
In Silico, In Vitro, and In Vivo Investigations on Adapalene as Repurposed Third Generation Retinoid against Multiple Myeloma and Leukemia

Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Virtual Drug Screening and Molecular Docking Analyses
2.2. Microscale Thermophoresis
2.3. MYC Cignal Reporter Assay
2.4. Cell Lines
2.5. Cell Viability Assessment
2.6. Cell Cycle Analysis
2.7. Confocal Microscopy of p-H3(Ser10)
2.8. Fluorescence Microscopy Imaging of Microtubules Structure
2.9. Apoptosis Assessment
2.10. Autophagy Examination
2.11. Western Blot
2.12. T-ALL Xenograft Zebrafish Model
3. Results
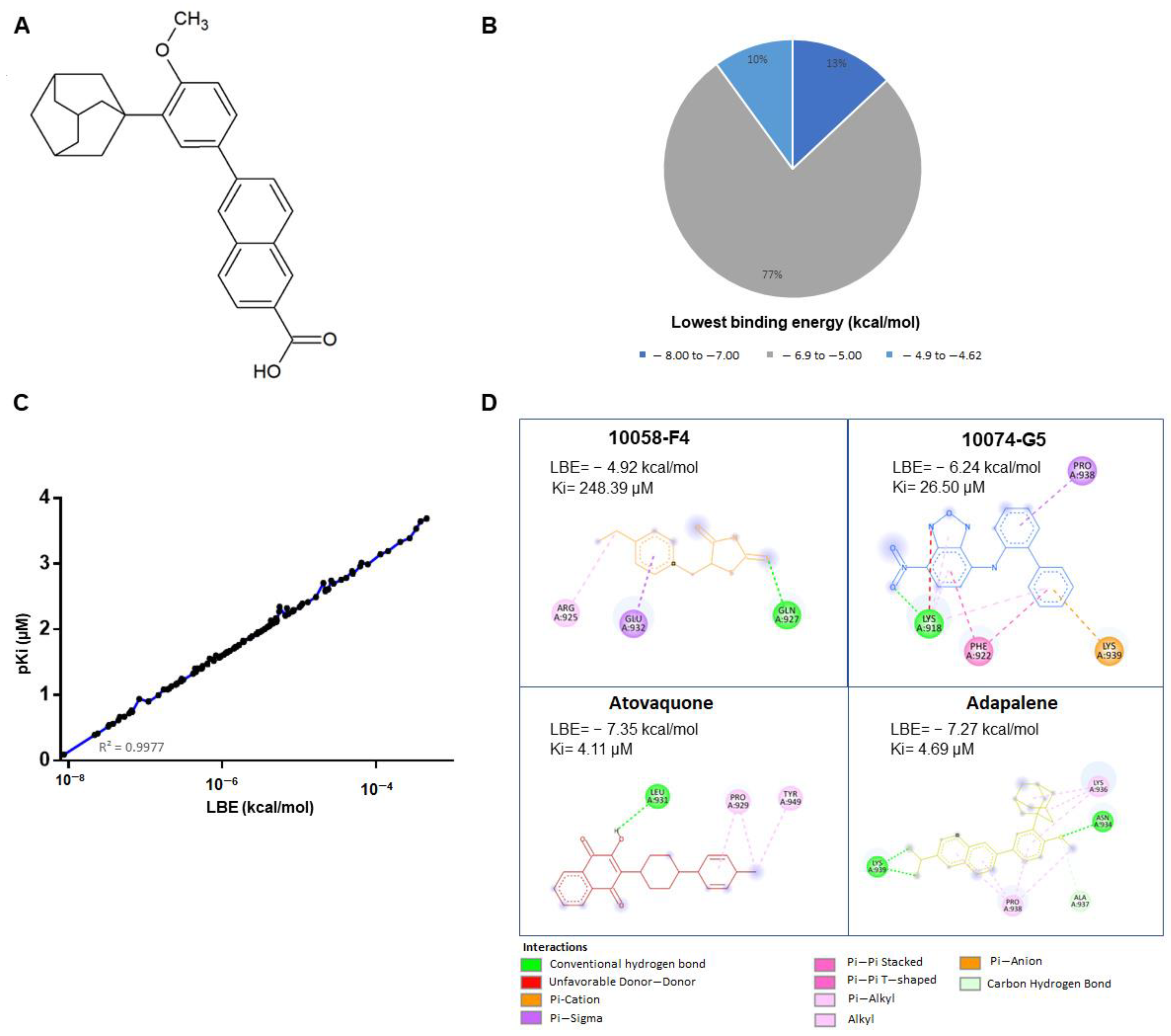
3.1. In Silico Drug Screening
3.2. Effectiveness of Atovaquone and Adapalene against HL60 and U266 Cells

3.3. Adapalene Strongly Bound to c-MYC
3.4. Adapalene Reduced the Viability of Hematological Cancer Cells
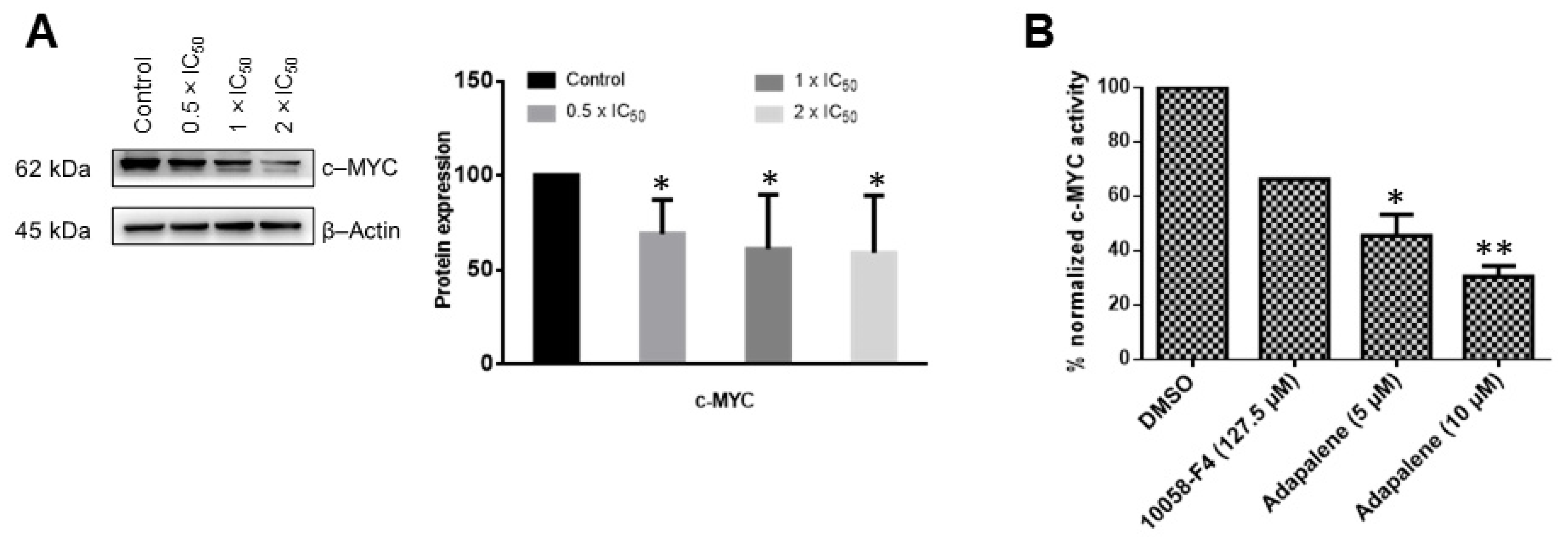
3.5. Adapalene Inhibited c-MYC Expression and Transcriptional Activity In Vitro
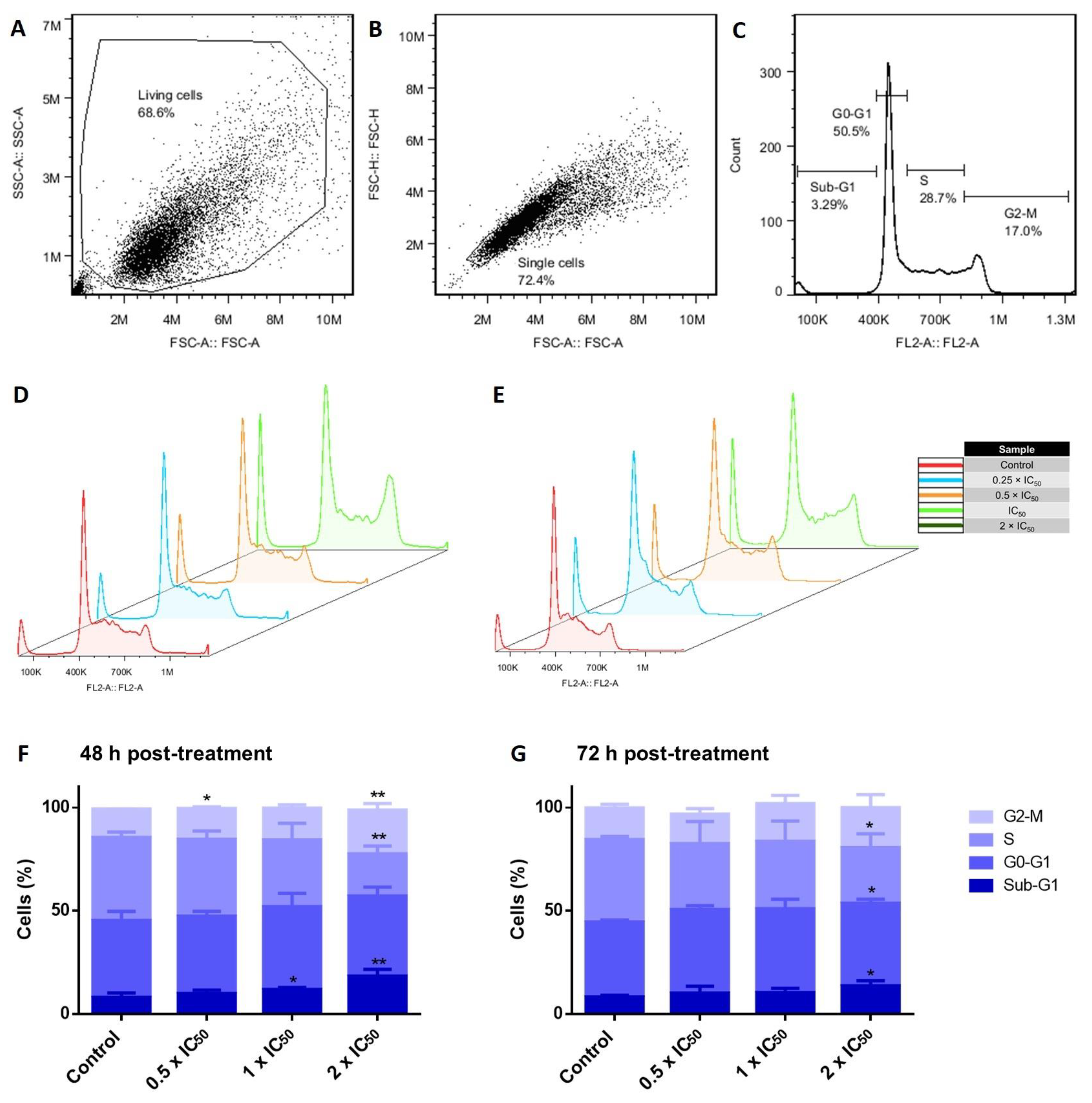
3.6. Cell Cycle Investigations in Adapalene-Treated AMO1 Cells
3.7. Adapalene Induced Mitotic Arrest as Detected by Phospho-Histone-3 (Ser10) Immunofluorescence
3.8. Adapalene Mitotic Arrest Involved the Distortion of the Tubulin Network as Detected by Immunofluorescence
3.9. Adapalene Induced Apoptosis, Autophagy, and DNA Damage in AMO1 Cells
3.10. Anticancer Activity of Adapalene In Vivo
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Banerjee, S.; Parasramka, M.A.; Paruthy, S.B. Garcinol: Preclinical perspective underpinning chemo-and radiosensitization of cancer. In Role of Nutraceuticals in Cancer Chemosensitization; Elsevier: Amsterdam, The Netherlands, 2018; pp. 297–324. [Google Scholar]
- Pinto, V.; Bergantim, R.; Caires, H.R.; Seca, H.; Guimarães, J.E.; Vasconcelos, M.H. Multiple myeloma: Available therapies and causes of drug resistance. Cancers 2020, 12, 407. [Google Scholar] [CrossRef] [PubMed]
- Van de Donk, N.W.C.J.; Pawlyn, C.; Yong, K.L. Multiple myeloma. Lancet 2021, 397, 410–427. [Google Scholar] [CrossRef] [PubMed]
- Bergsagel, P.L.; Kuehl, W.M. Chromosome translocations in multiple myeloma. Oncogene 2001, 20, 5611–5622. [Google Scholar] [CrossRef]
- Anguiano, A.; Tuchman, S.A.; Acharya, C.; Salter, K.; Gasparetto, C.; Zhan, F.; Dhodapkar, M.; Nevins, J.; Barlogie, B.; Shaughnessy, J.D., Jr. Gene expression profiles of tumor biology provide a novel approach to prognosis and may guide the selection of therapeutic targets in multiple myeloma. J. Clin. Oncol. 2009, 27, 4197. [Google Scholar] [CrossRef]
- Holien, T.; Misund, K.; Olsen, O.E.; Baranowska, K.A.; Buene, G.; Børset, M.; Waage, A.; Sundan, A. Myc amplifications in myeloma cell lines: Correlation with myc-inhibitor efficacy. Oncotarget 2015, 6, 22698. [Google Scholar] [CrossRef] [PubMed]
- Affer, M.; Chesi, M.; Chen, W.D.; Keats, J.J.; Demchenko, Y.N.; Tamizhmani, K.; Garbitt, V.M.; Riggs, D.L.; Brents, L.A.; Roschke, A.V. Promiscuous myc locus rearrangements hijack enhancers but mostly super-enhancers to dysregulate myc expression in multiple myeloma. Leukemia 2014, 28, 1725–1735. [Google Scholar] [CrossRef]
- Chng, W.J.; Huang, G.F.; Chung, T.H.; Ng, S.B.; Gonzalez-Paz, N.; Troska-Price, T.; Mulligan, G.; Chesi, M.; Bergsagel, P.L.; Fonseca, R. Clinical and biological implications of myc activation: A common difference between mgus and newly diagnosed multiple myeloma. Leukemia 2011, 25, 1026–1035. [Google Scholar] [CrossRef]
- Holien, T.; Våtsveen, T.K.; Hella, H.; Rampa, C.; Brede, G.; Grøseth, L.A.G.; Rekvig, M.; Børset, M.; Standal, T.; Waage, A. Bone morphogenetic proteins induce apoptosis in multiple myeloma cells by smad-dependent repression of myc. Leukemia 2012, 26, 1073–1080. [Google Scholar] [CrossRef]
- Kang, B.; Park, H.; Kim, B. Anticancer activity and underlying mechanism of phytochemicals against multiple myeloma. Int. J. Mol. Sci. 2019, 20, 2302. [Google Scholar] [CrossRef]
- Martínez-Martín, S.; Soucek, L. Myc inhibitors in multiple myeloma. Cancer Drug Resist. 2021, 4, 842. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, J.; Yin, J.; Gan, Y.; Xu, S.; Gu, Y.; Huang, W. Alternative approaches to target myc for cancer treatment. Signal Transduct. Target. Ther. 2021, 6, 117. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Shan, H.; Liu, M.; Liu, J.; Zhang, Z.; Xu, X.; Liu, Y.; Xu, H.; Lei, H.; Yu, M. Directly targeting c-myc contributes to the anti-multiple myeloma effect of anlotinib. Cell Death Dis. 2021, 12, 396. [Google Scholar] [CrossRef] [PubMed]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Boulos, J.C.; Saeed, M.E.M.; Chatterjee, M.; Bülbül, Y.; Crudo, F.; Marko, D.; Munder, M.; Klauck, S.M.; Efferth, T. Repurposing of the alk inhibitor crizotinib for acute leukemia and multiple myeloma cells. Pharmaceuticals 2021, 14, 1126. [Google Scholar] [CrossRef]
- Tolaymat, L.; Dearborn, H.; Zito, P.M. Adapalene. In StatPearls; StatPearls Publishing: St. Petersburg, FL, USA, 2022. [Google Scholar]
- Ocker, M.; Herold, C.; Ganslmayer, M.; Zopf, S.; Hahn, E.G.; Schuppan, D. Potentiated anticancer effects on hepatoma cells by the retinoid adapalene. Cancer Lett. 2004, 208, 51–58. [Google Scholar] [CrossRef]
- Rusu, A.; Tanase, C.; Pascu, G.-A.; Todoran, N. Recent advances regarding the therapeutic potential of adapalene. Pharmaceuticals 2020, 13, 217. [Google Scholar] [CrossRef]
- Shi, X.N.; Li, H.; Yao, H.; Liu, X.; Li, L.; Leung, K.S.; Kung, H.F.; Lin, M.C.M. Adapalene inhibits the activity of cyclin-dependent kinase 2 in colorectal carcinoma. Mol. Med. Rep. 2015, 12, 6501–6508. [Google Scholar] [CrossRef]
- Li, H.; Wang, C.; Li, L.; Bu, W.; Zhang, M.; Wei, J.; Tao, L.; Qian, K.; Ma, P. Adapalene suppressed the proliferation of melanoma cells by s-phase arrest and subsequent apoptosis via induction of DNA damage. Eur. J. Pharmacol. 2019, 851, 174–185. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, Q.; Luan, S.; Yang, K.; Zheng, M.; Li, K.; Chen, L.; Li, H. Adapalene inhibits ovarian cancer es-2 cells growth by targeting glutamic-oxaloacetic transaminase 1. Bioorg. Chem. 2019, 93, 103315. [Google Scholar] [CrossRef]
- Nong, H.-B.; Zhang, Y.-N.; Bai, Y.-G.; Zhang, Q.; Liu, M.-F.; Zhou, Q.; Shi, Z.-H.; Zeng, G.-F.; Zong, S.-H. Adapalene inhibits prostate cancer cell proliferation in vitro and in vivo by inducing DNA damage, s-phase cell cycle arrest, and apoptosis. Front. Pharmacol. 2022, 13, 801624. [Google Scholar] [CrossRef]
- Mehraj, U.; Wani, N.A.; Hamid, A.; Alkhanani, M.; Almilaibary, A.; Mir, M.A. Adapalene inhibits the growth of triple-negative breast cancer cells by s-phase arrest and potentiates the antitumor efficacy of gdc-0941. Front. Pharmacol. 2022, 13, 958443. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. Autodock4 and autodocktools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Clausen, D.M.; Guo, J.; Parise, R.A.; Beumer, J.H.; Egorin, M.J.; Lazo, J.S.; Prochownik, E.V.; Eiseman, J.L. In vitro cytotoxicity and in vivo efficacy, pharmacokinetics, and metabolism of 10074-g5, a novel small-molecule inhibitor of c-myc/max dimerization. J. Pharmacol. Exp. Ther. 2010, 335, 715–727. [Google Scholar] [CrossRef] [PubMed]
- Yap, J.L.; Wang, H.; Hu, A.; Chauhan, J.; Jung, K.-Y.; Gharavi, R.B.; Prochownik, E.V.; Fletcher, S. Pharmacophore identification of c-myc inhibitor 10074-g5. Bioorg. Med. Chem. Lett. 2013, 23, 370–374. [Google Scholar] [CrossRef]
- Elbadawi, M.; Boulos, J.C.; Dawood, M.; Zhou, M.; Gul, W.; ElSohly, M.A.; Klauck, S.M.; Efferth, T. The novel artemisinin dimer isoniazide eli-xxiii-98-2 induces c-myc inhibition, DNA damage, and autophagy in leukemia cells. Pharmaceutics 2023, 15, 1107. [Google Scholar] [CrossRef] [PubMed]
- Boulos, J.C.; Omer, E.A.; Rigano, D.; Formisano, C.; Chatterjee, M.; Leich, E.; Klauck, S.M.; Shan, L.-T.; Efferth, T. Cynaropicrin disrupts tubulin and c-myc-related signaling and induces parthanatos-type cell death in multiple myeloma. Acta Pharmacol. Sin. 2023, 1–17. [Google Scholar] [CrossRef]
- Geske, L.; Baier, J.; Boulos, J.C.; Efferth, T.; Opatz, T. Xylochemical synthesis and biological evaluation of the orchidaceous natural products isoarundinin i, bleochrin f, blestanol k, and pleionol. J. Nat. Prod. 2022, 86, 131–137. [Google Scholar] [CrossRef]
- Zhou, M.; Boulos, J.C.; Klauck, S.M.; Efferth, T. The cardiac glycoside zinc253504760 induces parthanatos-type cell death and g2/m arrest via downregulation of mek1/2 phosphorylation in leukemia cells. Cell Biol. Toxicol. 2023, 1–27. [Google Scholar] [CrossRef]
- Zhou, M.; Boulos, J.C.; Omer, E.A.; Klauck, S.M.; Efferth, T. Modes of action of a novel c-myc inhibiting 1,2,4-oxadiazole derivative in leukemia and breast cancer cells. Molecules 2023, 28, 5658. [Google Scholar] [CrossRef]
- Khalid, S.A.; Dawood, M.; Boulos, J.C.; Wasfi, M.; Drif, A.; Bahramimehr, F.; Shahhamzehei, N.; Shan, L.; Efferth, T. Identification of gedunin from a phytochemical depository as a novel multidrug resistance-bypassing tubulin inhibitor of cancer cells. Molecules 2022, 27, 5858. [Google Scholar] [CrossRef]
- Saeed, M.E.M.; Boulos, J.C.; Muecklich, S.B.; Leich, E.; Chatterjee, M.; Klauck, S.M.; Efferth, T. Disruption of lipid raft microdomains, regulation of cd38, tp53, and myc signaling, and induction of apoptosis by lomitapide in multiple myeloma cells. Cancer Genom. Proteom. 2022, 19, 540–555. [Google Scholar] [CrossRef] [PubMed]
- Rykowski, S.; Gurda-Woźna, D.; Orlicka-Płocka, M.; Fedoruk-Wyszomirska, A.; Giel-Pietraszuk, M.; Wyszko, E.; Kowalczyk, A.; Stączek, P.; Bak, A.; Kiliszek, A. Design, synthesis, and evaluation of novel 3-carboranyl-1, 8-naphthalimide derivatives as potential anticancer agents. Int. J. Mol. Sci. 2021, 22, 2772. [Google Scholar] [CrossRef] [PubMed]
- Saeed, M.E.M.; Boulos, J.C.; Machel, K.; Andabili, N.; Marouni, T.; Roth, W.; Efferth, T. Expression of the stem cell marker abcb5 in normal and tumor tissues. In Vivo 2022, 36, 1651–1666. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Yan, G.; Klauck, S.M.; Fleischer, E.; Klinger, A.; Sugimoto, Y.; Shan, L.; Efferth, T. Cytotoxicity of 4-hydroxy-n-(naphthalen-1-yl)-2-oxo-2h-chromene-3-carboxamide in multidrug-resistant cancer cells through activation of perk/eif2α/atf4 pathway. Biochem. Pharmacol. 2021, 193, 114788. [Google Scholar] [CrossRef] [PubMed]
- Neves, B.J.; Braga, R.C.; Melo-Filho, C.C.; Moreira-Filho, J.T.; Muratov, E.N.; Andrade, C.H. Qsar-based virtual screening: Advances and applications in drug discovery. Front. Pharmacol. 2018, 9, 1275. [Google Scholar] [CrossRef]
- Holien, T.; Våtsveen, T.K.; Hella, H.; Waage, A.; Sundan, A. Addiction to c-myc in multiple myeloma. Blood J. Am. Soc. Hematol. 2012, 120, 2450–2453. [Google Scholar] [CrossRef]
- Wienken, C.J.; Baaske, P.; Rothbauer, U.; Braun, D.; Duhr, S. Protein-binding assays in biological liquids using microscale thermophoresis. Nat. Commun. 2010, 1, 100. [Google Scholar] [CrossRef]
- Delmore, J.E.; Issa, G.C.; Lemieux, M.E.; Rahl, P.B.; Shi, J.; Jacobs, H.M.; Kastritis, E.; Gilpatrick, T.; Paranal, R.M.; Qi, J. Bet bromodomain inhibition as a therapeutic strategy to target c-myc. Cell 2011, 146, 904–917. [Google Scholar] [CrossRef]
- Ozawa, K. Reduction of phosphorylated histone h3 serine 10 and serine 28 cell cycle marker intensities after DNA damage. Cytom. Part A J. Int. Soc. Anal. Cytol. 2008, 73, 517–527. [Google Scholar] [CrossRef]
- Park, C.; Cha, H.-J.; Lee, H.; Hwang-Bo, H.; Ji, S.Y.; Kim, M.Y.; Hong, S.H.; Jeong, J.-W.; Han, M.H.; Choi, S.H. Induction of g2/m cell cycle arrest and apoptosis by genistein in human bladder cancer t24 cells through inhibition of the ros-dependent pi3k/akt signal transduction pathway. Antioxidants 2019, 8, 327. [Google Scholar] [CrossRef] [PubMed]
- Nagai, Y.; Mimura, N.; Rizq, O.; Isshiki, Y.; Oshima, M.; Rizk, M.; Saraya, A.; Koide, S.; Nakajima-Takagi, Y.; Miyota, M. The combination of the tubulin binding small molecule ptc596 and proteasome inhibitors suppresses the growth of myeloma cells. Sci. Rep. 2021, 11, 2074. [Google Scholar] [CrossRef]
- Rashan, L.J.; Özenver, N.; Boulos, J.C.; Dawood, M.; Roos, W.P.; Franke, K.; Papasotiriou, I.; Wessjohann, L.A.; Fiebig, H.-H.; Efferth, T. Molecular modes of action of an aqueous nerium oleander extract in cancer cells in vitro and in vivo. Molecules 2023, 28, 1871. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, M.; Hirata, N.; Tanaka, T.; Suizu, F.; Nakajima, H.; Chiorini, J.A. Autophagy as a modulator of cell death machinery. Cell Death Dis. 2020, 11, 517. [Google Scholar] [CrossRef]
- Li, Z.; Owonikoko, T.K.; Sun, S.-Y.; Ramalingam, S.S.; Doetsch, P.W.; Xiao, Z.-Q.; Khuri, F.R.; Curran, W.J.; Deng, X. C-myc suppression of DNA double-strand break repair. Neoplasia 2012, 14, 1190–1202, IN32–IN35. [Google Scholar] [CrossRef]
- Valdiglesias, V.; Giunta, S.; Fenech, M.; Neri, M.; Bonassi, S. Γh2ax as a marker of DNA double strand breaks and genomic instability in human population studies. Mutat. Res./Rev. Mutat. Res. 2013, 753, 24–40. [Google Scholar] [CrossRef] [PubMed]
- Sacchi, S.; Russo, D.; Avvisati, G.; Dastoli, G.; Lazzarino, M.; Pelicci, P.G.; Bonora, M.R.; Visani, G.; Grassi, C.; Iacona, I. All-trans retinoic acid in hematological malignancies, an update. Ger (gruppo ematologico retinoidi). Haematologica 1997, 82, 106–121. [Google Scholar]
- Chen, S.; Hu, Q.; Tao, X.; Xia, J.; Wu, T.; Cheng, B.; Wang, J. Retinoids in cancer chemoprevention and therapy: Meta-analysis of randomized controlled trials. Front. Genet. 2022, 13, 1065320. [Google Scholar] [CrossRef]
- Jin, Y.; Teh, S.S.; Lau, H.L.N.; Xiao, J.; Mah, S.H. Retinoids as anti-cancer agents and their mechanisms of action. Am. J. Cancer Res. 2022, 12, 938. [Google Scholar]
- Tang, X.-H.; Gudas, L.J. Retinoids, retinoic acid receptors, and cancer. Annu. Rev. Pathol. Mech. Dis. 2011, 6, 345–364. [Google Scholar] [CrossRef]
- Ni, X.; Hu, G.; Cai, X. The success and the challenge of all-trans retinoic acid in the treatment of cancer. Crit. Rev. Food Sci. Nutr. 2019, 59, S71–S80. [Google Scholar] [CrossRef] [PubMed]
- Cunliffe, W.J.; Caputo, R.; Dreno, B.; Förström, L.; Heenen, M.; Orfanos, C.E.; Privat, Y.; Aguilar, A.R.; Poncet, M.; Verschoore, M. Efficacy and safety comparison of adapalene (cd271) gel and tretinoin gel in the topical treatment of acne vulgaris. A european multicentre trial. J. Dermatol. Treat. 1997, 8, 173–178. [Google Scholar] [CrossRef]
- Boulos, J.C.; Idres, M.R.Y.; Efferth, T. Investigation of cancer drug resistance mechanisms by phosphoproteomics. Pharmacol. Res. 2020, 160, 105091. [Google Scholar] [CrossRef] [PubMed]
- Efferth, T.; Konkimalla, V.B.; Wang, Y.-F.; Sauerbrey, A.; Meinhardt, S.; Zintl, F.; Mattern, J.R.; Volm, M. Prediction of broad spectrum resistance of tumors towards anticancer drugs. Clin. Cancer Res. 2008, 14, 2405–2412. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.M.; Thomas, S.D.; Islam, A.; Muench, D.; Sedoris, K. C-myc and cancer metabolism. Clin. Cancer Res. 2012, 18, 5546–5553. [Google Scholar] [CrossRef]
- Dang, C.V. C-myc target genes involved in cell growth, apoptosis, and metabolism. Mol. Cell. Biol. 1999, 19, 1–11. [Google Scholar] [CrossRef]
- Dalla-Favera, R.; Bregni, M.; Erikson, J.; Patterson, D.; Gallo, R.C.; Croce, C.M. Human c-myc onc gene is located on the region of chromosome 8 that is translocated in burkitt lymphoma cells. Proc. Natl. Acad. Sci. USA 1982, 79, 7824–7827. [Google Scholar] [CrossRef]
- Vennstrom, B.; Sheiness, D.; Zabielski, J.; Bishop, J.M. Isolation and characterization of c-myc, a cellular homolog of the oncogene (v-myc) of avian myelocytomatosis virus strain 29. J. Virol. 1982, 42, 773–779. [Google Scholar] [CrossRef]
- Duffy, M.J.; O’Grady, S.; Tang, M.; Crown, J. Myc as a target for cancer treatment. Cancer Treat. Rev. 2021, 94, 102154. [Google Scholar] [CrossRef]
- Whitfield, J.R.; Soucek, L. The long journey to bring a myc inhibitor to the clinic. J. Cell Biol. 2021, 220, e202103090. [Google Scholar] [CrossRef]
- Jovanović, K.K.; Roche-Lestienne, C.; Ghobrial, I.M.; Facon, T.; Quesnel, B.; Manier, S. Targeting myc in multiple myeloma. Leukemia 2018, 32, 1295–1306. [Google Scholar] [CrossRef] [PubMed]
- Bretones, G.; Delgado, M.D.; León, J. Myc and cell cycle control. Biochim. Biophys. Acta (BBA)-Gene Regul. Mech. 2015, 1849, 506–516. [Google Scholar] [CrossRef] [PubMed]
- Boichuk, S.; Galembikova, A.; Zykova, S.; Ramazanov, B.; Khusnutdinov, R.; Dunaev, P.; Khaibullina, S.; Lombardi, V. Ethyl-2-amino-pyrrole-3-carboxylates are novel potent anticancer agents that affect tubulin polymerization, induce g2/m cell-cycle arrest, and effectively inhibit soft tissue cancer cell growth in vitro. Anti-Cancer Drugs 2016, 27, 620–634. [Google Scholar] [CrossRef] [PubMed]
- Mc Gee, M.M. Targeting the mitotic catastrophe signaling pathway in cancer. Mediat. Inflamm. 2015, 2015, 146282. [Google Scholar] [CrossRef]
- Alexandrova, N.; Niklinski, J.; Bliskovsky, V.; Otterson, G.A.; Blake, M.; Kaye, F.J.; Zajac-Kaye, M. The n-terminal domain of c-myc associates with alpha-tubulin and microtubules in vivo and in vitro. Mol. Cell. Biol. 1995, 15, 5188–5195. [Google Scholar] [CrossRef] [PubMed]
- Bressin, C.; Bourgarel-Rey, V.; Carré, M.; Pourroy, B.; Arango, D.; Braguer, D.; Barra, Y. Decrease in c-myc activity enhances cancer cell sensitivity to vinblastine. Anti-Cancer Drugs 2006, 17, 181–187. [Google Scholar] [CrossRef]
- Agut, R.; Falomir, E.; Murga, J.; Martín-Beltrán, C.; Gil-Edo, R.; Pla, A.; Carda, M.; Díaz, J.F.; Josa-Prado, F.; Marco, J.A. Synthesis of n-acyl derivatives of aminocombretastatin a-4 and study of their interaction with tubulin and downregulation of c-myc. Med. Chem. 2021, 17, 1129–1139. [Google Scholar] [CrossRef]
- Jiang, M.-R.; Li, Y.-C.; Yang, Y.; Wu, J.-R. C-myc degradation induced by DNA damage results in apoptosis of cho cells. Oncogene 2003, 22, 3252–3259. [Google Scholar] [CrossRef]
- Popov, N.; Herold, S.; Llamazares, M.; Schülein, C.; Eilers, M. Fbw7 and usp28 regulate myc protein stability in response to DNA damage. Cell Cycle 2007, 6, 2327–2331. [Google Scholar] [CrossRef]
- Watson, N.C.; Di, Y.M.; Orr, M.S.; Fornari, J.K., Jr.; Randolph, K.J.; Magnet, P.T.J.; Gewirtz, D.A. Influence of ionizing radiation on proliferation, c-myc expression and the induction of apoptotic cell death in two breast tumour cell lines differing in p53 status. Int. J. Radiat. Biol. 1997, 72, 547–559. [Google Scholar]
- Lu, H.-R.; Meng, L.-H.; Huang, M.; Zhu, H.; Miao, Z.-H.; Ding, J. DNA damage, c-myc suppression and apoptosis induced by the novel topoisomerase ii inhibitor, salvicine, in human breast cancer mcf-7 cells. Cancer Chemother. Pharmacol. 2005, 55, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Cui, F.; Fan, R.; Chen, Q.; He, Y.; Song, M.; Shang, Z.; Zhang, S.; Zhu, W.; Cao, J.; Guan, H. The involvement of c-myc in the DNA double-strand break repair via regulating radiation-induced phosphorylation of atm and DNA-pkcs activity. Mol. Cell. Biochem. 2015, 406, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Ahmadi, S.E.; Rahimi, S.; Zarandi, B.; Chegeni, R.; Safa, M. Myc: A multipurpose oncogene with prognostic and therapeutic implications in blood malignancies. J. Hematol. Oncol. 2021, 14, 121. [Google Scholar] [CrossRef] [PubMed]
- Siu, K.T.; Ramachandran, J.; Yee, A.J.; Eda, H.; Santo, L.; Panaroni, C.; Mertz, J.A.; Sims Iii, R.J.; Cooper, M.R.; Raje, N. Preclinical activity of cpi-0610, a novel small-molecule bromodomain and extra-terminal protein inhibitor in the therapy of multiple myeloma. Leukemia 2017, 31, 1760–1769. [Google Scholar] [CrossRef]
- Ghosalkar, J.; Iyer, S.; Malhotra, G.; Joshi, K. In vitro and in vivo anticancer potential of adapalene, a third-generation retinoid against bladder cancer. Cancer Res. 2018, 78, 2316. [Google Scholar] [CrossRef]
- Ocker, M.; Herold, C.; Ganslmayer, M.; Hahn, E.G.; Schuppan, D. The synthetic retinoid adapalene inhibits proliferation and induces apoptosis in colorectal cancer cells in vitro. Int. J. Cancer 2003, 107, 453–459. [Google Scholar] [CrossRef]
- Patergnani, S.; Missiroli, S.; Morciano, G.; Perrone, M.; Mantovani, C.M.; Anania, G.; Fiorica, F.; Pinton, P.; Giorgi, C. Understanding the role of autophagy in cancer formation and progression is a real opportunity to treat and cure human cancers. Cancers 2021, 13, 5622. [Google Scholar] [CrossRef]
- Jiang, Z.-F.; Shao, L.-J.; Wang, W.-M.; Yan, X.-B.; Liu, R.-Y. Decreased expression of beclin-1 and lc3 in human lung cancer. Mol. Biol. Rep. 2012, 39, 259–267. [Google Scholar] [CrossRef]
- Hwang, S.-K.; Jeong, Y.-J.; Shin, J.-M.; Magae, J.; Kim, C.-H.; Chang, Y.-C. Mac inhibits c-myc and induces autophagy by downregulation of cip2a in leukemia cells. Mol. Cell. Toxicol. 2018, 14, 417–424. [Google Scholar] [CrossRef]
- Li, H.; Liu, J.; Cao, W.; Xiao, X.; Liang, L.; Liu-Smith, F.; Wang, W.; Liu, H.; Zhou, P.; Ouyang, R. C-myc/mir-150/epg5 axis mediated dysfunction of autophagy promotes development of non-small cell lung cancer. Theranostics 2019, 9, 5134. [Google Scholar] [CrossRef]
- Fan, Y.-J.; Zong, W.-X. The cellular decision between apoptosis and autophagy. Chin. J. Cancer 2013, 32, 121. [Google Scholar] [PubMed]
- Kang, R.; Zeh, H.J.; Lotze, M.T.; Tang, D. The beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011, 18, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Konantz, M.; Balci, T.B.; Hartwig, U.F.; Dellaire, G.; André, M.C.; Berman, J.N.; Lengerke, C. Zebrafish xenografts as a tool for in vivo studies on human cancer. Ann. N. Y. Acad. Sci. 2012, 1266, 124–137. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Type | Cell Line | IC50 (µM) |
|---|---|---|
| MM | AMO1 | 1.76 ± 0.39 |
| KMS12BM | 2.61 ± 0.36 | |
| MOLP8 | 2.69 ± 0.29 | |
| NCI-H929 | 4.95 ± 1.21 | |
| RPMI8226 | 4.97 ± 1.33 | |
| OPM2 | 5.82 ± 2.07 | |
| KMS11 | 7.22 ± 1.20 | |
| L363 | 7.25 ± 0.73 | |
| JJN3 | 9.10 ± 1.85 | |
| Leukemia | CCRF-CEM | 1.83 ± 0.46 |
| CEM/ADR5000 | 2.30 ± 0.09 | |
| Normal leukocytes | PBMCs | 36.72 ± 0.64 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boulos, J.C.; Chatterjee, M.; Shan, L.; Efferth, T. In Silico, In Vitro, and In Vivo Investigations on Adapalene as Repurposed Third Generation Retinoid against Multiple Myeloma and Leukemia. Cancers 2023, 15, 4136. https://doi.org/10.3390/cancers15164136
Boulos JC, Chatterjee M, Shan L, Efferth T. In Silico, In Vitro, and In Vivo Investigations on Adapalene as Repurposed Third Generation Retinoid against Multiple Myeloma and Leukemia. Cancers. 2023; 15(16):4136. https://doi.org/10.3390/cancers15164136
Chicago/Turabian StyleBoulos, Joelle C., Manik Chatterjee, Letian Shan, and Thomas Efferth. 2023. "In Silico, In Vitro, and In Vivo Investigations on Adapalene as Repurposed Third Generation Retinoid against Multiple Myeloma and Leukemia" Cancers 15, no. 16: 4136. https://doi.org/10.3390/cancers15164136