

Optical Genome Mapping Reveals the Complex Genetic Landscape of Myeloma
 , and
, and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Selection
2.2. Sample Preparation, CD138 Plasma Cell Purification and Preservation
2.3. FISH Analysis
2.4. Optical Genome Mapping
2.5. OGM Analysis
3. Results and Discussion
3.1. Patients and Disease Characteristics
3.2. Metrics of OGM Technique on CD138+ Plasma Cells
3.3. Overall OGM Results
3.4. Successful Detection of Classical Primary Abnormalities in Myeloma by OGM
3.5. Limits Associated with Ploidy Levels in OGM
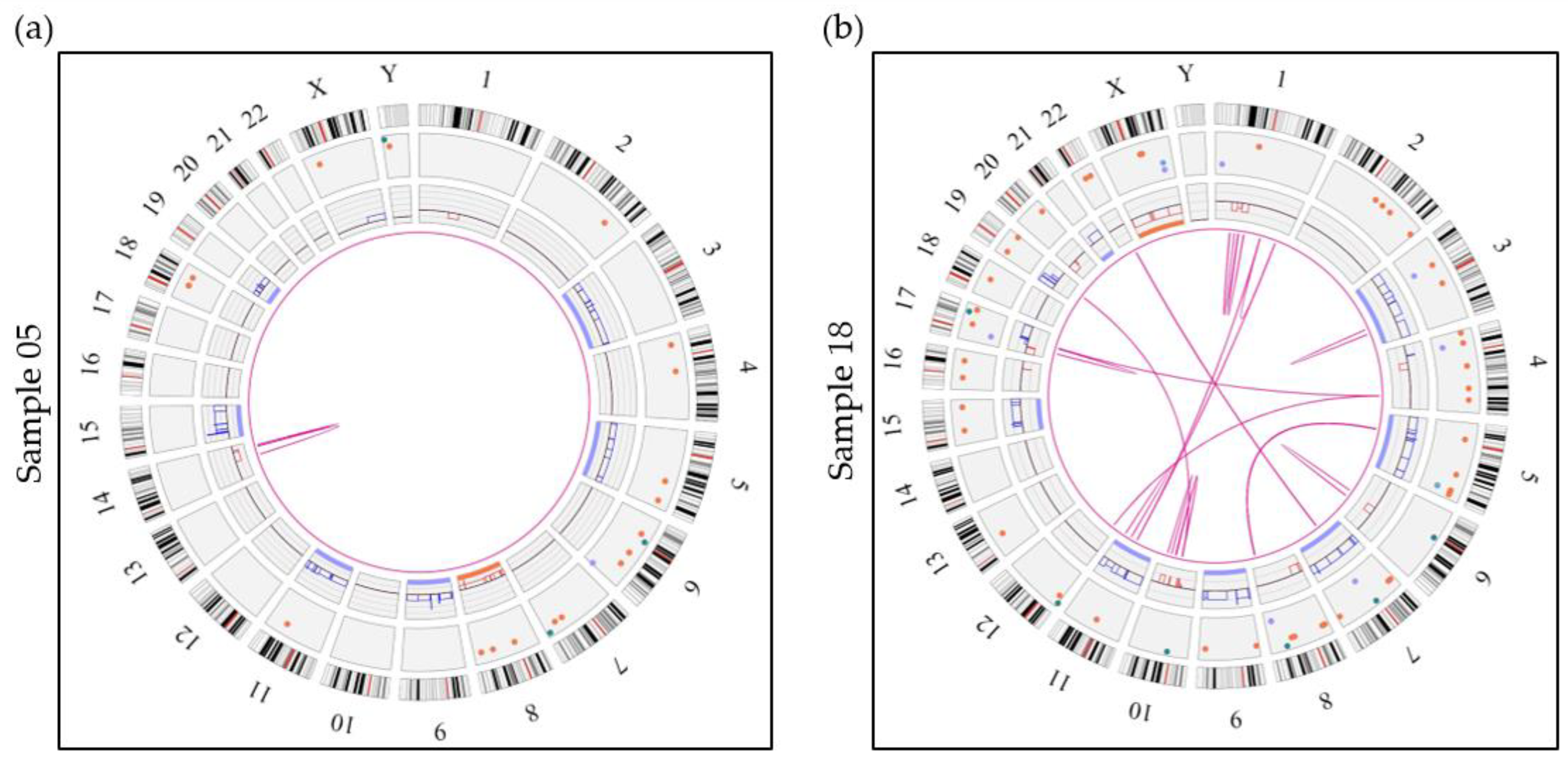
3.6. Detection of Secondary Abnormalities with OGM
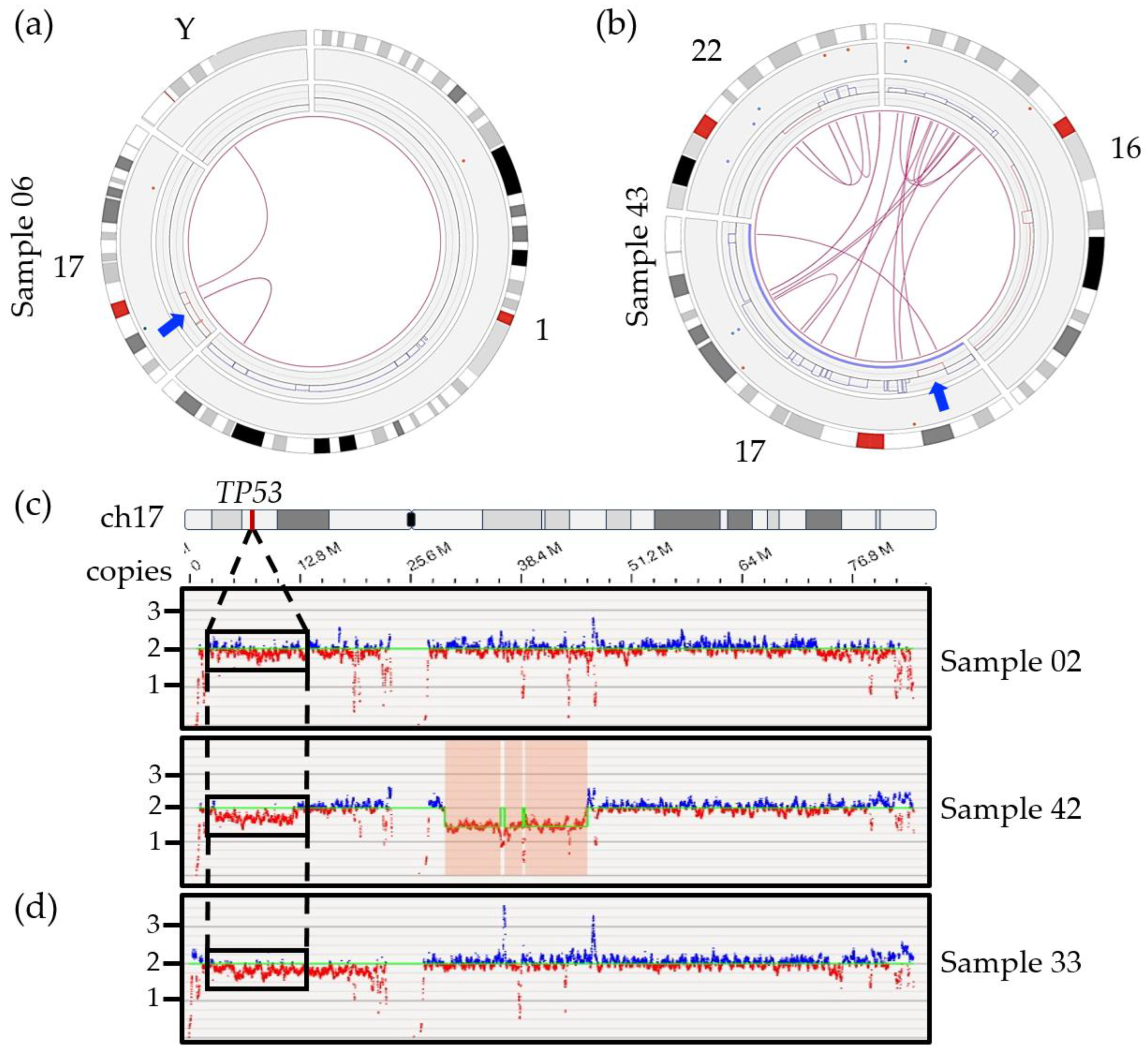
3.6.1. Detection of 17p/TP53 Deletion
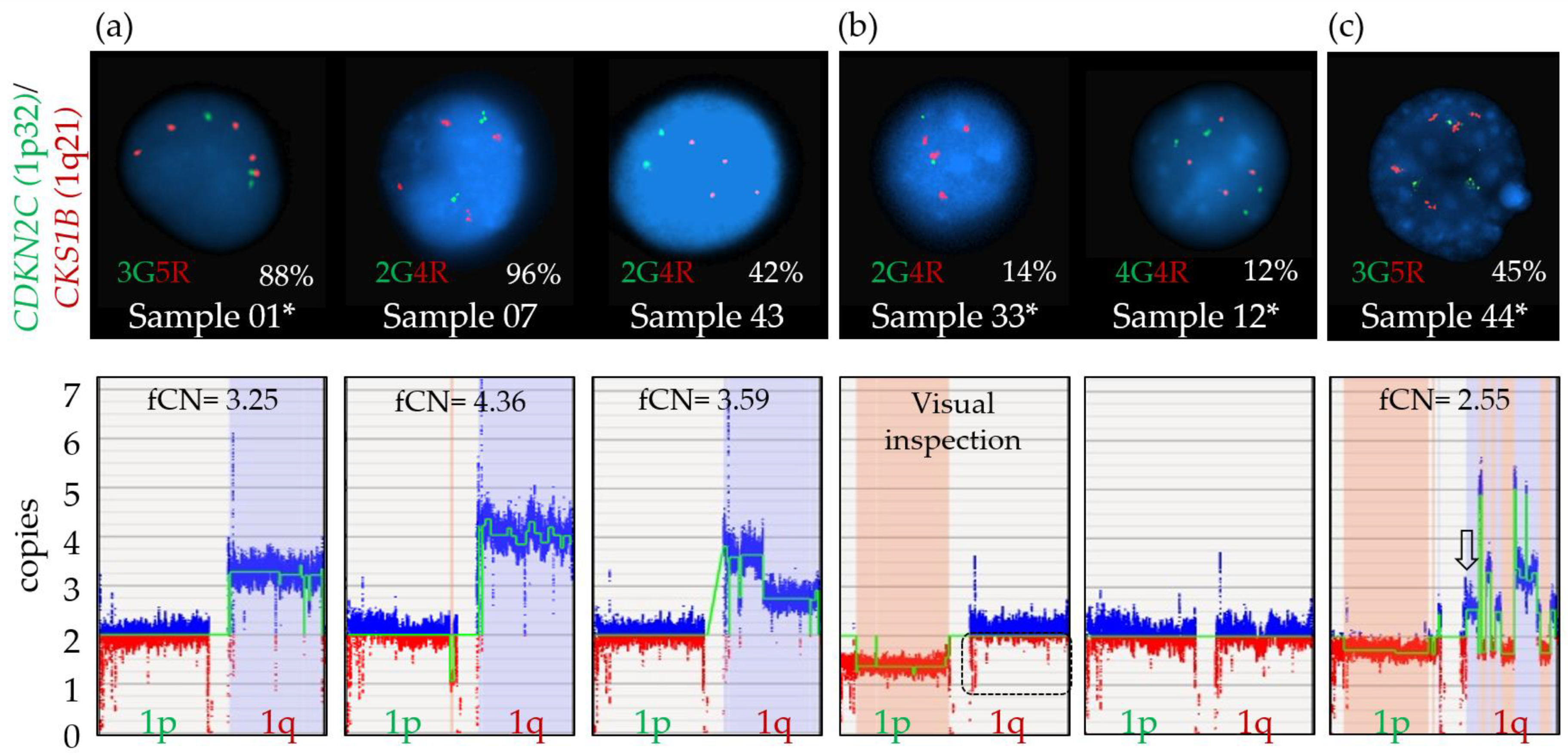
3.6.2. Chromosome 1 Abnormalities
3.6.3. 8q24.21/MYC Abnormalities
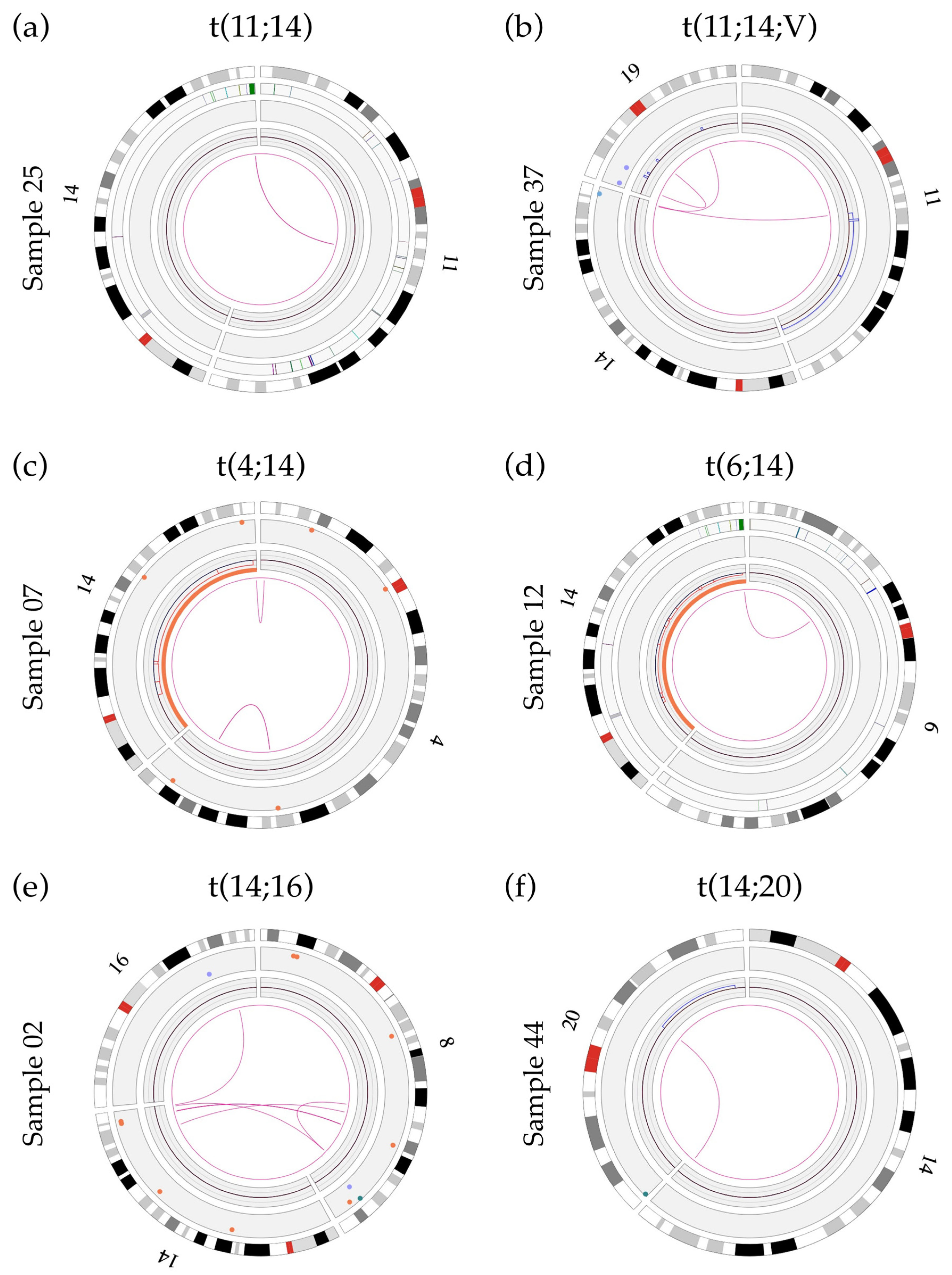
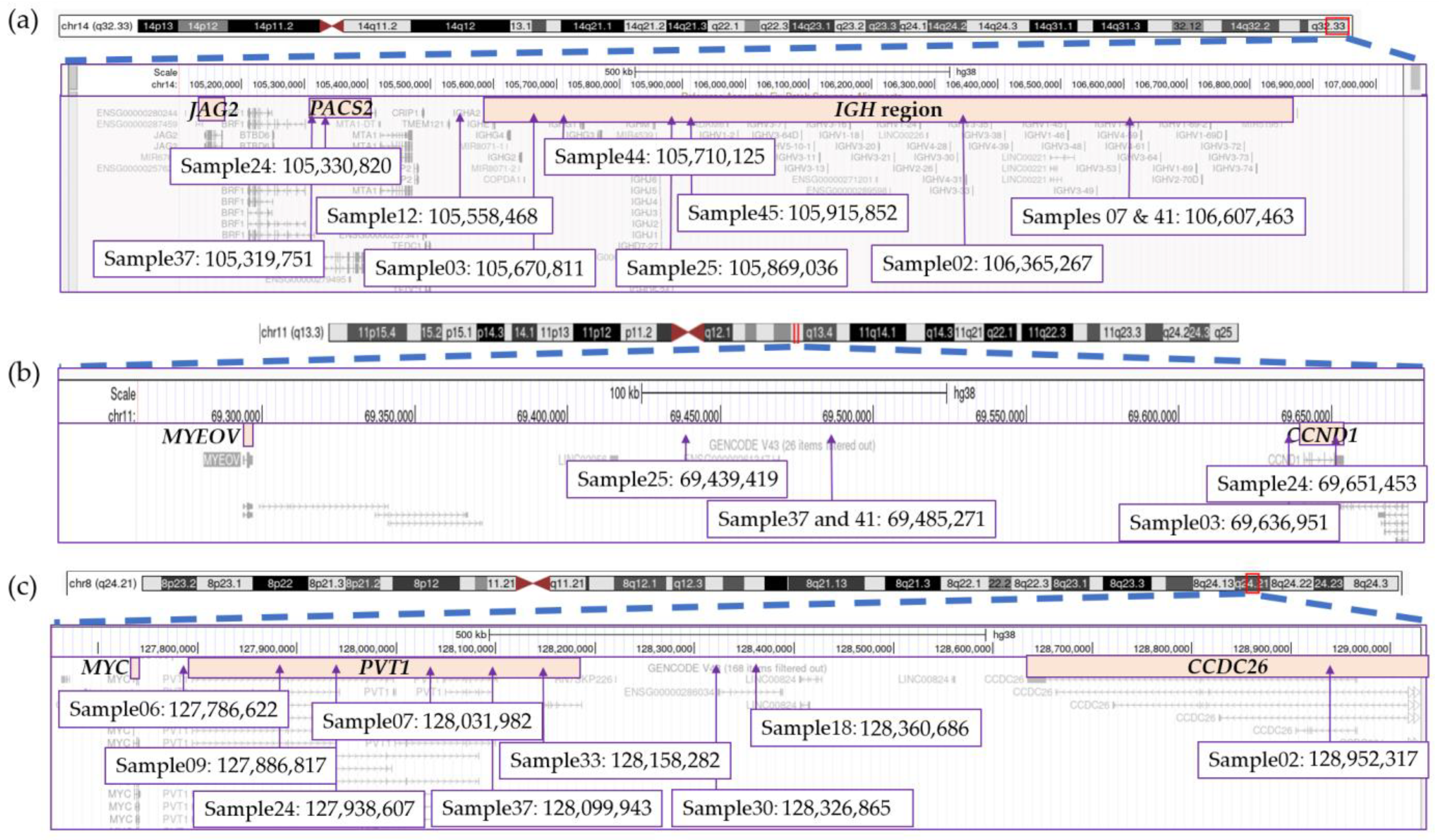
3.7. Significant Variability Found in Translocation Breakpoints
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Palumbo, A.; Anderson, K. Multiple Myeloma. N. Engl. J. Med. 2011, 364, 1046–1060. [Google Scholar] [CrossRef]
- Cardona-benavides, I.J.; de Ramón, C.; Gutiérrez, N.C. Genetic Abnormalities in Multiple Myeloma: Prognostic and Therapeutic Implications. Cells 2021, 10, 336. [Google Scholar] [CrossRef]
- Smadja, N.V.; Bastard, C.; Brigaudeau, C.; Leroux, D.; Fruchart, C. Hypodiploidy Is a Major Prognostic Factor in Multiple Myeloma. Blood 2001, 98, 2229–2238. [Google Scholar] [CrossRef] [PubMed]
- Gabrea, A.; Leif Bergsagel, P.; Michael Kuehl, W. Distinguishing Primary and Secondary Translocations in Multiple Myeloma. DNA Repair 2006, 5, 1225–1233. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Su, T.; Zhou, X.; Hu, W.-X.; Hu, J. Chromosome 1 Instability in Multiple Myeloma: Aberrant Gene Expression, Pathogenesis, and Potential Therapeutic Target. FASEB J. 2022, 36, e22341. [Google Scholar] [CrossRef] [PubMed]
- Flynt, E.; Bisht, K.; Sridharan, V.; Ortiz, M.; Towfic, F.; Thakurta, A. Prognosis, Biology, and Targeting of TP53 Dysregulation in Multiple Myeloma. Cells 2020, 9, 287. [Google Scholar] [CrossRef] [PubMed]
- Bolli, N.; Avet-Loiseau, H.; Wedge, D.C.; Van Loo, P.; Alexandrov, L.B.; Martincorena, I.; Dawson, K.J.; Iorio, F.; Nik-Zainal, S.; Bignell, G.R.; et al. Heterogeneity of Genomic Evolution and Mutational Profiles in Multiple Myeloma. Nat. Commun. 2014, 5, 2997. [Google Scholar] [CrossRef]
- Walker, B.A.; Boyle, E.M.; Wardell, C.P.; Murison, A.; Begum, D.B.; Dahir, N.M.; Proszek, P.Z.; Johnson, D.C.; Kaiser, M.F.; Melchor, L.; et al. Mutational Spectrum, Copy Number Changes, and Outcome: Results of a Sequencing Study of Patients with Newly Diagnosed Myeloma. J. Clin. Oncol. 2015, 33, 3911–3920. [Google Scholar] [CrossRef]
- Cleynen, A.; Szalat, R.; Kemal Samur, M.; Robiou du Pont, S.; Buisson, L.; Boyle, E.; Chretien, M.L.; Anderson, K.; Minvielle, S.; Moreau, P.; et al. Expressed Fusion Gene Landscape and Its Impact in Multiple Myeloma. Nat. Commun. 2017, 8, 1893. [Google Scholar] [CrossRef]
- Maura, F.; Boyle, E.M.; Rustad, E.H.; Ashby, C.; Kaminetzky, D.; Bruno, B.; Braunstein, M.; Bauer, M.; Blaney, P.; Wang, Y.; et al. Chromothripsis as a Pathogenic Driver of Multiple Myeloma. Semin. Cell Dev. Biol. 2022, 123, 115–123. [Google Scholar] [CrossRef]
- Palumbo, A.; Avet-Loiseau, H.; Oliva, S.; Lokhorst, H.M.; Goldschmidt, H.; Rosinol, L.; Richardson, P.; Caltagirone, S.; Lahuerta, J.J.; Facon, T.; et al. Revised International Staging System for Multiple Myeloma: A Report from International Myeloma Working Group. J. Clin. Oncol. 2015, 33, 2863–2869. [Google Scholar] [CrossRef] [PubMed]
- D’agostino, M.; Cairns, D.A.; Lahuerta, J.J.; Wester, R.; Bertsch, U.; Waage, A.; Zamagni, E.; Mateos, M.V.; Dall’olio, D.; Van De Donk, N.W.C.J.; et al. Second Revision of the International Staging System (R2-ISS) for Overall Survival in Multiple Myeloma: A European Myeloma Network (EMN) Report within the HARMONY Project. J. Clin. Oncol. 2022, 40, 3406–3418. [Google Scholar] [CrossRef]
- Rajkumar, S.V. Multiple Myeloma: 2022 Update on Diagnosis, Risk Stratification, and Management. Am. J. Hematol. 2022, 97, 1086–1107. [Google Scholar] [CrossRef] [PubMed]
- Sonneveld, P.; Avet-Loiseau, H.; Lonial, S.; Usmani, S.; Siegel, D.; Anderson, K.C.; Chng, W.-J.J.; Moreau, P.; Attal, M.; Kyle, R.A.; et al. Treatment of Multiple Myeloma with High-Risk Cytogenetics: A Consensus of the International Myeloma Working Group. Blood 2016, 127, 2955–2962. [Google Scholar] [CrossRef] [PubMed]
- Abdallah, N.; Baughn, L.B.; Rajkumar, S.V.; Kapoor, P.; Gertz, M.A.; Dispenzieri, A.; Lacy, M.Q.; Hayman, S.R.; Buadi, F.K.; Dingli, D.; et al. Implications of MYC Rearrangements in Newly Diagnosed Multiple Myeloma. Clin. Cancer Res. 2020, 26, 6581–6588. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, R.; Bhutani, D.; Lentzsch, S. How Do We Manage t(11;14) Plasma Cell Disorders with Venetoclax? Br. J. Haematol. 2022, 199, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Puertas, B.; González-Calle, V.; Sobejano-Fuertes, E.; Escalante, F.; Rey-Bua, B.; Padilla, I.; García-Sanz, R.; Puig, N.; Gutiérrez, N.C.; Mateos, M.-V. Multiple Myeloma with t(11;14): Impact of Novel Agents on Outcome. Blood Cancer J. 2023, 13, 40. [Google Scholar] [CrossRef]
- Fonseca, R.; Bergsagel, P.L.; Drach, J.; Shaughnessy, J.; Gutierrez, N.; Stewart, A.K.; Morgan, G.; Van Ness, B.; Chesi, M.; Minvielle, S.; et al. International Myeloma Working Group Molecular Classification of Multiple Myeloma: Spotlight Review. Leukemia 2009, 23, 2210–2221. [Google Scholar] [CrossRef]
- Fonseca, R.; Barlogie, B.; Bataille, R.; Bastard, C.; Bergsagel, P.L.; Chesi, M.; Davies, F.E.; Drach, J.; Greipp, P.R.; Kirsch, I.R.; et al. Genetics and Cytogenetics of Multiple Myeloma: A Workshop Report. Cancer Res. 2004, 64, 1546–1558. [Google Scholar] [CrossRef]
- He, H.; Fu, W.; Jiang, H.; Du, J.; Zhou, L.; Zhang, C.; Xi, H.; Li, R.; Hou, J. The Clinical Characteristics and Prognosis of IGH Deletion in Multiple Myeloma. Leuk. Res. 2015, 39, 515–519. [Google Scholar] [CrossRef]
- Smith, S.C.; Althof, P.A.; Dave, B.J.; Sanmann, J.N. High-Risk Cytogenetics in Multiple Myeloma: Further Scrutiny of Deletions within the IGH Gene Region Enhances Risk Stratification. Genes. Chromosomes Cancer 2020, 59, 569–574. [Google Scholar] [CrossRef] [PubMed]
- Rabani, H.; Ziv, M.; Lavi, N.; Aviv, A.; Suriu, C.; Shalata, A.; Haddid, Y.; Tadmor, T. Deletions and Amplifications of the IGH Variable and Constant Regions: A Novel Prognostic Parameter in Patients with Multiple Myeloma. Leuk. Res. 2020, 99, 106476. [Google Scholar] [CrossRef]
- Coccaro, N.; Anelli, L.; Zagaria, A.; Tarantini, F.; Cumbo, C.; Tota, G.; Minervini, C.F.; Minervini, A.; Conserva, M.R.; Redavid, I.; et al. Feasibility of Optical Genome Mapping in Cytogenetic Diagnostics of Hematological Neoplasms: A New Way to Look at DNA. Diagnostics 2023, 13, 1841. [Google Scholar] [CrossRef]
- Valkama, A.; Vorimo, S.; Kumpula, T.A.; Räsänen, H.; Savolainen, E.-R.; Pylkäs, K.; Mantere, T. Optical Genome Mapping as an Alternative to FISH-Based Cytogenetic Assessment in Chronic Lymphocytic Leukemia. Cancers 2023, 15, 1294. [Google Scholar] [CrossRef] [PubMed]
- Lühmann, J.L.; Stelter, M.; Wolter, M.; Kater, J.; Lentes, J.; Bergmann, A.K.; Schieck, M.; Göhring, G.; Möricke, A.; Cario, G.; et al. The Clinical Utility of Optical Genome Mapping for the Assessment of Genomic Aberrations in Acute Lymphoblastic Leukemia. Cancers 2021, 13, 4388. [Google Scholar] [CrossRef] [PubMed]
- Suttorp, J.; Lühmann, J.L.; Behrens, Y.L.; Göhring, G.; Steinemann, D.; Reinhardt, D.; von Neuhoff, N.; Schneider, M. Optical Genome Mapping as a Diagnostic Tool in Pediatric Acute Myeloid Leukemia. Cancers 2022, 14, 2058. [Google Scholar] [CrossRef] [PubMed]
- Soler, G.; Ouedraogo, Z.G.; Goumy, C.; Lebecque, B.; Aspas Requena, G.; Ravinet, A.; Kanold, J.; Véronèse, L.; Tchirkov, A. Optical Genome Mapping in Routine Cytogenetic Diagnosis of Acute Leukemia. Cancers 2023, 15, 2131. [Google Scholar] [CrossRef]
- Gerding, W.M.; Tembrink, M.; Nilius-Eliliwi, V.; Mika, T.; Dimopoulos, F.; Ladigan-Badura, S.; Eckhardt, M.; Pohl, M.; Wünnenberg, M.; Farshi, P.; et al. Optical Genome Mapping Reveals Additional Prognostic Information Compared to Conventional Cytogenetics in AML/MDS Patients. Int. J. Cancer 2022, 150, 1998–2011. [Google Scholar] [CrossRef]
- Kriegova, E.; Fillerova, R.; Minarik, J.; Savara, J.; Manakova, J.; Petrackova, A.; Dihel, M.; Balcarkova, J.; Krhovska, P.; Pika, T.; et al. Whole-Genome Optical Mapping of Bone-Marrow Myeloma Cells Reveals Association of Extramedullary Multiple Myeloma with Chromosome 1 Abnormalities. Sci. Rep. 2021, 11, 14671. [Google Scholar] [CrossRef]
- Hassan, H.; Szalat, R. Genetic Predictors of Mortality in Patients with Multiple Myeloma. Appl. Clin. Genet. 2021, 14, 241–254. [Google Scholar] [CrossRef]
- Marzocchi, G.; Castagnetti, F.; Luatti, S.; Baldazzi, C.; Stacchini, M.; Gugliotta, G.; Amabile, M.; Specchia, G.; Sessarego, M.; Giussani, U.; et al. Variant Philadelphia Translocations: Molecular-Cytogenetic Characterization and Prognostic Influence on Frontline Imatinib Therapy, a GIMEMA Working Party on CML Analysis. Blood 2011, 117, 6793–6800. [Google Scholar] [CrossRef] [PubMed]
- Nishida, K.; Yashige, H.; Maekawa, T.; Fujii, H.; Taniwaki, M.; Horiike, S.; Misawa, S.; Inazawa, J.; Abe, T. Chromosome Rearrangement, t(6;14) (P21.1;Q32.3), in Multiple Myeloma. Br. J. Haematol. 1989, 71, 295–296. [Google Scholar] [CrossRef] [PubMed]
- Sonoki, T.; Harder, L.; Horsman, D.E.; Karran, L.; Taniguchi, I.; Willis, T.G.; Gesk, S.; Steinemann, D.; Zucca, E.; Schlegelberger, B.; et al. Cyclin D3 Is a Target Gene of t(6;14)(P21.1;Q32.3) of Mature B-Cell Malignancies. Blood 2001, 98, 2837–2844. [Google Scholar] [CrossRef] [PubMed]
- Shaughnessy, J.; Gabrea, A.; Qi, Y.; Brents, L.; Zhan, F.; Tian, E.; Sawyer, J.; Barlogie, B.; Leif Bergsagel, P.; Kuehl, M. Cyclin D3 at 6p21 Is Dysregulated by Recurrent Chromosomal Translocations to Immunoglobulin Loci in Multiple Myeloma. Blood 2001, 98, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Saxe, D.; Seo, E.-J.; Bergeron, M.B.; Han, J.-Y. Recent Advances in Cytogenetic Characterization of Multiple Myeloma. Int. J. Lab. Hematol. 2019, 41, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Fonseca, R.; Ketterling, R.P.; Dispenzieri, A.; Lacy, M.Q.; Gertz, M.A.; Hayman, S.R.; Buadi, F.K.; Dingli, D.; Knudson, R.A.; et al. Trisomies in Multiple Myeloma: Impact on Survival in Patients with High-Risk Cytogenetics. Blood 2012, 119, 2100–2105. [Google Scholar] [CrossRef] [PubMed]
- Avet-Loiseau, H.; Attal, M.; Moreau, P.; Charbonnel, C.; Garban, F.; Hulin, C.; Leyvraz, S.; Michallet, M.; Yakoub-Agha, I.; Garderet, L.; et al. Genetic Abnormalities and Survival in Multiple Myeloma: The Experience of the Intergroupe Francophone Du Myélome. Blood 2007, 109, 3489–3495. [Google Scholar] [CrossRef] [PubMed]
- Wuilleme, S.; Robillard, N.; Lodé, L.; Magrangeas, F.; Beris, H.; Harousseau, J.-L.; Proffitt, J.; Minvielle, S.; Avet-Loiseau, H.; Minvielle, S.; et al. Ploidy, as Detected by Fluorescence in Situ Hybridization, Defines Different Subgroups in Multiple Myeloma. Leukemia 2005, 19, 275–278. [Google Scholar] [CrossRef]
- Avet-Louseau, H.; Daviet, A.; Sauner, S.; Bataille, R. Chromosome 13 Abnormalities in Multiple Myeloma Are Mostly Monosomy 13. Br. J. Haematol. 2000, 111, 1116–1117. [Google Scholar] [CrossRef]
- Locher, M.; Jukic, E.; Vogi, V.; Keller, M.A.; Kröll, T.; Schwendinger, S.; Oberhuber, K.; Verdorfer, I.; Mühlegger, B.E.; Witsch-Baumgartner, M.; et al. Amp(1q) and Tetraploidy Are Commonly Acquired Chromosomal Abnormalities in Relapsed Multiple Myeloma. Eur. J. Haematol. 2023, 110, 296–304. [Google Scholar] [CrossRef]
- Sidana, S.; Jevremovic, D.; Ketterling, R.P.; Tandon, N.; Greipp, P.T.; Baughn, L.B.; Dispenzieri, A.; Gertz, M.A.; Rajkumar, S.V.; Kumar, S.K. Tetraploidy Is Associated with Poor Prognosis at Diagnosis in Multiple Myeloma. Am. J. Hematol. 2019, 94, E117–E120. [Google Scholar] [CrossRef] [PubMed]
- Rücker, F.G.; Schlenk, R.F.; Bullinger, L.; Kayser, S.; Teleanu, V.; Kett, H.; Habdank, M.; Kugler, C.-M.; Holzmann, K.; Gaidzik, V.I.; et al. TP53 Alterations in Acute Myeloid Leukemia with Complex Karyotype Correlate with Specific Copy Number Alterations, Monosomal Karyotype, and Dismal Outcome. Blood 2012, 119, 2114–2121. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Kim, H.T.; Kasar, S.; Benien, P.; Du, W.; Hoang, K.; Aw, A.; Tesar, B.; Improgo, R.; Fernandes, S.; et al. Survival of Del17p CLL Depends on Genomic Complexity and Somatic Mutation. Clin. Cancer Res. 2017, 23, 735–745. [Google Scholar] [CrossRef] [PubMed]
- Haferlach, C.; Dicker, F.; Herholz, H.; Schnittger, S.; Kern, W.; Haferlach, T. Mutations of the TP53 Gene in Acute Myeloid Leukemia Are Strongly Associated with a Complex Aberrant Karyotype. Leukemia 2008, 22, 1539–1541. [Google Scholar] [CrossRef] [PubMed]
- Seifert, H.; Mohr, B.; Thiede, C.; Oelschlägel, U.; Schäkel, U.; Illmer, T.; Soucek, S.; Ehninger, G.; Schaich, M. The Prognostic Impact of 17p (P53) Deletion in 2272 Adults with Acute Myeloid Leukemia. Leukemia 2009, 23, 656–663. [Google Scholar] [CrossRef] [PubMed]
- Pang, A.W.C.; Kosco, K.; Sahajpal, N.; Sridhar, A.; Hauenstein, J.; Clifford, B.; Estabrook, J.; Chitsazan, A.; Sahoo, T.; Iqbal, A.; et al. Clinical Validation of Optical Genome Mapping for the Detection of Structural Variations in Hematological Malignancies. medRxiv 2022. [Google Scholar] [CrossRef]
- Levy, B.; Baughn, L.B.; Akkari, Y.; Chartrand, S.; LaBarge, B.; Claxton, D.; Lennon, P.A.; Cujar, C.; Kolhe, R.; Kroeger, K.; et al. Optical Genome Mapping in Acute Myeloid Leukemia: A Multicenter Evaluation. Blood Adv. 2023, 7, 1297–1307. [Google Scholar] [CrossRef] [PubMed]
- Puiggros, A.; Ramos-Campoy, S.; Kamaso, J.; de la Rosa, M.; Salido, M.; Melero, C.; Rodríguez-Rivera, M.; Bougeon, S.; Collado, R.; Gimeno, E.; et al. Optical Genome Mapping: A Promising New Tool to Assess Genomic Complexity in Chronic Lymphocytic Leukemia (CLL). Cancers 2022, 14, 3376. [Google Scholar] [CrossRef]
- Sahajpal, N.S.; Mondal, A.K.; Tvrdik, T.; Hauenstein, J.; Shi, H.; Deeb, K.K.; Saxe, D.; Hastie, A.R.; Chaubey, A.; Savage, N.M.; et al. Clinical Validation and Diagnostic Utility of Optical Genome Mapping for Enhanced Cytogenomic Analysis of Hematological Neoplasms. J. Mol. Diagn. 2022, 24, 1279–1291. [Google Scholar] [CrossRef]
- Corre, J.; Perrot, A.; Caillot, D.; Belhadj, K.; Hulin, C.; Leleu, X.; Mohty, M.; Facon, T.; Buisson, L.; Do Souto, L.; et al. Del(17p) without TP53 Mutation Confers a Poor Prognosis in Intensively Treated Newly Diagnosed Patients with Multiple Myeloma. Blood 2021, 137, 1192–1195. [Google Scholar] [CrossRef]
- Marcon, C.; Simeon, V.; Deias, P.; Facchin, G.; Corso, A.; Derudas, D.; Montefusco, V.; Offidani, M.; Petrucci, M.T.; Zambello, R.; et al. Experts’ Consensus on the Definition and Management of High Risk Multiple Myeloma. Front. Oncol. 2022, 12, 1096852. [Google Scholar] [CrossRef] [PubMed]
- Thakurta, A.; Ortiz, M.; Blecua, P.; Towfic, F.; Corre, J.; Serbina, N.V.; Flynt, E.; Yu, Z.; Yang, Z.; Palumbo, A.; et al. High Subclonal Fraction of 17p Deletion Is Associated with Poor Prognosis in Multiple Myeloma. Blood 2019, 133, 1217–1221. [Google Scholar] [CrossRef] [PubMed]
- Martello, M.; Poletti, A.; Borsi, E.; Solli, V.; Dozza, L.; Barbato, S.; Zamagni, E.; Tacchetti, P.; Pantani, L.; Mancuso, K.; et al. Clonal and Subclonal TP53 Molecular Impairment Is Associated with Prognosis and Progression in Multiple Myeloma. Blood Cancer J. 2022, 12, 15. [Google Scholar] [CrossRef] [PubMed]
- Mikhael, J.R.; Dingli, D.; Roy, V.; Reeder, C.B.; Buadi, F.K.; Hayman, S.R.; Dispenzieri, A.; Fonseca, R.; Sher, T.; Kyle, R.A.; et al. Management of Newly Diagnosed Symptomatic Multiple Myeloma: Updated Mayo Stratification of Myeloma and Risk-Adapted Therapy (MSMART) Consensus Guidelines 2013. Mayo Clin. Proc. 2013, 88, 360–376. [Google Scholar] [CrossRef] [PubMed]
- Zweegman, S.; van de Donk, N.W.C.J. Deletion 17p: A Matter of Size and Number? Blood 2021, 137, 1135–1136. [Google Scholar] [CrossRef]
- Boyd, K.D.; Ross, F.M.; Walker, B.A.; Wardell, C.P.; Tapper, W.J.; Chiecchio, L.; Dagrada, G.; Konn, Z.J.; Gregory, W.M.; Jackson, G.H.; et al. Mapping of Chromosome 1p Deletions in Myeloma Identifies FAM46C at 1p12 and CDKN2C at 1p32.3 as Being Genes in Regions Associated with Adverse Survival. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2011, 17, 7776–7784. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.A.; Leone, P.E.; Chiecchio, L.; Dickens, N.J.; Jenner, M.W.; Boyd, K.D.; Johnson, D.C.; Gonzalez, D.; Dagrada, G.P.; Protheroe, R.K.M.; et al. A Compendium of Myeloma-Associated Chromosomal Copy Number Abnormalities and Their Prognostic Value. Blood 2010, 116, e56–e65. [Google Scholar] [CrossRef]
- Mulligan, G.; Lichter, D.I.; Di Bacco, A.; Blakemore, S.J.; Berger, A.; Koenig, E.; Bernard, H.; Trepicchio, W.; Li, B.; Neuwirth, R.; et al. Mutation of NRAS but Not KRAS Significantly Reduces Myeloma Sensitivity to Single-Agent Bortezomib Therapy. Blood 2014, 123, 632–639. [Google Scholar] [CrossRef]
- Shirazi, F.; Jones, R.J.; Singh, R.K.; Zou, J.; Kuiatse, I.; Berkova, Z.; Wang, H.; Lee, H.C.; Hong, S.; Dick, L.; et al. Activating KRAS, NRAS, and BRAF Mutants Enhance Proteasome Capacity and Reduce Endoplasmic Reticulum Stress in Multiple Myeloma. Proc. Natl. Acad. Sci. USA 2020, 117, 20004–20014. [Google Scholar] [CrossRef]
- Panopoulou, A.; Cairns, D.A.; Holroyd, A.; Nichols, I.; Cray, N.; Pawlyn, C.; Cook, G.; Drayson, M.; Boyd, K.; Davies, F.E.; et al. Optimizing the Value of Lenalidomide Maintenance by Extended Genetic Profiling: An Analysis of 556 Patients in the Myeloma XI Trial. Blood 2023, 141, 1666–1674. [Google Scholar] [CrossRef]
- Walker, B.A.; Mavrommatis, K.; Wardell, C.P.; Ashby, T.C.; Bauer, M.; Davies, F.; Rosenthal, A.; Wang, H.; Qu, P.; Hoering, A.; et al. A High-Risk, Double-Hit, Group of Newly Diagnosed Myeloma Identified by Genomic Analysis. Leukemia 2019, 33, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The Human Genome Browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.A.; Wardell, C.P.; Johnson, D.C.; Kaiser, M.F.; Begum, D.B.; Dahir, N.B.; Ross, F.M.; Davies, F.E.; Gonzalez, D.; Morgan, G.J. Characterization of IGH Locus Breakpoints in Multiple Myeloma Indicates a Subset of Translocations Appear to Occur in Pregerminal Center B Cells. Blood 2013, 121, 3413–3419. [Google Scholar] [CrossRef] [PubMed]
- Misund, K.; Keane, N.; Stein, C.K.; Asmann, Y.W.; Day, G.; Welsh, S.; Van Wier, S.A.; Riggs, D.L.; Ahmann, G.; Chesi, M.; et al. MYC Dysregulation in the Progression of Multiple Myeloma. Leukemia 2020, 34, 322–326. [Google Scholar] [CrossRef] [PubMed]
- Chong, L.C.; Ben-Neriah, S.; Slack, G.W.; Freeman, C.; Ennishi, D.; Mottok, A.; Collinge, B.; Abrisqueta, P.; Farinha, P.; Boyle, M.; et al. High-Resolution Architecture and Partner Genes of MYC Rearrangements in Lymphoma with DLBCL Morphology. Blood Adv. 2018, 2, 2755–2765. [Google Scholar] [CrossRef] [PubMed]
- Stong, N.; Ortiz-Estévez, M.; Towfic, F.; Samur, M.; Agarwal, A.; Corre, J.; Flynt, E.; Munshi, N.; Avet-Loiseau, H.; Thakurta, A. The Location of the t(4;14) Translocation Breakpoint within the NSD2 Gene Identifies a Subset of Patients with High-Risk NDMM. Blood 2023, 141, 1574–1583. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Age * | Disease | Prior Lines of Therapy | Isotype | BM Plasma Cells/cPC (%) | R-ISS | HD-MEL | Interval from HD-MEL to Sample (Months) | Other Characteristics |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 61 | NDMM | IgG kappa | 26/0 | 1 | No | |||
| 2 | 59 | RRMM | 3 | IgA kappa | 60/0 | NA | Yes | 7 | |
| 3 | 56 | RRMM | 1 | FLC kappa | 28/- | NA | Yes | 19 | |
| 5 | 67 | NDMM | IgG kappa | 70/0 | 2 | No | |||
| 6 | 72 | RRMM | 6 | IgA kappa | 36/0 | 3 | No | Radiation therapy for prostate cancer 11 years before sample collection | |
| 7 | 68 | sPCL | 4 | IgG lambda | 94/13 | 3 | Yes | 48 | |
| 9 | 58 | NDMM | IgG kappa | 19/0 | 1 | No | |||
| 12 | 82 | SMM | IgG lambda | 10/0 | - | No | Recent diagnosis of MDS at sample collection | ||
| 18 | 71 | RRMM | 6 | IgG kappa | 35/0 | 2 | Yes | 81 | ASCT tandem |
| 24 | 82 | NDMM | FLC lambda | 25/0 | 2 | No | |||
| 25 | 72 | NDMM | IgG kappa | 60/- | 2 | No | |||
| 30 | 65 | NDMM | IgG lambda | 65/- | 2 | No | |||
| 33 | 62 | NDMM | FLC lambda | 80/1 | 3 | No | |||
| 37 | 76 | NDMM | FLC kappa | 25/0 | 2 | No | |||
| 38 | 69 | NDMM | IgG kappa | 97/0 | 3 | No | |||
| 41 | 84 | RRMM | 4 | IgA kappa | 33/- | NA | No | ||
| 42 | 79 | NDMM | IgG kappa | 65/0 | NA | No | |||
| 43 | 83 | NDMM | IgA kappa | 48/0 | 3 | No | |||
| 44 | 88 | NDMM | IgG kappa | 14/0 | 2 | No | |||
| 45 | 59 | RRMM | 1 | IgA kappa | 44/3 | NA | Yes | 6 |
| Samples | FISH (% of Abnormal Cells) | OGM (fCN/VAF) | Concordance |
|---|---|---|---|
| 1 (non-diploid) | TP53/D17Z1 (4 × 81%) | Gain of chromosome 17 | Yes |
| FGFR3 (3 × 90%) | Gain of chromosomes 5, 6, 7, 9, 15, 19 | Ploidy * | |
| IGH (3 × 45%, 4 × 46%) | Gain of chromosome 14 | Yes | |
| MAFB (3 × 94%) | Ploidy * | ||
| 1p32/CDKN2C (3 × 88%) | Ploidy * | ||
| 1q21/CKS1B amplification (5 × 88%) | 1q21/CKS1B amplification (3.25/0.63) | Yes | |
| 2 | Deletion 17p/TP53 (1 × 6%) | Deletion 17p/TP53 (visual inspection) | Yes |
| t(14;16) (2F 98%) | t(14;16) (0.48) | Yes | |
| Deletion 1p32/CDKN2C (1 × 22%) | Deletion 1p32/CDKN2C (1.73/0.135) | Yes | |
| 1q21/CKS1B gain (3 × 71%) | 1q21/CKS1B gain (2.72/0.36) | Yes | |
| 3 | Deletion 17p/TP53 (1 × 95%) | Deletion 17p/TP53 (0.91/0.54) | Yes |
| t(11;14) (2F 77%, 3F 17%) | t(11;14) (0.55) | Yes | |
| 5 | Normal | Yes | |
| Gain of chromosomes 3, 5, 9, 11, 15, 19 | N/A | ||
| 6 | Deletion 17p/TP53 (1 × 75%) | Deletion 17p/TP53 (0.81/0.59) | Yes |
| Gain of chromosomes 3, 7, 11, 15 | N/A | ||
| 1q21/CKS1B gain (3 × 70%) | 1q21/CKS1B gain (2.86/0.43) | Yes | |
| 7 | t(4;14) (1F 97%) | t(4;14) (0.51) | Yes |
| 1q21/CKS1B amplification (4 × 96%) | 1q21/CKS1B amplification (4.36/1.18) | Yes | |
| 9 | IGH (1 × 6%) | Gain of chromosomes 3, 5, 9, 11, 15, 19, 21 | No |
| 12 (non-diploid) | TP53/D17Z1 (4 × 14%) | t(6;14) (0.52) | Ploidy * |
| FGFR3 (4 × 16%) | Ploidy * | ||
| IGH (3x 18%, 4 × 13%) | Yes | ||
| MAF (4 × 14%) | Ploidy * | ||
| MAFB (4 × 10%) | Ploidy * | ||
| 1p32/CDKN2C (4 × 12%) | Ploidy * | ||
| 1q21/CKS1B amplification (4 × 12%) | 1q amplification not detected | Ploidy * | |
| 18 | Deletion 17p/TP53 (1 × 98%) | Deletion 17p/TP53 (1.02/0.49) | Yes |
| Deletion 1p32/CDKN2C (1 × 96%) | Deletion 1p32/CDKN2C (1.05/0.47) | Yes | |
| Gain of chromosomes 3, 5, 7, 9, 11, 15, 21 | N/A | ||
| 24 | t(11;14) (2F 87%) | t(11;14) (0.24) | Yes |
| 1q21/CKS1B gain (3 × 95%) | 1q21/CKS1B gain (2.92/0.46) | Yes | |
| 25 | t(11;14) (2F 97%) | t(11;14) (0.69) | Yes |
| 30 | Gain of chromosomes 3, 5, 7, 15, 18, 19 | N/A | |
| 1q21/CKS1B gain (3 × 60%) | 1q21/CKS1B gain (2.41/0.20) | Yes | |
| 33 (non-diploid) | TP53/D17Z1 (2 × 17.5%, 3 × 70%) | Deletion 1p32/CDKN2C (1.37/0.32) | Ploidy * |
| FGFR3 (3 × 64%) | Gain of chromosomes 3, 5, 7, 9, 15 | Ploidy * | |
| MAFB (3 × 91%) | Ploidy * | ||
| 1q21/CKS1B (3 × 81%, 4 × 14%) | 1q21/CKS1B gain by visual inspection | Yes | |
| 37 | t(11;14) (1F 89%) | t(11;14) (0.45) | Yes |
| 1q21/CKS1B gain (3 × 49%) | 1q21/CKS1B gain (2.32/0.16) | Yes | |
| 38 | Gain of chromosomes 9, 11, 15, 19 | N/A | |
| 1q21/CKS1B gain (3 × 91%) | 1q21/CKS1B gain (3.05/0.53) | Yes | |
| 41 | t(11;14) (2F 96%) | t(11;14) (0.25) | Yes |
| 1q21/CKS1B gain (3 × 95%) | 1q21/CKS1B gain (2.99/0.49) | Yes | |
| 42 | Gain of chromosomes 5, 9, 11, 15, 18, 19 | N/A | |
| Deletion 17p/TP53 (1 × 45%) | Deletion 17p/TP53 by visual inspection | Yes | |
| Deletion 1p32/CDKN2C (1 × 78%, 0 × 12%) | Deletion 1p32 CNV (1.08/0.46), SV targeted deletion CDKN2C (0.9/0.22) | Yes | |
| 43 | Gain of chromosomes 3, 5, 7, 9, 11, 17, 18, 19 | N/A | |
| Deletion 17p/TP53 (1 × 97%%) | Deletion 17p/TP53 (1.04/0.48) | Yes | |
| 1q21/CKS1B amplification (3 × 29%, 4 × 42%; 5 × 19%) | 1q21/CKS1B amplification (3.59/0.8) | Yes | |
| 44 (non-diploid) | TP53/D17Z1 (3 × 5%, 4 × 17%, 5 × 41%) | Gain of chromosome 17 | Yes |
| FGFR3 (3 × 59%) | Ploidy * | ||
| MAF (3 × 35%) | Ploidy * | ||
| t(14;20) (2F 66%, 3F 43%) | t(14;20) (0.28) | Yes | |
| 1p32/CDKN2C (3 × 50,4%) | Deletion 1p32/CDKN2C (1.72/0.14) | Ploidy * | |
| 1q21/CKS1B amplification (3 × 4%, 4 × 21%, 5 × 45%) | 1q21/CKS1B gain (2.55/0.68) | Ploidy * | |
| 45 (non-diploid) | TP53/D17Z1 (3 × 67%) | Deletion 1p32/CDKN2C (1.42/0.29) | Ploidy * |
| t(4;14) (2F: 8%; 3F: 85%) | t(4;14) (0.28) | Yes | |
| 1q21/CKS1B amplification (5 × 4%, 6 × 28%, 7 × 49%, 8 × 12%) | 1q21/CSK1B amplification (4.15/1.08) | Ploidy * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giguère, A.; Raymond-Bouchard, I.; Collin, V.; Claveau, J.-S.; Hébert, J.; LeBlanc, R. Optical Genome Mapping Reveals the Complex Genetic Landscape of Myeloma. Cancers 2023, 15, 4687. https://doi.org/10.3390/cancers15194687
Giguère A, Raymond-Bouchard I, Collin V, Claveau J-S, Hébert J, LeBlanc R. Optical Genome Mapping Reveals the Complex Genetic Landscape of Myeloma. Cancers. 2023; 15(19):4687. https://doi.org/10.3390/cancers15194687
Chicago/Turabian StyleGiguère, Amélie, Isabelle Raymond-Bouchard, Vanessa Collin, Jean-Sébastien Claveau, Josée Hébert, and Richard LeBlanc. 2023. "Optical Genome Mapping Reveals the Complex Genetic Landscape of Myeloma" Cancers 15, no. 19: 4687. https://doi.org/10.3390/cancers15194687