
Revisiting Circulating Extracellular Matrix Fragments as Disease Markers in Myelofibrosis and Related Neoplasms
 , , , , ,
, , , , ,
Abstract
:Simple Summary
Abstract
1. Introduction

2. The Normal Bone Marrow Stroma

3. The Connective Tissue Response to Injury
3.1. Stroma Generation in Normal Wound Healing and Tumors
3.2. Bone Marrow Stroma Generation in Primary Myelofibrosis (PMF)
4. Histological Studies on Bone Marrow Stroma in MPNs
5. Biochemical and Immunohistochemical Studies of Bone Marrow
6. Studies of Extracellular Matrix Metabolism
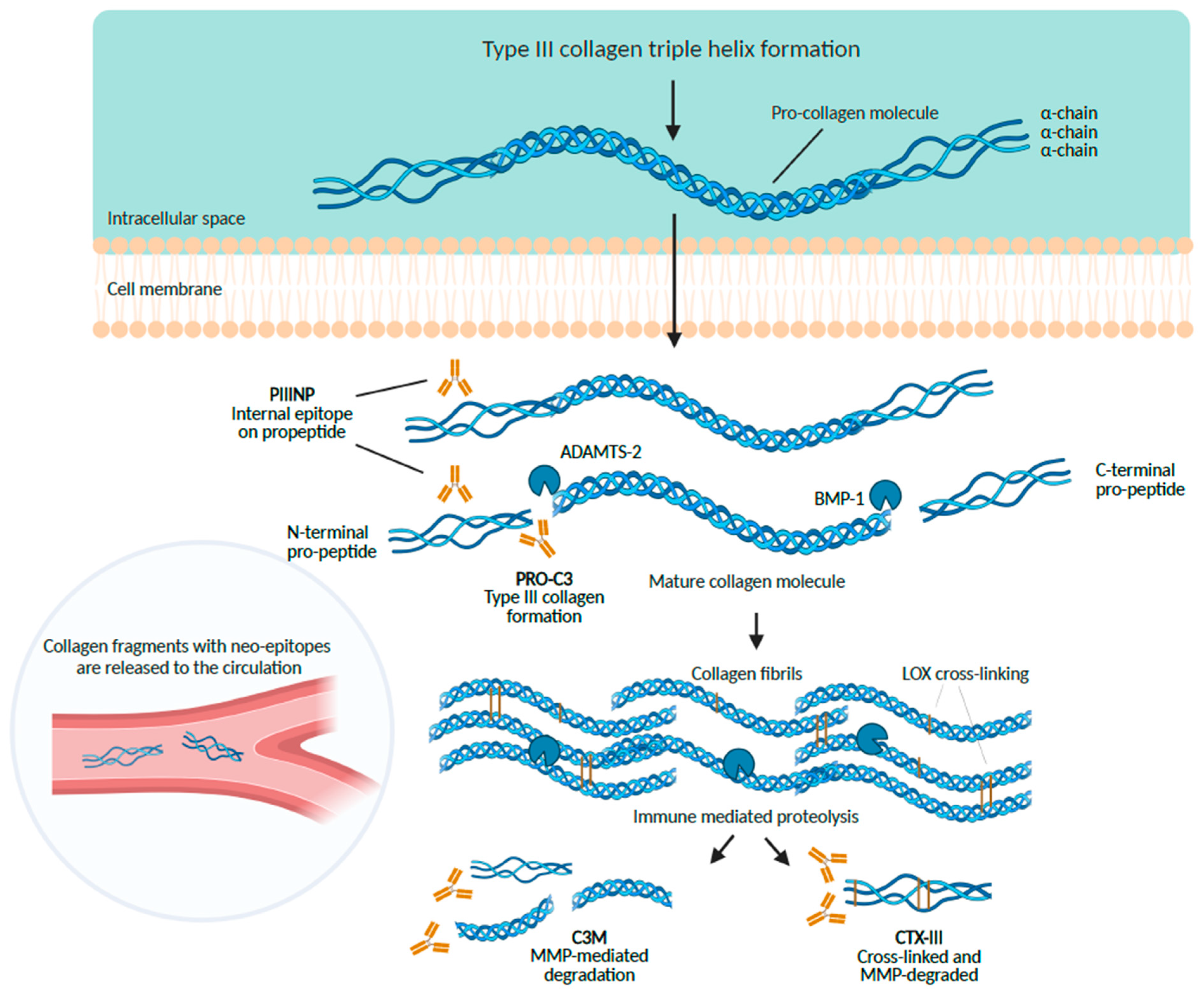
6.1. The Aminoterminal Propeptide of Type III Procollagen (PIIINP)
6.2. Biomarkers of Type I Collagen Metabolism
6.3. Basement Membrane Proteins
6.4. Hyaluronan
6.5. Fibronectin
6.6. Metalloproteinases (MMPs) and Tissue Inhibitors of Metalloproteinases (TIMPs)

{kind=link}
{kind=link}
| Biomarker | No. Patients | Summary of Results | Conclusions/Comments | Refs. |
|---|---|---|---|---|
| S-PIIINP | 441 | S-PIIINP values were increased in PV with even more elevated levels in post-PV-MF and strikingly elevated in patients with severe myelofibrosis [81]. | S-PIIINP associates significantly with the extent of reticulin fibrosis. S-PIIINP is a quantitative marker for myelofibrosis [81]; | [81,83,84,85,86, 96,98,261, 262,268,272,276] |
| S-PIIINP values were normal in patients without reticulin fibrosis; increased in PV and MF. S-PIIINP values above 25 ng/mL associated with MF of recent onset (less than or equal to 2 years) and values below 25 ng/mL with MF of more than 4 years’ duration [83]. | S-PIIINP is a non-invasive method for accurate assessment of bone marrow fibrosis. S-PIIIINP may be used to evaluate the efficacy of antifibrosing agents [83]. | |||
| S-PIIINP values were increased in PV and related to degree of reticulin fibrosis. S-PIIINP values were increased in spent phase of PV only treated with phlebotomy. S-PIIINP values were increased in patients transforming into post-PV-MF and increased in PMF [86]. | S-PIIINP is higher in myelofibrosis of recent onset (less than 2 years) than in myelofibrosis longer than 2 years. S-PIIINP is stable in PV patients treated with 32P or hydroxyurea [86]. | |||
| S-PIIINP values were increased in PV and even more in patients with TMD and MF; S-PIIIINP values were virtually normal in OMS (deposition of type I collagen) [84]. | S-PIIINP is a useful indicator of disease activity in MPNs S-PIIINP positively correlates to the degree of reticulin fibrosis. Near normal S-PIIINP values in OMS likely reflect stable disease without concurrent type III collagen synthesis. [84]. | |||
| S-PIIINP values were normal or elevated in PV and TMD. S-PIIINP values were normal or even low levels in OMS. S-PIIINP values were increased in MF and in CML associated with bone marrow MF. S-PIIINP and S-Type IV collagen correlated significantly with each other and with the leucocyte count [85]. | S-PIIINP is a useful indicator of disease activity in MPNs. Normal and even low S-PIIINP values in OMS may reflect stable disease without ongoing type III collagen synthesis Interstitial type III collagen and basement membrane metabolism are closely related [85]. | |||
| S-PIIINP values were strongly raised in MPNs. All three biomarkers (S-PIIINP, S-PICP and S-Laminin ) were significantly elevated in patients with active/transforming disease [98]. | S-PIIINP is a useful indicator of disease activity in MPNs. S-Laminin and S-PICP do not offer offer any advantage over S-PIIIP. Interstitial type III collagen and basement membrane metabolism are closely related [98]. | |||
| S-PIIINP values were slightly elevated in patients with stable disease and highly elevated in patients transforming into myelofibrosis. S-PIIINP values covariated closely with S-laminin, the leucocyte count and LDH [276]. | S-PIIINP is a useful indicator of disease activity in MPNs. Interstitial type III collagen and basement membrane metabolism are closely related [276]. | |||
| S-PIIINP values correlated significantly with the leukocyte count and with S-HU. S-PIIINP values decreased during cytotoxic treatment in concert with declining leukocyte counts and S-HU [96]. | S-PIIINP is a useful indicator of disease activity in MPNs. S-PIIINP may be useful in monitoring the efficacy of cytotoxic treatment in terms of inhibiting development and progression of bone marrow fibrosis [96]. | |||
| S-PIIINP values were increased in PMF. S-PIIINP values were only increased slightly in patients with stable disease (n = 3) compared to the single patient with more active disease. S-PIIINP values declined during treatment with acetylsalicylic acid (ASA), although normalization did not occur. Using gel filtration analysis the antigens related to S-PIIIINP were found to be heterogenous with at least two peaks, exhibiting molecular masses equal to and smaller than PIIINP [261]. | This study includes only four patients and accordingly does not allow robust conclusions [261]. Studies on the impact of ASA and anti-inflammatory treatment upon type III collagen metabolism are needed [261]. | |||
| S-PIIIP values were elevated in PMF. S-PIIIP values were normal in younger patients, having higher Hb- and platelet counts and lower S-ferritin values platelet count. S-PIIIP values were significantly higher in patients with active disease (fever, sweating, weight loss) than in patients with non-active disease S-PIIIP values correlated with decreasing Hb-concentration and platelet count and increasing WBC, serum ferritin and number of transfusions (univariate analysis). S-PIIINP values correlated independently with increasing WBC, serum ferritin and age (multivariate analysis). S-PIIINP values did not associate with morphometric grading of bone marrow fibrosis, megakaryocyte number, or lymphoid infiltration [262] | S-PIIIP values in PMF correlates more with overall disease activity than with the extent of bone marrow fibrosis [262]. The association between normal S-PIIINP and lower S-ferritin values in younger patients with higher HB-concentrations may likely reflect that S-PIIINP also is a biomarker of the chronic inflammatory state in PMF. Studies on the associations between S-PIIINP, biomarkers of chronic inflammation (e.g., CRP, ferritin, inflammatory cytokines), bone marrow megakaryocyte morphology and bone marrow fibrosis and the impact of cytotoxic (HU) or stem-cell targeting therapy ( pegylated interferon-alpha2 ) as monotherapies or in combination (e.g., with JAK1-2 inhibitor) are needed. | |||
| S-PIIINP values were elevated in PMF, S-PIIINP values decreased during treatment with anthracycline, which was given due to accelerated phase disease [268]. | S-PIIINP is a valuable biomarker in PMF. Cytotoxic treatment lowers elevated S-PIIINP values [268]. | |||
| S-PIIINP values were elevated in MPNs. S-PIIINP values were highest in patients with MF. S-PIIINP and S-ICTP correlated significantly. S-PIIIINP and P-suPAR correlated significantly. S-PIIINP values did not correlate with P-MMP-2 and MMP-9 [272]. | S-PIIINP is a useful indicator of disease activity in MPNs. Type III and type I collagen metabolism are closely associated, reflecting concurrent type III synthesis (PIIINP) and type I degradation (ICTP). Elevated S-ICTP values in MPN may not only reflect type I collagen degradation in the bone marrow but also increased bone resorption. Enzymes of the uPA system might participate in the bone marrow remodelling processes characteristic of MPN [272]. | |||
| S-PICP | 26 | S-PICP values were slightly elevated in MPNs, reflecting increased type I collagen synthesis. S-PICP values were significantly elevated in patients with active/transforming disease. S-PICP and S-laminin P1 values showed a strong correlation [98]. | S-PICP values do not offer any advantage over S-PIIIP for monitoring of disease activity. Increased type I collagen synthesis associates with progressive disease. Interstitial type I collagen and basement membrane metabolism are closely related [98]. | [98] |
| S-ICTP | 50 | S-ICTP values were elevated in MPN. S-ICTP values were only significantly higher among MF and PV patients. S-ICTP and S-PIIINP values correlated significantly. S-ICTP and P-suPAR values correlated significantly. S-ICTP and P-MMP-2/MMP-9 values did not correlate significantly. | Elevated S-ICTP values in MPNs reflect ongoing type I collagen degradation. Elevated S-ICTP values may not only reflect enhanced type I collagen degradation in the bone marrow but also increased type I collagen degradation in bone tissue (increased bone resorption). Increased bone resorption with the development of osteopenia/osteoporosis may be driven by chronic inflammation in MPNs. In this regard, the significant correlation between S-ICTP and P-suPAR may also reflect the chronic inflammatory state and not only the involvement of these biomarkers in the bone marrow remodeling processes in MPNs. | [272] |
| S-TIVC | 41 | S-TIVC values were normal or elevated in PV and TMD. S-TIVC values were elevated in MF and in CML associated with bone marrow MF. S-TIVC and S-PIIINP correlated significantly and with the leucocyte count. | Measurement of type IV collagen provides a noninvasive means for following the accumulation of basement membrane collagen in the bone marrow in patients with MPN. S-TIVC associates with disease activity as assessed by the leukocyte count. Interstitial (type III collagen ) and basement membrane metabolism (type IV collagen ) are tightly associated processes in MPNs. | [85] |
| S-Laminin1 | 58 | S-Laminin1 values were slightly elevated in MPNs S-Laminin1, S-PIIINP and S-PICP values were significantly elevated in patients with active/transforming disease. S-Laminin P1 and S-PICP levels showed a strong correlation [98]. | S-Laminin did not appear to offer any advantage over S-PIIIP for the monitoring of disease activity. Basement membrane (laminin) and interstitial collagen ( PIIINP, PICP) metabolism are closely related in MPNs [98]. | [98,276] |
| S-Laminin1 values were slightly elevated in patients with stable disease. S-Laminin1 values were highly elevated in patients with progressive disease transforming into myelofibrosis. S-Laminin1 covariated closely with S-PIIINP, the leucocyte count and LDH [276]. | S-Laminin1 values were significantly lower in patients with a huge spleen as compared with patients, having a normal spleen size or previously being splenectomized.The above observation may reflect that the aminin uptake/degradation is increased in the enlarged spleen in MPNs. | |||
| S-HYA | 59 | S-HYA values were normal in patients with stable disease and increased in patients with active disease. S-HYA values correlated significantly with the leukocyte count and with S-PIIINP. S-HYA values decreased during cytotoxic treatment in concert with declining leukocyte counts and S-PIIINP. | S-HYA values displayed only slight changes in MPNs with with frequent overlaps between patient categories and HC. The clinical utility of S-HYA may be restrained, although sequential measurements of S-HYA may provide a reflection of the MPN process in individual patients. | [96] |
| P-Fibronectin | 69 | P-Fibronectin values were normal in ET. P-Fibronectin values were significantly reduced in PV and MF. P-Fibronectin values were lowest in in patients with marked splenomegaly. P-Fibronectin values rose from less than 100 mg/L to 177 mg/L after splenectomy in a patients with MF [82]. | Low P-fibronectin values in MPNs may be attributed to increased consumption of P-Fibronectin in the expanded mononuclear phagocyte system in the liver and spleen, reduced hepatic synthesis, and/or fibronectin taking part in the clearance of circulating immune complexes. Low P-Fibronectin values in patients with MPNs may contribute to an increased risk of infections. | [82,88,90] |
| P-Fibronectin values were significantly lower in patients with PMF. P-Fibronectin values in MF patients differed significantly, when compared with patients with PV, TMD or CML. P-Fibronectin values were lowest in patients with large spleens [88]. | Low P-fibronectin concentrations in splenomegalic patients may be due to enhanced consumption of the opsonin in the expanded splenic mononuclear-macrophage system [88]. | |||
| P-Fibronectin values correlated inversely with CIC, which were highly elevated in 11 of 20 with MF secondary to MPN. The CIC contained fibronectin, IgG and C3. P-Fibronectin levels increased after therapeutic plasmapheresis, which efficiently removed CIC [90]. | The findings suggest that fibronectin as a major non-specific opsonin is important for the normal clearance of immune complexes [90]. | |||
| P-Fibronectin (EDA) | 122 | P-EDA FN values were significantly elevated in PMF as compared to HCs. P-EDA F values did not differ between PV/ET patients and HCs. P-EDA FN values differed among patients with different degrees of BM fibrosis with a trend towards increasing P-EDA FN levels with increasing BM fibrosis grades (not significant). P-EDA FN differed significantly between patients with pre-fibrotic myelofibrosis (BM fibrosis grade 0) + those with early myelofibrosis (BM fibrosis grade 1) as compared to those with BM fibrosis grade 2 + those with BM fibrosis grade 3 (advanced fibrosis) [297]. | Patients with PMF exhibited higher levels of the EDA FN isoform as compared to HCs. | [297,298] |
| P-EDA FN values were higher in patients with a homozygous JAK2V617F genotype Increased P-EDA-FN values were associated with anemia, elevated high-sensitivity C-reactive protein, bone marrow fibrosis and splanchnic vein thrombosis at diagnosis. Elevated P-EDA-FN at diagnosis was a predictor of large splenomegaly [298]. | P-EDA-FN in primary myelofibrosis may represent a marker of disease progression, and a novel target to treat splenomegaly [298]. | |||
| P-YKL-40 | 48 | P-YKL-40 values were significantly elevated in PMF vs. control subjects. P-YKL-40 values were increased from ET over PV to PMF [302]. | P-YKL-40 may be a novel biomarker of chronic inflammation, tissue remodelling and atherosclerotic inflammation in MPN [302,303]. | [302,303] |
| P-YKL-40 values were significantly elevated in PMF vs. control subjects. P-YKL-40 values were increased from ET over PV to PMF [302]. | P-YKL-40 might be a novel marker of disease burden and progression in MPN [303]. | |||
| S-YKL-40 | 111 | S-YKL-40 values were significantly higher in post-ET MF, PV, post-PV MF and PMF patients, when compared to HC. S-YKL-40 values were associated with biomarkers of an increased inflammatory state (higher C-reactive protein, poor performance status, presence of constitutional symptoms and cardiovascular risk factors).Higher S-YKL-40 values in MF patients were also associated with blast phase disease, lower hemoglobin and higher Dynamic International Prognostic Scoring System score. Higher S-YKL-40 values were independently associated with an increased risk of thrombosis and impaired survival in MF patients [304]. | Higher S-YKL-40 might have a pathophysiological role in disease progression and thrombosis development. Assessing S-YKL-40 could help in identification of ET and PV patients at a high risk of future cardiovascular events and has a good potential for improving prognostication of MF patients [304]. | [304] |
| S-CHIT1 | 91 | S-CHIT1 values were significantly higher in PV and post-PV myelofibrosis transformation (post-PV MF). S-CHIT1 values were not significantly higher in ET, post-ET MF transformation, and PMF patients, when compared to healthy controls. S-CHIT1 values in PV were positively correlated with hemoglobin, hematocrit, absolute basophil count and the presence of reticulin fibrosis in the bone marrow. | A positive correlation between S-CHIT1 and the hemoglobin, hematocrit, and absolute basophil count in PV might imply macrophages closely related to clonal erythropoiesis as cells of CHIT1 origin. A positive association between S-CHIT1 and reticulin fibrosis might indicate its potential role in PV progression. S-CHIT1 might a circulating biomarker of macrophage activation with an important role in inflammation-induced tissue remodeling and fibrosis in PV. | [305] |
| P-Pentraxin-3 (P-PTX3) | 244/477/140 | P-PTX3 and P-hs-CRP were measured in 244 consecutive ET and PV patients. After a median follow up of 5.3 years (range 0–24), 68 CV events were diagnosed. Major thrombosis rate was higher in the highest hsCRP and lower at the highest PTX3 levels. These associations remained significant in multivariate analyses [306]. | P-hs-CRP and P-PTXT3 independently and in opposite ways modulate the intrinsic risk of CV events in patients with MPN [306]. | [306,307] [308] |
| P-PTX3 levels in 477 ET and PV patients were significantly increased in carriers of homozygous JAK2V617F mutation compared to all the other genotypes and triple negative ET patients, while hs-CRP levels were independent of the mutational profile. The risk of hematological evolution and death from any cause was about 2- and 1.5-fold increased in individuals with high PTX-3 levels, while the thrombosis rate tended to be lower. High hs-CRP levels were associated with risk of haematological evolution, death and also major thrombosis. After sequential adjustment for potential confounders (age, gender, diagnosis and treatments) and the presence of JAK2V617F homozygous status, high hs-CRP levels remained significant for all outcomes, while JAK2V617F homozygous status as well as treatments were the factors independently accounting for adverse outcomes among patients with high PTX3 levels [307]. | The JAK2V617F mutation influences MPN-associated inflammation with a strong correlation between allele burden and PTX3 levels. P-hs-CRP and P-PTX3 might be of prognostic value for patients with ET and PV, but their validation in future prospective studies is needed [307]. | |||
| P-PTX3 values were significantly higher in PMF patients than in HC. High PTX3 values (≥70 ng/mL) associated with an unfavourable overall survival. P-PTX3 values independently predicted PMF patients’ overall survival. P-PTX3 values correlated with parameters of tumor burden, including total leucocyte count, mutated JAK2 allele burden, lactate dehydrogenase levels, and spleen size [308]. | PTX3is released from macrophages and endothelial cells, and promotes the transition of monocytes to fibrocytes. P-PTX3 levels constitute an independent indicator of disease burden, clonal expansion and overall survival in patients with PMF. Monitoring of PTX3 plasma levels might be a useful tool in clinical decision making [308]. | |||
| P-suPAR | 50 | P-suPAR correlated significantly with serum markers of collagen metabolism (S-PIIINP and S-ICTP) [272] | Enzymes of the uPA system might participate in the bone marrow remodelling processes characteristic of MPN [272]. | [272,273] |
| P-suPAR values were significantly higher in MPN patients. P-suPAR values differed significantly between MPN-subgroups, the highest levels being found in patients with MF and PV. P-suPAR values were only significantly increased in PV and MF patients. P-suPAR significantly correlated to P-LDH. P-uPA did not differ between patients and controls [273]. | Increased P-suPAR levels in patients with MPN may reflect increased uPAR production in the bone marrow, leading to enhanced bone marrow remodeling [273]. | |||
| P-TIMP-1 P-MMP-1 P-MMP-2 P-MMP-3 P-MMP-9 | 67 | P-MMP-3 levels were decreased in patients with advanced MF. P-MMP-1, P-MMP-2, and P-MMP-9 levels were not significantly different from HC. P-TIMP-1 levels were elevated in ET, PV and MF patients and in particular in advanced MF. P-MMPs levels were not elevated in ET and PV patients | The abnormal accumulation of ECM is dependent upon the balance between matrix metalloproteinases (MMPs) and tisse inhibitors of metalloproteinases (TIMPs). Accumulation of connective tissue in the bone marrow is associated with reduced MMP activity together with increased TIMP-1 activity, which may be important in fibrosis formation in the bone marrow in MPNs. | [299,300] |
| P-TIMP-1 | 50 | Plasma levels of total-, free- and complexed TIMP-1, TIMP-2, MMP-2 and MMP-9 were measured in 50 patients with MPN. P-TIMP-1 levels were significantly higher in MPN patients. P-TIMP-1 levels significantly correlated with P-suPAR and P-uPAR. P-TIMP-2 and P-MMP-2 levels did not differ beween patients and controls. P-TIMP-1 and P-TIMP-2 levels correlated significantly. P-MMP-9 levels significantly higher among PV patients. P-TIMP-1/MMP-9 ratio was significantly higher in patients with MF. | The family of MMPs and TIMPs facilitate and inhibit matrix degradation processes, respectively. A disturbed TIMP-1/MMP ratio may reflect an imbalance of the extracellular homeostasis towards an increased matrix deposition promoting fibrosis. | [275] |
| U-HYPRL | 47 | U-HYPRL was normal in 16 patients with PMF and in 5 out of 6 patients with acute myelofibrosis. In patients with OMS (n = 8) values for U-HYPRL were insignificantly higher than those PMF. U-HYPRL increased in 10 patients (1 AMF patient, 3 OMS patients and 6 patients with CML in the accelerated phase of the disease). All but 1 of these patients had been treated, or were being treated, with cytotoxic agents at the time of investigation [89]. | The findings of normal U-HYPRL may be explained by impaired degradation of bone marrow collagen which, together with enhanced collagen synthesis from bone marrow fibroblasts, accounts for progressive accumulation of connective tissue in the bone marrow in myelofibrosis patients. This process is influenced by cytotoxic treatment as reflected in increased urinary hydroxyproline excretion in those patients receiving cytotoxic agents [89]. | [80,89] |
| U-HYPRL was normal in PMF patients. U-HYPRL was increased in patients with metastasis, the highest levels being recorded in those with concomitant bone marrow fibrosis [80]. | The result suggests differences in the pathogenesis of “MPN-myelofibrosis” (normal U-HYPRL) as compared to myelofibrosis consequent to bone marrow metastasis (increased U-HYPRL) [80]. |
7. Reasons to Revisit Blood-Based Extracellular Matrix Biomarkers in Patients with Myeloproliferative Neoplasms
8. Perspectives and Future Research Directions
8.1. Zinpentraxin alfa
8.2. Colchicine
8.3. N-Acetylcysteine (NAC)
8.4. Statins and Metformin
| Pancreatic Cancer | Myelofibrosis | |
|---|---|---|
| Stroma Composition | Type I, III and IV collagens | Early stage: Type III and IV collagen Advanced stage: In addition Type I collagen |
| Stroma Changes | The normal basement membrane architecture is lost during PC progression and cancer cells are typically in direct contact with the interstitial matrix, i.e., type I and type III collagens. | The normal basement membrane architecture is changed during progression of MPNs with the formation of continuous sheets below basement membranes; megakaryocytes are typically in direct contact with the interstitial matrix, i.e., type I and type III collagens and transmigrate together with CD34+ and progenitors the endothelial lining into the circulation to seed in the spleen, liver, lungs and elsewhere (“metastasis”). |
| Biomarkers of Collagen Synthesis | Type III procollagen-peptides: Elevated | S-PIIINP, S-Type IV, S-Laminin1: Using ELISAs, levels of S-PIIINP, type IV antigen and laminin1 have been measured in MF and related neoplasms (please see text). These ELISAs measure type III collagen synthesis and synthesis of basement membrane constituents rather than degradation. |
| Biomarkers of Collagen Degradation | C1M, C3M, C4M, C4M12a1: Elevated | C1M, C3M, C4M, C4M12a1: to be performed |
| Therapies: Stem-cell targeting; Immunomodulating; Anti-inflammatory Fibrosis-resolving. | Stem-cell targeting: IFN-alpha2 or IFN-beta? Immunomodulators: Immune-check inhibitors, IFN-alpha2 or IFN-beta?; inhibitors of TGF-beta signalling Anti-inflammatory agents (e.g., Jak1-2 inhibitors)? Fibrosis-resolving agents (e.g., colchicine, metformin, angiotensin II blocker, statins, zinpentraxin alfa)? | Stem-cell targeting: IFN-alpha2 or IFN-beta? Immunomodulators: Immune-check inhibitors, IFN-alpha2 or IFN-beta?; inhibitors of TGF-beta signalling. Anti-inflammatory agents (e.g., Jak1-2 inhibitors)? Fibrosis-resolving agents (e.g., colchicine, metformin, angiotensin II blocker, statins, zinpentraxin alfa)? |
| Research Questions |
|---|
| Does measurement of circulating ECM peptides have the potential to differentiate genuine ET from prefibrotic myelofibrosis, prefibrotic myelofibrosis from classic myelofibrosis and predict leukemic transformation in MPNs? |
| Do distinct circulating ECM peptides or ECM peptide signatures depict disease activity/burden as measured by conventional parameters (leukocyte and platelet counts, JAK2V617F allelic burden, plasma lactic dehydrogenase level) |
| Do circulating ECM peptides or ECM peptide signatures associate with antecedent thrombosis and/or predict thrombosis risk |
| How does artificial intelligence-based morphological fingerprinting of megakaryocytes and “Continuous Indexing of Fibrosis” correlate with circulating ECM peptide signatures? |
| Do circulating ECM peptides reflect comorbidity burden in MPNs? |
| Do circulating ECM peptides or ECM peptide signatures predict treatment response (hydroxyurea, IFN-alpha2, JAK1-2 inhibitor (ruxolitinib or fedratanib), statin (atorvastatin), combination therapy (IFN-alpha2 + ruxolitinib), epitope -vaccination)? |
| Do CHIP-JAK2V617F positive citizens have a distinct ECM peptide signature as compared to CHIP-JAK2V617F negative citizens? |
| Does measurement of circulating ECM peptides in the CHIP stage have the potential to diagnosis early development of MPNs? |
| Does measurement of circulating ECM peptides in the CHIP stage have the potential to diagnosis early development of inflammation-mediated comorbidities and associated organ dysfunction and incipient organ fibrosis in diseases such as dementia, age-related macular degeneration, aneurysms, myocardial fibrosis with heart failure (HFpEF), pulmonary dysfunction/failure, chronic kidney failure or osteopenia? |
| Does measurement of circulating ECM peptides in the CHIP stage have the potential of early detection of development of MPNs or another cancer? |
| Do circulating ECM peptides or ECM peptide signatures associate with distinct immune cell profiles, including a T-cell exhaustion signature? |
| Do circulating ECM peptides or ECM peptide signatures predict overall survival in the CHIP stage and in MPNs? |
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dameshek, W. Some speculations on the myeloproliferative syndromes. Blood 1951, 6, 372–375. [Google Scholar] [CrossRef] [PubMed]
- Spivak, J.L. Myeloproliferative Neoplasms. N. Engl. J. Med. 2017, 377, 895–896. [Google Scholar] [CrossRef]
- Mead, A.J.; Mullally, A. Myeloproliferative neoplasm stem cells. Blood 2017, 129, 1607–1616. [Google Scholar] [CrossRef]
- Lundberg, P.; Karow, A.; Nienhold, R.; Looser, R.; Hao-Shen, H.; Nissen, I.; Girsberger, S.; Lehmann, T.; Passweg, J.; Stern, M.; et al. Clonal evolution and clinical correlates of somatic mutations in myeloproliferative neoplasms. Blood 2014, 123, 2220–2228. [Google Scholar] [CrossRef] [PubMed]
- Grinfeld, J.; Nangalia, J.; Baxter, E.J.; Wedge, D.C.; Angelopoulos, N.; Cantrill, R.; Godfrey, A.L.; Papaemmanuil, E.; Gundem, G.; MacLean, C.; et al. Classification and personalized prognosis in myeloproliferative neoplasms. N. Engl. J. Med. 2018, 379, 1416–1430. [Google Scholar] [CrossRef]
- Skov, V. Next generation sequencing in MPNs. Lessons from the past and prospects for use as predictors of prognosis and treatment responses. Cancers 2020, 12, 2194. [Google Scholar] [CrossRef] [PubMed]
- Feenstra, J.D.M.; Nivarthi, H.; Gisslinger, H.; Leroy, E.; Rumi, E.; Chachoua, I.; Bagienski, K.; Kubesova, B.; Pietra, D.; Gisslinger, B.; et al. Whole-exome sequencing identifies novel MPL and JAK2 mutations in triple-negative myeloproliferative neoplasms. Blood 2016, 127, 325–332. [Google Scholar] [CrossRef]
- Angona, A.; Fernandez-Rodrıguez, C.; Alvarez-Larran, A.; Camacho, L.; Longarón, R.; Torres, E.; Pairet, S.; Besses, C.; Bellosillo, B. Molecular characterisation of triple negative essential thrombocythaemia patients by platelet analysis and targeted sequencing. Blood Cancer J. 2016, 6, e463. [Google Scholar] [CrossRef]
- Kjær, L. Clonal Hematopoiesis and Mutations of Myeloproliferative Neoplasms. Cancers 2020, 12, 2100. [Google Scholar] [CrossRef]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef]
- Marchioli, R.; Finazzi, G.; Landolfi, R.; Kutti, J.; Gisslinger, H.; Patrono, C.; Marilus, R.; Villegas, A.; Tognoni, G.; Barbui, T. Vascular and neoplastic risk in a large cohort of patients with polycythemia vera. J. Clin. Oncol. 2005, 23, 2224–2232. [Google Scholar] [CrossRef]
- Hultcrantz, M.; Kristinsson, S.Y.; Andersson, T.M.-L.; Landgren, O.; Eloranta, S.; Derolf, R.; Dickman, P.W.; Björkholm, M. Patterns of survival among patients with myeloproliferative neoplasms diagnosed in Sweden from 1973 to 2008: A population-based study. J. Clin. Oncol. 2012, 30, 2995–3001. [Google Scholar] [CrossRef] [PubMed]
- Barbui, T.; Finazzi, G.; Falanga, A. Myeloproliferative neoplasms and thrombosis. Blood 2013, 122, 2176–2184. [Google Scholar] [CrossRef]
- Moliterno, A.R.; Ginzburg, Y.Z.; Hoffman, R. Clinical insights into the origins of thrombosis in myeloproliferative neoplasms. Blood 2021, 137, 1145–1153. [Google Scholar] [CrossRef]
- Hasselbalch, H.C.; Elvers, M.; Schafer, A.I. The pathobiology of thrombosis, microvascular disease, and hemorrhage in the myeloproliferative neoplasms. Blood 2021, 137, 2152–2160. [Google Scholar] [CrossRef] [PubMed]
- Solli, C.N.; Chamat-Hedemand, S.; Elming, H.; Ngo, A.; Kjær, L.; Skov, V.; Sørensen, A.L.; Ellervik, C.; Fuchs, A.; Sigvardsen, P.E.; et al. Coronary artery- and aortic valve calcifications in patients with Philadelphia-negative myeloproliferative neoplasms. Int. J. Cardiol. 2022, 364, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Solli, C.N.; Chamat-Hedemand, S.; Elming, H.; Ngo, A.; Kjær, L.; Skov, V.; Sørensen, A.L.; Ellervik, C.; Hasselbalch, H.; Bruun, N.E. High JAK2V617F variant allele frequency is associated with coronary artery but not aortic valve calcifications in patients with Philadelphia-negative myeloproliferative neoplasms. Eur. J. Haematol. 2023, 111, 400–406. [Google Scholar] [CrossRef]
- Jaiswal, S.; Natarajan, P.; Silver, A.J.; Gibson, C.J.; Bick, A.G.; Shvartz, E.; McConkey, M.; Gupta, N.; Gabriel, S.; Ardissino, D.; et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N. Engl. J. Med. 2017, 377, 111–121. [Google Scholar] [CrossRef]
- Jaiswal, S.; Libby, P. Clonal haematopoiesis: Connecting ageing and inflammation in cardiovascular disease. Nat. Rev. Cardiol. 2020, 17, 137–144, Correction in Nat. Rev. Cardiol. 2020, 17, 828. [Google Scholar] [CrossRef]
- Hasselbalch, H.C. Perspectives on chronic inflammation in essential thrombocythemia, polycythemia vera, and myelofibrosis: Is chronic inflammation a trigger and driver of clonal evolution and development of accelerated atherosclerosis and second cancer? Blood 2012, 119, 3219–3225. [Google Scholar] [CrossRef] [PubMed]
- Hasselbalch, H.C. Chronic inflammation as a promotor of mutagenesis in essential thrombocythemia, polycythemia vera and myelofibrosis. A human inflammation model for cancer development? Leuk. Res. 2013, 37, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Hasselbalch, H.C. The role of cytokines in the initiation and progression of myelofibrosis. Cytokine Growth Factor. Rev. 2013, 24, 133–145. [Google Scholar] [CrossRef]
- Hasselbalch, H.C.; Bjørn, M.E. MPNs as Inflammatory Diseases: The Evidence, Consequences, and Perspectives. Mediat. Inflamm. 2015, 2015, 102476. [Google Scholar] [CrossRef] [PubMed]
- Fleischman, A.G. Inflammation as a Driver of Clonal Evolution in Myeloproliferative Neoplasm. Mediat. Inflamm. 2015, 2015, 606819. [Google Scholar] [CrossRef] [PubMed]
- Koschmieder, S.; Mughal, T.I.; Hasselbalch, H.C.; Barosi, G.; Valent, P.; Kiladjian, J.-J.; Jeryczynski, G.; Gisslinger, H.; Jutzi, J.S.; Pahl, H.L.; et al. Myeloproliferative neoplasms and inflammation: Whether to target the malignant clone or the inflammatory process or both. Leukemia 2016, 30, 1018–1024. [Google Scholar] [CrossRef]
- Mendez Luque, L.F.; Blackmon, A.L.; Ramanathan, G.; Fleischman, A.G. Key Role of Inflammation in Myeloproliferative Neoplasms: Instigator of Disease Initiation, Progression and Symptoms. Curr. Hematol. Malig. Rep. 2019, 14, 145–153. [Google Scholar] [CrossRef]
- Koschmieder, S.; Chatain, N. Role of inflammation in the biology of myeloproliferative neoplasms. Blood Rev. 2020, 42, 100711. [Google Scholar] [CrossRef]
- Nasillo, V.; Riva, G.; Paolini, A.; Forghieri, F.; Roncati, L.; Lusenti, B.; Maccaferri, M.; Messerotti, A.; Pioli, V.; Gilioli, A.; et al. Inflammatory Microenvironment and Specific T Cells in Myeloproliferative Neoplasms: Immunopathogenesis and Novel Immunotherapies. Int. J. Mol. Sci. 2021, 22, 1906. [Google Scholar] [CrossRef]
- Frederiksen, H.; Farkas, D.K.; Christiansen, C.F.; Hasselbalch, H.C.; Sørensen, H.T.; Sorensen, H.T. Chronic myeloproliferative neoplasms and subsequent cancer risk: A Danish population-based cohort study. Blood 2011, 118, 6515–6520. [Google Scholar] [CrossRef]
- Frederiksen, H.; Farkas, D.K.; Christiansen, C.F.; Larsen, T.S.; Hasselbalch, H.C.; Stentoft, J.; Sørensen, H.T. Survival of patients with chronic myeloproliferative neoplasms and new primary cancers: A population-based cohort study. Lancet Haematol. 2015, 2, e289–e296. [Google Scholar] [CrossRef]
- Pettersson, H.; Knutsen, H.; Holmberg, E.; Andréasson, B. Increased incidence of another cancer in myeloproliferative neoplasms patients at the time of diagnosis. Eur. J. Haematol. 2015, 94, 152–156. [Google Scholar] [CrossRef] [PubMed]
- Brabrand, M.; Frederiksen, H. Risks of Solid and Lymphoid Malignancies in Patients with Myeloproliferative Neoplasms: Clinical Implications. Cancers 2020, 12, 3061. [Google Scholar] [CrossRef] [PubMed]
- Cumbo, C.; Anelli, L.; Zagaria, A.; Coccaro, N.; Tarantini, F.; Specchia, G.; Musto, P.; Albano, F. Second Cancer Onset in Myeloproliferative Neoplasms: What, When, Why? Int. J. Mol. Sci. 2022, 23, 3177. [Google Scholar] [CrossRef]
- Hasselbalch, H.C. Perspectives on the increased risk of second cancer in patients with essential thrombocythemia, polycythemia vera and myelofibrosis. Eur. J. Haematol. 2015, 94, 96–98. [Google Scholar] [CrossRef] [PubMed]
- Barosi, G. An immune dysregulation in MPN. Curr. Hematol. Malig. Rep. 2014, 9, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Holmström, M.O.; Hasselbalch, H.C.; Andersen, M.H. Cancer Immune Therapy for Philadelphia Chromosome-Negative Chronic Myeloproliferative Neoplasms. Cancers 2020, 12, 1763. [Google Scholar] [CrossRef] [PubMed]
- Liisborg, C.; Hasselbalch, H.C.; Sørensen, T.L. Ocular Manifestations in Patients with Philadelphia-Negative Myeloproliferative Neoplasms. Cancers 2020, 12, 573. [Google Scholar] [CrossRef] [PubMed]
- Liisborg, C.; Skov, V.; Kjær, L.; Hasselbalch, H.C.; Sørensen, T.L. Retinal drusen in patients with chronic myeloproliferative blood cancers are associated with an increased proportion of senescent T cells and signs of an aging immune system. Aging 2021, 13, 25763–25777. [Google Scholar] [CrossRef] [PubMed]
- Liisborg, C.; Nielsen, M.K.; Hasselbalch, H.C.; Sørensen, T.L. Patients with myeloproliferative neoplasms and high levels of systemic inflammation develop age-related macular degeneration. Eclinicalmedicine 2020, 26, 100526. [Google Scholar] [CrossRef]
- Liisborg, C.; Skov, V.; Kjær, L.; Hasselbalch, H.C.; Sørensen, T.L. Patients with MPNs and retinal drusen show signs of complement system dysregulation and a high degree of chronic low-grade inflammation. Eclinicalmedicine 2022, 43, 101248. [Google Scholar] [CrossRef] [PubMed]
- Liisborg, C.; Skov, V.; Kjær, L.; Hasselbalch, H.C.; Sørensen, T.L. Lower CXCR3 expression in both patients with neovascular AMD and advanced stages of chronic myeloproliferative blood cancers. PLoS ONE 2022, 17, e0269960. [Google Scholar] [CrossRef]
- Liisborg, C. Age-related macular degeneration and myeloproliferative neoplasms—A common pathway. Acta Ophthalmol. 2022, 100, 3–35. [Google Scholar] [CrossRef]
- Farmer, S.; Horváth-Puhó, E.; Vestergaard, H.; Hermann, A.P.; Frederiksen, H. Chronic myeloproliferative neoplasms and risk of osteoporotic fractures; a nationwide population-based cohort study. Br. J. Haematol. 2013, 163, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Johansson, P.; Kristjansdottir, H.L.; Johansson, H.; Jakir, A.; Mellström, D.; Lewerin, C. Highly increased risk of fracture in patients with myeloproliferative neoplasm. Leuk. Lymphoma. 2021, 62, 211–217. [Google Scholar] [CrossRef]
- Farmer, S.; Vestergaard, H.; Hansen, S.; Shanbhogue, V.V.; Stahlberg, C.I.; Hermann, A.P.; Frederiksen, H. Bone geometry, bone mineral density, and micro-architecture in patients with myelofibrosis: A cross-sectional study using DXA, HR-pQCT, and bone turnover markers. Int. J. Hematol. 2015, 102, 67–75. [Google Scholar] [CrossRef]
- Farmer, S.; Ocias, L.F.; Vestergaard, H.; Broesby-Olsen, S.; Hermann, A.P.; Frederiksen, H. Bone morbidity in chronic myeloproliferative neoplasms. Expert Rev. Hematol. 2015, 8, 447–456. [Google Scholar] [CrossRef]
- Oikonomidou, P.R.; Casu, C.; Yang, Z.; Crielaard, B.; Shim, J.H.; Rivella, S.; Vogiatzi, M.G. Polycythemia is associated with bone loss and reduced osteoblast activity in mice. Osteoporos. Int. 2016, 27, 1559–1568. [Google Scholar] [CrossRef]
- Farmer, S.; Shanbhogue, V.V.; Hansen, S.; Stahlberg, C.I.; Vestergaard, H.; Hermann, A.P.; Frederiksen, H. Bone mineral density and microarchitecture in patients with essential thrombocythemia and polycythemia vera. Osteoporos. Int. 2017, 28, 677–685. [Google Scholar] [CrossRef]
- Karagianni, A.; Matsuura, S.; Gerstenfeld, L.C.; Ravid, K. Inhibition of Osteoblast Differentiation by JAK2V617F Megakaryocytes Derived from Male Mice with Primary Myelofibrosis. Front. Oncol. 2022, 12, 929498. [Google Scholar] [CrossRef]
- Karagianni, A.; Ravid, K. Myeloproliferative disorders and their effects on bone homeostasis: The role of megakaryocytes. Blood 2022, 139, 3127–3137. [Google Scholar] [CrossRef]
- Kristinsson, S.Y.; Landgren, O.; Samuelsson, J.; Bjorkholm, M.; Goldin, L.R. Autoimmunity and the risk of myeloproliferative neoplasms. Haematologica 2010, 95, 1216–1220. [Google Scholar] [CrossRef] [PubMed]
- Kuriakose, E.; Lascu, E.; Wang, Y.L.; Gjoni, S.; Cross, N.; Baumann, R.; Tam, K.; Scherl, E.; Longman, R.; Silver, R.T. The JAK2 V617F Mutation Seen in Myeloproliferative Neoplasms (MPNs) Occurs in Patients with Inflammatory Bowel Disease: Implications of a Pilot Study. Int. J. Clin. Med. 2013, 4, 10–15. [Google Scholar] [CrossRef]
- Bak, M.; Jess, T.; Flachs, E.M.; Zwisler, A.-D.; Juel, K.; Frederiksen, H. Risk of Inflammatory Bowel Disease in Patients with Chronic Myeloproliferative Neoplasms: A Danish Nationwide Cohort Study. Cancers 2020, 12, 2700. [Google Scholar] [CrossRef] [PubMed]
- Hesselø, H.; Bak, M.; Boysen, T.; Bytzer, P.; Hasselbalch, H.C. Myeloproliferative neoplasms and chronic inflammatory bowel disease. Ugeskr Laeger. 2020, 182, V09190483. [Google Scholar] [PubMed]
- Said, S.M.; Leung, N.; Sethi, S.; Cornell, L.D.; Fidler, M.E.; Grande, J.P.; Herrmann, S.; Tefferi, A.; D’Agati, V.D.; Nasr, S.H. Myeloproliferative neoplasms cause glomerulopathy. Kidney Int. 2011, 80, 753–759. [Google Scholar] [CrossRef]
- Christensen, A.S.; Møller, J.B.; Hasselbalch, H.C. Chronic kidney disease in patients with the Philadelphia-negative chronic myeloproliferative neoplasms. Leuk. Res. 2014, 38, 490–495. [Google Scholar] [CrossRef]
- Maruyama, K.; Nakagawa, N.; Suzuki, A.; Kabara, M.; Matsuki, M.; Shindo, M.; Iwasaki, S.; Ogawa, Y.; Hasebe, N. Novel Detection of CALR-Mutated Cells in Myeloproliferative Neoplasm-Related Glomerulopathy with Interstitial Extramedullary Hematopoiesis: A Case Report. Am. J. Kidney Dis. 2019, 74, 844–848. [Google Scholar] [CrossRef]
- Lu, A.; Pallero, M.A.; Owusu, B.Y.; Borovjagin, A.V.; Lei, W.; Sanders, P.W.; Murphy-Ullrich, J.E. Calreticulin is important for the development of renal fibrosis and dysfunction in diabetic nephropathy. Matrix Biol. Plus. 2020, 8, 100034. [Google Scholar] [CrossRef]
- Krečak, I.; Holik, H.; Martina, M.P.; Zekanović, I.; Coha, B.; Gverić-Krečak, V. Chronic kidney disease could be a risk factor for thrombosis in essential thrombocythemia and polycythemia vera. Int. J. Hematol. 2020, 112, 377–384. [Google Scholar] [CrossRef]
- Lucijanic, M.; Galusic, D.; Krecak, I.; Sedinic, M.; Holik, H.; Perisa, V.; Peric, M.M.; Zekanovic, I.; Stoos-Veic, T.; Kusec, R. Reduced renal function strongly affects survival and thrombosis in patients with myelofibrosis. Ann. Hematol. 2020, 99, 2779–2785. [Google Scholar] [CrossRef]
- Person, F.; Meyer, S.C.; Hopfer, H.; Menter, T. Renal post-mortem findings in myeloproliferative and myelodysplastic/myeloproliferative neoplasms. Virchows Arch. 2021, 479, 1013–1020. [Google Scholar] [CrossRef]
- Buttner-Herold, M.; Sticht, C.; Wiech, T.; Porubsky, S. Renal disease associated with myeloproliferative and myelodysplastic/myeloproliferative neoplasia. Histopathology 2021, 78, 738–748. [Google Scholar] [CrossRef]
- Gecht, J.; Tsoukakis, I.; Kricheldorf, K.; Stegelmann, F.; Klausmann, M.; Griesshammer, M.; Schulz, H.; Hollburg, W.; Göthert, J.R.; Sockel, K.; et al. Kidney Dysfunction Is Associated with Thrombosis and Disease Severity in Myeloproliferative Neoplasms: Implications from the German Study Group for MPN Bioregistry. Cancers 2021, 13, 4086. [Google Scholar] [CrossRef]
- Larsen, M.K.; Skov, V.; Kjær, L.; Møller-Palacino, N.A.; Pedersen, R.K.; Andersen, M.; Ottesen, J.T.; Cordua, S.; Poulsen, H.E.; Dahl, M.; et al. Clonal haematopoiesis of indeterminate potential and impaired kidney function-A Danish general population study with 11 years follow-up. Eur. J. Haematol. 2022, 109, 576–585. [Google Scholar] [CrossRef]
- Dawoud, A.A.Z.; Gilbert, R.D.; Tapper, W.J.; Cross, N.C.P. Clonal myelopoiesis promotes adverse outcomes in chronic kidney disease. Leukemia 2022, 36, 507–515. [Google Scholar] [CrossRef]
- Hasselbalch, H.C. Perspectives on the impact of JAK-inhibitor therapy upon inflammation-mediated comorbidities in myelofibrosis and related neoplasms. Expert Rev. Hematol. 2014, 7, 203–216. [Google Scholar] [CrossRef] [PubMed]
- García-Fortes, M.; Hernández-Boluda, J.C.; Álvarez-Larrán, A.; Raya, J.M.; Angona, A.; Estrada, N.; Fox, L.; Cuevas, B.; García-Hernández, M.C.; Gómez-Casares, M.T.; et al. Impact of Individual Comorbidities on Survival of Patients with Myelofibrosis. Cancers 2022, 14, 2331. [Google Scholar] [CrossRef]
- Geyer, H.L.; Dueck, A.C.; Scherber, R.M.; Mesa, R.A. Impact of Inflammation on Myeloproliferative Neoplasm Symptom Development. Mediat. Inflamm. 2015, 2015, 284706. [Google Scholar] [CrossRef] [PubMed]
- Frederiksen, H.; Szépligeti, S.K.; Bak, M.; Ghanima, W.; Hasselbalch, H.C.; Christiansen, C.F. Vascular Diseases in Patients with Chronic Myeloproliferative Neoplasms—Impact of Comorbidity. Clin. Epidemiol. 2019, 11, 955–967. [Google Scholar] [CrossRef] [PubMed]
- Lataillade, J.-J.; Pierre-Louis, O.; Hasselbalch, H.; Uzan, G.; Jasmin, C.; Martyré, M.-C.; Le Bousse-Kerdilès, M.-C.; on behalf of the French INSERM and the European EUMNET Networks on Myelofibrosis. Does primary myelofibrosis involve a defective stem cell niche? From concept to evidence. Blood 2008, 112, 3026–3035. [Google Scholar] [CrossRef] [PubMed]
- Hasselbalch, H.C. Myelofibrosis with myeloid metaplasia: The advanced phase of an untreated disseminated hematological cancer. Time to change our therapeutic attitude with early upfront treatment? Leuk. Res. 2009, 33, 11–18. [Google Scholar] [CrossRef]
- Le Bousse-Kerdilès, M.C. Primary myelofibrosis and the “bad seeds in bad soil” concept. Fibrogenesis Tissue Repair. 2012, 5 (Suppl. S1). [Google Scholar] [CrossRef]
- Desterke, C.; Martinaud, C.; Ruzehaji, N.; Le Bousse-Kerdilès, M.-C. Inflammation as a Keystone of Bone Marrow Stroma Alterations in Primary Myelofibrosis. Mediat. Inflamm. 2015, 2015, 415024. [Google Scholar] [CrossRef]
- Čokić, V.P.; Mitrović-Ajtić, O.; Beleslin-Čokić, B.B.; Marković, D.; Buač, M.; Diklić, M.; Kraguljac-Kurtović, N.; Damjanović, S.; Milenković, P.; Gotić, M.; et al. Proinflammatory Cytokine IL-6 and JAK-STAT Signaling Pathway in Myeloproliferative Neoplasms. Mediat. Inflamm. 2015, 2015, 453020. [Google Scholar] [CrossRef]
- Hoermann, G.; Greiner, G.; Valent, P. Cytokine Regulation of Microenvironmental Cells in Myeloproliferative Neoplasms. Mediat. Inflamm. 2015, 2015, 869242. [Google Scholar] [CrossRef] [PubMed]
- Hermouet, S.; Bigot-Corbel, E.; Gardie, B. Pathogenesis of Myeloproliferative Neoplasms: Role and Mechanisms of Chronic Inflammation. Mediat. Inflamm. 2015, 2015, 145293. [Google Scholar] [CrossRef] [PubMed]
- Mondet, J.; Hussein, K.; Mossuz, P. Circulating Cytokine Levels as Markers of Inflammation in Philadelphia Negative Myeloproliferative Neoplasms: Diagnostic and Prognostic Interest. Mediat. Inflamm. 2015, 2015, 670580. [Google Scholar] [CrossRef]
- Zahr, A.A.; Salama, M.E.; Carreau, N.; Tremblay, D.; Verstovsek, S.; Mesa, R.; Hoffman, R.; Mascarenhas, J. Bone marrow fibrosis in myelofibrosis: Pathogenesis, prognosis and targeted strategies. Haematologica 2016, 101, 660–671. [Google Scholar] [CrossRef] [PubMed]
- Jutzi, J.S.; Mullally, A. Remodeling the Bone Marrow Microenvironment—A Proposal for Targeting Pro-inflammatory Contributors in MPN. Front. Immunol. 2020, 11, 2093. [Google Scholar] [CrossRef]
- Wang, J.C.; Aung, M.K.; Tobin, M.S. Urinary hydroxyproline excretion in myelofibrosis. Blood 1980, 55, 383–385. [Google Scholar] [CrossRef]
- Hochweiss, S.; Fruchtman, S.; Hahn, E.G.; Gilbert, H.; Donovan, P.B.; Johnson, J.; Goldberg, J.D.; Berk, P.D. Increased serum procollagen III aminoterminal peptide in myelofibrosis. Am. J. Hematol. 1983, 15, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Norfolk, D.R.; Bowen, M.; Roberts, B.E.; Child, J.A. Plasma fibronectin in myeloproliferative disorders and chronic granulocytic leukaemia. Br. J. Haematol. 1983, 55, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Arrago, J.P.; Poirier, O.; Najean, Y. Evolution des polyglobulies primitives vers la myélofibrose. Surveillance par le dosage de l’amino-propeptide du procollagène III [Development of polycythemia to myelofibrosis. Monitoring by the assay of the procollagen III amino-propeptide]. Presse Med. 1984, 13, 2429–2432. [Google Scholar] [PubMed]
- Hasselbalch, H.; Junker, P.; Lisse, I.; Bentsen, K.D. Serum procollagen III peptide in chronic myeloproliferative disorders. Scand. J. Haematol. 1985, 35, 550–557. [Google Scholar] [CrossRef] [PubMed]
- Hasselbalch, H.; Junker, P.; Lisse, I.; Bentsen, K.D.; Risteli, L.; Risteli, J. Serum markers for type IV collagen and type III procollagen in the myelofibrosis-osteomyelosclerosis syndrome and other chronic myeloproliferative disorders. Am. J. Hematol. 1986, 23, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Arrago, J.P.; Poirier, O.; Chomienne, C.; D’Agay, M.F.; Najean, Y. Type III aminoterminal propeptide of procollagen in some haematological malignancies. Scand. J. Haematol. 1986, 36, 288–294. [Google Scholar] [CrossRef]
- Apaja-Sarkkinen, M.; Autio-Harmainen, H.; Alavaikko, M.; Risteli, J.; Risteli, L. Immunohistochemical study of basement membrane proteins and type III procollagen in myelofibrosis. Br. J. Haematol. 1986, 63, 571–580. [Google Scholar] [CrossRef]
- Hasselbalch, H.; Clemmensen, I. Plasma fibronectin in idiopathic myelofibrosis and related chronic myeloproliferative disorders. Scand. J. Clin. Lab. Investig. 1987, 47, 429–433. [Google Scholar] [CrossRef]
- Hasselbalch, H. Urinary hydroxyproline excretion in the myelofibrosis-osteomyelosclerosis syndrome and related diseases. Eur. J. Haematol. 1987, 39, 447–451. [Google Scholar] [CrossRef]
- Baglin, T.P.; Simpson, A.W.; Price, S.M.; Boughton, B.J. Composition of immune complexes and their relation to plasma fibronectin in chronic myeloproliferative disorders. J. Clin. Pathol. 1987, 40, 1468–1471. [Google Scholar] [CrossRef]
- Hasselbalch, H.; Junker, P.; Hørslev-Petersen, K.; Lisse, I.; Bentsen, K.D. Procollagen type III aminoterminal peptide in serum in idiopathic myelofibrosis and allied conditions: Relation to disease activity and effect of chemotherapy. Am. J. Hematol. 1990, 33, 18–26. [Google Scholar] [CrossRef]
- Baglin, T.P.; Price, S.M.; Boughton, B.J. Circulating high molecular weight IgG fibronectin complexes in myeloproliferative disorders. J. Clin. Pathol. 1990, 43, 102–105. [Google Scholar] [CrossRef]
- Hasselbalch, H. On the pathogenesis of angiogenesis in idiopathic myelofibrosis. Am. J. Hematol. 1990, 33, 151. [Google Scholar] [CrossRef]
- Hasselbalch, H. Idiopathic myelofibrosis: A review. Eur. J. Haematol. 1990, 45, 65–72. [Google Scholar] [CrossRef]
- Lisse, I.; Hasselbalch, H.; Junker, P. Bone marrow stroma in idiopathic myelofibrosis and other haematological diseases. An immunohistochemical study. APMIS 1991, 99, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Hasselbalch, H.; Junker, P.; Lisse, I.; Lindqvist, U.; Engström Laurent, A. Circulating hyaluronan in the myelofibrosis/osteomyelosclerosis syndrome and other myeloproliferative disorders. Am. J. Hematol. 1991, 36, 1–8. [Google Scholar] [CrossRef]
- Hasselbalch, H.C. Idiopathic myelofibrosis. Clinical aspects and studies on extracellular matrix metabolism. Dan. Med. Bull. 1993, 40, 39–55. [Google Scholar] [PubMed]
- Reilly, J.T.; Brindley, L.; Kay, M.; Fielding, S.; Kennedy, A.; Dolan, G.; Smith, A. Bone marrow and serum connective tissue polypeptides in idiopathic myelofibrosis. Clin. Lab. Haematol. 1995, 17, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Karsdal, M.; Henriksen, K.; Leeming, D.; Mitchell, P.; Duffin, K.; Barascuk, N.; Klickstein, L.; Aggarwal, P.; Nemirovskiy, O.; Byrjalsen, I.; et al. Biochemical markers and the FDA Critical Path: How biomarkers may contribute to the understanding of pathophysiology and provide unique and necessary tools for drug development. Biomarkers 2009, 14, 181–202. [Google Scholar] [CrossRef] [PubMed]
- Karsdal, M.A.; Henriksen, K.; Leeming, D.J.; Woodworth, T.; Vassiliadis, E.; Bay-Jensen, A.C. Novel combinations of Post-Translational Modification (PTM) neo-epitopes provide tissue-specific biochemical markers—Are they the cause or the consequence of the disease? Clin. Biochem. 2010, 43, 793–804. [Google Scholar] [CrossRef] [PubMed]
- Skjøt-Arkil, H.; Barascuk, N.; Register, T.; Karsdal, M.A.; Guo, X.; Higgs, B.W.; Yao, Y.; Roskos, L.K.; White, W.I.; Heegaard, N.H.H.; et al. Macrophage-Mediated Proteolytic Remodeling of the Extracellular Matrix in Atherosclerosis Results in Neoepitopes: A Potential New Class of Biochemical Markers. ASSAY Drug Dev. Technol. 2010, 8, 542–552. [Google Scholar] [CrossRef] [PubMed]
- Leeming, D.J.; Bay-Jensen, A.C.; Vassiliadis, E.; Larsen, M.R.; Henriksen, K.; Karsdal, M.A. Post-translational modifications of the extracellular matrix are key events in cancer progression: Opportunities for biochemical marker development. Biomarkers 2011, 16, 193–205. [Google Scholar] [CrossRef]
- Karsdal, M.A.; Nielsen, M.J.; Sand, J.M.; Henriksen, K.; Genovese, F.; Bay-Jensen, A.-C.; Smith, V.; Adamkewicz, J.I.; Christiansen, C.; Leeming, D.J. Extracellular Matrix Remodeling: The Common Denominator in Connective Tissue Diseases Possibilities for Evaluation and Current Understanding of the Matrix as More Than a Passive Architecture, but a Key Player in Tissue Failure. ASSAY Drug Dev. Technol. 2013, 11, 70–92. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Willumsen, N.; Zheng, Q.; Xue, Y.; Karsdal, M.A.; Bay-Jensen, A.C.; Marsh, S.; King, C.R.; Van Booven, D.J.; Revollo, J.Y.; et al. Bringing cancer serological diagnosis to a new level: Focusing on HER2, protein ectodomain shedding and neoepitope technology. Futur. Oncol. 2013, 9, 35–44. [Google Scholar] [CrossRef]
- Nielsen, M.J.; Nedergaard, A.F.; Sun, S.; Veidal, S.S.; Larsen, L.; Zheng, Q.; Suetta, C.; Henriksen, K.; Christiansen, C.; Karsdal, M.A.; et al. The neo-epitope specific PRO-C3 ELISA measures true formation of type III collagen associated with liver and muscle parameters. Am. J. Transl. Res. 2013, 5, 303–315. [Google Scholar] [PubMed]
- Willumsen, N.; Bager, C.L.; Leeming, D.J.; Smith, V.; Karsdal, M.A.; Dornan, D.; Bay-Jensen, A.-C. Extracellular matrix specific protein fingerprints measured in serum can separate pancreatic cancer patients from healthy controls. BMC Cancer 2013, 13, 554. [Google Scholar] [CrossRef]
- Willumsen, N.; Bager, C.L.; Leeming, D.J.; Smith, V.; Christiansen, C.; Karsdal, M.A.; Dornan, D.; Bay-Jensen, A. Serum biomarkers reflecting specific tumor tissue remodeling processes are valuable diagnostic tools for lung cancer. Cancer Med. 2014, 3, 1136–1145. [Google Scholar] [CrossRef]
- Bager, C.; Willumsen, N.; Leeming, D.; Smith, V.; Karsdal, M.; Dornan, D.; Bay-Jensen, A. Collagen degradation products measured in serum can separate ovarian and breast cancer patients from healthy controls: A preliminary study. Cancer Biomark. 2015, 15, 783–788. [Google Scholar] [CrossRef]
- Kristensen, J.H.; Karsdal, M.A.; Sand, J.M.; Willumsen, N.; Diefenbach, C.; Svensson, B.; Hägglund, P.; Oersnes-Leeming, D.J. Serological assessment of neutrophil elastase activity on elastin during lung ECM remodeling. BMC Pulm. Med. 2015, 15, 53. [Google Scholar] [CrossRef]
- Kristensen, J.; Larsen, L.; Dasgupta, B.; Brodmerkel, C.; Curran, M.; Karsdal, M.; Sand, J.; Willumsen, N.; Knox, A.; Bolton, C.; et al. Levels of circulating MMP-7 degraded elastin are elevated in pulmonary disorders. Clin. Biochem. 2015, 48, 1083–1088. [Google Scholar] [CrossRef]
- Hansen, N.U.; Willumsen, N.; Sand, J.M.; Larsen, L.; Karsdal, M.A.; Leeming, D.J. Type VIII collagen is elevated in diseases associated with angiogenesis and vascular remodeling. Clin. Biochem. 2016, 49, 903–908. [Google Scholar] [CrossRef]
- Bager, C.L.; Willumsen, N.; Kehlet, S.N.; Hansen, H.B.; Bay-Jensen, A.-C.; Leeming, D.J.; Dragsbaek, K.; Neergaard, J.S.; Christiansen, C.; Høgdall, E.; et al. Remodeling of the Tumor Microenvironment Predicts Increased Risk of Cancer in Postmenopausal Women: The Prospective Epidemiologic Risk Factor (PERF I) Study. Cancer Epidemiol. Biomark. Prev. 2016, 25, 1348–1355. [Google Scholar] [CrossRef]
- Bager, C.; Gudmann, N.; Willumsen, N.; Leeming, D.; Karsdal, M.; Bay-Jensen, A.; Høgdall, E.; Balslev, I.; He, Y. Quantification of fibronectin as a method to assess ex vivo extracellular matrix remodeling. Biochem. Biophys. Res. Commun. 2016, 478, 586–591. [Google Scholar] [CrossRef] [PubMed]
- Genovese, F.; Karsdal, M.A. Protein degradation fragments as diagnostic and prognostic biomarkers of connective tissue diseases: Understanding the extracellular matrix message and implication for current and future serological biomarkers. Expert Rev. Proteom. 2016, 13, 213–225. [Google Scholar] [CrossRef] [PubMed]
- Willumsen, N.; Bager, C.L.; Leeming, D.J.; Bay-Jensen, A.-C.; Karsdal, M.A. Nidogen-1 Degraded by Cathepsin S can be Quantified in Serum and is Associated with Non–Small Cell Lung Cancer. Neoplasia 2017, 19, 271–278. [Google Scholar] [CrossRef]
- Willumsen, N.; Bager, C.L.; Bay-Jensen, A.-C.; Kehlet, S.N.; Harling, H.; Leeming, D.J.; Karsdal, M.A.; Jorgensen, L.N. Unique insight into microenvironmental changes in colorectal cancer: Ex. vivo assessment of matrix metalloprotease-mediated molecular changes in human colorectal tumor tissue and corresponding non-neoplastic adjacent tissue. Oncol. Lett. 2017, 13, 3774–3780. [Google Scholar] [CrossRef] [PubMed]
- Willumsen, N.; Bager, C.L.; Kehlet, S.N.; Dragsbaek, K.; Neergaard, J.S.; Hansen, H.B.; Bay-Jensen, A.C.; Leeming, D.J.; Lipton, A.; Christiansen, C.; et al. Excessive matrix metalloprotease-mediated degradation of interstitial tissue (type I collagen) independently predicts short-term survival in an observational study of postmenopausal women diagnosed with cancer. Oncotarget 2017, 8, 52501–52510. [Google Scholar] [CrossRef]
- Kehlet, S.; Bager, C.; Willumsen, N.; Dasgupta, B.; Brodmerkel, C.; Curran, M.; Brix, S.; Leeming, D.; Karsdal, M.A. Cathepsin-S degraded decorin are elevated in fibrotic lung disorders—Development and biological validation of a new serum biomarker. BMC Pulm. Med. 2017, 17, 110. [Google Scholar] [CrossRef]
- Willumsen, N.; Thomsen, L.B.; Bager, C.L.; Jensen, C.; Karsdal, M.A. Quantification of altered tissue turnover in a liquid biopsy: A proposed precision medicine tool to assess chronic inflammation and desmoplasia associated with a pro-cancerous niche and response to immuno-therapeutic anti-tumor modalities. Cancer Immunol. Immunother. 2018, 67, 1–12. [Google Scholar] [CrossRef]
- Kehlet, S.N.; Manon-Jensen, T.; Sun, S.; Brix, S.; Leeming, D.J.; Karsdal, M.A.; Willumsen, N. A fragment of SPARC reflecting increased collagen affinity shows pathological relevance in lung cancer—Implications of a new collagen chaperone function of SPARC. Cancer Biol. Ther. 2018, 19, 904–912. [Google Scholar] [CrossRef]
- Kehlet, S.N.; Willumsen, N.; Armbrecht, G.; Dietzel, R.; Brix, S.; Henriksen, K.; Karsdal, M.A. Age-related collagen turnover of the interstitial matrix and basement membrane: Implications of age- and sex-dependent remodeling of the extracellular matrix. PLoS ONE 2018, 13, e0194458. [Google Scholar] [CrossRef]
- Nielsen, S.H.; Willumsen, N.; Brix, S.; Sun, S.; Manon-Jensen, T.; Karsdal, M.; Genovese, F. Tumstatin, a Matrikine Derived from Collagen Type IVα3, is Elevated in Serum from Patients with Non-Small Cell Lung Cancer. Transl. Oncol. 2018, 11, 528–534. [Google Scholar] [CrossRef]
- Jensen, C.; Nielsen, S.H.; Mortensen, J.H.; Kjeldsen, J.; Klinge, L.G.; Krag, A.; Harling, H.; Jørgensen, L.N.; Karsdal, M.A.; Willumsen, N. Serum type XVI collagen is associated with colorectal cancer and ulcerative colitis indicating a pathological role in gastrointestinal disorders. Cancer Med. 2018, 7, 4619–4626. [Google Scholar] [CrossRef]
- Lipton, A.; Leitzel, K.; Ali, S.M.; Polimera, H.V.; Nagabhairu, V.; Marks, E.; Richardson, A.E.; Krecko, L.; Ali, A.; Koestler, W.; et al. High turnover of extracellular matrix reflected by specific protein fragments measured in serum is associated with poor outcomes in two metastatic breast cancer cohorts. Int. J. Cancer 2018, 143, 3027–3034. [Google Scholar] [CrossRef] [PubMed]
- Jensen, C.; Madsen, D.H.; Hansen, M.; Schmidt, H.; Svane, I.M.; Karsdal, M.A.; Willumsen, N. Non-invasive biomarkers derived from the extracellular matrix associate with response to immune checkpoint blockade (anti-CTLA-4) in metastatic melanoma patients. J. Immunother. Cancer 2018, 6, 152. [Google Scholar] [CrossRef] [PubMed]
- Willumsen, N.; Jorgensen, L.N.; Karsdal, M.A. Vastatin (the NC1 domain of human type VIII collagen a1 chain) is linked to stromal reactivity and elevated in serum from patients with colorectal cancer. Cancer Biol. Ther. 2019, 20, 692–699. [Google Scholar] [CrossRef] [PubMed]
- Thorlacius-Ussing, J.; Kehlet, S.N.; Rønnow, S.R.; Karsdal, M.A.; Willumsen, N. Non-invasive profiling of protease-specific elastin turnover in lung cancer: Biomarker potential. J. Cancer Res. Clin. Oncol. 2019, 145, 383–392. [Google Scholar] [CrossRef]
- Nielsen, S.H.; Willumsen, N.; Leeming, D.J.; Daniels, S.J.; Brix, S.; Karsdal, M.A.; Genovese, F.; Nielsen, M.J. Serological Assessment of Activated Fibroblasts by alpha-Smooth Muscle Actin (α-SMA): A Noninvasive Biomarker of Activated Fibroblasts in Lung Disorders. Transl. Oncol. 2019, 12, 368–374. [Google Scholar] [CrossRef]
- Leeming, D.; Willumsen, N.; Sand, J.; Nielsen, S.H.; Dasgupta, B.; Brodmerkel, C.; Curran, M.; Bager, C.; Karsdal, M. A serological marker of the N-terminal neoepitope generated during LOXL2 maturation is elevated in patients with cancer or idiopathic pulmonary fibrosis. Biochem. Biophys. Rep. 2018, 17, 38–43. [Google Scholar] [CrossRef]
- Nissen, N.I.; Karsdal, M.; Willumsen, N. Collagens and Cancer associated fibroblasts in the reactive stroma and its relation to Cancer biology. J. Exp. Clin. Cancer Res. 2019, 38, 115. [Google Scholar] [CrossRef]
- Willumsen, N.; Bager, C.; Karsdal, M.A. Matrix Metalloprotease Generated Fragments of Type VI Collagen Have Serum Biomarker Potential in Cancer—A Proof of Concept Study. Transl. Oncol. 2019, 12, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Bager, C.L.; Willumsen, N.; Christiansen, C.; Bay-Jensen, A.C.; Nielsen, H.B.; Karsdal, M. Bone and Soft Tissue Turnover in Relation to All-cause Mortality in Postmenopausal Women. J. Gerontol. A Biol. Sci. Med. Sci. 2019, 74, 1098–1104. [Google Scholar] [CrossRef] [PubMed]
- Banys-Paluchowski, M.; Loibl, S.; Witzel, I.; Mundhenke, C.; Lederer, B.; Solbach, C.; Karn, T.; Marmé, F.; Nekljudova, V.; Schem, C.; et al. Clinical Relevance of Collagen Protein Degradation Markers C3M and C4M in the Serum of Breast Cancer Patients Treated with Neoadjuvant Therapy in the GeparQuinto Trial. Cancers 2019, 11, 1186. [Google Scholar] [CrossRef]
- Nissen, N.I.; Karsdal, M.; Willumsen, N. Post-translational modifications of vimentin reflect different pathological processes associated with non-small cell lung cancer and chronic obstructive pulmonary disease. Oncotarget 2019, 10, 6829–6841. [Google Scholar] [CrossRef] [PubMed]
- Willumsen, N.; Ali, S.M.; Leitzel, K.; Drabick, J.J.; Yee, N.; Polimera, H.V.; Nagabhairu, V.; Krecko, L.; Ali, A.; Maddukuri, A.; et al. Collagen fragments quantified in serum as measures of desmoplasia associate with survival outcome in patients with advanced pancreatic cancer. Sci. Rep. 2019, 9, 19761. [Google Scholar] [CrossRef] [PubMed]
- Karsdal, M.A. Biochemistry of collagens, laminins and elastin: Structure, function and biomarkers. In Biochemistry of Collagens, Laminins Elastin; Elsevier: Amsterdam, The Netherlands, 2019. [Google Scholar]
- Nielsen, S.H.; Mortensen, J.H.; Willumsen, N.; Rasmussen, D.G.K.; Mogensen, D.J.; Di Sabatino, A.; Mazza, G.; Jørgensen, L.N.; Giuffrida, P.; Pinzani, M.; et al. A Fragment of Collagen Type VI alpha-3 chain is Elevated in Serum from Patients with Gastrointestinal Disorders. Sci. Rep. 2020, 10, 5910. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.M.; Willumsen, N.; Dehlendorff, C.; Johansen, A.Z.; Jensen, B.V.; Hansen, C.P.; Hasselby, J.P.; Bojesen, S.E.; Pfeiffer, P.; Nielsen, S.E.; et al. Clinical value of serum hyaluronan and propeptide of type III collagen in patients with pancreatic cancer. Int. J. Cancer 2020, 146, 2913–2922. [Google Scholar] [CrossRef] [PubMed]
- Thorlacius-Ussing, J.; Manon-Jensen, T.; Sun, S.; Leeming, D.J.; Sand, J.M.; Karsdal, M.; Willumsen, N. Serum Type XIX Collagen is Significantly Elevated in Non-Small Cell Lung Cancer: A Preliminary Study on Biomarker Potential. Cancers 2020, 12, 1510. [Google Scholar] [CrossRef] [PubMed]
- Jensen, C.; Sinkeviciute, D.; Madsen, D.H.; Önnerfjord, P.; Hansen, M.; Schmidt, H.; Karsdal, M.A.; Svane, I.M.; Willumsen, N. Granzyme B Degraded Type IV Collagen Products in Serum Identify Melanoma Patients Responding to Immune Checkpoint Blockade. Cancers 2020, 12, 2786. [Google Scholar] [CrossRef]
- Hurkmans, D.P.; Jensen, C.; Koolen, S.L.W.; Aerts, J.; Karsdal, M.A.; Mathijssen, R.H.J.; Willumsen, N. Blood-based extracellular matrix biomarkers are correlated with clinical outcome after PD-1 inhibition in patients with metastatic melanoma. J. Immunother. Cancer 2020, 8, e001193. [Google Scholar] [CrossRef]
- Jensen, C.; Holm Nielsen, S.; Eslam, M.; Genovese, F.; Nielsen, M.J.; Vongsuvanh, R.; Uchila, R.; van der Poorten, D.; George, J.; Karsdal, M.A.; et al. Cross-Linked Multimeric Pro-Peptides of Type III Collagen (PC3X) in Hepatocellular Carcinoma—A Biomarker That Provides Additional Prognostic Value in AFP Positive Patients. J. Hepatocell. Carcinoma. 2020, 7, 301–313. [Google Scholar] [CrossRef] [PubMed]
- Nissen, N.I.; Kehlet, S.; Boisen, M.K.; Liljefors, M.; Jensen, C.; Johansen, A.Z.; Johansen, J.S.; Erler, J.T.; Karsdal, M.; Mortensen, J.H.; et al. Prognostic value of blood-based fibrosis biomarkers in patients with metastatic colorectal cancer receiving chemotherapy and bevacizumab. Sci. Rep. 2021, 11, 865. [Google Scholar] [CrossRef] [PubMed]
- Türlü, C.; Willumsen, N.; Marando, D.; Schjerling, P.; Biskup, E.; Hannibal, J.; Jorgensen, L.N.; Ågren, M.S. A Human Cellular Model for Colorectal Anastomotic Repair: The Effect of Localization and Transforming Growth Factor-β1 Treatment on Collagen Deposition and Biomarkers. Int. J. Mol. Sci. 2021, 22, 1616. [Google Scholar] [CrossRef]
- Ramos, M.I.P.; Tian, L.; de Ruiter, E.J.; Song, C.; Paucarmayta, A.; Singh, A.; Elshof, E.; Vijver, S.V.; Shaik, J.; Bosiacki, J.; et al. Cancer immunotherapy by NC410, a LAIR-2 Fc protein blocking human LAIR-collagen interaction. Elife 2021, 10, e62927. [Google Scholar] [CrossRef]
- Nissen, N.I.; Kehlet, S.; Johansen, A.Z.; Chen, I.M.; Karsdal, M.; Johansen, J.S.; Diab, H.M.H.; Jørgensen, L.N.; Sun, S.; Manon-Jensen, T.; et al. Noninvasive prognostic biomarker potential of quantifying the propeptides of Type XI collagen alpha-1 chain (PRO-C11) in patients with pancreatic ductal adenocarcinoma. Int. J. Cancer 2021, 149, 228–238. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.L.; Bager, C.L.; Willumsen, N.; Ramchandani, D.; Kornhauser, N.; Ling, L.; Cobham, M.; Andreopoulou, E.; Cigler, T.; Moore, A.; et al. Tetrathiomolybdate (TM)-associated copper depletion influences collagen remodeling and immune response in the pre-metastatic niche of breast cancer. NPJ Breast Cancer 2021, 7, 108. [Google Scholar] [CrossRef] [PubMed]
- Reese-Petersen, A.L.; Willumsen, N.; Palau, P.; Nunez, J.; Sun, S.; Jensen, T.M.; Kamall, B.; Karsdal, M.A.; Genovese, F. Evaluation of a novel biomarker of type XXVIII collagen formation, PRO-C28, in samples from cancer and heart failure with preserved ejection fraction patients. J. Pharm. Biomed. Anal. 2021, 204, 114272. [Google Scholar] [CrossRef]
- Jensen, C.; Nissen, N.I.; Von Arenstorff, C.S.; Karsdal, M.A.; Willumsen, N. Serological assessment of collagen fragments and tumor fibrosis may guide immune checkpoint inhibitor therapy. J. Exp. Clin. Cancer Res. 2021, 40, 326. [Google Scholar] [CrossRef] [PubMed]
- Karsdal, M.; Genovese, F.; Rasmussen, D.; Bay-Jensen, A.; Mortensen, J.; Nielsen, S.H.; Willumsen, N.; Jensen, C.; Manon-Jensen, T.; Jennings, L.; et al. Considerations for understanding protein measurements: Identification of formation, degradation and more pathological relevant epitopes. Clin. Biochem. 2021, 97, 11–24. [Google Scholar] [CrossRef]
- O’Connor, A.; Pannee, J.; Poole, T.; Arber, C.; Portelius, E.; Swift, I.J.; Heslegrave, A.J.; Abel, E.; Willumsen, N.; Rice, H.; et al. Plasma amyloid-β ratios in autosomal dominant Alzheimer’s disease: The influence of genotype. Brain 2021, 144, 2964–2970. [Google Scholar] [CrossRef]
- Thorseth, M.-L.; Carretta, M.; Jensen, C.; Mølgaard, K.; Jürgensen, H.J.; Engelholm, L.H.; Behrendt, N.; Willumsen, N.; Madsen, D.H. Uncovering mediators of collagen degradation in the tumor microenvironment. Matrix Biol. Plus 2022, 13, 100101. [Google Scholar] [CrossRef]
- Nissen, N.I.; Johansen, A.Z.; Chen, I.; Johansen, J.S.; Pedersen, R.S.; Hansen, C.P.; Karsdal, M.A.; Willumsen, N. Collagen Biomarkers Quantify Fibroblast Activity In Vitro and Predict Survival in Patients with Pancreatic Ductal Adenocarcinoma. Cancers 2022, 14, 819. [Google Scholar] [CrossRef] [PubMed]
- Willumsen, N.; Jensen, C.; Green, G.; Nissen, N.I.; Neely, J.; Nelson, D.M.; Pedersen, R.S.; Frederiksen, P.; Chen, I.M.; Boisen, M.K.; et al. Fibrotic activity quantified in serum by measurements of type III collagen pro-peptides can be used for prognosis across different solid tumor types. Cell Mol. Life Sci. 2022, 79, 204. [Google Scholar] [CrossRef]
- Thorlacius-Ussing, J.; Jensen, C.; Madsen, E.A.; Nissen, N.I.; Manon-Jensen, T.; Chen, I.M.; Johansen, J.S.; Diab, H.M.H.; Jørgensen, L.N.; Karsdal, M.A.; et al. Type XX Collagen Is Elevated in Circulation of Patients with Solid Tumors. Int. J. Mol. Sci. 2022, 23, 4144. [Google Scholar] [CrossRef]
- Pedersen, R.S.; Nissen, N.I.; Jensen, C.; Thorlacius-Ussing, J.; Manon-Jensen, T.; Olesen, M.L.; Langholm, L.L.; Diab, H.M.H.; Jorgensen, L.N.; Hansen, C.P.; et al. Plasma Kallikrein-Activated TGF-β Is Prognostic for Poor Overall Survival in Patients with Pancreatic Ductal Adenocarcinoma and Associates with Increased Fibrogenesis. Biomolecules 2022, 12, 1315. [Google Scholar] [CrossRef]
- Madsen, E.A.; Thorlacius-Ussing, J.; Nissen, N.I.; Jensen, C.; Chen, I.M.; Johansen, J.S.; Diab, H.M.H.; Jørgensen, L.N.; Hansen, C.P.; Karsdal, M.A.; et al. Type XXII Collagen Complements Fibrillar Collagens in the Serological Assessment of Tumor Fibrosis and the Outcome in Pancreatic Cancer. Cells 2022, 11, 3763. [Google Scholar] [CrossRef] [PubMed]
- Prockop, D.J.; Kivirikko, K.I.; Tuderman, L.; Guzman, N.A. The biosynthesis of collagen and its disorders (first of two parts). N. Engl. J. Med. 1979, 301, 13–23. [Google Scholar] [CrossRef]
- Prockop, D.J.; Kivirikko, K.I.; Tuderman, L.; Guzman, N.A. The biosynthesis of collagen and its disorders (second of two parts). N. Engl. J. Med. 1979, 301, 77–85. [Google Scholar] [CrossRef]
- Miller, E.J.; Gay, S. The collagens: An overview and update. Methods Enzymol. 1987, 144, 3–41. [Google Scholar] [PubMed]
- Gay, S.; Miller, E.J. Collagen in the Physiology and Pathology of Connective Tissue; Gustav Fischer: Stuttgart, Germany; New York, NY, USA, 1978. [Google Scholar]
- Dvorak, H.F. Tumors: Wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 1986, 315, 1650–1659. [Google Scholar]
- Dvorak, H.F. Tumors: Wounds that do not heal-redux. Cancer Immunol. Res. 2015, 3, 1–164. [Google Scholar] [CrossRef]
- Dvorak, H.F. Tumors: Wounds That Do Not Heal—A Historical Perspective with a Focus on the Fundamental Roles of Increased Vascular Permeability and Clotting. Semin. Thromb. Hemost. 2019, 45, 576–592. [Google Scholar] [CrossRef]
- Dvorak, H.F. Reconciling VEGF with VPF: The Importance of Increased Vascular Permeability for Stroma Formation in Tumors, Healing Wounds, and Chronic Inflammation. Front. Cell Dev. Biol. 2021, 9, 660609. [Google Scholar] [CrossRef]
- Ross, R.; Raines, E.W.; Bowen-Pope, D.F. The biology of platelet-derived growth factor. Cell 1986, 46, 155–169. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.J.; Afshar-Kharghan, V.; Schafer, A.I. Paraneoplastic thrombocytosis: The secrets of tumor self-promotion. Blood 2014, 124, 184–187. [Google Scholar] [CrossRef]
- Hasselbalch, H.C. The platelet-cancer loop in myeloproliferative cancer. Is thrombocythemia an enhancer of cancer invasiveness and metastasis in essential thrombocythemia, polycythemia vera and myelofibrosis? Leuk. Res. 2014, 38, 1230–1236. [Google Scholar] [CrossRef]
- Schlesinger, M. Role of platelets and platelet receptors in cancer metastasis. J. Hematol. Oncol. 2018, 11, 125. [Google Scholar] [CrossRef]
- Litwiniuk, M.; Krejner, A.; Speyrer, M.S.; Gauto, A.R.; Grzela, T. Hyaluronic Acid in Inflammation and Tissue Regeneration. Wounds 2016, 28, 78–88. [Google Scholar] [PubMed]
- Abatangelo, G.; Vindigni, V.; Avruscio, G.; Pandis, L.; Brun, P. Hyaluronic Acid: Redefining Its Role. Cells 2020, 9, 1743. [Google Scholar] [CrossRef] [PubMed]
- Burkhardt, R.; Bartl, R.; Beil, E.; Hoffman, E.; Kronseder, A. Myelofibrosis-Osteomyelosclerosis Syndrome—Review of literature and histomorphology. In Advances in the Biosciences, Dahlem Workshop on Myelofibrosis-Osteomyelosclerosis Syndrome; Elsevier: Berlin, Germany, 1974; Volume 16, pp. 9–56. [Google Scholar]
- Mesa, R.A.; Hanson, C.A.; Rajkumar, S.V.; Schroeder, G.; Tefferi, A. Evaluation and clinical correlations of bone marrow angiogenesis in myelofibrosis with myeloid metaplasia. Blood 2000, 96, 3374–3380. [Google Scholar] [CrossRef] [PubMed]
- Panteli, K.; Zagorianakou, N.; Bai, M.; Katsaraki, A.; Agnantis, N.J.; Bourantas, K. Angiogenesis in chronic myeloproliferative diseases detected by CD34 expression. Eur. J. Haematol. 2004, 72, 410–415. [Google Scholar] [CrossRef]
- Panteli, K.; Zagorianakou, N.; Agnantis, N.; Bourantas, K.; Bai, M. Clinical correlation of bone marrow microvessel density in essential thrombocythemia. Acta Haematol. 2005, 114, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Boveri, E.; Passamonti, F.; Rumi, E.; Pietra, D.; Elena, C.; Arcaini, L.; Pascutto, C.; Castello, A.; Cazzola, M.; Magrini, U.; et al. Bone marrow microvessel density in chronic myeloproliferative disorders: A study of 115 patients with clinicopathological and molecular correlations. Br. J. Haematol. 2008, 140, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Medinger, M.; Skoda, R.; Gratwohl, A.; Theocharides, A.; Buser, A.; Heim, D.; Dirnhofer, S.; Tichelli, A.; Tzankov, A. Angiogenesis and vascular endothelial growth factor-/receptor expression in myeloproliferative neoplasms: Correlation with clinical parameters and JAK2-V617F mutational status. Br. J. Haematol. 2009, 146, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Lennert, K.; Nakai, K.; Schwarze, E.W. Patoanatomical features of the bone marrow. Clin. Haematol. 1975, 39, 233–239. [Google Scholar]
- Reilly, J.T.; Nash, J.R.; Mackie, M.J.; McVerry, B.A. Immuno-enzymatic detection of fibronectin in normal and pathological haematopoietic tissue. Br. J. Haematol. 1985, 59, 497–504. [Google Scholar] [CrossRef]
- Reilly, J.T.; Nash, J.R.; Mackie, M.J.; McVerry, B.A. Endothelial cell proliferation in myelofibrosis. Br. J. Haematol. 1985, 60, 625–630. [Google Scholar] [CrossRef]
- Assoian, R.K.; Grotendorst, G.R.; Miller, D.M.; Sporn, M.B. Cellular transformation by coordinated action of three peptide growth factors from human platelets. Nature 1984, 309, 804–806. [Google Scholar] [CrossRef]
- Miyazono, K.; Okabe, T.; Urabe, A.; Takaku, F.; Heldin, C.H. Purification and properties of an endothelial cell growth factor from human platelets. J. Biol. Chem. 1987, 262, 4098–4103. [Google Scholar] [CrossRef]
- Stone, A.P.; Nascimento, T.F.; Barrachina, M.N. The bone marrow niche from the inside out: How megakaryocytes are shaped by and shape hematopoiesis. Blood 2022, 139, 483–491. [Google Scholar] [CrossRef]
- Zhan, H.; Kaushansky, K. Megakaryocytes as the Regulator of the Hematopoietic Vascular Niche. Front. Oncol. 2022, 12, 912060. [Google Scholar] [CrossRef]
- Mitroulis, I.; Kalafati, L.; Bornhäuser, M.; Hajishengallis, G.; Chavakis, T. Regulation of the Bone Marrow Niche by Inflammation. Front. Immunol. 2020, 11, 1540. [Google Scholar] [CrossRef] [PubMed]
- Takizawa, H.; Manz, M.G. Impact of inflammation on early hematopoiesis and the microenvironment. Int. J. Hematol. 2017, 106, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Leimkühler, N.B.; Schneider, R.K. Inflammatory bone marrow microenvironment. Hematol. Am. Soc. Hematol. Educ. Program. 2019, 2019, 294–302. [Google Scholar] [CrossRef] [PubMed]
- Zhan, H.; Ma, Y.; Lin, C.H.S.; Kaushansky, K. JAK2V617F-mutant megakaryocytes contribute to hematopoietic stem/progenitor cell expansion in a model of murine myeloproliferation. Leukemia 2016, 30, 2332–2341. [Google Scholar] [CrossRef]
- Zhang, Y.; Lin, C.H.S.; Kaushansky, K.; Zhan, H. JAK2V617F Megakaryocytes Promote Hematopoietic Stem/Progenitor Cell Expansion in Mice through Thrombopoietin/MPL Signaling. Stem Cells 2018, 36, 1676–1684. [Google Scholar] [CrossRef]
- Lee, S.; Wong, H.; Castiglione, M.; Murphy, M.; Kaushansky, K.; Zhan, H. JAK2V617F Mutant Megakaryocytes Contribute to Hematopoietic Aging in a Murine Model of Myeloproliferative Neoplasm. Stem Cells 2022, 40, 359–370. [Google Scholar] [CrossRef]
- Kurkinen, M.; Vaheri, A.; Roberts, P.J.; Stenman, S. Sequential appearance of fibronectin and collagen in experimental granulation tissue. Lab. Invest. 1980, 43, 47–51. [Google Scholar]
- Andreasen, A.P. Myelofibrosis; Munksgaard: Copenhagen, Denmark, 1958. [Google Scholar]
- Pitcock, J.A.; Reinhard, E.H.; Justus, B.W.; Mendelsohn, R.S. A clinical and pathological study of seventy cases of myelofibrosis. Ann. Intern. Med. 1962, 57, 73–84. [Google Scholar] [CrossRef]
- Bouroncle, B.A.; Doan, C.A. Myelofibrosis. Clinical, hematologic and pathologic study of 110 patients. Am. J. Med. Sci. 1962, 243, 697–715. [Google Scholar] [CrossRef]
- Buyssens, N.; Bourgeois, N.H. Chronic myelolytic leukemia versus idiopathic myelofibrosis: A diagnostic problem in bone marrow biopsies. Cancer 1977, 40, 1548–1561. [Google Scholar] [CrossRef] [PubMed]
- Ward, H.P.; Block, M.H. The natural history of agnogenic myeloid metaplasia (AMM) and a critical evaluation of its relationship with the myeloproliferative syndrome. Medicine 1971, 50, 357–420. [Google Scholar] [CrossRef] [PubMed]
- Manoharan, A.; Smart, R.C.; Pitney, W.R. Prognostic factors in myelofibrosis. Pathology 1982, 14, 455–461. [Google Scholar] [CrossRef]
- Lohman, T.P.; Beckman, E.N. Progressive myelofibrosis in agnogenic myeloid metaplasia. Arch. Pathol. Lab. Med. 1983, 107, 593–594. [Google Scholar]
- Thiele, J.; Kvasnicka, H.M.; Müllauer, L.; Buxhofer-Ausch, V.; Gisslinger, B.; Gisslinger, H. Essential thrombocythemia versus early primary myelofibrosis: A multicenter study to validate the WHO classification. Blood 2011, 117, 5710–5718. [Google Scholar] [CrossRef]
- Barbui, T.; Thiele, J.; Vannucchi, A.M.; Tefferi, A. Problems and pitfalls regarding WHO-defined diagnosis of early/prefibrotic primary myelofibrosis versus essential thrombocythemia. Leukemia 2013, 27, 1953–1958. [Google Scholar] [CrossRef]
- Madelung, A.B.; Bondo, H.; Stamp, I.; Loevgreen, P.; Nielsen, S.L.; Falensteen, A.; Knudsen, H.; Ehinger, M.; Dahl-Sørensen, R.; Mortensen, N.B.; et al. World Health Organization-defined classification of myeloproliferative neoplasms: Morphological reproducibility and clinical correlations--the Danish experience. Am. J. Hematol. 2013, 88, 1012–1016. [Google Scholar] [CrossRef]
- Thiele, J.; Kvasnicka, H.M.; Orazi, A.; Gianelli, U.; Gangat, N.; Vannucchi, A.M.; Barbui, T.; Arber, D.A.; Tefferi, A. The international consensus classification of myeloid neoplasms and acute leukemias: Myeloproliferative neoplasms. Am. J. Hematol. 2022, 98, 166–179. [Google Scholar] [CrossRef]
- Buhr, T.; Hebeda, K.; Kaloutsi, V.; Porwit, A.; Van der Walt, J.; Kreipe, H. European Bone Marrow Working Group trial on reproducibility of World Health Organization criteria to discriminate essential thrombocythemia from prefibrotic primary myelofibrosis. Haematologica 2012, 97, 360–365. [Google Scholar] [CrossRef] [PubMed]
- Manoharan, A.; Pitney, W.R. Chemotherapy resolves symptoms and reverses marrow fibrosis in myelofibrosis. Scand. J. Haematol. 1984, 33, 453–459. [Google Scholar] [CrossRef]
- Wolf, B.C.; Neiman, R.S. Myelofibrosis with myeloid metaplasia: Pathophysiologic implications of the correlation between bone marrow changes and progression of splenomegaly. Blood 1985, 65, 803–809. [Google Scholar] [CrossRef] [PubMed]
- Manoharan, A.; Chen, C.F.; Wilson, L.S.; Griffiths, K.A.; Robinson, D.E. Ultrasonic characterization of splenic tissue in myelofibrosis: Further evidence for reversal of fibrosis with chemotherapy. Eur. J. Haematol. 1988, 40, 149–154. [Google Scholar] [CrossRef]
- Thiele, J.; Simon, K.G.; Fischer, R.; Zankovich, R. Follow-up studies with sequential bone marrow biopsies in chronic myeloid leukaemia and so-called primary (idiopathic) osteo-myelofibrosis. Evolution of histopathological lesions and clinical course in 40 patients. Pathol. Res. Pract. 1988, 183, 434–445. [Google Scholar] [CrossRef] [PubMed]
- Zankovich, R.; Thiele, J.; Mödder, B.; Steinberg, T.; Simon, K.G.; Fischer, R.; Diehl, V. Die sogenannte primäre (idiopathische) Osteomyelofibrose/-sklerose (OMF) im Rahmen chronischer myeloproliferativer Erkrankungen. I. Initiale klinische und histologische Befunde bei 102 Patienten mit besonderer Berücksichtigung der frühen zellreichen und der fortgeschrittenen fibrosklerotischen Stadien [So-called primary (idiopathic) osteomyelofibrosis/-sclerosis (OMF) within the scope of chronic myeloproliferative diseases. I. Initial clinical and histologic findings in 102 patients with special reference to early hyperplastic and advanced fibrosclerotic stages]. Med. Klin. 1988, 83, 617–628. [Google Scholar]
- Hasselbalch, H.; Lisse, I. A sequential histological study of bone marrow fibrosis in idiopathic myelofibrosis. Eur. J. Haematol. 1991, 46, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Bentley, S.A.; Herman, C.J. Bone marrow fibre production in myelofibrosis: A quantitative study. Br. J. Haematol. 1979, 42, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Thiele, J.; Kvasnicka, H.M.; Boeltken, B.; Zanko-vich, R.; Diehl, V.; Fischer, R. Initial (prefibrotic) stages of idiopathic (primary) myelofibrosis (IMF)—A clinicopathological study. Leukemia 1999, 13, 1741–1748. [Google Scholar] [CrossRef]
- Barbui, T.; Thiele, J.; Passamonti, F.; Rumi, E.; Boveri, E.; Ruggeri, M.; Rodeghiero, F.; d’Amore, E.S.; Randi, M.L.; Bertozzi, I.; et al. Survival and disease progression in essential thrombocythemia are significantly influenced by accurate morphologic diagnosis: An international study. J. Clin. Oncol. 2011, 29, 3179–3184. [Google Scholar] [CrossRef]
- Barbui, T.; Thiele, J.; Carobbio, A.; Passamonti, F.; Rumi, E.; Randi, M.L.; Bertozzi, I.; Vannucchi, A.M.; Gisslinger, H.; Gisslinger, B.; et al. Disease characteristics and clinical outcome in young adults with essential thrombocythemia versus early/prefibrotic primary myelofibrosis. Blood 2012, 120, 569–571. [Google Scholar] [CrossRef] [PubMed]
- Guglielmelli, P.; Pacilli, A.; Rotunno, G.; Rumi, E.; Rosti, V.; Delaini, F.; Maffioli, M.; Fanelli, T.; Pancrazzi, A.; Pietra, D.; et al. Presentation and outcome of patients with 2016 WHO diagnosis of prefibrotic and overt primary myelofibrosis. Blood 2017, 129, 3227–3236. [Google Scholar] [CrossRef]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef] [PubMed]
- Barbui, T.; Thiele, J.; Gisslinger, H.; Kvasnicka, H.M.; Vannucchi, A.M.; Guglielmelli, P.; Orazi, A.; Tefferi, A. The 2016 WHO classification and diagnostic criteria for myeloproliferative neoplasms: Document summary and in-depth discussion. Blood Cancer J. 2018, 8, 15. [Google Scholar] [CrossRef]
- Arber, D.A.; Orazi, A.; Hasserjian, R.P.; Borowitz, M.J.; Calvo, K.R.; Kvasnicka, H.-M.; Wang, S.A.; Bagg, A.; Barbui, T.; Branford, S.; et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: Integrating morphologic, clinical, and genomic data. Blood 2022, 140, 1200–1228. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, B.S.; Erber, W.N.; Bareford, D.; Buck, G.; Wheatley, K.; East, C.L.; Paul, B.; Harrison, C.N.; Green, A.R.; Campbell, P.J. Bone marrow pathology in essential thrombocythemia: Interobserver reliability and utility for identifying disease subtypes. Blood 2008, 111, 60–70. [Google Scholar] [CrossRef]
- Curto-Garcia, N.; Ianotto, J.C.; Harrison, C.N. What is pre-fibrotic myelofibrosis and how should it be managed in 2018? Br. J. Haematol. 2018, 183, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Barbui, T.; Thiele, J.; Gisslinger, H.; Orazi, A.; Vannucchi, A.M.; Gianelli, U.; Beham-Schmid, C.; Tefferi, A. Comments on pre-fibrotic myelofibrosis and how should it be managed. Br. J. Haematol. 2019, 186, 358–360. [Google Scholar] [CrossRef]
- Pereira, A.; Cervantes, F.; Brugues, R.; Rozman, C. Bone marrow histopathology in primary myelofibrosis: Clinical and haematologic correlations and prognostic evaluation. Eur. J. Haematol. 1990, 44, 95–99. [Google Scholar] [CrossRef]
- Shreiner, D.P. Spontaneous hematologic remission in agnogenic myeloid metaplasia. Am. J. Med. 1976, 60, 1014–1018. [Google Scholar] [CrossRef]
- Lau, S.O.; Ramsay, N.K.; Smith, C.M.; McKenna, R.; Kersey, J.H. Spontaneous resolution of severe childhood myelofibrosis. J. Pediatr. 1981, 98, 585–588. [Google Scholar] [CrossRef]
- Parmentier, C.; Charbord, P.; Tibi, M.; Tubiana, M. Splenic irradiation in myelofibrosis. Clinical findings and ferrokinetics. Int. J. Radiat. Oncol. Biol. Phys. 1977, 2, 1075–1081. [Google Scholar] [CrossRef]
- Pettit, J.E.; Lewis, S.M.; Goolden, A.W.G. Polycythaemia vera-transformation to myelofibrosis and subsequent reversal. Scand. J. Haematol. 1978, 20, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Söderström, N.; Bandmann, U.; Lundh, B. Patho-anatomical features of the spleen and liver. Clin. Haematol. 1975, 4, 309–329. [Google Scholar] [CrossRef]
- Barosi, G.; Baraldi, A.; Cazzola, M.; Fortunato, A.; Palestra, P.; Polino, G.; Ramella, S.; Spriano, P. Polycythaemia following splenectomy in myelofibrosis with myeloid metaplasia. A reorganization of erythropoiesis. Scand. J. Haematol. 1984, 32, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, K.; Hotta, T. Spontaneous remission of agnogenic myeloid metaplasia in a splenectomized patient: A case report with erythrokinetic studies. Acta Haematol. 1990, 83, 45–48. [Google Scholar] [CrossRef]
- Smith, J.W.; Shulman, H.M.; Thomas, E.D.; Fefer, A.; Buckner, C.D. Bone marrow transplantation for acute myelosclerosis. Cancer 1981, 48, 2198–2203. [Google Scholar] [CrossRef]
- Wolf, J.L.; Spruce, W.E.; Bearman, R.M.; Forman, S.J.; Scott, E.P.; Fahey, J.L.; Farbstein, M.J.; Rappaport, H.; Blume, K.G. Reversal of acute (“malignant”) myelosclerosis by allogeneic bone marrow transplantation. Blood 1982, 59, 191–193. [Google Scholar] [CrossRef] [PubMed]
- Rozman, C.; Grañena, A.; Hernández-Prieto, M.; Vela, E.; Brugués, R. Bone-marrow transplantation for acute myelofibrosis. Lancet 1982, 1, 618. [Google Scholar] [CrossRef]
- Kroener, J.F.; McMillan, R.; Beutler, E. Acute myelofibrosis. Treatment with allogeneic bone marrow transplantation. JAMA 1983, 249, 1189–1190. [Google Scholar] [CrossRef]
- Mehta, A.B.; Baughan, A.S.; Catovsky, D.; Goldman, J.M.; Johnson, S.A.; Galton, D.A. Reversal of marrow fibrosis in acute megakaryoblastic leukaemia after remission-induction and consolidation chemotherapy followed by bone marrow transplantation. Br. J. Haematol. 1983, 53, 445–449. [Google Scholar] [CrossRef]
- Dokal, I.; Jones, L.; Deenmamode, M.; Lewis, S.M.; Goldman, J.M. Allogeneic bone marrow transplantation for primary myelofibrosis. Br. J. Haematol. 1989, 71, 158–160. [Google Scholar] [CrossRef]
- Alchalby, H.; Yunus, D.-R.; Zabelina, T.; Ayuk, F.; Kröger, N. Incidence and risk factors of poor graft function after allogeneic stem cell transplantation for myelofibrosis. Bone Marrow Transplant. 2016, 51, 1223–1227. [Google Scholar] [CrossRef] [PubMed]
- Chatain, N.; Koschmieder, S.; Jost, E. Role of Inflammatory Factors during Disease Pathogenesis and Stem Cell Transplantation in Myeloproliferative Neoplasms. Cancers 2020, 12, 2250. [Google Scholar] [CrossRef]
- Charbord, P.; Neel, H.; Caillou, B.; Kahn, E.; Parmentier, C. Effect of alkylating agents on hematopoiesis in myelofibrosis. 4 case report. Nouv. Rev. Fr. Hematol. 1978, 26, 361–369. [Google Scholar]
- Silver, R.T.; Vandris, K.; Goldman, J.J. Recombinant interferon-α may retard progression of early primary myelofibrosis: A preliminary report. Blood 2011, 117, 6669–6672. [Google Scholar] [CrossRef]
- Pizzi, M.; Silver, R.T.; Barel, A.; Orazi, A. Recombinant interferon-α in myelofibrosis reduces bone marrow fibrosis, improves its morphology and is associated with clinical response. Mod. Pathol. 2015, 28, 1315–1323. [Google Scholar] [CrossRef]
- Bjørn, M.E.; Hasselbalch, H.C. Minimal residual disease or cure in MPNs? Rationales and perspectives on combination therapy with interferon-alpha2 and ruxolitinib. Expert. Rev. Hematol. 2017, 10, 393–404. [Google Scholar] [CrossRef] [PubMed]
- Hasselbalch, H.C.; Holmström, M.O. Perspectives on interferon-alpha in the treatment of polycythemia vera and related myeloproliferative neoplasms: Minimal residual disease and cure? Semin. Immunopathol. 2019, 41, 5–19. [Google Scholar] [CrossRef]
- Mikkelsen, S.U.; Kjaer, L.; Bjørn, M.E.; Knudsen, T.A.; Sørensen, A.L.; Andersen, C.B.L.; Bjerrum, O.W.; Brochmann, N.; El Fassi, D.; Kruse, T.A.; et al. Safety and efficacy of combination therapy of interferon-α2 and ruxolitinib in polycythemia vera and myelofibrosis. Cancer Med. 2018, 7, 3571–3581. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, A.L.; Mikkelsen, S.U.; Knudsen, T.A.; Bjørn, M.E.; Andersen, C.L.; Bjerrum, O.W.; Brochmann, N.; Patel, D.A.; Gjerdrum, L.M.R.; El Fassi, D.; et al. Ruxolitinib and interferon-α2 combination therapy for patients with polycythemia vera or myelofibrosis: A phase II study. Haematologica 2020, 105, 2262–2272. [Google Scholar] [CrossRef]
- Hasselbalch, H.C.; Silver, R.T. New Perspectives of Interferon-alpha2 and Inflammation in Treating Philadelphia-negative Chronic Myeloproliferative Neoplasms. Hemasphere 2021, 5, e645. [Google Scholar] [CrossRef]
- Ozsoylu, S.; Ruacan, S. High-dose intravenous corticosteroid treatment in childhood idiopathic myelofibrosis. Acta Haematol. 1986, 75, 49–51. [Google Scholar] [CrossRef] [PubMed]
- Hasselbalch, H.; Jans, H.; Nielsen, P.L. A distinct subtype of idiopathic myelofibrosis with bone marrow features mimicking hairy cell leukemia: Evidence of an autoimmune pathogenesis. Am. J. Hematol. 1987, 25, 225–229. [Google Scholar] [CrossRef]
- Ozsoylu, S. High-dose intravenous methylprednisolone for idiopathic myelofibrosis. Lancet 1988, 1, 766. [Google Scholar] [CrossRef] [PubMed]
- Ozsoylu, S. High dose intravenous methylprednisolone for idiopathic myelofibrosis. Br. J. Haematol. 1988, 70, 388. [Google Scholar] [CrossRef]
- Inoue, Y.; Matsubara, A.; Okuya, S.; Okafuji, K.; Kaku, K.; Kaneko, T. Myelofibrosis and systemic lupus erythematosus: Reversal of fibrosis with high-dose corticosteroid therapy. Acta Haematol. 1992, 88, 32–36. [Google Scholar] [CrossRef]
- Rizzi, R.; Pastore, D.; Liso, A.; Liuzzi, G.M.; Dalena, A.M.; Specchia, G.; Ricco, R.; Liso, V. Autoimmune myelofibrosis: Report of three cases and review of the literature. Leuk. Lymphoma 2004, 45, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Paquette, R.L.; Meshkinpour, A.; Rosen, P.J. Autoimmune myelofibrosis. A steroid-responsive cause of bone marrow fibrosis associated with systemic lupus erythematosus. Medicine 1994, 73, 145–152. [Google Scholar] [CrossRef]
- Masarova, L.; Wang, W.; Newberry, K.J.; Kantarjian, H.; Verstovsek, S. Complete remission in a patient with JAK2- and IDH2-positive myelofibrosis. Blood 2016, 128, 877–880. [Google Scholar] [CrossRef]
- Charron, D.; Robert, L.; Couty, M.-C.; Binet, J.-L. Biochemical and histological analysis of bone marrow collagen in myelofibrosis. Br. J. Haematol. 1979, 41, 151–161. [Google Scholar] [CrossRef]
- Bentley, S.A.; Alabaster, O.; Foidart, J.M. Collagen heterogeneity in normal human bone marrow. Br. J. Haematol. 1981, 48, 287–291. [Google Scholar] [CrossRef]
- Gay, S.; Gay, R.E.; Prchal, J.T. Immunohistochemical studies of bone marrow collagen. In Myelofibrosis and the Biology of Connective Tissue; Berk, P.D., Castro-Malaspina, H., Wasserman, L.R., Eds.; Alan R, Liss, Inc.: New York, NY, USA, 1984; pp. 291–306. [Google Scholar]
- Fleischmajer, R.; Perlish, J.S.; Timpl, R. Collagen fibrillogenesis in human skin. Ann. N. Y. Acad. Sci. 1985, 460, 246–257. [Google Scholar] [CrossRef]
- Schaffner, F.; Poper, H. Capillarization of hepatic sinusoids in man. Gastroenterology 1963, 44, 239–242. [Google Scholar] [CrossRef] [PubMed]
- Risteli, L.; Risteli, J. Radioimmunoassays for monitoring connective tissue metabolism. Rheumatol 1986, 10, 216–245. [Google Scholar]
- Risteli, L.; Risteli, J. Noninvasive methods for detection of organ fibrosis. In Focus on Connective Tissue in Health and Disease; Rojkind, M., Ed.; CRC Press Inc.: Boca Raton, FL, USA, 1990; Volume 1. [Google Scholar]
- Fessler, J.H.; Fessler, L.I. Biosynthesis of procollagen. Annu. Rev. Biochem. 1978, 47, 129–162. [Google Scholar] [CrossRef] [PubMed]
- Vellenga, E.; Mulder, N.H.; van Zanten, A.K.; Nieweg, H.O.; Woldring, M.G. The significance of the aminoterminal propeptide of type III procollagen in paroxysmal nocturnal haemoglobinuria and myelofibrosis. Eur. J. Nucl. Med. 1983, 8, 499–501. [Google Scholar] [CrossRef] [PubMed]
- Barosi, G.; Costa, A.; Liberato, L.N.; Polino, G.; Spriano, P.; Magrini, U. Serum procollagen-III-peptide level correlates with disease activity in myelofibrosis with myeloid metaplasia. Br. J. Haematol. 1989, 72, 16–20. [Google Scholar] [CrossRef]
- Rohde, H.; Vargas, L.; Kalbfleisch, H.; Bruguera, M.; Timpl, R. Radioimmunoassay for type III procollagen peptide and its application to human liver disease. Eur. J. Clin. Investig. 1979, 9, 451–459. [Google Scholar] [CrossRef]
- Niemelä, O.; Risteli, L.; Sotaniemi, E.A.; Risteli, J. Heterogeneity of the antigens related to the aminoterminal propeptide of type III procollagen in human serum. Clin. Chim. Acta 1982, 124, 39–44. [Google Scholar] [CrossRef]
- Hørslev-Petersen, K.; Bentsen, K.D.; Junker, P.; Lorenzen, I. Serum amino-terminal type III procollagen peptide in rheumatoid arthritis. Relationship to disease activity, treatment, and development of joint erosions. Arthritis Rheum. 1986, 29, 592–599. [Google Scholar] [CrossRef]
- Hørslev-Petersen, K.; Bentsen, K.D.; Engström-Laurent, A.; Junker, P.; Halberg, P.; Lorenzen, I. Serum amino terminal type III procollagen peptide and serum hyaluronan in rheumatoid arthritis: Relation to clinical and serological parameters of inflammation during 8 and 24 months’ treatment with levamisole, penicillamine, or azathioprine. Ann. Rheum. Dis. 1988, 47, 116–126. [Google Scholar] [CrossRef]
- Hørslev-Petersen, K.; Saxne, T.; Haar, D.; Thomsen, B.S.; Bentsen, K.D.; Junker, P.; Lorenzen, I. The aminoterminal-type-III procollagen peptide and proteoglycans in serum and synovial fluid of patients with rheumatoid arthritis or reactive arthritis. Rheumatol. Int. 1988, 8, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Parmentier, C.; Schlumberger, M.; Caillou, B.; Boaziz, C. Serum procollagen-III-peptide as a marker for primary myelofibrosis: Effect of anthracyclin. Eur. J. Haematol. 1987, 39, 475. [Google Scholar] [CrossRef]
- Bentsen, K.D.; Boesby, S.; Kirkegaard, P.; Hansen, C.P.; Jensen, S.L.; Hørslev-Petersen, K.; Lorenzen, I. Is the aminoterminal propeptide of type III procollagen degraded in the liver? A study of type III procollagen peptide in serum during liver transplantation in pigs. J. Hepatol. 1988, 6, 144–150. [Google Scholar] [CrossRef]
- Niemelä, O.; Risteli, L.; Sotaniemi, E.A.; Risteli, J. Aminoterminal propeptide of type III procollagen in serum in alcoholic liver disease. Gastroenterology 1983, 85, 254–259. [Google Scholar] [CrossRef]
- Bentsen, K.D.; Horn, T.; Risteli, J.; Risteli, L.; Engström-Laurent, A.; Hørslev-Petersen, K.; Lorenzen, I. Serum aminoterminal type III procollagen peptide and the 7S domain of type IV collagen in patients with alcohol abuse. Relation to ultrastructural fibrosis in the acinar zone 3 and to serum hyaluronan. Liver 1987, 7, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.K.; Riisbro, R.; Holten-Andersen, M.N.; Brown, P.d.N.; Junker, P.; Brünner, N.; Hasselbalch, H.C. Collagen metabolism and enzymes of the urokinase plasminogen activator system in chronic myeloproliferative disorders: Correlation between plasma-soluble urokinase plasminogen activator receptor and serum markers for collagen metabolism. Eur. J. Haematol. 2003, 71, 276–282. [Google Scholar] [CrossRef]
- Jensen, M.K.; Riisbro, R.; de Nully Brown, P.; Brünner, N.; Hasselbalch, H.C. Elevated soluble urokinase plasminogen activator receptor in plasma from patients with idiopathic myelofibrosis or polycythaemia vera. Eur. J. Haematol. 2002, 69, 43–49. [Google Scholar] [CrossRef]
- Christensen, T.D.; Jensen, C.; Larsen, O.; Leerhøy, B.; Hansen, C.P.; Madsen, K.; Høgdall, D.; Karsdal, M.A.; Chen, I.M.; Nielsen, D.; et al. Blood-based tumor fibrosis markers as diagnostic and prognostic biomarkers in patients with biliary tract cancer. Int. J. Cancer 2023, 152, 1036–1049. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.K.; Holten-Andersen, M.N.; Riisbro, R.; de Nully Brown, P.; Larsen, M.B.; Kjeldsen, L.; Heickendorff, L.; Brünner, N.; Hasselbalch, H.C. Elevated plasma levels of TIMP-1 correlate with plasma suPAR/uPA in patients with chronic myeloproliferative disorders. Eur. J. Haematol. 2003, 71, 377–384. [Google Scholar] [CrossRef]
- Hasselbalch, H.; Junker, P. Serum laminin P1 in idiopathic myelofibrosis and related diseases. Leuk. Res. 1994, 18, 623–628. [Google Scholar] [CrossRef]
- Niemelä, O.; Risteli, L.; Sotaniemi, E.A.; Risteli, J. Type IV collagen and laminin-related antigens in human serum in alcoholic liver disease. Eur. J. Clin. Investig. 1985, 15, 132–137. [Google Scholar] [CrossRef]
- Horn, T.; Junge, J.; Chris-Toffersen, P. Early alcoholic liver injury: Changes of the Disse space in acinar zone 3. Liver 1985, 5, 301–310. [Google Scholar] [CrossRef]
- Hørslev-Petersen, K. Circulating Extracellular Matrix Components as Markers for Connective Tissue Response to Inflammation. A Clinical and Experimental Study with Special Emphasis on Serum Aminoterminal Type III Procollagen Peptide in Rheumatic Diseases. Ph.D. Thesis, Lægeforeningens Forlag, Copenhagen, Denmark, 1990. [Google Scholar]
- Mason, R.M. Recent advances in the biochemistry of hyaluronic acid in cartilage. Prog. Clin. Biol. Res. 1981, 54, 87–112. [Google Scholar]
- Engström-Laurent, A.; Hällgren, R. Circulating hyaluronate in rheumatoid arthritis: Relationship to inflammatory activity and the effect of corticosteroid therapy. Ann. Rheum. Dis. 1985, 44, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Engström-Laurent, A.; Feltelius, N.; Hällgren, R.; Wasteson, A. Raised serum hyaluronate levels in scleroderma: An effect of growth factor induced activation of connective tissue cells? Ann. Rheum. Dis. 1985, 44, 614–620. [Google Scholar] [CrossRef] [PubMed]
- Engström-Laurent, A.; Lööf, L.; Nyberg, A.; Schröder, T. Increased serum levels of hyaluronate in liver disease. Hepatology 1985, 5, 638–642. [Google Scholar] [CrossRef]
- Groopman, J.E. The pathogenesis of myelofibrosis in myeloproliferative disorders. Ann. Intern. Med. 1980, 92, 857–858. [Google Scholar] [CrossRef] [PubMed]
- Castro-Malaspina, H.; Rabellino, E.M.; Yen, A.; Nachman, R.L.; Moore, M.A. Human megakaryocyte stimulation of proliferation of bone marrow fibroblasts. Blood 1981, 57, 781–787. [Google Scholar] [CrossRef]
- Castro-Malaspina, H.; Moore, M.A. Pathophysiological mechanisms operating in the development of myelofibrosis: Role of megakaryocytes. Nouv. Rev. Fr. Hematol. 1982, 24, 221–226. [Google Scholar]
- Castro-Malaspina, H. Pathogenesis of myelofibrosis: Role of ineffective megakaryopoiesis and megakaryocyte components. In Myelofibrosis and the Biology of Connective Tissue; Berk, P.D., Castro-Malaspina, H., Wasseman, L.R., Eds.; Alan R. Liss, Inc.: New York, NY, USA, 1984. [Google Scholar]
- Fraser, J.R.; Laurent, T.C.; Pertoft, H.; Baxter, E. Plasma clearance, tissue distribution and metabolism of hyaluronic acid injected intravenously in the rabbit. Biochem. J. 1981, 200, 415–424. [Google Scholar] [CrossRef]
- Fraser, J.R.; Appelgren, L.E.; Laurent, T.C. Tissue uptake of circulating hyaluronic acid. A whole body autoradiographic study. Cell Tissue Res. 1983, 233, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, S.; Fraser, J.R.; Laurent, T.C.; Pertoft, H.; Smedsrød, B. Endothelial cells are a site of uptake and degradation of hyaluronic acid in the liver. Exp. Cell Res. 1983, 144, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Fraser, J.R.; Laurent, T.C.; Engström-Laurent, A.; Laurent, U.G. Elimination of hyaluronic acid from the blood stream in the human. Clin. Exp. Pharmacol. Physiol. 1984, 11, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Fraser, J.R.; Kimpton, W.G.; Laurent, T.C.; Cahill, R.N.; Vakakis, N. Uptake and degradation of hyaluronan in lymphatic tissue. Biochem. J. 1988, 256, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Laurent, U.B.G.; Laurent, T.C. On the origin of hylaronate in blood. Biochem. Int. 1981, 2, 195–199. [Google Scholar]
- Parmley, R.T.; Hurst, R.E.; Takagi, M.; Spicer, S.S.; Austin, R.L. Glycosaminoglycans in human neutrophils and leukemic myeloblasts: Ultrastructural, cytochemical, immunologic, and biochemical characterization. Blood 1983, 61, 257–266. [Google Scholar] [CrossRef]
- Minguell, J.J.; Tavassoli, M. Proteoglycan synthesis by hematopoietic progenitor cells. Blood 1989, 73, 1821–1827. [Google Scholar] [CrossRef]
- Lemańska-Perek, A.; Adamik, B. Fibronectin and its soluble EDA-FN isoform as biomarkers for inflammation and sepsis. Adv. Clin. Exp. Med. 2019, 28, 1561–1567. [Google Scholar] [CrossRef]
- Malara, A.; Gruppi, C.; Abbonante, V.; Cattaneo, D.; De Marco, L.; Massa, M.; Iurlo, A.; Gianelli, U.; Balduini, C.L.; Tira, M.E.; et al. EDA fibronectin-TLR4 axis sustains megakaryocyte expansion and inflammation in bone marrow fibrosis. J. Exp. Med. 2019, 216, 587–604. [Google Scholar] [CrossRef] [PubMed]
- Malara, A.; Gruppi, C.; Massa, M.; Tira, M.E.; Rosti, V.; Balduini, A.; Barosi, G. Elevated plasma EDA fibronectin in primary myelofibrosis is determined by high allele burden of JAK2V617F mutation and strongly predicts splenomegaly progression. Front. Oncol. 2022, 12, 987643. [Google Scholar] [CrossRef]
- Wang, J.C.; Novetsky, A.; Chen, C.; Novetsky, A.D. Plasma matrix metalloproteinase and tissue inhibitor of metalloproteinase in patients with agnogenic myeloid metaplasia or idiopathic primary myelofibrosis. Br. J. Haematol. 2002, 119, 709–712. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.C. Importance of plasma matrix metalloproteinases (MMP) and tissue inhibitors of metalloproteinase (TIMP) in development of fibrosis in agnogenic myeloid metaplasia. Leuk. Lymphoma 2005, 46, 1261–1268. [Google Scholar] [CrossRef] [PubMed]
- Skov, V.; Thomassen, M.; Kjær, L.; Riley, C.; Larsen, T.S.; Bjerrum, O.W.; Kruse, T.A.; Hasselbalch, H.C. Extracellular Matrix-Related Genes Are Deregulated in Peripheral Blood from Patients with Myelofibrosis and Related Neoplasms. Blood 2018, 132 (Suppl. S1), 5491. [Google Scholar] [CrossRef]
- Bjørn, M.E.; Andersen, C.L.; Jensen, M.K.; Hasselbalch, H.C. Circulating YKL-40 in myelofibrosis a potential novel biomarker of disease activity and the inflammatory state. Eur. J. Haematol. 2014, 93, 224–228. [Google Scholar] [CrossRef] [PubMed]
- Andersen, C.L.; Bjørn, M.E.; McMullin, M.F.; Harrison, C.; Samuelsson, J.; Ejerblad, E.; Zweegman, S.; Fernandes, S.; Bareford, D.; Knapper, S.; et al. Circulating YKL-40 in patients with essential thrombocythemia and polycythemia vera treated with the novel histone deacetylase inhibitor vorinostat. Leuk. Res. 2014, 38, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Krečak, I.; Gverić-Krečak, V.; Lapić, I.; Rončević, P.; Gulin, J.; Fumić, K.; Krečak, F.; Holik, H.; Duraković, N. Circulating YKL-40 in Philadelphia-negative myeloproliferative neoplasms. Acta Clin. Belg. 2021, 76, 32–39. [Google Scholar] [CrossRef]
- Krecak, I.; Gveric-Krecak, V.; Roncevic, P.; Basic-Kinda, S.; Gulin, J.; Lapic, I.; Fumic, K.; Ilic, I.; Horvat, I.; Zadro, R.; et al. Serum chitotriosidase: A circulating biomarker in polycythemia vera. Hematology 2018, 23, 793–802. [Google Scholar] [CrossRef]
- Barbui, T.; Carobbio, A.; Finazzi, G.; Vannucchi, A.M.; Barosi, G.; Antonioli, E.; Guglielmelli, P.; Pancrazzi, A.; Salmoiraghi, S.; Zilio, P.; et al. Inflammation and thrombosis in essential thrombocythemia and polycythemia vera: Different role of C-reactive protein and pentraxin 3. Haematologica 2011, 96, 315–318. [Google Scholar] [CrossRef]
- Lussana, F.; Carobbio, A.; Salmoiraghi, S.; Guglielmelli, P.; Vannucchi, A.M.; Bottazzi, B.; Leone, R.; Mantovani, A.; Barbui, T.; Rambaldi, A. Driver mutations (JAK2V617F, MPLW515L/K or CALR), pentraxin-3 and C-reactive protein in essential thrombocythemia and polycythemia vera. J. Hematol. Oncol. 2017, 10, 54. [Google Scholar] [CrossRef] [PubMed]
- Verstovsek, S.; Foltz, L.; Gupta, V.; Hasserjian, R.; Manshouri, T.; Mascarenhas, J.; Mesa, R.; Pozdnyakova, O.; Ritchie, E.; Veletic, I.; et al. Safety and efficacy of zinpentraxin as monotherapy or in combination with ruxolitinib in myelofibrosis. A stage I of a Phase II trial. Haematologica 2023. epub ahead of print. [Google Scholar] [CrossRef]
- Chirinos, J.A.; Zhao, L.; Reese-Petersen, A.L.; Cohen, J.B.; Genovese, F.; Richards, A.M.; Doughty, R.N.; Diez, J.; Gonzales, A.; Querejeta, R.; et al. Endotrophin, a Collagen VI Formation–Derived Peptide, in Heart Failure. NEJM Evid. 2022, 1, 1–12. [Google Scholar] [CrossRef]
- Paulus, W.J.; Tschöpe, C. A novel paradigm for heart failure with preserved ejection fraction: Comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J. Am. Coll. Cardiol. 2013, 62, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Zile, M.R.; Baicu, C.F.; Ikonomidis, J.S.; Stroud, R.E.; Nietert, P.J.; Bradshaw, A.D.; Slater, R.; Palmer, B.M.; Van Buren, P.; Meyer, M.; et al. Myocardial stiffness in patients with heart failure and a preserved ejection fraction: Contributions of collagen and titin. Circulation 2015, 131, 1247–1259. [Google Scholar] [CrossRef]
- Gomberg-Maitland, M.; Shah, S.J.; Guazzi, M. Inflammation in Heart Failure with Preserved Ejection Fraction: Time to put out the Fire. JACC Heart Fail. 2016, 4, 325–328. [Google Scholar] [CrossRef]
- Franssen, C.; Chen, S.; Unger, A.; Korkmaz, H.I.; De Keulenaer, G.W.; Tschöpe, C.; Leite-Moreira, A.F.; Musters, R.; Niessen, H.W.; Linke, W.A.; et al. Myocardial Microvascular Inflammatory Endothelial Activation in Heart Failure with Preserved Ejection Fraction. JACC Heart Fail. 2016, 4, 312–324. [Google Scholar] [CrossRef]
- Sweeney, M.; Corden, B.; Cook, S.A. Targeting cardiac fibrosis in heart failure with preserved ejection fraction: Mirage or miracle? EMBO Mol. Med. 2020, 12, e10865. [Google Scholar] [CrossRef]
- Paulus, W.J.; Zile, M.R. From Systemic Inflammation to Myocardial Fibrosis: The Heart Failure with Preserved Ejection Fraction Paradigm Revisited. Circ. Res. 2021, 128, 1451–1467. [Google Scholar] [CrossRef]
- Ling, S.; Xu, J.-W. NETosis as a Pathogenic Factor for Heart Failure. Oxid. Med. Cell Longev. 2021, 2021, 6687096. [Google Scholar] [CrossRef] [PubMed]
- Sanhueza-Olivares, F.; Troncoso, M.F.; la Fuente, F.P.-D.; Martinez-Bilbao, J.; Riquelme, J.A.; Norambuena-Soto, I.; Villa, M.; Lavandero, S.; Castro, P.F.; Chiong, M. A potential role of autophagy-mediated vascular senescence in the pathophysiology of HFpEF. Front. Endocrinol. 2022, 13, 1057349. [Google Scholar] [CrossRef] [PubMed]
- Courau, T.; Nehar-Belaid, D.; Florez, L.; Levacher, B.; Vazquez, T.; Brimaud, F.; Bellier, B.; Klatzmann, D. TGF-β and VEGF cooperatively control the immunotolerant tumor environment and the efficacy of cancer immunotherapies. JCI Insight. 2016, 1, e85974. [Google Scholar] [CrossRef]
- Kuczek, D.E.; Larsen, A.M.H.; Thorseth, M.-L.; Carretta, M.; Kalvisa, A.; Siersbæk, M.S.; Simões, A.M.C.; Roslind, A.; Engelholm, L.H.; Noessner, E.; et al. Collagen density regulates the activity of tumor-infiltrating T cells. J. Immunother. Cancer 2019, 7, 68. [Google Scholar] [CrossRef] [PubMed]
- Grauslund, J.H.; Holmström, M.O.; Jørgensen, N.G.; Klausen, U.; Weis-Banke, S.E.; El Fassi, D.; Schöllkopf, C.; Clausen, M.B.; Gjerdrum, L.M.R.; Breinholt, M.F.; et al. Therapeutic Cancer Vaccination with a Peptide Derived from the Calreticulin Exon 9 Mutations Induces Strong Cellular Immune Responses in Patients with CALR-Mutant Chronic Myeloproliferative Neoplasms. Front. Oncol. 2021, 11, 225. [Google Scholar] [CrossRef]
- Grauslund, J.H.; Holmström, M.O.; Martinenaite, E.; Lisle, T.L.; Glöckner, H.J.; El Fassi, D.; Klausen, U.; Mortensen, R.E.J.; Jørgensen, N.; Kjær, L.; et al. An arginase1- and PD-L1-derived peptide-based vaccine for myeloproliferative neoplasms: A first-in-man clinical trial. Front. Immunol. 2023, 14, 1117466. [Google Scholar] [CrossRef]
- Barbui, T.; Carobbio, A.; Finazzi, G.; Guglielmelli, P.; Salmoiraghi, S.; Rosti, V.; Rambaldi, A.; Vannucchi, A.M.; Barosi, G. Elevated C-reactive protein is associated with shortened leukemia-free survival in patients with myelofibrosis. Leukemia 2013, 27, 2084–2086. [Google Scholar] [CrossRef] [PubMed]
- Castaño, A.P.; Lin, S.L.; Surowy, T.; Nowlin, B.T.; Turlapati, S.A.; Patel, T.; Singh, A.; Li, S.; Lupher, M.L.; Duffield, J.S. Serum amyloid P inhibits fibrosis through FcγR-dependent monocyte-macrophage regulation in vivo. Sci. Transl. Med. 2009, 1, 5ra13. [Google Scholar] [CrossRef]
- Mantovani, A.; Garlanda, C.; Doni, A.; Bottazzi, B. Pentraxins in innate immunsity: From C-reactive protein to the long pentraxin PTX3. J. Clin. Immunol. 2008, 28, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Pilling, D.; Gomer, R.H. The Development of Serum Amyloid P as a Possible Therapeutic. Front. Immunol. 2018, 9, 2328. [Google Scholar] [CrossRef]
- Veletic, I.; Manshouri, T.; Newberry, K.J.; Garnett, J.; Verstovsek, S.; Estrov, Z. Pentraxin-3 plasma levels correlate with tumour burden and overall survival in patients with primary myelofibrosis. Br. J. Haematol. 2019, 185, 382–386. [Google Scholar] [CrossRef]
- Murray, L.A.; Rosada, R.; Moreira, A.P.; Joshi, A.; Kramer, M.S.; Hesson, D.P.; Argentieri, R.L.; Mathai, S.; Gulati, M.; Herzog, E.L.; et al. Serum amyloid P therapeutically attenuates. murine bleomycin-induced pulmonary fibrosis via its effects on macrophages. PLoS ONE 2010, 5, e9683. [Google Scholar] [CrossRef]
- Nakagawa, N.; Barron, L.; Gomez, I.G.; Johnson, B.G.; Roach, A.M.; Kameoka, S.; Jack, R.M.; Lupher, M.L.; Gharib, S.A.; Duffield, J.S. Pentraxin-2 suppresses c-Jun/AP-1 signaling to inhibit progressive fibrotic disease. JCI Insight 2016, 1, e87446. [Google Scholar] [CrossRef]
- Verstovsek, S.; Manshouri, T.; Pilling, D.; Bueso-Ramos, C.E.; Newberry, K.J.; Prijic, S.; Knez, L.; Bozinovic, K.; Harris, D.M.; Spaeth, E.L.; et al. Role of neoplastic monocyte-derived fibrocytes in primary myelofibrosis. J. Exp. Med. 2016, 213, 1723–1740. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; van den Blink, B.; Hamblin, M.J.; Brown, A.W.; Golden, J.A.; Ho, L.A.; Wijsenbeek, M.S.; Vasakova, M.; Pesci, A.; Antin-Ozerkis, D.E.; et al. Effect of Recombinant Human Pentraxin 2 vs. Placebo on Change in Forced Vital Capacity in Patients with Idiopathic Pulmonary Fibrosis: A Randomized Clinical Trial. JAMA 2018, 319, 2299–2307. [Google Scholar] [CrossRef]
- Raghu, G.; van den Blink, B.; Hamblin, M.J.; Brown, A.W.; Golden, J.A.; Ho, L.A.; Wijsenbeek, M.S.; Vasakova, M.; Pesci, A.; Antin-Ozerkis, D.E.; et al. Long-term treatment with recombinant human pentraxin 2 protein in patients with idiopathic pulmonary fibrosis: An open-label extension study. Lancet Respir. Med. 2019, 7, 657–664. [Google Scholar] [CrossRef]
- Raghu, G.; Hamblin, M.J.; Brown, A.W.; Golden, J.A.; Ho, L.A.; Wijsenbeek, M.S.; Vasakova, M.; Pesci, A.; Antin-Ozerkis, D.E.; Meyer, K.C.; et al. Long-term evaluation of the safety and efficacy of recombinant human pentraxin-2 (rhPTX-2) in patients with idiopathic pulmonary fibrosis (IPF): An open-label extension study. Respir. Res. 2022, 23, 129. [Google Scholar] [CrossRef]
- Leung, Y.Y.; Yao, H.L.L.; Kraus, V.B. Colchicine—Update on mechanisms of action and therapeutic uses. Semin. Arthritis Rheum. 2015, 45, 341–350. [Google Scholar] [CrossRef]
- Slobodnick, A.; Shah, B.; Pillinger, M.H.; Krasnokutsky, S. Colchicine: Old and new. Am. J. Med. 2015, 128, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Opstal, T.S.J.; Hoogeveen, R.M.; Fiolet, A.T.L.; Silvis, M.J.M.; The, S.H.K.; Bax, W.A.; de Kleijn, D.P.V.; Mosterd, A.; Stroes, E.S.G.; Cornel, J.H. Colchicine Attenuates Inflammation beyond the Inflammasome in Chronic Coronary Artery Disease: A LoDoCo2 Proteomic Substudy. Circulation 2020, 142, 1996–1998. [Google Scholar] [CrossRef]
- Martínez, G.J.; Celermajer, D.S.; Patel, S. The NLRP3 inflammasome and the emerging role of colchicine to inhibit atherosclerosis-associated inflammation. Atherosclerosis 2018, 269, 262–271. [Google Scholar] [CrossRef] [PubMed]
- Deftereos, S.; Giannopoulos, G.; Papoutsidakis, N.; Panagopoulou, V.; Kossyvakis, C.; Raisakis, K.; Cleman, M.W.; Stefanadis, C. Colchicine and the heart: Pushing the envelope. J. Am. Coll Cardiol. 2013, 62, 1817–1825. [Google Scholar] [CrossRef] [PubMed]
- Casanova, P.; Artola, R.T.; Mihos, C.G.; Pineda, A.M.; Santana, O. The cardiovascular effects of colchicine: A comprehensive review. Cardiol. Rev. 2015, 23, 317–322. [Google Scholar] [CrossRef]
- Chaldakov, G.N.; Ghenev, P.I. Colchicine, Inflammation and Fibrosis in Cardiovascular Disease: Merging Three Classical Tales. Biomed. Rev. 2017, 28, 105–110. [Google Scholar] [CrossRef]
- Nidorf, S.M.; Thompson, P.L. Why Colchicine Should Be Considered for Secondary Prevention of Atherosclerosis: An Overview. Clin. Ther. 2019, 41, 41–48. [Google Scholar] [CrossRef]
- Tardif, J.-C.; Kouz, S.; Waters, D.D.; Bertrand, O.F.; Diaz, R.; Maggioni, A.P.; Pinto, F.J.; Ibrahim, R.; Gamra, H.; Kiwan, G.S.; et al. Efficacy and Safety of low-dose colchicine after myocardial infarction. N. Engl. J. Med. 2019, 381, 2497–2505. [Google Scholar] [CrossRef]
- Nidorf, S.M.; Fiolet, A.T.L.; Mosterd, A.; Eikelboom, J.W.; Schut, A.; Opstal, T.S.J.; The, S.H.K.; Xu, X.-F.; Ireland, M.A.; Lenderink, T.; et al. Colchicine in patients with chronic coronary disease. N. Engl. J. Med. 2020, 383, 1838–1847. [Google Scholar] [CrossRef] [PubMed]
- Kraler, S.; Wenzl, F.A.; Lüscher, T.F. Repurposing Colchicine to Combat Residual Cardiovascular Risk: The LoDoCo2 Trial. Eur. J. Clin. Investig. 2020, 50, e13424. [Google Scholar] [CrossRef] [PubMed]
- Fiolet, A.T.L.; Silvis, M.J.M.; Opstal, T.S.J.; Bax, W.A.; van der Horst, F.A.L.; Mosterd, A.; de Kleijn, D.; Cornel, J.H. Short-term effect of low-dose colchicine on inflammatory biomarkers, lipids, blood count and renal function in chronic coronary artery disease and elevated high-sensitivity C-reactive protein. PLoS ONE 2020, 15, e0237665. [Google Scholar] [CrossRef]
- Bouabdallaoui, N.; Tardif, J.-C. Colchicine in the Management of Acute and Chronic Coronary Artery Disease. Curr. Cardiol. Rep. 2021, 23, 120. [Google Scholar] [CrossRef]
- Fiolet, A.T.L.; Opstal, T.S.J.; Mosterd, A.; Eikelboom, J.W.; Jolly, S.S.; Keech, A.C.; Kelly, P.; Tong, D.C.; Layland, J.; Nidorf, S.M.; et al. Efficacy and safety of low-dose colchicine in patients with coronary disease: A systematic review and metaanalysis of randomized trials. Eur. Heart J. 2021, 42, 2765–2775. [Google Scholar] [CrossRef] [PubMed]
- Siak, J.; Flint, N.; Shmueli, H.G.; Siegel, R.J.; Rader, F. The Use of Colchicine in Cardiovascular Diseases: A Systematic Review. Am. J. Med. 2021, 134, 735–744. [Google Scholar] [CrossRef]
- Vaidya, K.; Tucker, B.; Kurup, R.; Khandkar, C.; Pandzic, E.; Barraclough, J.; Machet, J.; Misra, A.; Kavurma, M.; Martinez, G.; et al. Colchicine inhibits neutrophil extracellular trap Formation in patients with acute coronary syndrome after percutaneous coronary intervention. J. Am. Heart Assoc. 2021, 10, e018993. [Google Scholar] [CrossRef]
- Imazio, M.; Nidorf, M. Colchicine and the heart. Eur. Heart J. 2021, 42, 2745–2760. [Google Scholar] [CrossRef] [PubMed]
- Cantrell, M.S.; Soto-Avellaneda, A.; Wall, J.D.; Ajeti, A.D.; Morrison, B.E.; Warner, L.R.; McDougal, O.M. Repurposing Drugs to Treat Heart and Brain Illness. Pharmaceuticals 2021, 14, 573. [Google Scholar] [CrossRef]
- Deftereos, S.G.; Beerkens, F.J.; Shah, B.; Giannopoulos, G.; Vrachatis, D.A.; Giotaki, S.G.; Siasos, G.; Nicolas, J.; Arnott, C.; Patel, S.; et al. Colchicine in Cardiovascular Disease: In-Depth Review. Circulation 2022, 145, 61–78. [Google Scholar]
- Robinson, P.C.; Terkeltaub, R.; Pillinger, M.H.; Shah, B.; Karalis, V.; Karatza, E.; Liew, D.; Imazio, M.; Cornel, J.H.; Thompson, P.L.; et al. Consensus Statement Regarding the Efficacy and Safety of Long-Term Low-Dose Colchicine in Gout and Cardiovascular Disease. Am. J. Med. 2022, 135, 32–38. [Google Scholar] [CrossRef]
- Becatti, M.; Abbate, A.; Fiorillo, C.; Carnevale, R.; Kumar, S. Editorial: New Insights into Oxidative Stress and Inflammation in the Pathophysiology and Treatment of Cardiovascular Diseases. Front. Mol. Biosci. 2022, 9, 940465. [Google Scholar] [CrossRef]
- Skov, V.; Larsen, T.S.; Thomassen, M.; Riley, C.H.; Jensen, M.K.; Bjerrum, O.W.; Kruse, T.A.; Hasselbalch, H.C. Whole-blood transcriptional profiling of interferon-inducible genes identifies highly upregulated IFI27 in primary myelofibrosis. Eur. J. Haematol. 2011, 87, 54–60. [Google Scholar] [CrossRef]
- Skov, V.; Larsen, T.S.; Thomassen, M.; Riley, C.H.; Jensen, M.K.; Bjerrum, O.W.; Kruse, T.A.; Hasselbalch, H.C. Molecular profiling of peripheral blood cells from patients with polycythemia vera and related neoplasms: Identification of deregulated genes of significance for inflammation and immune surveillance. Leuk. Res. 2012, 36, 1387–1392. [Google Scholar] [CrossRef] [PubMed]
- Hasselbalch, H.C.; Skov, V.; Larsen, T.S.; Thomassen, M.; Riley, C.H.; Jensen, M.K.; Bjerrum, O.W.; Kruse, T.A. High expression of carcinoembryonic antigen-related cell adhesion molecule (CEACAM) 6 and 8 in primary myelofibrosis. Leuk. Res. 2011, 35, 1330–1334. [Google Scholar] [CrossRef] [PubMed]
- Skov, V.; Thomassen, M.; Riley, C.H.; Jensen, M.K.; Bjerrum, O.W.; Kruse, T.A.; Hasselbalch, H.C.; Larsen, T.S. Gene expression profiling with principal component analysis depicts the biological continuum from essential thrombocythemia over polycythemia vera to myelofibrosis. Exp. Hematol. 2012, 40, 771–780. [Google Scholar] [CrossRef]
- Hasselbalch, H.C.; Skov, V.; Larsen, T.S.; Thomassen, M.; Riley, C.H.; Jensen, M.K.; Bjerrum, O.W.; Kruse, T.A. Transcriptional profiling of whole blood identifies a unique 5-gene signature for myelofibrosis and imminent myelofibrosis transformation. PLoS ONE 2014, 9, e85567. [Google Scholar] [CrossRef] [PubMed]
- Skov, V.; Burton, M.; Thomassen, M.; Larsen, T.S.; Riley, C.H.; Madelung, A.B.; Kjær, L.; Bondo, H.; Stamp, I.; Ehinger, M.; et al. A 7-Gene Signature Depicts the Biochemical Profile of Early Prefibrotic Myelofibrosis. PLoS ONE 2016, 11, e0161570. [Google Scholar] [CrossRef] [PubMed]
- Hasselbalch, H.C.; Thomassen, M.; Riley, C.H.; Kjær, L.; Larsen, T.S.; Jensen, M.K.; Bjerrum, O.W.; Kruse, T.A.; Skov, V. Whole blood transcriptional profiling reveals deregulation of oxidative and antioxidative defence genes in myelofibrosis and related neoplasms. Potential implications of downregulation of Nrf2 for genomic instability and disease progression. PLoS ONE 2014, 9, e112786, Erratum in: PLoS ONE 2015, 10, e0118049. [Google Scholar] [CrossRef]
- Skov, V.; Thomassen, M.; Kjaer, L.; Ellervik, C.; Larsen, M.K.; Knudsen, T.A.; Kruse, T.A.; Hasselbalch, H.C. Interferon-alpha2 treatment of patients with polycythemia vera and related neoplasms favorably impacts deregulation of oxidative stress genes and antioxidative defense mechanisms. PLoS ONE 2022, 17, e0270669. [Google Scholar] [CrossRef] [PubMed]
- Skov, V.; Thomassen, M.; Kjaer, L.; Larsen, M.K.; Ellervik, C.; Knudsen, T.A.; Kruse, T.A.; Hasselbalch, H.C. Interferon-alpha2 Favorably Impacts Deregulated Atherosclerosis Genes in Polycythemia Vera and Related Neoplasms. Blood 2022, 140, 9604–9605. [Google Scholar] [CrossRef]
- Skov, V.; Thomassen, M.; Kjaer, L.; Larsen, M.K.; Knudsen, T.A.; Ellervik, C.; Junker, P.; Kruse, T.A.; Hasselbalch, H.C. Transcriptional Profiling of Extracellular Matrix-Related Genes in Peripheral Blood from Patients with Polycythemia Vera and Related Neoplasms. Impact of Interferon-Alpha2. Poster ASH. In Proceedings of the 14th International Congress on Myeloproliferative Neoplasms Program Guide, New York, NY, USA, 27–28 October 2022. [Google Scholar]
- Skov, V.; Riley, C.H.; Thomassen, M.; Kjær, L.; Stauffer Larsen, T.; Bjerrum, O.W.; Kruse, T.A.; Hasselbalch, H.C. Significantly upregulated thrombo-inflammatory genes are normoregulated or significantly downregulated during treatment with interferon-alpha2 in patients with Philadelphia-negative chronic myeloproliferative neoplasms. Blood 2019, 134 (Suppl. S1), 2978. [Google Scholar] [CrossRef]
- Wolach, O.; Sellar, R.S.; Martinod, K.; Cherpokova, D.; McConkey, M.; Chappell, R.J.; Silver, A.J.; Adams, D.; Castellano, C.A.; Schneider, R.K.; et al. Increased neutrophil extracellular trap formation promotes thrombosis in myeloproliferative neoplasms. Sci. Transl. Med. 2018, 10, eaan8292. [Google Scholar] [CrossRef]
- Massarenti, L.; Knudsen, T.A.; Enevold, C.; Skov, V.; Kjær, L.; Larsen, M.K.; Nielsen, C.H.; Hasselbalch, H.C. Interferon-alpha2 Treatment Reduces Circulating Neutrophil Extracellular Trap Levels in Myeloproliferative Neoplasms. Br. J. Haematol. 2023, in press. [Google Scholar] [CrossRef]
- Chrysanthopoulou, A.; Mitroulis, I.; Apostolidou, E.; Arelaki, S.; Mikroulis, D.; Konstantinidis, T.; Sivridis, E.; Koffa, M.; Giatromanolaki, A.; Boumpas, D.T.; et al. Neutrophil extracellular traps promote differentiation and function of fibroblasts. J. Pathol. 2014, 233, 294–307. [Google Scholar] [CrossRef]
- Ehrlich, H.P.; Ross, R.; Bornstein, P. Effects of antimicrotubular agents on the secretion of collagen. A biochemical and morphological study. J. Cell Biol. 1974, 62, 390–405. [Google Scholar] [CrossRef] [PubMed]
- Harris, E.D., Jr.; Krane, S.M. Effects of colchicine on collagenase in cultures of rheumatoid synovium. Arthritis Rheum. 1971, 14, 669–684. [Google Scholar] [CrossRef]
- Rennard, S.I.; Bitterman, P.B.; Ozaki, T.; Rom, W.N.; Crystal, R.G. Colchicine suppresses the release of fibroblast growth factors from alveolar macrophages in vitro. The basis of a possible therapeutic approach ot the fibrotic disorders. Am. Rev. Respir. Dis. 1988, 137, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Peters, S.G.; McDougall, J.C.; Douglas, W.W.; Coles, D.T.; DeRemee, R.A. Colchicine in the treatment of pulmonary fibrosis. Chest 1993, 103, 101–104. [Google Scholar] [CrossRef] [PubMed]
- Entzian, P.; Schlaak, M.; Seitzer, U.; Bufe, A.; Acil, Y.; Zabel, P. Antiinflammatory and antifibrotic properties of colchicine: Implications for idiopathic pulmonary fibrosis. Lung 1997, 175, 41–51. [Google Scholar] [CrossRef]
- Davies, H.R.; Richeldi, L.; Walters, E.H. Immunomodulatory agents for idiopathic pulmonary fibrosis. Cochrane Database Syst. Rev. 2003, 3, CD003134, Update in: Cochrane Database Syst. Rev. 2010, 9, CD003134. [Google Scholar] [CrossRef]
- Bjørn, M.E.; Hasselbalch, H.C. The Role of Reactive Oxygen Species in Myelofibrosis and Related Neoplasms. Mediat. Inflamm. 2015, 2015, 648090. [Google Scholar] [CrossRef] [PubMed]
- Marty, C.; Lacout, C.; Droin, N.; Le Couédic, J.-P.; Ribrag, V.; Solary, E.; Vainchenker, W.; Villeval, J.-L.; Plo, I. A role for reactive oxygen species in JAK2V617F myeloproliferative neoplasm progression. Leukemia 2013, 27, 2187–2195. [Google Scholar] [CrossRef]
- Sørensen, A.L.; Hasselbalch, H.C.; Bjørn, M.E.; Nielsen, C.H.; Cordua, S.; Skov, V.; Kjær, L.; Poulsen, H.E.; Ellervik, C. Elevated levels of oxidized nucleosides in individuals with the JAK2V617F mutation from a general population study. Redox Biol. 2021, 41, 101895. [Google Scholar] [CrossRef]
- Craver, B.M.; Ramanathan, G.; Hoang, S.; Chang, X.; Luque, L.F.M.; Brooks, S.; Lai, H.Y.; Fleischman, A.G. N-acetylcysteine inhibits thrombosis in a murine model of myeloproliferative neoplasm. Blood Adv. 2020, 4, 312–321. [Google Scholar] [CrossRef] [PubMed]
- Demedts, M.; Behr, J.; Buhl, R.; Costabel, U.; Dekhuijzen, R.; Jansen, H.M.; MacNee, W.; Thomeer, M.; Wallaert, B.; Laurent, F.; et al. High-dose acetylcysteine in idiopathic pulmonary fibrosis. N. Engl. J. Med. 2005, 353, 2229–2242. [Google Scholar] [CrossRef] [PubMed]
- Hunninghake, G.W. Antioxidant therapy for idiopathic pulmonary fibrosis. N. Engl. J. Med. 2005, 353, 2285–2287. [Google Scholar] [CrossRef]
- Sun, T.; Liu, J.; Zhao, W. Efficacy of N-Acetylcysteine in Idiopathic Pulmonary Fibrosis: A Systematic Review and Meta-Analysis. Medicine 2016, 95, e3629. [Google Scholar] [CrossRef]
- Idiopathic Pulmonary Fibrosis Clinical Research Network; Martinez, F.J.; de Andrade, J.A.; Anstrom, K.J.; King, T.E., Jr.; Raghu, G. Randomized trial of acetylcysteine in idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014, 370, 2093–2101. [Google Scholar] [PubMed]
- Podolanczuk, A.J.; Kim, J.S.; Cooper, C.B.; Lasky, J.A.; Murray, S.; Oldham, J.M.; Raghu, G.; Flaherty, K.R.; Spino, C.; Noth, I.; et al. Design and rationale for the prospective treatment efficacy in IPF using genotype for NAC selection (PRECISIONS) clinical trial. BMC Pulm. Med. 2022, 22, 475. [Google Scholar] [CrossRef]
- Pedersen, N.T. Occurrence of megakaryocytes in various vessels and their retention in the pulmonary capillaries in man. Scand. J. Haematol. 1978, 21, 369–375. [Google Scholar] [CrossRef]
- Lefrançais, E.; Ortiz-Muñoz, G.; Caudrillier, A.; Mallavia, B.; Liu, F.; Sayah, D.M.; Thornton, E.E.; Headley, M.B.; David, T.; Coughlin, S.R.; et al. The lung is a site of platelet biogenesis and a reservoir for haematopoietic progenitors. Nature 2017, 544, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Borges, I.; Sena, I.; Azevedo, P.; Andreotti, J.; Almeida, V.; Paiva, A.; Santos, G.; Guerra, D.; Prazeres, P.; Mesquita, L.L.; et al. Lung as a Niche for Hematopoietic Progenitors. Stem Cell Rev. Rep. 2017, 13, 567–574. [Google Scholar] [CrossRef]
- Bacci, B.; Gobbo, F.; Roccaro, M.; Peli, A.; Zingariella, M.; Martelli, F.; Verachi, P.; Bardelli, C.; Mazzarini, M.; Sarli, G.; et al. Altered Megakaryocytes Are Associated with Development of Pulmonary Fibrosis in Mice Carrying the Hypomorphic Gata1low Mutation. Blood 2019, 134 (Suppl. S1), 2336. [Google Scholar] [CrossRef]
- Sparks, J.A.; He, X.; Huang, J.; Fletcher, E.A.; Zaccardelli, A.; Friedlander, H.M.; Gill, R.R.; Hatabu, H.; Nishino, M.; Murphy, D.J.; et al. Rheumatoid Arthritis Disease Activity Predicting Incident Clinically Apparent Rheumatoid Arthritis-Associated Interstitial Lung Disease: A Prospective Cohort Study. Arthritis Rheumatol. 2019, 71, 1472–1482. [Google Scholar] [CrossRef]
- Robles-Pérez, A.; Luburich, P.; Bolivar, S.; Dorca, J.; Nolla, J.M.; Molina-Molina, M.; Narváez, J. A prospective study of lung disease in a cohort of early rheumatoid arthritis patients. Sci. Rep. 2020, 10, 15640. [Google Scholar] [CrossRef]
- Florescu, A.; Gherghina, F.L.; Mușetescu, A.E.; Pădureanu, V.; Roșu, A.; Florescu, M.M.; Criveanu, C.; Florescu, L.-M.; Bobircă, A. Novel Biomarkers, Diagnostic and Therapeutic Approach in Rheumatoid Arthritis Interstitial Lung Disease—A Narrative Review. Biomedicines 2022, 10, 1367. [Google Scholar] [CrossRef] [PubMed]
- Vermant, M.; Goos, T.; Gogaert, S.; De Cock, D.; Verschueren, P.; Wuyts, W.A. Are genes the missing link to detect and prognosticate RA-ILD? Rheumatol. Adv. Pract. 2023, 7, rkad023. [Google Scholar] [CrossRef]
- Pedersen, K.M.; Çolak, Y.; Hasselbalch, H.C.; Ellervik, C.; Nordestgaard, B.G.; Bojesen, S.E. Two-fold risk of pneumonia and respiratory mortality in individuals with myeloproliferative neoplasm: A population-based cohort study. Eclinicalmedicine 2020, 21, 100295. [Google Scholar] [CrossRef]
- Ridker, P.M.; Cannon, C.P.; Morrow, D.; Rifai, N.; Rose, L.M.; McCabe, C.H.; Pfeffer, M.A.; Braunwald, E. Pravastatin or Atorvastatin, I. Infection Therapy-Thrombolysis in Myocardial Infarction, C-reactive protein levels and outcomes after statin therapy. N. Engl. J. Med. 2005, 352, 20–28. [Google Scholar] [CrossRef]
- Cholesterol Treatment Trialists’ (CTT) Collaboration; Baigent, C.; Blackwell, L.; Emberson, J.; Holland, L.E.; Reith, C.; Bhala, N.; Peto, R.; Barnes, E.H.; Keech, A.; et al. Efficacy and safety of more intensive lowering of LDL cholesterol: A meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet 2010, 376, 1670–1681. [Google Scholar] [PubMed]
- Oesterle, A.; Laufs, U.; Liao, J.K. Pleiotropic effects of statins on the cardiovascular system. Circ. Res. 2017, 120, 229–243. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, A.L.; Hasselbalch, H.C.; Nielsen, C.H.; Poulsen, H.E.; Ellervik, C. Statin treatment, oxidative stress and inflammation in a Danish population. Redox Biol. 2019, 21, 101088. [Google Scholar] [CrossRef]
- Hasselbalch, H.C.; Riley, C.H. Statins in the treatment of polycythaemia vera and allied disorders: An antithrombotic and cytoreductive potential? Leuk. Res. 2006, 30, 1217–1225. [Google Scholar] [CrossRef] [PubMed]
- Hasselbalch, H.C.; Riley, C.H. The mevalonate pathway as a therapeutic target in the Ph-negative chronic myeloproliferative disorders. Curr. Drug Targets 2007, 8, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, A.L.; Kallenbach, K.; Hasselbalch, H.C. A remarkable hematological and molecular response pattern in a patient with polycythemia vera during combination therapy with simvastatin and alendronate. Leuk. Res. Rep. 2016, 6, 20–23. [Google Scholar] [CrossRef]
- Griner, L.N.; McGraw, K.L.; Johnson, J.O.; List, A.F.; Reuther, G.W. JAK2-V617F-mediated signalling is dependent on lipid rafts and statins inhibit JAK2-V617F-dependent cell growth. Br. J. Haematol. 2013, 160, 177–187. [Google Scholar] [CrossRef]
- Pisanti, S.; Picardi, P.; Ciaglia, E.; D’alessandro, A.; Bifulco, M. Novel prospects of statins a therapeutic agents in cancer. Pharmacol. Res. 2014, 88, 84–98. [Google Scholar] [CrossRef]
- Nielsen, S.F.; Nordestgaard, B.G.; Bojesen, S.E. Statin use and reduced cancer-related mortality. N. Engl. J. Med. 2013, 368, 576–577. [Google Scholar] [CrossRef]
- Kristensen, D.T.; Øvlisen, A.K.; Jakobsen, L.H.K.; Severinsen, M.T.; Hannig, L.H.; Starklint, J.; Hilsøe, M.H.; Vallentin, A.P.; Brabrand, M.; Hasselbalch, H.C.; et al. Use of statins and risk of myeloproliferative neoplasms—A Danish nationwide case-control study. Blood Adv. 2023, 7, 3450–3457. [Google Scholar] [CrossRef]
- Vancura, A.; Bu, P.; Bhagwat, M.; Zeng, J.; Vancurova, I. Metformin as an Anticancer Agent. Trends Pharmacol. Sci. 2018, 39, 867–878. [Google Scholar] [CrossRef]
- Du, Y.; Zhu, Y.-J.; Zhou, Y.-X.; Ding, J.; Liu, J.-Y. Metformin in therapeutic applications in human diseases: Its mechanism of action and clinical study. Mol. Biomed. 2022, 3, 41. [Google Scholar] [CrossRef] [PubMed]
- Naseri, A.; Sanaie, S.; Hamzehzadeh, S.; Seyedi-Sahebari, S.; Hosseini, M.-S.; Gholipour-Khalili, E.; Majidazar, R.; Seraji, P.; Daneshvar, S.; Rezazadeh-Gavgani, E. Metformin: New applications for an old drug. J. Basic Clin. Physiol. Pharmacol. 2022, 34, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Lan, W.-H.; Lin, T.-Y.; Yeh, J.-A.; Feng, C.-L.; Hsu, J.-T.; Lin, H.-J.; Kuo, C.-J.; Lai, C.-H. Mechanism Underlying Metformin Action and Its Potential to Reduce Gastric Cancer Risk. Int. J. Mol. Sci. 2022, 23, 14163. [Google Scholar] [CrossRef]
- Feng, Y.M.; Jia, B.B.; Shen, Z. Metformin and bladder cancer: Drug repurposing as a potential tool for novel therapy: A review. Medicine 2022, 101, e31635. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Hou, Y.; Chen, X.; Zhang, P.; Kang, M.; Jin, Q.; Ji, J.; Gao, M. Metformin-induced stromal depletion to enhance the penetration of gemcitabine-loaded magnetic nanoparticles for pancreatic cancer targeted therapy. J. Am. Chem. Soc. 2020, 142, 4944–4954. [Google Scholar] [CrossRef]
- Larsen, M.K.K.; Skov, V.; Kjær, L.; Poulsen, H.E.; Eickhardt-Dalbøge, C.S.; Knudsen, T.A.; Christensen, S.F.; Sørensen, A.L.; Kristiansen, M.H.; Wienecke, T.; et al. Neutrophil-to-Lymphocyte Ratio as a Predictor of All-Cause Mortality in the General Population and in Patients with Myeloproliferative Neoplasms. Blood 2022, 140, 6842–6843. [Google Scholar] [CrossRef]
- Kristensen, D.T.; Øvlisen, A.K.; Jakobsen, L.K.B.; Severinsen, M.T.; Hanning, L.H.; Starklint, J.; Hilsøe, M.H.; Vallentin, A.P.; Brabrand, M.; Hasselbalch, H.C.; et al. Metformin Use and the Risk of Myeloproliferative Neoplasms in a Danish Population Based Cohort. Blood 2022, 140, 9673–9674. [Google Scholar] [CrossRef]
- Cordua, S.; Kjaer, L.; Skov, V.; Pallisgaard, N.; Hasselbalch, H.C.; Ellervik, C. Prevalence and phenotypes of JAK2 V617F and calreticulin mutations in a Danish general population. Blood 2019, 134, 469–479. [Google Scholar] [CrossRef] [PubMed]
- Cordua, S.; Kjaer, L.; Skov, V.; Pallisgaard, N.; Kefala, M.; Gjerdrum, L.M.R.; Hasselbalch, H.C.; Ellervik, C. Early detection of myeloproliferative neoplasms in a Danish general population study. Leukemia 2021, 35, 2706–2709. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, C.; Birgens, H.S.; Nordestgaard, B.G.; Bojesen, S.E. Diagnostic value of JAK2 V617F somatic mutation for myeloproliferative cancer in 49,488 individuals from the general population. Br. J. Haematol. 2013, 160, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Rockey, D.C.; Bell, P.D.; Hill, J.A. Fibrosis—A Common Pathway to Organ Injury and Failure. N. Engl. J. Med. 2015, 372, 1138–1149. [Google Scholar] [CrossRef]
- Moliterno, A.R.; Kaizer, H.; Reeves, B.N. JAK2 V617F allele burden in polycythemia vera: Burden of proof. Blood 2023, 141, 1934–1942. [Google Scholar] [PubMed]
- Egyed, M.; Kajtar, B.; Foldesi, C.; Skov, V.; Kjær, L.; Hasselbalch, H.C. Ropeginterferon-alfa2b resolves angina pectoris and reduces JAK2V617F in a patient with clonal hematopoiesis of indeterminate potential: A case report. Front. Hematol. 2022, 1, 1005666. [Google Scholar] [CrossRef]
- Karsdal, M.A.; Nielsen, S.H.; Leeming, D.J.; Langholm, L.L.; Nielsen, M.J.; Manon-Jensen, T.; Siebuhr, A.; Gudmann, N.S.; Ronnow, S.; Sand, J.M.; et al. The good and the bad collagens of fibrosis—Their role in signaling and organ function. Adv. Drug Deliv. Rev. 2017, 121, 43–56. [Google Scholar] [CrossRef]
- Brisson, B.K.; Mauldin, E.A.; Lei, W.; Vogel, L.K.; Power, A.M.; Lo, A.; Dopkin, D.; Khanna, C.; Wells, R.G.; Puré, E.; et al. Type III Collagen Directs Stromal Organization and Limits Metastasis in a Murine Model of Breast Cancer. Am. J. Pathol. 2015, 185, 1471–1486. [Google Scholar] [CrossRef]
- Beck, A.H.; Espinosa, I.; Gilks, C.B.; van de Rijn, M.; West, R.B. The fibromatosis signature defines a robust stromal response in breast carcinoma. Lab. Invest. 2008, 88, 591–601. [Google Scholar] [CrossRef]
- Di Martino, J.S.; Nobre, A.R.; Mondal, C.; Taha, I.; Farias, E.F.; Fertig, E.J.; Naba, A.; Aguirre-Ghiso, J.A.; Bravo-Cordero, J.J. A tumor-derived type III collagen-rich ECM niche regulates tumor cell dormancy. Nat. Cancer 2022, 3, 90–107. [Google Scholar] [CrossRef]
- Piao, X.-M.; Hwang, B.; Jeong, P.; Byun, Y.J.; Kang, H.W.; Seo, S.P.; Kim, W.T.; Lee, J.-Y.; Ha, Y.-S.; Lee, Y.-S.; et al. Collagen type VI-α1 and 2 repress the proliferation, migration and invasion of bladder cancer cells. Int. J. Oncol. 2021, 59, 37. [Google Scholar] [CrossRef]
- Özdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef] [PubMed]
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersall, I.W.; et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 2014, 25, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, S.; Hamberger, F.; Ravichandra, A.; Miller, M.; Nair, A.; Affo, S.; Filliol, A.; Chin, L.; Savage, T.M.; Yin, D.; et al. Tumor restriction by type I collagen opposes tumor-promoting effects of cancer-associated fibroblasts. J. Clin. Investig. 2021, 131, e146987. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Kim, J.; Yang, S.; Wang, H.; Wu, C.-J.; Sugimoto, H.; LeBleu, V.S.; Kalluri, R. Type I collagen deletion in αSMA+ myofibroblasts augments immune suppression and accelerates progression of pancreatic cancer. Cancer Cell 2021, 39, 548–565. [Google Scholar] [CrossRef]
- Sirinukunwattana, K.; Aberdeen, A.; Theissen, H.; Sousos, N.; Psaila, B.; Mead, A.J.; Turner, G.D.H.; Rees, G.; Rittscher, J.; Royston, D. Artificial intelligence-based morphological fingerprinting of megakaryocytes: A new tool for assessing disease in MPN patients. Blood Adv. 2020, 4, 3284–3294. [Google Scholar] [CrossRef]
- Ryou, H.; Sirinukunwattana, K.; Aberdeen, A.; Grindstaff, G.; Stolz, B.J.; Byrne, H.; Harrington, H.A.; Sousos, N.; Godfrey, A.L.; Harrison, C.N.; et al. Continuous Indexing of Fibrosis (CIF): Improving the assessment and classification of MPN patients. Leukemia 2023, 37, 348–358. [Google Scholar] [CrossRef]
- Balduini, A.; Badalucco, S.; Pugliano, M.T.; Baev, D.; De Silvestri, A.; Cattaneo, M.; Rosti, V.; Barosi, G. In vitro megakaryocyte differentiation and proplatelet formation in Ph-negative classical myeloproliferative neoplasms: Distinct patterns in the different clinical phenotypes. PLoS ONE 2011, 6, e21015. [Google Scholar] [CrossRef]
- Migliaccio, A.R.; Balduini, A.; Zhan, H. Editorial: Megakaryocytes as regulators of tumor microenvironments. Front. Oncol. 2022, 12, 1090658. [Google Scholar] [CrossRef]
- Varricchio, L.; Hoffman, R. Megakaryocytes Are Regulators of the Tumor Microenvironment and Malignant Hematopoietic Progenitor Cells in Myelofibrosis. Front. Oncol. 2022, 12, 906698. [Google Scholar] [CrossRef]
- Barosi, G.; Campanelli, R.; Massa, M.; Catarsi, P.; Carolei, A.; Abbà, C.; Villani, L.; Magrini, U.; Gregato, G.; Bertolini, F.; et al. Clonal Megakaryocyte Dysplasia with Isolated Thrombocytosis Is a Distinct Myeloproliferative Neoplasm Phenotype. Acta Haematol. 2023, 146, 14–25. [Google Scholar] [CrossRef]
- Barosi, G.; Rosti, V.; Gale, R.P. Myelofibrosis-type megakaryocyte dysplasia (MTMD) as a distinct category of BCR::ABL-negative myeloproliferative neoplasms. Challenges and perspectives. Leukemia 2023, 37, 725–727. [Google Scholar] [CrossRef] [PubMed]
- Di Buduo, C.A.; Miguel, C.P.; Balduini, A. Inside-to-outside and back to the future of megakaryopoiesis. Res. Pract. Thromb. Haemost. 2023, 7, 100197. [Google Scholar] [CrossRef] [PubMed]
- Barosi, G.; Rosti, V.; Bonetti, E.; Campanelli, R.; Carolei, A.; Catarsi, P.; Isgrò, A.M.; Lupo, L.; Massa, M.; Poletto, V.; et al. Evidence that prefibrotic myelofibrosis is aligned along a clinical and biological continuum featuring primary myelofibrosis. PLoS ONE 2012, 7, e35631. [Google Scholar] [CrossRef] [PubMed]
- Malara, A.; Di Buduo, C.A.; Abbonante, V.; Balduini, A. Bone marrow microenvironment of MPN cells. Int. Rev. Cell Mol. Biol. 2021, 365, 71–96. [Google Scholar] [CrossRef] [PubMed]
- Leiva, O.; Ng, S.K.; Chitalia, S.; Balduini, A.; Matsuura, S.; Ravid, K. The role of the extracellular matrix in primary myelofibrosis. Blood Cancer J. 2017, 7, e525. [Google Scholar] [CrossRef]
- Papadantonakis, N.; Matsuura, S.; Ravid, K. Megakaryocyte pathology and bone marrow fibrosis: The lysyl oxidase connection. Blood 2012, 120, 1774–1781. [Google Scholar] [CrossRef] [PubMed]
- Kagan, H.M.; Li, W. Lysyl oxidase: Properties, specificity, and biological roles inside and outside of the cell. J. Cell Biochem. 2003, 88, 660–672. [Google Scholar] [CrossRef]
- Eliades, A.; Papadantonakis, N.; Bhupatiraju, A.; Burridge, K.A.; Johnston-Cox, H.A.; Migliaccio, A.R.; Crispino, J.D.; Lucero, H.A.; Trackman, P.C.; Ravid, K. Control of megakaryocyte expansion and bone marrow fibrosis by lysyl oxidase. J. Biol. Chem. 2011, 286, 27630–27638. [Google Scholar] [CrossRef]
- Piasecki, A.; Leiva, O.; Ravid, K. Lysyl oxidase inhibition in primary myelofibrosis: A Renewed strategy. Arch. Stem Cell Ther. 2020, 1, 23–27. [Google Scholar] [PubMed]
- Leiva, O.; Hobbs, G.; Ravid, K.; Libby, P. Cardiovascular Disease in Myeloproliferative Neoplasms: JACC: CardioOncology State-of-the-Art Review. JACC CardioOncol. 2022, 4, 166–182. [Google Scholar] [CrossRef] [PubMed]
- Leiva, O.; Ng, S.K.; Matsuura, S.; Chitalia, V.; Lucero, H.; Findlay, A.; Turner, C.; Jarolimek, W.; Ravid, K. Novel lysyl oxidase inhibitors attenuate hallmarks of primary myelofibrosis in mice. Int. J. Hematol. 2019, 110, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Verstovsek, S.; Savona, M.R.; Mesa, R.A.; Dong, H.; Maltzman, J.D.; Sharma, S.; Silverman, J.; Oh, S.T.; Gotlib, J. A phase 2 study of simtuzumab in patients with primary, post-polycythaemia vera or post-essential thrombocythaemia myelofibrosis. Br. J. Haematol. 2017, 176, 939–949. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Streuer, A.; Jann, J.-C.; Altrock, E.; Schmitt, N.; Flach, J.; Sens-Albert, C.; Rapp, F.; Wolf, J.; Nowak, V.; et al. Inhibition of lysyl oxidases synergizes with 5-azacytidine to restore erythropoiesis in myelodysplastic and myeloid malignancies. Nat. Commun. 2023, 14, 1497. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Wang, L.; Wang, M.; Zhou, S.; Lu, Y.; Cui, H.; Racanelli, A.C.; Zhang, L.; Ye, T.; Ding, B.; et al. Targeting fibrosis, mechanisms and clinical trials. Signal Transduct. Target. Ther. 2022, 7, 206. [Google Scholar] [CrossRef]
- Yao, Y.; Findlay, A.; Stolp, J.; Rayner, B.; Ask, K.; Jarolimek, W. Pan-Lysyl Oxidase Inhibitor PXS-5505 Ameliorates Multiple-Organ Fibrosis by Inhibiting Collagen Crosslinks in Rodent Models of Systemic Sclerosis. Int. J. Mol. Sci. 2022, 23, 5533. [Google Scholar] [CrossRef]
- Tadmor, T.; Bejar, J.; Attias, D.; Mischenko, E.; Sabo, E.; Neufeld, G.; Vadasz, Z. The expression of lysyl-oxidase gene family members in myeloproliferative neoplasms. Am. J. Hematol. 2013, 88, 355–358. [Google Scholar] [CrossRef]
- Abbonante, V.; Chitalia, V.; Rosti, V.; Leiva, O.; Matsuura, S.; Balduini, A.; Ravid, K. Upregulation of lysyl oxidase and adhesion to collagen of human megakaryocytes and platelets in primary myelofibrosis. Blood 2017, 130, 829–831. [Google Scholar] [CrossRef]
- Matsuura, S.; Mi, R.; Koupenova, M.; Eliades, A.; Patterson, S.; Toselli, P.; Thon, J.; Italiano, J.E., Jr.; Trackman, P.C.; Papadantonakis, N.; et al. Lysyl oxidase is associated with increased thrombosis and platelet reactivity. Blood 2016, 127, 1493–1501. [Google Scholar] [CrossRef]
- Leiva, O.; Leon, C.; Kah Ng, S.; Mangin, P.; Gachet, C.; Ravid, K. The role of extracellular matrix stiffness in megakaryocyte and platelet development and function. Am. J. Hematol. 2018, 93, 430–441. [Google Scholar] [CrossRef] [PubMed]
- Lucero, H.A.; Ravid, K.; Grimsby, J.L.; Lucero, H.A.; Rich, C.B.; DiCamillo, S.J.; Mäki, J.M.; Myllyharju, J.; Kagan, H.M. Lysyl oxidase oxidizes cell membrane proteins and enhances the chemotactic response of vascularsmooth muscle cells. J. Biol. Chem. 2008, 283, 24103–24117. [Google Scholar] [CrossRef] [PubMed]
- Hasselbalch, H.; Skov, V.; Kjær, L.; Larsen, M.K.; Knudsen, T.A.; Lucijanić, M.; Kusec, R. Recombinant Interferon-β in the Treatment of Polycythemia Vera and Related Neoplasms: Rationales and Perspectives. Cancers 2022, 14, 5495. [Google Scholar] [CrossRef]
- Vitale, G.; van Eijck, C.H.; van Koetsveld Ing, P.M.; Erdmann, J.I.; Speel, E.J.; van der Wansem Ing, K.; Mooij, D.M.; Colao, A.; Lombardi, G.; Croze, E.; et al. Type I interferons in the treatment of pancreatic cancer: Mechanisms of action and role of related receptors. Ann. Surg. 2007, 246, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Yomimaru, Y.; Eguchi, H.; Wada, H.; Tomokuni, A.; Kobayashi, S.; Marubashi, S.; Takeda, Y.; Tanemura, M.; Umeshita, K.; Mori, M.; et al. Synergistic antitumor effect of interferon-ß with gemcitabine in interferon-α-non-responsive pancreatic cancer cells. Int. J. Oncol. 2011, 38, 1237–1243. [Google Scholar]
- Booy, S.; Eijck, C.H.J.; Dogan, F.; Koetsveld, P.M.; Hofland, L.J. Influence of type-I Interferon receptor expression level on the response to type-I Interferons in human pancreatic cancer cells. J. Cell Mol. Med. 2014, 18, 492–502. [Google Scholar] [CrossRef] [PubMed]
- Booy, S.; Hofland, L.; van Eijck, C. Potentials of interferon therapy in the treatment of pancreatic cancer. J. Interferon Cytokine Res. 2015, 35, 327–339. [Google Scholar] [CrossRef]
- Blaauboer, A.; Booy, S.; van Koetsveld, P.M.; Karels, B.; Dogan, F.; van Zwienen, S.; van Eijck, C.H.J.; Hofland, L.J. Interferon-beta enhances sensitivity to gemcitabine in pancreatic cancer. BMC Cancer 2020, 20, 913. [Google Scholar] [CrossRef]
- Zhang, J.; Li, R.; Huang, S. The immunoregulation effect of tumor microenvironment in pancreatic ductal adenocarcinoma. Front. Oncol. 2022, 12, 951019. [Google Scholar] [CrossRef]
- Blaauboer, A.; Van Koetsveld, P.M.; Mustafa, D.A.; Dumas, J.; Dogan, F.; Van Zwienen, S.; Van Eijck, C.H.; Hofland, L.J. Immunomodulatory antitumor effect of interferon-beta combined with gemcitabine in pancreatic cancer. Int. J. Oncol. 2022, 61, 97. [Google Scholar] [CrossRef]
- Doherty, M.R.; Cheon, H.; Junk, D.J.; Vinayak, S.; Varadan, V.; Telli, M.L.; Ford, J.M.; Stark, G.R.; Jackson, M.W. Interferon-beta represses cancer stem cell properties in triple-negative breast cancer. Proc. Natl. Acad. Sci. USA 2017, 114, 13792–13797. [Google Scholar] [CrossRef]
- Doherty, M.R.; Parvani, J.G.; Tamagno, I.; Junk, D.J.; Bryson, B.L.; Cheon, H.J.; Stark, G.R.; Jackson, M.W. The opposing effects of interferon-beta and oncostatin-M as regulators of cancer stem cell plasticity in triple-negative breast cancer. Breast Cancer Res. 2019, 21, 54. [Google Scholar] [CrossRef] [PubMed]
- Aricò, E.; Castiello, L.; Capone, I.; Gabriele, L.; Belardelli, F. Type I Interferons and Cancer: An Evolving Story Demanding Novel Clinical Applications. Cancers 2019, 11, 1943. [Google Scholar] [CrossRef] [PubMed]
- Larsen, A.M.H.; Kuczek, D.E.; Kalvisa, A.; Siersbæk, M.S.; Thorseth, M.-L.; Johansen, A.Z.; Carretta, M.; Grøntved, L.; Vang, O.; Madsen, D.H. Collagen Density Modulates the Immunosuppressive Functions of Macrophages. J. Immunol. 2020, 205, 1461–1472. [Google Scholar] [CrossRef] [PubMed]
- Vijver, S.V.; Singh, A.; Mommers-Elshof, E.T.A.M.; Meeldijk, J.; Copeland, R.; Boon, L.; Langermann, S.; Flies, D.; Meyaard, L.; Ramos, M.I.P. Collagen Fragments Produced in Cancer Mediate T Cell Suppression through Leukocyte-Associated Immunoglobulin-like Receptor 1. Front. Immunol. 2021, 12, 733561. [Google Scholar] [CrossRef] [PubMed]
- Tougaard, N.H.; Møller, A.L.; Rønn, P.F.; Hansen, T.W.; Genovese, F.; Karsdal, M.A.; Rasmussen, D.G.K.; Rossing, P. Endotrophin as a Marker of Complications in a Type 2 Diabetes Cohort. Diabetes Care 2022, 45, 2746–2748. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hasselbalch, H.C.; Junker, P.; Skov, V.; Kjær, L.; Knudsen, T.A.; Larsen, M.K.; Holmström, M.O.; Andersen, M.H.; Jensen, C.; Karsdal, M.A.; et al. Revisiting Circulating Extracellular Matrix Fragments as Disease Markers in Myelofibrosis and Related Neoplasms. Cancers 2023, 15, 4323. https://doi.org/10.3390/cancers15174323
Hasselbalch HC, Junker P, Skov V, Kjær L, Knudsen TA, Larsen MK, Holmström MO, Andersen MH, Jensen C, Karsdal MA, et al. Revisiting Circulating Extracellular Matrix Fragments as Disease Markers in Myelofibrosis and Related Neoplasms. Cancers. 2023; 15(17):4323. https://doi.org/10.3390/cancers15174323
Chicago/Turabian StyleHasselbalch, Hans Carl, Peter Junker, Vibe Skov, Lasse Kjær, Trine A. Knudsen, Morten Kranker Larsen, Morten Orebo Holmström, Mads Hald Andersen, Christina Jensen, Morten A. Karsdal, and et al. 2023. "Revisiting Circulating Extracellular Matrix Fragments as Disease Markers in Myelofibrosis and Related Neoplasms" Cancers 15, no. 17: 4323. https://doi.org/10.3390/cancers15174323