L.J.; Cillessen, S.A.G.M.; Bende, R.J.; Van Noesel, C.J.M.; Kersten, M.J.; Pals, S.T.; Spaargaren, M. MALT1-Dependent Cleavage of CYLD Promotes NF-κB Signaling and Growth of Aggressive B-Cell Receptor-Dependent Lymphomas. Blood Cancer J. 2023, 13, 37. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Wang, S.; Li, H.-L.; Luo, H.; Wu, X.; Lu, J.; Wang, H.-W.; Chen, Y.; Chen, D.; Wu, W.-T.; et al. FOSL1 Promotes Proneural-to-Mesenchymal Transition of Glioblastoma Stem Cells via UBC9/CYLD/NF-κB Axis. Mol. Ther. 2022, 30, 2568–2583. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Lei, Y.; Huang, Y.; Zhang, J.; Chen, Y.; Chen, K.; Lin, J.; Sun, S.; Lin, X. CYLD Expression in Dendritic Cells Involved in the Immunoregulation of Pulmonary Adenocarcinoma via NF-κB Pathway. Artif. Cells Nanomed. Biotechnol. 2020, 48, 137–142. [Google Scholar] [CrossRef]
- Zhou, S.; Yin, D.; Hu, Z.; Luo, C.; Zhou, Z.; Xin, H.; Yang, X.; Shi, Y.; Wang, Z.; Huang, X.; et al. A Positive Feedback Loop Between Cancer Stem-Like Cells and Tumor-Associated Neutrophils Controls Hepatocellular Carcinoma Progression. Hepatology 2019, 70, 1214–1230. [Google Scholar] [CrossRef]
- Xu, D.; Zhou, P.; Wang, Y.; Zhang, L.; Fu, W.; Ruan, B.; Xu, H.; Hu, C.; Tian, L.; Qin, J.; et al. Reciprocal Activation between STAT3 and miR-181b Regulates the Proliferation of Esophageal Cancer Stem-like Cells via the CYLD Pathway. Mol. Cancer 2016, 15, 40. [Google Scholar] [CrossRef]
- Trompouki, E.; Tsagaratou, A.; Kosmidis, S.K.; Dollé, P.; Qian, J.; Kontoyiannis, D.L.; Cardoso, W.V.; Mosialos, G. Truncation of the Catalytic Domain of the Cylindromatosis Tumor Suppressor Impairs Lung Maturation. Neoplasia 2009, 11, 469–476. [Google Scholar] [CrossRef]
- Karatzas, D.N.; Xanthopoulos, K.; Kotantaki, P.; Pseftogas, A.; Teliousis, K.; Hatzivassiliou, E.G.; Kontoyiannis, D.L.; Poutahidis, T.; Mosialos, G. Inactivation of CYLD in Intestinal Epithelial Cells Exacerbates Colitis-Associated Colorectal Carcinogenesis—A Short Report. Cell. Oncol. 2016, 39, 287–293. [Google Scholar] [CrossRef]
- Papathanasiou, M.; Tsiftsoglou, S.A.; Polyzos, A.P.; Papadopoulou, D.; Valakos, D.; Klagkou, E.; Karagianni, P.; Pliatska, M.; Talianidis, I.; Agelopoulos, M.; et al. Identification of a Dynamic Gene Regulatory Network Required for Pluripotency Factor-induced Reprogramming of Mouse Fibroblasts and Hepatocytes. EMBO J. 2021, 40, e102236. [Google Scholar] [CrossRef]
- Aranda, P.S.; LaJoie, D.M.; Jorcyk, C.L. Bleach Gel: A Simple Agarose Gel for Analyzing RNA Quality. Electrophoresis 2012, 33, 366–369. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.; Marumoto, T.; Yamaguchi, S.; Okano, S.; Takeda, N.; Sakamoto, C.; Kawano, H.; Nii, T.; Miyamoto, S.; Nagai, Y.; et al. Inhibition of PTEN Tumor Suppressor Promotes the Generation of Induced Pluripotent Stem Cells. Mol. Ther. 2013, 21, 1242–1250. [Google Scholar] [CrossRef] [PubMed]
- Demichev, V.; Messner, C.B.; Vernardis, S.I.; Lilley, K.S.; Ralser, M. DIA-NN: Neural Networks and Interference Correction Enable Deep Proteome Coverage in High Throughput. Nat. Methods 2020, 17, 41–44. [Google Scholar] [CrossRef] [PubMed]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus Computational Platform for Comprehensive Analysis of (Prote)Omics Data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Perez-Riverol, Y.; Bai, J.; Bandla, C.; Hewapathirana, S.; García-Seisdedos, D.; Kamatchinathan, S.; Kundu, D.; Prakash, A.; Frericks-Zipper, A.; Eisenacher, M.; et al. The PRIDE database resources in 2022: A Hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2022, 50, D543–D552. [Google Scholar] [CrossRef]
- Wang, W.; Yang, J.; Liu, H.; Lu, D.; Chen, X.; Zenonos, Z.; Campos, L.S.; Rad, R.; Guo, G.; Zhang, S.; et al. Rapid and Efficient Reprogramming of Somatic Cells to Induced Pluripotent Stem Cells by Retinoic Acid Receptor Gamma and Liver Receptor Homolog 1. Proc. Natl. Acad. Sci. USA 2011, 108, 18283–18288. [Google Scholar] [CrossRef]
- Wang, J.; Yu, H.; Ma, Q.; Zeng, P.; Wu, D.; Hou, Y.; Liu, X.; Jia, L.; Sun, J.; Chen, Y.; et al. Phase Separation of OCT4 Controls TAD Reorganization to Promote Cell Fate Transitions. Cell Stem Cell 2021, 28, 1868–1883. [Google Scholar] [CrossRef] [PubMed]
- Samavarchi-Tehrani, P.; Golipour, A.; David, L.; Sung, H.; Beyer, T.A.; Datti, A.; Woltjen, K.; Nagy, A.; Wrana, J.L. Functional Genomics Reveals a BMP-Driven Mesenchymal-to-Epithelial Transition in the Initiation of Somatic Cell Reprogramming. Cell Stem Cell 2010, 7, 64–77. [Google Scholar] [CrossRef]
- Li, R.; Liang, J.; Ni, S.; Zhou, T.; Qing, X.; Li, H.; He, W.; Chen, J.; Li, F.; Zhuang, Q.; et al. A Mesenchymal-to-Epithelial Transition Initiates and Is Required for the Nuclear Reprogramming of Mouse Fibroblasts. Cell Stem Cell 2010, 7, 51–63. [Google Scholar] [CrossRef]
- Polo, J.M.; Anderssen, E.; Walsh, R.M.; Schwarz, B.A.; Nefzger, C.M.; Lim, S.M.; Borkent, M.; Apostolou, E.; Alaei, S.; Cloutier, J.; et al. A Molecular Roadmap of Reprogramming Somatic Cells into iPS Cells. Cell 2012, 151, 1617–1632. [Google Scholar] [CrossRef]
- Muhl, L.; Genové, G.; Leptidis, S.; Liu, J.; He, L.; Mocci, G.; Sun, Y.; Gustafsson, S.; Buyandelger, B.; Chivukula, I.V.; et al. Single-Cell Analysis Uncovers Fibroblast Heterogeneity and Criteria for Fibroblast and Mural Cell Identification and Discrimination. Nat. Commun. 2020, 11, 3953. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Evans, T. Activation-Induced Cytidine Deaminase Regulates Fibroblast Growth Factor/Extracellular Signal-Regulated Kinases Signaling to Achieve the Naïve Pluripotent State During Reprogramming. Stem Cells 2019, 37, 1003–1017. [Google Scholar] [CrossRef] [PubMed]
- Ngondo, R.P.; Cirera-Salinas, D.; Yu, J.; Wischnewski, H.; Bodak, M.; Vandormael-Pournin, S.; Geiselmann, A.; Wettstein, R.; Luitz, J.; Cohen-Tannoudji, M.; et al. Argonaute 2 Is Required for Extra-Embryonic Endoderm Differentiation of Mouse Embryonic Stem Cells. Stem Cell Rep. 2018, 10, 461–476. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Zhao, W.; Huang, Y.; Tong, H.; Xia, Y.; Jiang, Q.; Qin, J. Loss of Polycomb Group Protein Pcgf1 Severely Compromises Proper Differentiation of Embryonic Stem Cells. Sci. Rep. 2017, 7, 46276. [Google Scholar] [CrossRef]
- David, L.; Samavarchi-Tehrani, P.; Golipour, A.; Wrana, J.L. Looking into the Black Box: Insights into the Mechanisms of Somatic Cell Reprogramming. Genes 2011, 2, 81–106. [Google Scholar] [CrossRef]
- Nagasaka, R.; Matsumoto, M.; Okada, M.; Sasaki, H.; Kanie, K.; Kii, H.; Uozumi, T.; Kiyota, Y.; Honda, H.; Kato, R. Visualization of Morphological Categories of Colonies for Monitoring of Effect on Induced Pluripotent Stem Cell Culture Status. Regen. Ther. 2017, 6, 41–51. [Google Scholar] [CrossRef]
- Jo, J.; Hwang, S.; Kim, H.J.; Hong, S.; Lee, J.E.; Lee, S.-G.; Baek, A.; Han, H.; Lee, J.I.; Lee, I.; et al. An Integrated Systems Biology Approach Identifies Positive Cofactor 4 as a Factor That Increases Reprogramming Efficiency. Nucleic Acids Res. 2016, 44, 1203–1215. [Google Scholar] [CrossRef]
- Tai, C.-I.; Ying, Q.-L. Gbx2, a LIF/Stat3 Target, Promotes Reprogramming to and Retention of the Pluripotent Ground State. J. Cell Sci. 2013, 126, 1093–1098. [Google Scholar] [CrossRef]
- Yang, C.-S.; Chang, K.-Y.; Rana, T.M. Genome-Wide Functional Analysis Reveals Factors Needed at the Transition Steps of Induced Reprogramming. Cell Rep. 2014, 8, 327–337. [Google Scholar] [CrossRef]
- O’Malley, J.; Skylaki, S.; Iwabuchi, K.A.; Chantzoura, E.; Ruetz, T.; Johnsson, A.; Tomlinson, S.R.; Linnarsson, S.; Kaji, K. High-Resolution Analysis with Novel Cell-Surface Markers Identifies Routes to iPS Cells. Nature 2013, 499, 88–91. [Google Scholar] [CrossRef]
- Kim, S.-I.; Oceguera-Yanez, F.; Hirohata, R.; Linker, S.; Okita, K.; Yamada, Y.; Yamamoto, T.; Yamanaka, S.; Woltjen, K. KLF4 N-Terminal Variance Modulates Induced Reprogramming to Pluripotency. Stem Cell Rep. 2015, 4, 727–743. [Google Scholar] [CrossRef]
- Li, X.; Pei, D.; Zheng, H. Transitions between Epithelial and Mesenchymal States during Cell Fate Conversions. Protein Cell 2014, 5, 580–591. [Google Scholar] [CrossRef] [PubMed]
- Shao, X.; Taha, I.N.; Clauser, K.R.; Gao, Y.; Naba, A. MatrisomeDB: The ECM-Protein Knowledge Database. Nucleic Acids Res. 2020, 48, D1136–D1144. [Google Scholar] [CrossRef] [PubMed]
- Moore, N.H.; Costa, L.G.; Shaffer, S.A.; Goodlett, D.R.; Guizzetti, M. Shotgun Proteomics Implicates Extracellular Matrix Proteins and Protease Systems in Neuronal Development Induced by Astrocyte Cholinergic Stimulation. J. Neurochem. 2009, 108, 891–908. [Google Scholar] [CrossRef] [PubMed]
- Apóstolo, N.; Smukowski, S.N.; Vanderlinden, J.; Condomitti, G.; Rybakin, V.; Ten Bos, J.; Trobiani, L.; Portegies, S.; Vennekens, K.M.; Gounko, N.V.; et al. Synapse Type-Specific Proteomic Dissection Identifies IgSF8 as a Hippocampal CA3 Microcircuit Organizer. Nat. Commun. 2020, 11, 5171. [Google Scholar] [CrossRef]
- Jaber, M.; Radwan, A.; Loyfer, N.; Abdeen, M.; Sebban, S.; Khatib, A.; Yassen, H.; Kolb, T.; Zapatka, M.; Makedonski, K.; et al. Comparative Parallel Multi-Omics Analysis during the Induction of Pluripotent and Trophectoderm States. Nat. Commun. 2022, 13, 3475. [Google Scholar] [CrossRef]
- Bansho, Y.; Lee, J.; Nishida, E.; Nakajima-Koyama, M. Identification and Characterization of Secreted Factors That Are Upregulated during Somatic Cell Reprogramming. FEBS Lett. 2017, 591, 1584–1600. [Google Scholar] [CrossRef]
- Chondronasiou, D.; Martínez De Villarreal, J.; Melendez, E.; Lynch, C.J.; Pozo, N.D.; Kovatcheva, M.; Aguilera, M.; Prats, N.; Real, F.X.; Serrano, M. Deciphering the Roadmap of in Vivo Reprogramming toward Pluripotency. Stem Cell Rep. 2022, 17, 2501–2517. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape Provides a Biologist-Oriented Resource for the Analysis of Systems-Level Datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Hansson, J.; Rafiee, M.R.; Reiland, S.; Polo, J.M.; Gehring, J.; Okawa, S.; Huber, W.; Hochedlinger, K.; Krijgsveld, J. Highly Coordinated Proteome Dynamics during Reprogramming of Somatic Cells to Pluripotency. Cell Rep. 2012, 2, 1579–1592. [Google Scholar] [CrossRef]
- Jiao, J.; Dang, Y.; Yang, Y.; Gao, R.; Zhang, Y.; Kou, Z.; Sun, X.-F.; Gao, S. Promoting Reprogramming by FGF2 Reveals That the Extracellular Matrix Is a Barrier for Reprogramming Fibroblasts to Pluripotency. Stem Cells 2013, 31, 729–740. [Google Scholar] [CrossRef]
- Vasaikar, S.V.; Deshmukh, A.P.; Den Hollander, P.; Addanki, S.; Kuburich, N.A.; Kudaravalli, S.; Joseph, R.; Chang, J.T.; Soundararajan, R.; Mani, S.A. EMTome: A Resource for Pan-Cancer Analysis of Epithelial-Mesenchymal Transition Genes and Signatures. Br. J. Cancer 2021, 124, 259–269. [Google Scholar] [CrossRef]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database Hallmark Gene Set Collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef]
- Urbanik, T. CYLD Deletion Triggers Nuclear Factor-κB-Signaling and Increases Cell Death Resistance in Murine Hepatocytes. World J. Gastroenterol. 2014, 20, 17049. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.-X.; Huang, Z.; Yang, X.; Wang, X.; Zhao, L.-P.; Wang, P.-X.; Zhang, X.-J.; Alves-Bezerra, M.; Cai, L.; Zhang, P.; et al. The Deubiquitinating Enzyme Cylindromatosis Mitigates Nonalcoholic Steatohepatitis. Nat. Med. 2018, 24, 213–223. [Google Scholar] [CrossRef]
- Hellerbrand, C.; Massoumi, R. Cylindromatosis-A Protective Molecule against Liver Diseases: CYLD IN LIVER DISEASE. Med. Res. Rev. 2016, 36, 342–359. [Google Scholar] [CrossRef]
- Qi, L.; Zang, H.; Wu, W.; Nagarkatti, P.; Nagarkatti, M.; Liu, Q.; Robbins, J.; Wang, X.; Cui, T. CYLD Exaggerates Pressure Overload-Induced Cardiomyopathy via Suppressing Autolysosome Efflux in Cardiomyocytes. J. Mol. Cell. Cardiol. 2020, 145, 59–73. [Google Scholar] [CrossRef]
- Wang, H.; Lai, Y.; Mathis, B.J.; Wang, W.; Li, S.; Qu, C.; Li, B.; Shao, L.; Song, H.; Janicki, J.S.; et al. Deubiquitinating Enzyme CYLD Mediates Pressure Overload-Induced Cardiac Maladaptive Remodeling and Dysfunction via Downregulating Nrf2. J. Mol. Cell. Cardiol. 2015, 84, 143–153. [Google Scholar] [CrossRef]
- Zhou, Y.-J.; Duan, D.-Q.; Lu, L.-Q.; Tang, L.-J.; Zhang, X.-J.; Luo, X.-J.; Peng, J. The SPATA2/CYLD Pathway Contributes to Doxorubicin-Induced Cardiomyocyte Ferroptosis via Enhancing Ferritinophagy. Chem.-Biol. Interact. 2022, 368, 110205. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Gao, S.; Yang, Y.; Li, H.; Zhou, J.; Chen, M.; Yang, S.; Zhang, Y.; Zhang, L.; Meng, X.; et al. CYLD Deubiquitinates Plakoglobin to Promote Cx43 Membrane Targeting and Gap Junction Assembly in the Heart. Cell Rep. 2022, 41, 111864. [Google Scholar] [CrossRef] [PubMed]
- Zajicek, A.S.; Ruan, H.; Dai, H.; Skolfield, M.C.; Phillips, H.L.; Burnette, W.J.; Javidfar, B.; Sun, S.-C.; Akbarian, S.; Yao, W.-D. Cylindromatosis Drives Synapse Pruning and Weakening by Promoting Macroautophagy through Akt-mTOR Signaling. Mol. Psychiatry 2022, 27, 2414–2424. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Li, D.; Yang, L.; Long, C. Deficiency of the CYLD Impairs Fear Memory of Mice and Disrupts Neuronal Activity and Synaptic Transmission in the Basolateral Amygdala. Front. Cell. Neurosci. 2021, 15, 740165. [Google Scholar] [CrossRef]
- Pirooznia, S.K.; Wang, H.; Panicker, N.; Kumar, M.; Neifert, S.; Dar, M.A.; Lau, E.; Kang, B.G.; Redding-Ochoa, J.; Troncoso, J.C.; et al. Deubiquitinase CYLD Acts as a Negative Regulator of Dopamine Neuron Survival in Parkinson’s Disease. Sci. Adv. 2022, 8, eabh1824. [Google Scholar] [CrossRef] [PubMed]
- Ganjam, G.K.; Terpolilli, N.A.; Diemert, S.; Eisenbach, I.; Hoffmann, L.; Reuther, C.; Herden, C.; Roth, J.; Plesnila, N.; Culmsee, C. Cylindromatosis Mediates Neuronal Cell Death in Vitro and in Vivo. Cell Death Differ. 2018, 25, 1394–1407. [Google Scholar] [CrossRef]
- Nguyen, J.; Massoumi, R.; Alliston, T. CYLD, a Mechanosensitive Deubiquitinase, Regulates TGFβ Signaling in Load-Induced Bone Formation. Bone 2020, 131, 115148. [Google Scholar] [CrossRef] [PubMed]
- Läubli, H.; Borsig, L. Altered Cell Adhesion and Glycosylation Promote Cancer Immune Suppression and Metastasis. Front. Immunol. 2019, 10, 2120. [Google Scholar] [CrossRef]
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the Extracellular Matrix in Development and Disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786–801. [Google Scholar] [CrossRef]
- Cox, T.R. The Matrix in Cancer. Nat. Rev. Cancer 2021, 21, 217–238. [Google Scholar] [CrossRef]
- Pickup, M.W.; Mouw, J.K.; Weaver, V.M. The Extracellular Matrix Modulates the Hallmarks of Cancer. EMBO Rep. 2014, 15, 1243–1253. [Google Scholar] [CrossRef] [PubMed]
- Scott, L.E.; Weinberg, S.H.; Lemmon, C.A. Mechanochemical Signaling of the Extracellular Matrix in Epithelial-Mesenchymal Transition. Front. Cell Dev. Biol. 2019, 7, 135. [Google Scholar] [CrossRef]
- Ishida, T.; Nakao, S.; Ueyama, T.; Harada, Y.; Kawamura, T. Metabolic Remodeling during Somatic Cell Reprogramming to Induced Pluripotent Stem Cells: Involvement of Hypoxia-Inducible Factor 1. Inflamm. Regen. 2020, 40, 8. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, K.; Fukuda, A.; Hisatake, K. Mechanisms of the Metabolic Shift during Somatic Cell Reprogramming. Int. J. Mol. Sci. 2019, 20, 2254. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Kim, H.; Lee, S.; Youn, H.; Youn, B. Role of Metabolic Reprogramming in Epithelial–Mesenchymal Transition (EMT). Int. J. Mol. Sci. 2019, 20, 2042. [Google Scholar] [CrossRef]
- Panopoulos, A.D.; Yanes, O.; Ruiz, S.; Kida, Y.S.; Diep, D.; Tautenhahn, R.; Herrerías, A.; Batchelder, E.M.; Plongthongkum, N.; Lutz, M.; et al. The Metabolome of Induced Pluripotent Stem Cells Reveals Metabolic Changes Occurring in Somatic Cell Reprogramming. Cell Res. 2012, 22, 168–177. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Upregulated ECM-associated proteins in Δ9 D3 and D4 early iPSCs | ||||
| Bmp1 | Emilin1 | Igfbp7 | Nid1 | Tgfbi |
| Ccn2 | Emilin2 | Loxl2 | Npnt | Thbs1 |
| Ccn4 | Fbln1 | Loxl4 | Pcolce2 | Tinagl1 |
| Cd44 | Fbln2 | Ltbp1 | Plg | Tll1 |
| Col16a1 | Fbn1 | Ltbp2 | Sdc2 | Tnc |
| Col18a1 | Fbn2 | Mfap2 | Serpine1 | |
| Col8a1 | Fn1 | Mfge8 | Slit3 | |
| Efemp2 | Hspg2 | Mxra7 | Tgfb1 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bekas, N.; Samiotaki, M.; Papathanasiou, M.; Mokos, P.; Pseftogas, A.; Xanthopoulos, K.; Thanos, D.; Mosialos, G.; Dafou, D. Inactivation of Tumor Suppressor CYLD Inhibits Fibroblast Reprogramming to Pluripotency. Cancers 2023, 15, 4997. https://doi.org/10.3390/cancers15204997
Bekas N, Samiotaki M, Papathanasiou M, Mokos P, Pseftogas A, Xanthopoulos K, Thanos D, Mosialos G, Dafou D. Inactivation of Tumor Suppressor CYLD Inhibits Fibroblast Reprogramming to Pluripotency. Cancers. 2023; 15(20):4997. https://doi.org/10.3390/cancers15204997
Chicago/Turabian StyleBekas, Nikolaos, Martina Samiotaki, Maria Papathanasiou, Panagiotis Mokos, Athanasios Pseftogas, Konstantinos Xanthopoulos, Dimitris Thanos, George Mosialos, and Dimitra Dafou. 2023. "Inactivation of Tumor Suppressor CYLD Inhibits Fibroblast Reprogramming to Pluripotency" Cancers 15, no. 20: 4997. https://doi.org/10.3390/cancers15204997