
Differentially Expressed Genes, miRNAs and Network Models: A Strategy to Shed Light on Molecular Interactions Driving HNSCC Tumorigenesis
 , , and
, , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. RNAseq Data, from miRNAs to Differentially Expressed Genes (DEGs)
2.2. Extraction of miRNA Targets and Bipartite Network Reconstruction
2.3. Functional and Topological Analysis of PPI Networks Reconstructed from miRNA Targets and DEGs
2.4. Differentially Expressed miRNAs and Survival Analysis
3. Results
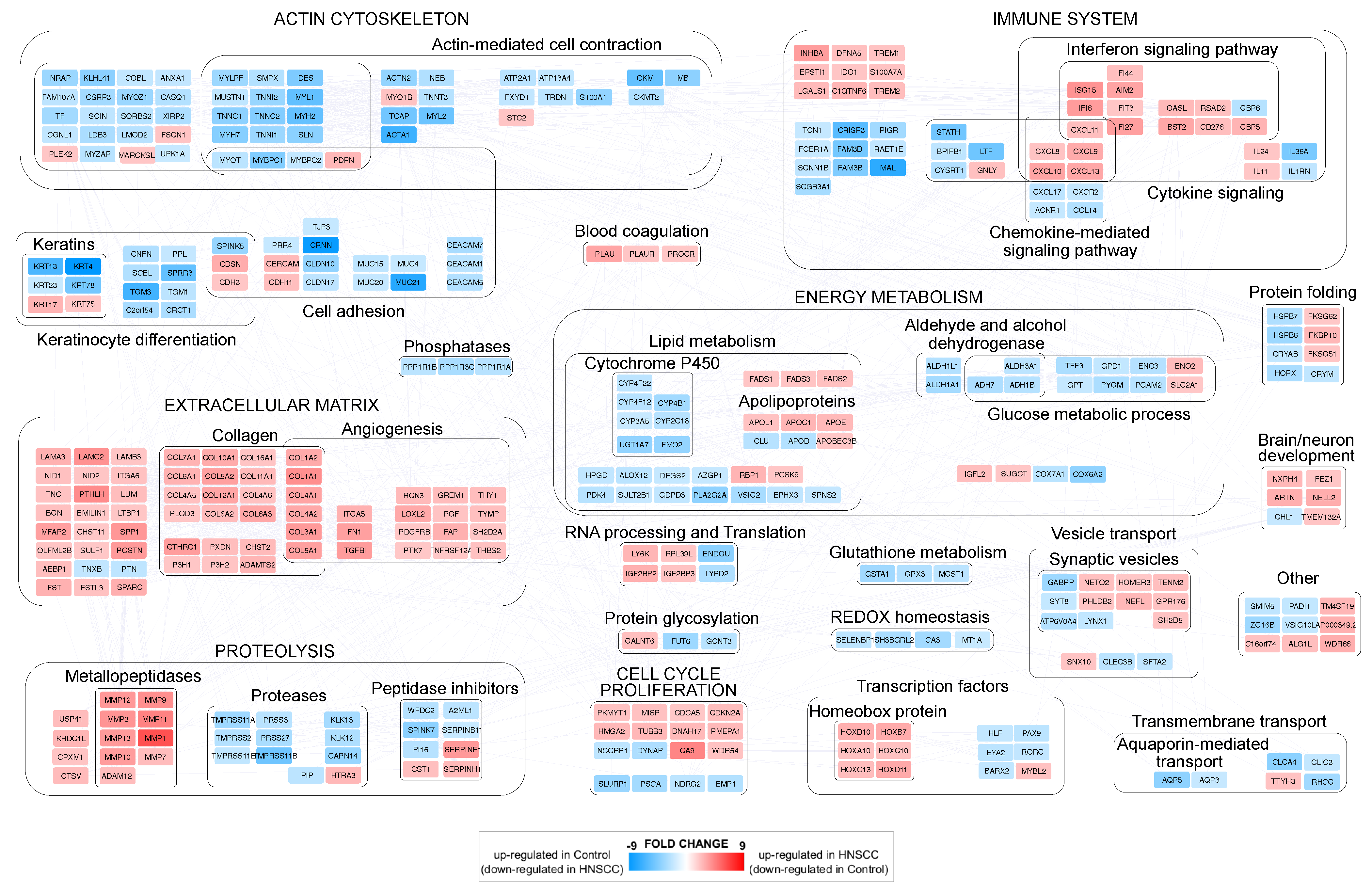
3.1. Differentially Expressed Genes (DEGs) in Head and Neck Cancer
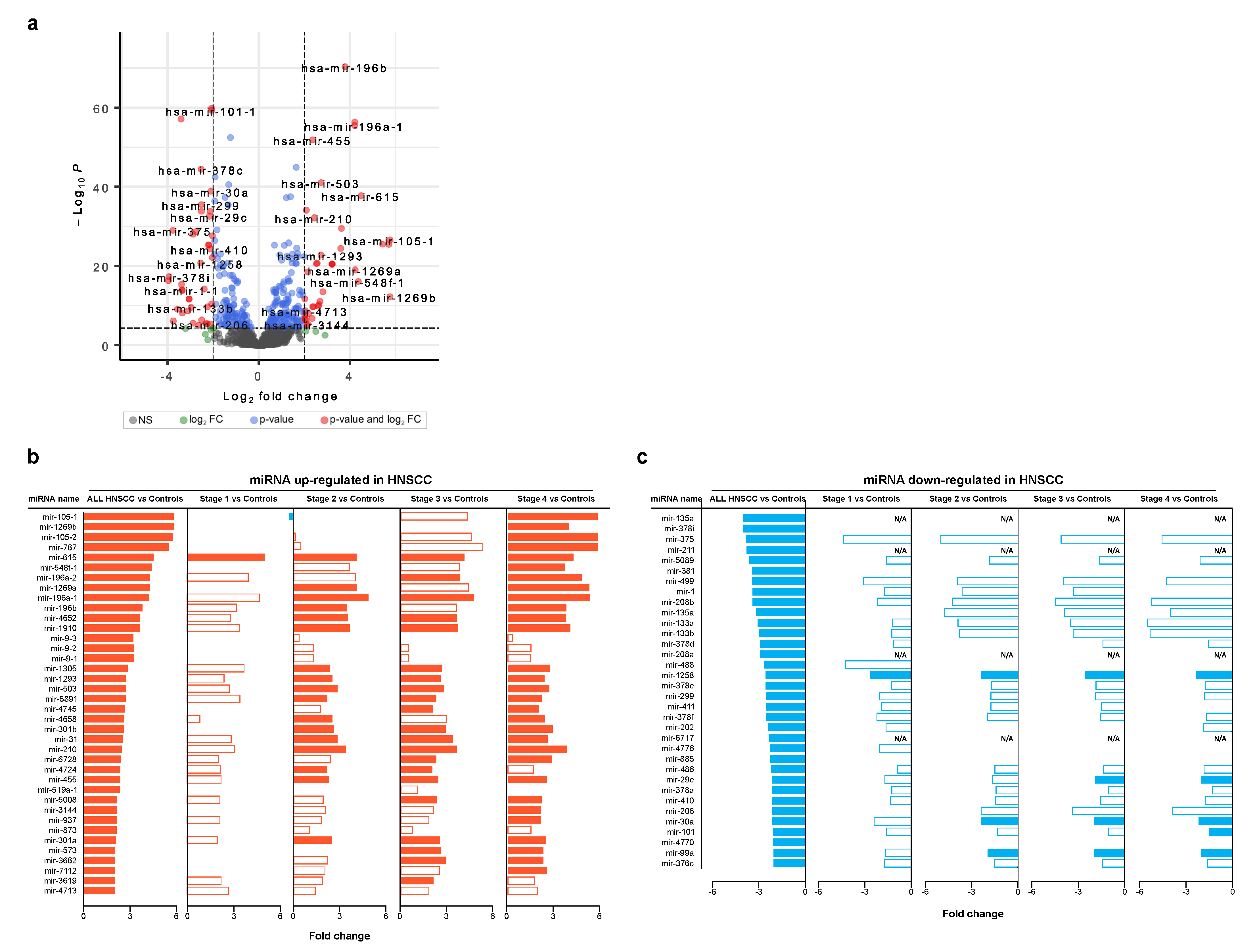
3.2. Differentially Expressed miRNAs in Head and Neck Cancer
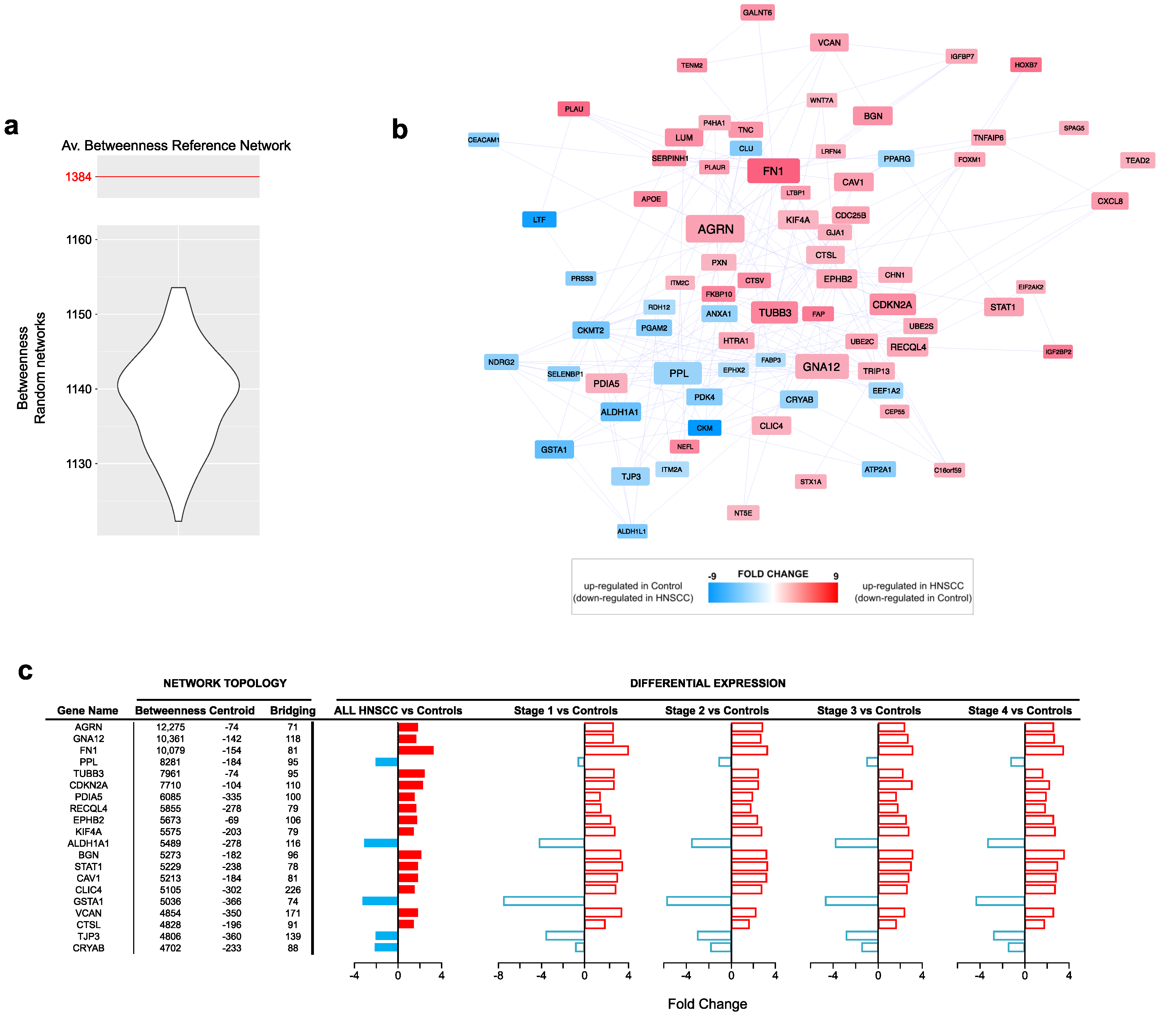
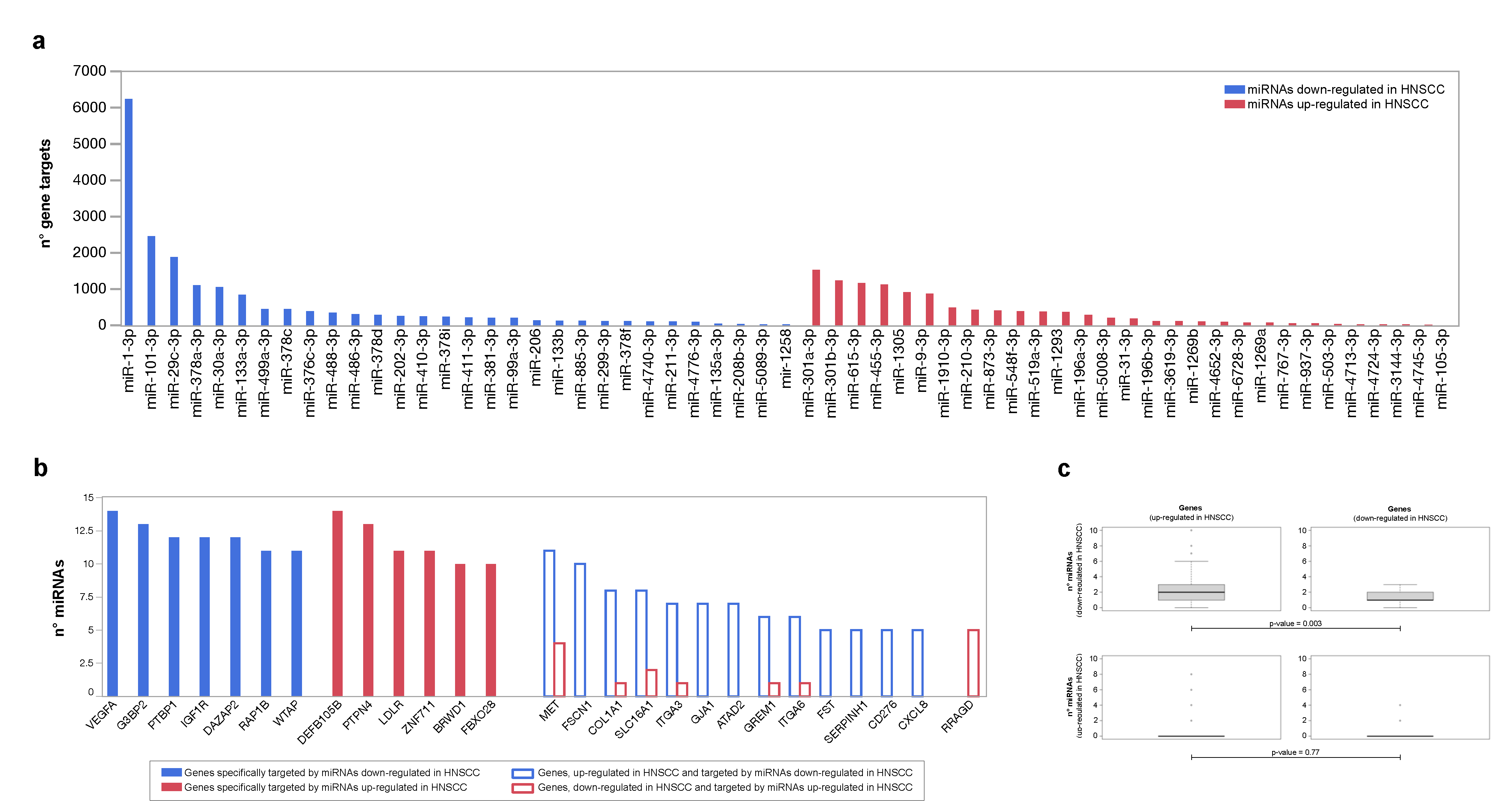
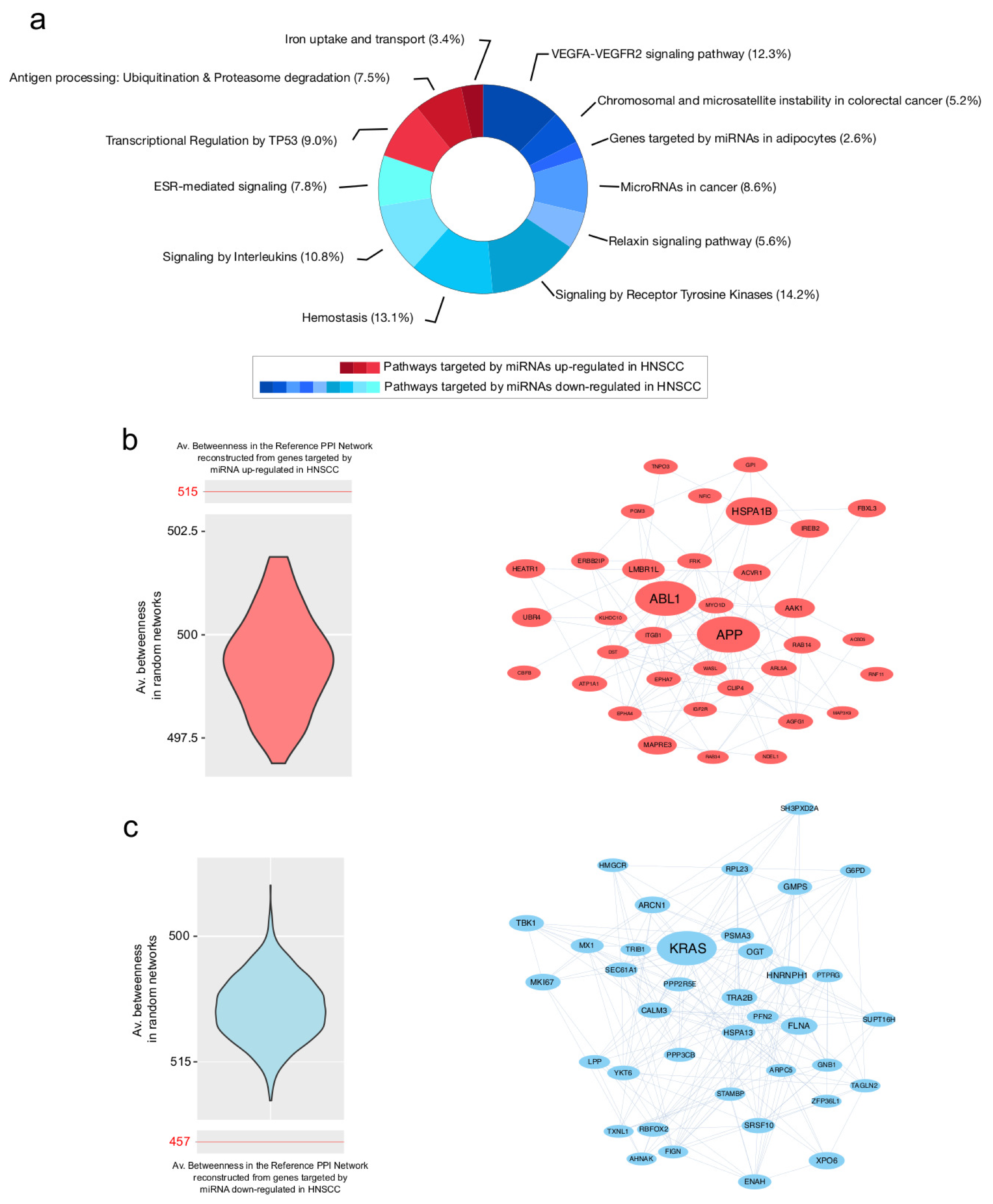
3.3. PPI Network Models of Genes Most Targeted by Up- and Downregulated miRNAs: From Modulated Pathways to Hubs
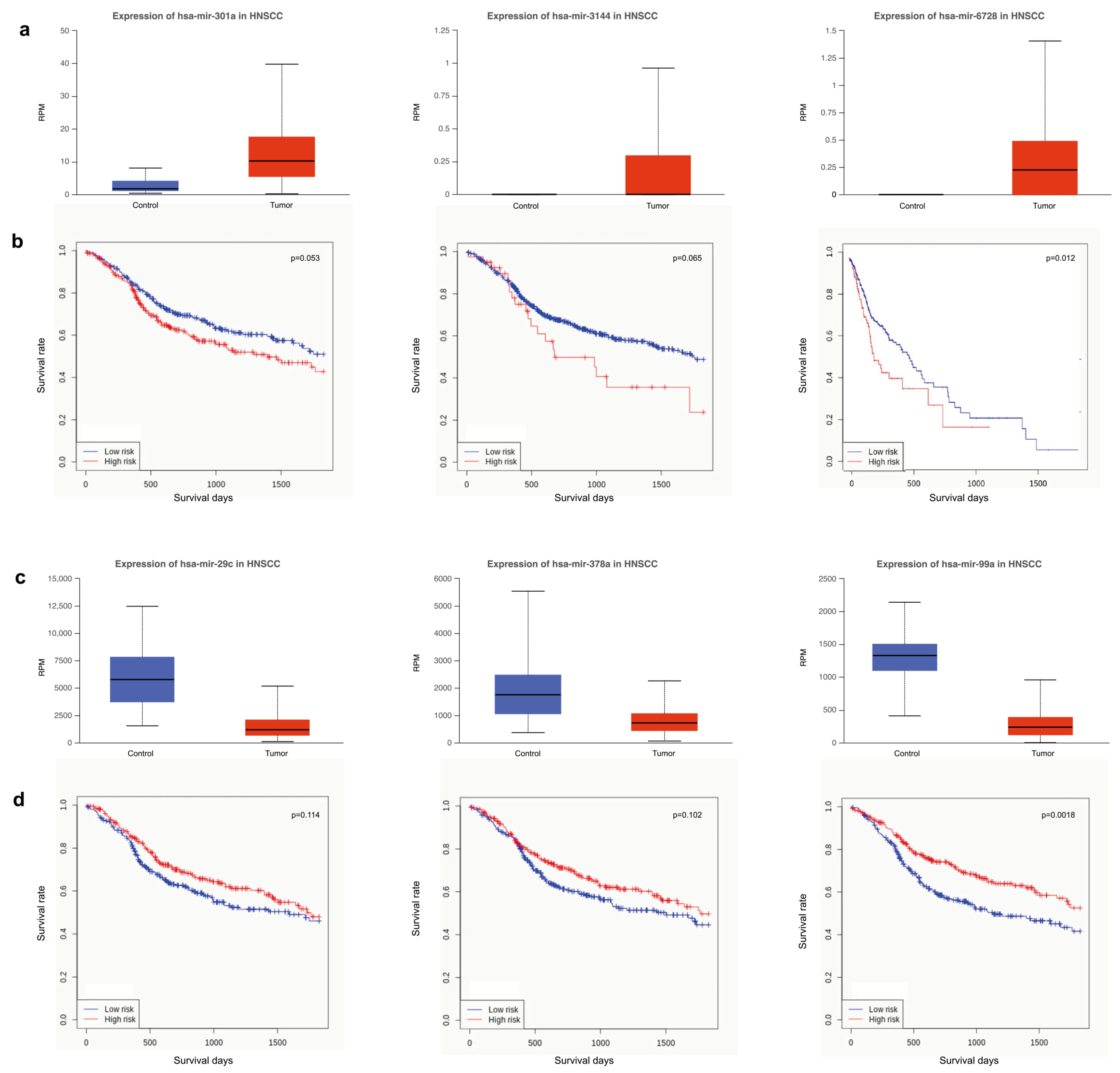
3.4. Survival Analysis Using miRNAs Targeting Key Tumor Suppressors and Oncogenes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| HNSCC | Head and Neck Squamous Cell Carcinoma |
| PPI | Protein–Protein Interaction |
| FC | Log2(Fold Change) |
| DEGs | Differentially Expressed Genes |
| STAR | Spliced Transcripts Alignment to a Reference |
| RPM | Reads Per Million |
| GLN | Generalized Linear Model |
| FDR | False Discovery Rate |
| ECM | Extracellular Matrix |
References
- Gormley, M.; Creaney, G.; Schache, A.; Ingarfield, K.; Conway, D.I. Reviewing the epidemiology of head and neck cancer: Definitions, trends and risk factors. Br. Dent. J. 2022, 233, 780–786. [Google Scholar] [CrossRef] [PubMed]
- Massa, S.T.; Cass, L.M.; Challapalli, S.; Zahirsha, Z.; Simpson, M.; Ward, G.; Osazuwa-Peters, N. Demographic predictors of head and neck cancer survival differ in the elderly. Laryngoscope 2019, 129, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Kanno, H.; Nagata, H.; Ishiguro, A.; Tsuzuranuki, S.; Nakano, S.; Nonaka, T.; Kiyohara, K.; Kimura, T.; Sugawara, A.; Okazaki, Y.; et al. Designation Products: Boron Neutron Capture Therapy for Head and Neck Carcinoma. Oncologist 2021, 26, e1250–e1255. [Google Scholar] [CrossRef] [PubMed]
- Hajmohammadi, E.; Molaei, T.; Mowlaei, S.H.; Alam, M.; Abbasi, K.; Khayatan, D.; Rahbar, M.; Tebyanian, H. Sonodynamic therapy and common head and neck cancers: In vitro and in vivo studies. Eur. Rev. Med. Pharmacol. Sci. 2021, 25, 5113–5121. [Google Scholar] [CrossRef]
- Shibata, H.; Saito, S.; Uppaluri, R. Immunotherapy for Head and Neck Cancer: A Paradigm Shift From Induction Chemotherapy to Neoadjuvant Immunotherapy. Front. Oncol. 2021, 11, 727433. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, A.; Burtness, B. Treating Head and Neck Cancer in the Age of Immunotherapy: A 2023 Update. Drugs 2023, 83, 217–248. [Google Scholar] [CrossRef]
- Mascolo, M.; Siano, M.; Ilardi, G.; Russo, D.; Merolla, F.; Rosa, G.D.; Staibano, S. Epigenetic disregulation in oral cancer. Int. J. Mol. Sci. 2012, 13, 2331–2353. [Google Scholar] [CrossRef] [PubMed]
- Pentenero, M.; Bowers, L.; Jayasinghe, R.; Cheong, S.C.; Farah, C.S.; Kerr, A.R.; Alevizos, I. World Workshop on Oral Medicine VII: Functional pathways involving differentially expressed lncRNAs in oral squamous cell carcinoma. Oral Dis. 2019, 25 (Suppl. 1), 79–87. [Google Scholar] [CrossRef] [PubMed]
- Hussain, A.; Tebyaniyan, H.; Khayatan, D. The Role of Epigenetic in Dental and Oral Regenerative Medicine by Different Types of Dental Stem Cells: A Comprehensive Overview. Stem Cells Int. 2022, 2022, 5304860. [Google Scholar] [CrossRef] [PubMed]
- Iorio, M.V.; Croce, C.M. Causes and consequences of microRNA dysregulation. Cancer J. 2012, 18, 215–222. [Google Scholar] [CrossRef]
- Fang, C.; Li, Y. Prospective applications of microRNAs in oral cancer. Oncol. Lett. 2019, 18, 3974–3984. [Google Scholar] [CrossRef] [PubMed]
- El-Mahdy, H.A.; Mohamadin, A.M.; Abulsoud, A.I.; Khidr, E.G.; El-Husseiny, A.A.; Ismail, A.; Elsakka, E.G.E.; Mokhlis, H.A.; El-Husseiny, H.M.; Doghish, A.S. miRNAs as potential game-changers in head and neck cancer: Future clinical and medicinal uses. Pathol. Res. Pract. 2023, 245, 154457. [Google Scholar] [CrossRef] [PubMed]
- Rosolen, D.; Nunes-Souza, E.; Marchi, R.; Tofolo, M.V.; Antunes, V.C.; Berti, F.C.B.; Fonseca, A.S.; Cavalli, L.R. MiRNAs Action and Impact on Mitochondria Function, Metabolic Reprogramming and Chemoresistance of Cancer Cells: A Systematic Review. Biomedicines 2023, 11, 693. [Google Scholar] [CrossRef]
- Ali Syeda, Z.; Langden, S.S.S.; Munkhzul, C.; Lee, M.; Song, S.J. Regulatory Mechanism of MicroRNA Expression in Cancer. Int. J. Mol. Sci. 2020, 21, 1723. [Google Scholar] [CrossRef]
- Kozaki, K.i.; Imoto, I.; Mogi, S.; Omura, K.; Inazawa, J. Exploration of tumor-suppressive microRNAs silenced by DNA hypermethylation in oral cancer. Cancer Res. 2008, 68, 2094–2105. [Google Scholar] [CrossRef]
- Taş, B.; Güre, A.O. The effect of Maras powder and smoking on the microRNA deregulation of oral mucosa. J. Appl. Oral Sci. Rev. FOB 2020, 28, e20190382. [Google Scholar] [CrossRef]
- Shiah, S.G.; Hsiao, J.R.; Chang, H.J.; Hsu, Y.M.; Wu, G.H.; Peng, H.Y.; Chou, S.T.; Kuo, C.C.; Chang, J.Y. MiR-30a and miR-379 modulate retinoic acid pathway by targeting DNA methyltransferase 3B in oral cancer. J. Biomed. Sci. 2020, 27, 46. [Google Scholar] [CrossRef]
- Paluszczak, J. The Significance of the Dysregulation of Canonical Wnt Signaling in Head and Neck Squamous Cell Carcinomas. Cells 2020, 9, 723. [Google Scholar] [CrossRef]
- Alotaibi, M.; Valova, V.; HÄnsel, T.; Stromberger, C.; Kofla, G.; Olze, H.; Piwonski, I.; Albers, A.; Ochsenreither, S.; Coordes, A. Impact of Smoking on the Survival of Patients With High-risk HPV-positive HNSCC: A Meta-analysis. In Vivo 2021, 35, 1017–1026. [Google Scholar] [CrossRef]
- Stephen, J.K.; Worsham, M.J. Human papilloma virus (HPV) modulation of the HNSCC epigenome. Methods Mol. Biol. 2015, 1238, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Ghiani, L.; Chiocca, S. High Risk-Human Papillomavirus in HNSCC: Present and Future Challenges for Epigenetic Therapies. Int. J. Mol. Sci. 2022, 23, 3483. [Google Scholar] [CrossRef]
- Subha, S.T.; Chin, J.W.; Cheah, Y.K.; Mohtarrudin, N.; Saidi, H.I. Multiple microRNA signature panel as promising potential for diagnosis and prognosis of head and neck cancer. Mol. Biol. Rep. 2022, 49, 1501–1511. [Google Scholar] [CrossRef]
- Dioguardi, M.; Caloro, G.A.; Laino, L.; Alovisi, M.; Sovereto, D.; Crincoli, V.; Aiuto, R.; Coccia, E.; Troiano, G.; Lo Muzio, L. Circulating miR-21 as a Potential Biomarker for the Diagnosis of Oral Cancer: A Systematic Review with Meta-Analysis. Cancers 2020, 12, 936. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Lu, J.; Wang, F.; Liu, X.; Peng, X.; Yu, B.; Zhao, F.; Li, X. Dynamic Changes in Plasma MicroRNAs Have Potential Predictive Values in Monitoring Recurrence and Metastasis of Nasopharyngeal Carcinoma. BioMed Res. Int. 2018, 2018, 7329195. [Google Scholar] [CrossRef] [PubMed]
- Yi, S.J.; Liu, P.; Chen, B.L.; Ou-Yang, L.; Xiong, W.M.; Su, J.P. Circulating miR-31-5p may be a potential diagnostic biomarker in nasopharyngeal carcinoma. Neoplasma 2019, 66, 825–829. [Google Scholar] [CrossRef]
- Shibata, T.; Cao, D.Y.; Dar, T.B.; Ahmed, F.; Bhat, S.A.; Veiras, L.C.; Bernstein, E.A.; Khan, A.A.; Chaum, M.; Shiao, S.L.; et al. miR766-3p and miR124-3p Dictate Drug Resistance and Clinical Outcome in HNSCC. Cancers 2022, 14, 5273. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Wang, J.; Liang, F.; Song, P.; Yan, X.; Wu, S.; Huang, X.; Han, P. Identification of key genes for HNSCC from public databases using bioinformatics analysis. Cancer Cell Int. 2021, 21, 549. [Google Scholar] [CrossRef]
- Zimmermann, M.; Zouhair, A.; Azria, D.; Ozsahin, M. The epidermal growth factor receptor (EGFR) in head and neck cancer: Its role and treatment implications. Radiat. Oncol. 2006, 1, 11. [Google Scholar] [CrossRef] [PubMed]
- Sigismund, S.; Avanzato, D.; Lanzetti, L. Emerging functions of the EGFR in cancer. Mol. Oncol. 2018, 12, 3–20. [Google Scholar] [CrossRef]
- Napolitano, V.; Russo, D.; Morra, F.; Merolla, F.; Varricchio, S.; Ilardi, G.; Di Crescenzo, R.M.; Martino, F.; Mascolo, M.; Celetti, A.; et al. Neuropilin-1 Expression Associates with Poor Prognosis in HNSCC and Elicits EGFR Activation upon CDDP-Induced Cytotoxic Stress. Cancers 2021, 13, 3822. [Google Scholar] [CrossRef]
- Ganci, F.; Sacconi, A.; Bossel Ben-Moshe, N.; Manciocco, V.; Sperduti, I.; Strigari, L.; Covello, R.; Benevolo, M.; Pescarmona, E.; Domany, E.; et al. Expression of TP53 mutation-associated microRNAs predicts clinical outcome in head and neck squamous cell carcinoma patients. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2013, 24, 3082–3088. [Google Scholar] [CrossRef] [PubMed]
- Vahabi, M.; Blandino, G.; Di Agostino, S. MicroRNAs in head and neck squamous cell carcinoma: A possible challenge as biomarkers, determinants for the choice of therapy and targets for personalized molecular therapies. Transl. Cancer Res. 2021, 10, 3090–3110. [Google Scholar] [CrossRef]
- Dong, F.; Su, L.; Tan, J.; Luo, H. The anticancer effect of EGFR-targeting artificial microRNA controlled by SLPI promoter in nasopharyngeal carcinoma. J. Clin. Lab. Anal. 2022, 36, e24729. [Google Scholar] [CrossRef]
- Vella, D.; Zoppis, I.; Mauri, G.; Mauri, P.; Di Silvestre, D. From protein-protein interactions to protein co-expression networks: A new perspective to evaluate large-scale proteomic data. EURASIP J. Bioinform. Syst. Biol. 2017, 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Sanati, N.; Iancu, O.D.; Wu, G.; Jacobs, J.E.; McWeeney, S.K. Network-Based Predictors of Progression in Head and Neck Squamous Cell Carcinoma. Front. Genet. 2018, 9, 183. [Google Scholar] [CrossRef]
- Ma, J.; Yao, Y.; Tao, D.T.; Yin, J.Y.; Chen, L.; Cui, Y.; Wang, Y.; Hu, C.L.; Tian, M.X.; Wang, Q. Comprehensive and Integrated Analysis Identifies ZEB1 as a Key Novel Gene in Oral Squamous Cell Carcinoma. Contrast Media Mol. Imaging 2022, 2022, 4486104. [Google Scholar] [CrossRef] [PubMed]
- Li, C.Y.; Cai, J.H.; Tsai, J.J.P.; Wang, C.C.N. Identification of Hub Genes Associated With Development of Head and Neck Squamous Cell Carcinoma by Integrated Bioinformatics Analysis. Front. Oncol. 2020, 10, 681. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Cheng, D.; Guo, J.; Chen, H.; Zhang, S.; Bao, Y. SOX9 and IL1A as the Potential Gene Biomarkers of the Oral Cancer. Comb. Chem. High Throughput Screen. 2023, 26, 1461–1479. [Google Scholar] [CrossRef]
- Fang, L.; Shi, L.; Wang, W.; Chen, Q.; Rao, X. Identifying key genes and small molecule compounds for nasopharyngeal carcinoma by various bioinformatic analysis. Medicine 2021, 100, e27257. [Google Scholar] [CrossRef]
- Di Silvestre, D.; Garavelli, S.; Procaccini, C.; Prattichizzo, F.; Passignani, G.; De Rosa, V.; Mauri, P.; Matarese, G.; de Candia, P. CD4, T-Cell Activation Prompts Suppressive Function by Extracellular Vesicle-Associated MicroRNAs. Front. Cell Dev. Biol. 2021, 9, 753884. [Google Scholar] [CrossRef] [PubMed]
- Procaccini, C.; Garavelli, S.; Carbone, F.; Di Silvestre, D.; La Rocca, C.; Greco, D.; Colamatteo, A.; Lepore, M.T.; Russo, C.; De Rosa, G.; et al. Signals of pseudo-starvation unveil the amino acid transporter SLC7A11 as key determinant in the control of Treg cell proliferative potential. Immunity 2021, 54, 1543–1560.e6. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Fernandez, S.; Brock, G. Power analysis for RNA-Seq differential expression studies using generalized linear mixed effects models. BMC Bioinform. 2020, 21, 198. [Google Scholar] [CrossRef]
- Xiao, F.; Zuo, Z.; Cai, G.; Kang, S.; Gao, X.; Li, T. miRecords: An integrated resource for microRNA-target interactions. Nucleic Acids Res. 2009, 37, D105–D110. [Google Scholar] [CrossRef]
- Huang, H.Y.; Lin, Y.C.D.; Cui, S.; Huang, Y.; Tang, Y.; Xu, J.; Bao, J.; Li, Y.; Wen, J.; Zuo, H.; et al. miRTarBase update 2022: An informative resource for experimentally validated miRNA-target interactions. Nucleic Acids Res. 2022, 50, D222–D230. [Google Scholar] [CrossRef]
- Karagkouni, D.; Paraskevopoulou, M.D.; Chatzopoulos, S.; Vlachos, I.S.; Tastsoglou, S.; Kanellos, I.; Papadimitriou, D.; Kavakiotis, I.; Maniou, S.; Skoufos, G.; et al. DIANA-TarBase v8: A decade-long collection of experimentally supported miRNA-gene interactions. Nucleic Acids Res. 2018, 46, D239–D245. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Scardoni, G.; Tosadori, G.; Faizan, M.; Spoto, F.; Fabbri, F.; Laudanna, C. Biological network analysis with CentiScaPe: Centralities and experimental dataset integration. F1000Research 2014, 3, 139. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed]
- Di Silvestre, D.; Brambilla, F.; Lavatelli, F.; Chirivì, M.; Canetti, D.; Bearzi, C.; Rizzi, R.; Bijzet, J.; Hazenberg, B.P.C.; Bellotti, V.; et al. The Protein Network in Subcutaneous Fat Biopsies from Patients with AL Amyloidosis: More Than Diagnosis? Cells 2023, 12, 699. [Google Scholar] [CrossRef]
- Tosadori, G.; Bestvina, I.; Spoto, F.; Laudanna, C.; Scardoni, G. Creating, generating and comparing random network models with NetworkRandomizer. F1000Research 2016, 5, 2524. [Google Scholar] [CrossRef]
- Vandin, F.; Papoutsaki, A.; Raphael, B.J.; Upfal, E. Accurate computation of survival statistics in genome-wide studies. PLoS Comput. Biol. 2015, 11, e1004071. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Wang, S.; Wang, Z. Identification of genetic mechanisms underlying lipid metabolism-mediated tumor immunity in head and neck squamous cell carcinoma. BMC Med. Genom. 2023, 16, 110. [Google Scholar] [CrossRef]
- Guo, Q.; Lu, T.; Xu, H.; Luo, Q.; Liu, Z.; Jiang, S.; Pan, J.; Lin, S.; Lin, M.; Guo, F. Identification of immune-related genes contributing to head and neck squamous cell carcinoma development using weighted gene co-expression network analysis. Cancer Rep. 2023, 6, e1808. [Google Scholar] [CrossRef] [PubMed]
- Clark, E.S.; Brown, B.; Whigham, A.S.; Kochaishvili, A.; Yarbrough, W.G.; Weaver, A.M. Aggressiveness of HNSCC tumors depends on expression levels of cortactin, a gene in the 11q13 amplicon. Oncogene 2009, 28, 431–444. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.H.; Hsu, J.Y.; Chu, C.T.; Chang, Y.W.; Fan, J.R.; Yang, M.H.; Chen, H.C. Loss of cell-cell adhesion triggers cell migration through Rac1-dependent ROS generation. Life Sci. Alliance 2023, 6, e202201529. [Google Scholar] [CrossRef]
- Antra; Parashar, P.; Hungyo, H.; Jain, A.; Ahmad, S.; Tandon, V. Unraveling molecular mechanisms of head and neck cancer. Crit. Rev. Oncol. 2022, 178, 103778. [Google Scholar] [CrossRef]
- Zou, M.; Zhang, C.; Sun, Y.; Wu, H.; Xiao, F.; Gao, W.; Zhao, F.; Fan, X.; Wu, G. Comprehensive analysis of matrix metalloproteinases and their inhibitors in head and neck squamous cell carcinoma. Acta Oncol. 2022, 61, 505–515. [Google Scholar] [CrossRef]
- Sun, G.G.; Lu, Y.F.; Fu, Z.Z.; Cheng, Y.J.; Hu, W.N. EMP1 inhibits nasopharyngeal cancer cell growth and metastasis through induction apoptosis and angiogenesis. Tumour. Biol. 2014, 35, 3185–3193. [Google Scholar] [CrossRef]
- de Nooij-van Dalen, A.G.; van Dongen, G.A.M.S.; Smeets, S.J.; Nieuwenhuis, E.J.C.; Stigter-van Walsum, M.; Snow, G.B.; Brakenhoff, R.H. Characterization of the human Ly-6 antigens, the newly annotated member Ly-6K included, as molecular markers for head-and-neck squamous cell carcinoma. Int. J. Cancer 2003, 103, 768–774. [Google Scholar] [CrossRef]
- Chen, X.L.; Lei, L.; Hong, L.L.; Ling, Z.Q. Potential role of NDRG2 in reprogramming cancer metabolism and epithelial-to-mesenchymal transition. Histol. Histopathol. 2018, 33, 655–663. [Google Scholar] [CrossRef]
- Lyukmanova, E.N.; Bychkov, M.L.; Sharonov, G.V.; Efremenko, A.V.; Shulepko, M.A.; Kulbatskii, D.S.; Shenkarev, Z.O.; Feofanov, A.V.; Dolgikh, D.A.; Kirpichnikov, M.P. Human secreted proteins SLURP-1 and SLURP-2 control the growth of epithelial cancer cells via interactions with nicotinic acetylcholine receptors. Br. J. Pharmacol. 2018, 175, 1973–1986. [Google Scholar] [CrossRef]
- Wu, X.; Li, J.; Yan, T.; Ke, X.; Li, X.; Zhu, Y.; Yang, J.; Li, Z. HOXB7 acts as an oncogenic biomarker in head and neck squamous cell carcinoma. Cancer Cell Int. 2021, 21, 393. [Google Scholar] [CrossRef]
- Platais, C.; Hakami, F.; Darda, L.; Lambert, D.W.; Morgan, R.; Hunter, K.D. The role of HOX genes in head and neck squamous cell carcinoma. J. Oral Pathol. Med. 2016, 45, 239–247. [Google Scholar] [CrossRef]
- Di Leva, G.; Garofalo, M.; Croce, C.M. MicroRNAs in cancer. Annu. Rev. Pathol. 2014, 9, 287–314. [Google Scholar] [CrossRef] [PubMed]
- Gong, W.; Donnelly, C.R.; Heath, B.R.; Bellile, E.; Donnelly, L.A.; Taner, H.F.; Broses, L.; Brenner, J.C.; Chinn, S.B.; Ji, R.R.; et al. Cancer-specific type-I interferon receptor signaling promotes cancer stemness and effector CD8+ T-cell exhaustion. Oncoimmunology 2021, 10, 1997385. [Google Scholar] [CrossRef] [PubMed]
- Nisar, S.; Yousuf, P.; Masoodi, T.; Wani, N.A.; Hashem, S.; Singh, M.; Sageena, G.; Mishra, D.; Kumar, R.; Haris, M.; et al. Chemokine-Cytokine Networks in the Head and Neck Tumor Microenvironment. Int. J. Mol. Sci. 2021, 22, 4584. [Google Scholar] [CrossRef]
- Liu, J.; Zhao, J.; Xu, J.; Sun, Q.; Qin, X.; Chen, G.; Gao, T.; Bai, G.; Guo, Z. SPINK5 is a Prognostic Biomarker Associated With the Progression and Prognosis of Laryngeal Squamous Cell Carcinoma Identified by Weighted Gene Co-Expression Network Analysis. Evol. Bioinform. Online 2022, 18, 11769343221077118. [Google Scholar] [CrossRef]
- Tang, X.; Tang, Q.; Yang, X.; Xiao, Z.A.; Zhu, G.; Yang, T.; Yang, Q.; Zhang, Y.; Li, S. FN1 promotes prognosis and radioresistance in head and neck squamous cell carcinoma: From radioresistant HNSCC cell line to integrated bioinformatics methods. Front. Genet. 2022, 13, 1017762. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Vasaikar, S.; Eskaros, A.; Kim, Y.; Lewis, J.S.; Zhang, B.; Zijlstra, A.; Weaver, A.M. EPHB2 carried on small extracellular vesicles induces tumor angiogenesis via activation of ephrin reverse signaling. JCI Insight 2019, 4, e132447. [Google Scholar] [CrossRef]
- Bai, Y.T.; Wang, X.; He, M.J.; Xie, J.R.; Chen, X.J.; Zhou, G. The potential of lipid droplet-associated genes as diagnostic and prognostic biomarkers in head and neck squamous cell carcinoma. Comb. Chem. High Throughput Screen 2023. [CrossRef]
- Su, K.; Wang, Y.; Gu, H.; Ma, L.; Xuan, G. Overexpression of fatty acid desaturase 3 predicts poor prognosis in head and neck squamous cell carcinoma. Medicine 2022, 101, e32119. [Google Scholar] [CrossRef]
- Chan, N.N.; Yamazaki, M.; Maruyama, S.; Abé, T.; Haga, K.; Kawaharada, M.; Izumi, K.; Kobayashi, T.; Tanuma, J.I. Cholesterol Is a Regulator of CAV1 Localization and Cell Migration in Oral Squamous Cell Carcinoma. Int. J. Mol. Sci. 2023, 24, 6035. [Google Scholar] [CrossRef]
- Miao, X.; Wang, B.; Chen, K.; Ding, R.; Wu, J.; Pan, Y.; Ji, P.; Ye, B.; Xiang, M. Perspectives of lipid metabolism reprogramming in head and neck squamous cell carcinoma: An overview. Front. Oncol. 2022, 12, 1008361. [Google Scholar] [CrossRef]
- Li, X.; Lu, Y.; Wen, P.; Yuan, Y.; Xiao, Z.; Shi, H.; Feng, E. Matrine restrains the development of colorectal cancer through regulating the AGRN/Wnt/β-catenin pathway. Environ. Toxicol. 2023, 38, 809–819. [Google Scholar] [CrossRef]
- Li, X.; Zhang, G.; Wang, Y.; Elgehama, A.; Sun, Y.; Li, L.; Gu, Y.; Guo, W.; Xu, Q. Loss of periplakin expression is associated with the tumorigenesis of colorectal carcinoma. Biomed. Pharmacother. 2017, 87, 366–374. [Google Scholar] [CrossRef]
- Okudela, K.; Woo, T.; Mitsui, H.; Suzuki, T.; Tajiri, M.; Sakuma, Y.; Miyagi, Y.; Tateishi, Y.; Umeda, S.; Masuda, M.; et al. Downregulation of ALDH1A1 expression in non-small cell lung carcinomas–its clinicopathologic and biological significance. Int. J. Clin. Exp. Pathol. 2013, 6, 1–12. [Google Scholar] [PubMed]
- Liu, X.; Sui, X.; Zhang, C.; Wei, K.; Bao, Y.; Xiong, J.; Zhou, Z.; Chen, Z.; Wang, C.; Zhu, H.; et al. Glutathione S-transferase A1 suppresses tumor progression and indicates better prognosis of human primary hepatocellular carcinoma. J. Cancer 2020, 11, 83–91. [Google Scholar] [CrossRef]
- Liu, C.; Yu, Z.; Huang, S.; Zhao, Q.; Sun, Z.; Fletcher, C.; Jiang, Y.; Zhang, D. Combined identification of three miRNAs in serum as effective diagnostic biomarkers for HNSCC. EBioMedicine 2019, 50, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Lopez, Y.O.N.; Victoria, B.; Golusinski, P.; Golusinski, W.; Masternak, M.M. Characteristic miRNA expression signature and random forest survival analysis identify potential cancer-driving miRNAs in a broad range of head and neck squamous cell carcinoma subtypes. Rep. Pract. Oncol. Radiother. 2018, 23, 6–20. [Google Scholar] [CrossRef] [PubMed]
- Koshizuka, K.; Kikkawa, N.; Hanazawa, T.; Yamada, Y.; Okato, A.; Arai, T.; Katada, K.; Okamoto, Y.; Seki, N. Inhibition of integrin β1-mediated oncogenic signalling by the antitumor, family in head and neck squamous cell carcinoma. Oncotarget 2018, 9, 3663–3676. [Google Scholar] [CrossRef] [PubMed]
- Centuori, S.M.; Bauman, J.E. c-Met Signaling as a Therapeutic Target in Head and Neck Cancer. Cancer J. 2022, 28, 346–353. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, S.; Tanimoto, N.; Nishida, R.; Matsui, A. Functional analysis of the miR-145/Fascin1 cascade in canine oral squamous cell carcinoma. Oral Dis. 2023, 29, 1495–1504. [Google Scholar] [CrossRef] [PubMed]
- Kuang, J.; Zhao, M.; Li, H.; Dang, W.; Li, W. Identification of potential therapeutic target genes and mechanisms in head and neck squamous cell carcinoma by bioinformatics analysis. Oncol. Lett. 2016, 11, 3009–3014. [Google Scholar] [CrossRef] [PubMed]
- Khammanivong, A.; Sorenson, B.S.; Ross, K.F.; Dickerson, E.B.; Hasina, R.; Lingen, M.W.; Herzberg, M.C. Involvement of calprotectin (S100A8/A9) in molecular pathways associated with HNSCC. Oncotarget 2016, 7, 14029–14047. [Google Scholar] [CrossRef]
- Montag, M.; Dyckhoff, G.; Lohr, J.; Helmke, B.M.; Herrmann, E.; Plinkert, P.K.; Herold-Mende, C. Angiogenic growth factors in tissue homogenates of HNSCC: Expression pattern, prognostic relevance, and interrelationships. Cancer Sci. 2009, 100, 1210–1218. [Google Scholar] [CrossRef]
- Alam, M.M.; Fermin, J.M.; Knackstedt, M.; Noonan, M.J.; Powell, T.; Goodreau, L.; Daniel, E.K.; Rong, X.; Moore-Medlin, T.; Khandelwal, A.R.; et al. Everolimus downregulates STAT3/HIF-1α/VEGF pathway to inhibit angiogenesis and lymphangiogenesis in mutant head and neck squamous cell carcinoma (HNSCC). Oncotarget 2023, 14, 85–95. [Google Scholar] [CrossRef]
- Aubrey, B.J.; Strasser, A.; Kelly, G.L. Tumor-Suppressor Functions of the TP53 Pathway. Cold Spring Harb. Perspect. Med. 2016, 6, a026062. [Google Scholar] [CrossRef]
- Yu, D.; Liu, X.; Han, G.; Liu, Y.; Zhao, X.; Wang, D.; Bian, X.; Gu, T.; Wen, L. The let-7 family of microRNAs suppresses immune evasion in head and neck squamous cell carcinoma by promoting PD-L1 degradation. Cell Commun. Signal. 2019, 17, 173. [Google Scholar] [CrossRef]
- Adyns, L.; Proost, P.; Struyf, S. Role of Defensins in Tumor Biology. Int. J. Mol. Sci. 2023, 24, 5268. [Google Scholar] [CrossRef]
- Golovine, K.; Abalakov, G.; Lian, Z.; Chatla, S.; Karami, A.; Chitrala, K.N.; Madzo, J.; Nieborowska-Skorska, M.; Huang, J.; Skorski, T. ABL1 kinase as a tumor suppressor in AML1-ETO and NUP98-PMX1 leukemias. Blood Cancer J. 2023, 13, 42. [Google Scholar] [CrossRef] [PubMed]
- Timar, J.; Kashofer, K. Molecular epidemiology and diagnostics of KRAS mutations in human cancer. Cancer Metastasis Rev. 2020, 39, 1029–1038. [Google Scholar] [CrossRef]
- Arantes, L.M.R.B.; De Carvalho, A.C.; Melendez, M.E.; Lopes Carvalho, A. Serum, plasma and saliva biomarkers for head and neck cancer. Expert Rev. Mol. Diagn. 2018, 18, 85–112. [Google Scholar] [CrossRef] [PubMed]
- Masood, Y.; Kqueen, C.Y.; Rajadurai, P. Role of miRNA in head and neck squamous cell carcinoma. Expert Rev. Anticancer Ther. 2015, 15, 183–197. [Google Scholar] [CrossRef]
- Wu, Z.H.; Zhong, Y.; Zhou, T.; Xiao, H.J. miRNA biomarkers for predicting overall survival outcomes for head and neck squamous cell carcinoma. Genomics 2021, 113, 135–141. [Google Scholar] [CrossRef]
- Yang, L.; Lu, P.; Yang, X.; Li, K.; Chen, X.; Qu, S. Excavating novel diagnostic and prognostic long non-coding RNAs (lncRNAs) for head and neck squamous cell carcinoma: An integrated bioinformatics analysis of competing endogenous RNAs (ceRNAs) and gene co-expression networks. Bioengineered 2021, 12, 12821–12838. [Google Scholar] [CrossRef] [PubMed]
- Okada, R.; Koshizuka, K.; Yamada, Y.; Moriya, S.; Kikkawa, N.; Kinoshita, T.; Hanazawa, T.; Seki, N. Regulation of Oncogenic Targets by (Passenger Strand of Duplex) in Head and Neck Squamous Cell Carcinoma. Cells 2019, 8, 1535. [Google Scholar] [CrossRef]
- Wu, C.; Tong, L.; Wu, C.; Chen, D.; Chen, J.; Li, Q.; Jia, F.; Huang, Z. Two miRNA prognostic signatures of head and neck squamous cell carcinoma: A bioinformatic analysis based on the TCGA dataset. Cancer Med. 2020, 9, 2631–2642. [Google Scholar] [CrossRef]
- Lu, L.; Wu, Y.; Feng, M.; Xue, X.; Fan, Y. A novel seven-miRNA prognostic model to predict overall survival in head and neck squamous cell carcinoma patients. Mol. Med. Rep. 2019, 20, 4340–4348. [Google Scholar] [CrossRef]
- Fekete, J.T.; Welker, Á.; Győrffy, B. miRNA Expression Signatures of Therapy Response in Squamous Cell Carcinomas. Cancers 2020, 13, 63. [Google Scholar] [CrossRef]
- Granda-Díaz, R.; Manterola, L.; Hermida-Prado, F.; Rodríguez, R.; Santos, L.; García-de-la Fuente, V.; Fernández, M.T.; Corte-Torres, M.D.; Rodrigo, J.P.; Álvarez Teijeiro, S.; et al. Targeting oncogenic functions of miR-301a in head and neck squamous cell carcinoma by PI3K/PTEN and MEK/ERK pathways. Biomed. Pharmacother. 2023, 161, 114512. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Sun, S.; Li, J.; Zhao, X.; Li, Z.; Sha, T.; Cui, Z. NCAPG, mediated by miR-378a-3p, regulates cell proliferation, cell cycle progression, and apoptosis of oral squamous cell carcinoma through the GSK-3β/β-catenin signaling. Neoplasma 2021, 68, 1201–1211. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Centrality | Average Value |
|---|---|
| Betweenness | 1384.6 |
| Centroid | −437 |
| Bridging | 68.4 |
| Degree | 14.2 |
| Radiality | 4.8 |
| Closeness | 0.000506 |
| Stress | 18,601 |
| Eccentricity | 0.185 |
| Eigenvector | 0.0161 |
| Diameter 1 | 7 |
| Average Distance 1 | 3.18 |
| Centrality | Average Value in PPI Network of Genes Targeted by Upregulated miRNAs | Average Value in PPI Network of Genes Targeted by Downregulated miRNAs |
|---|---|---|
| Betweenness | 515.4 | 457.5 |
| Centroid | −260.8 | −234.5 |
| Bridging | 12.8 | 7.5 |
| Degree | 28.7 | 44.6 |
| Radiality | 3.75 | 2.91 |
| Closeness | 0.00109 | 0.00115 |
| Stress | 7883 | 9399 |
| Eccentricity | 0.274 | 0.309 |
| Eigenvector | 0.0353 | 0.0369 |
| Diameter 1 | 5 | 4 |
| Average Distance 1 | 2.24 | 2.08 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arfin, S.; Kumar, D.; Lomagno, A.; Mauri, P.L.; Di Silvestre, D. Differentially Expressed Genes, miRNAs and Network Models: A Strategy to Shed Light on Molecular Interactions Driving HNSCC Tumorigenesis. Cancers 2023, 15, 4420. https://doi.org/10.3390/cancers15174420
Arfin S, Kumar D, Lomagno A, Mauri PL, Di Silvestre D. Differentially Expressed Genes, miRNAs and Network Models: A Strategy to Shed Light on Molecular Interactions Driving HNSCC Tumorigenesis. Cancers. 2023; 15(17):4420. https://doi.org/10.3390/cancers15174420
Chicago/Turabian StyleArfin, Saniya, Dhruv Kumar, Andrea Lomagno, Pietro Luigi Mauri, and Dario Di Silvestre. 2023. "Differentially Expressed Genes, miRNAs and Network Models: A Strategy to Shed Light on Molecular Interactions Driving HNSCC Tumorigenesis" Cancers 15, no. 17: 4420. https://doi.org/10.3390/cancers15174420