
Efficacy of Systemically Administered Retargeted Oncolytic Herpes Simplex Viruses—Clearance and Biodistribution in Naïve and HSV-Preimmune Mice
 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
3. Results
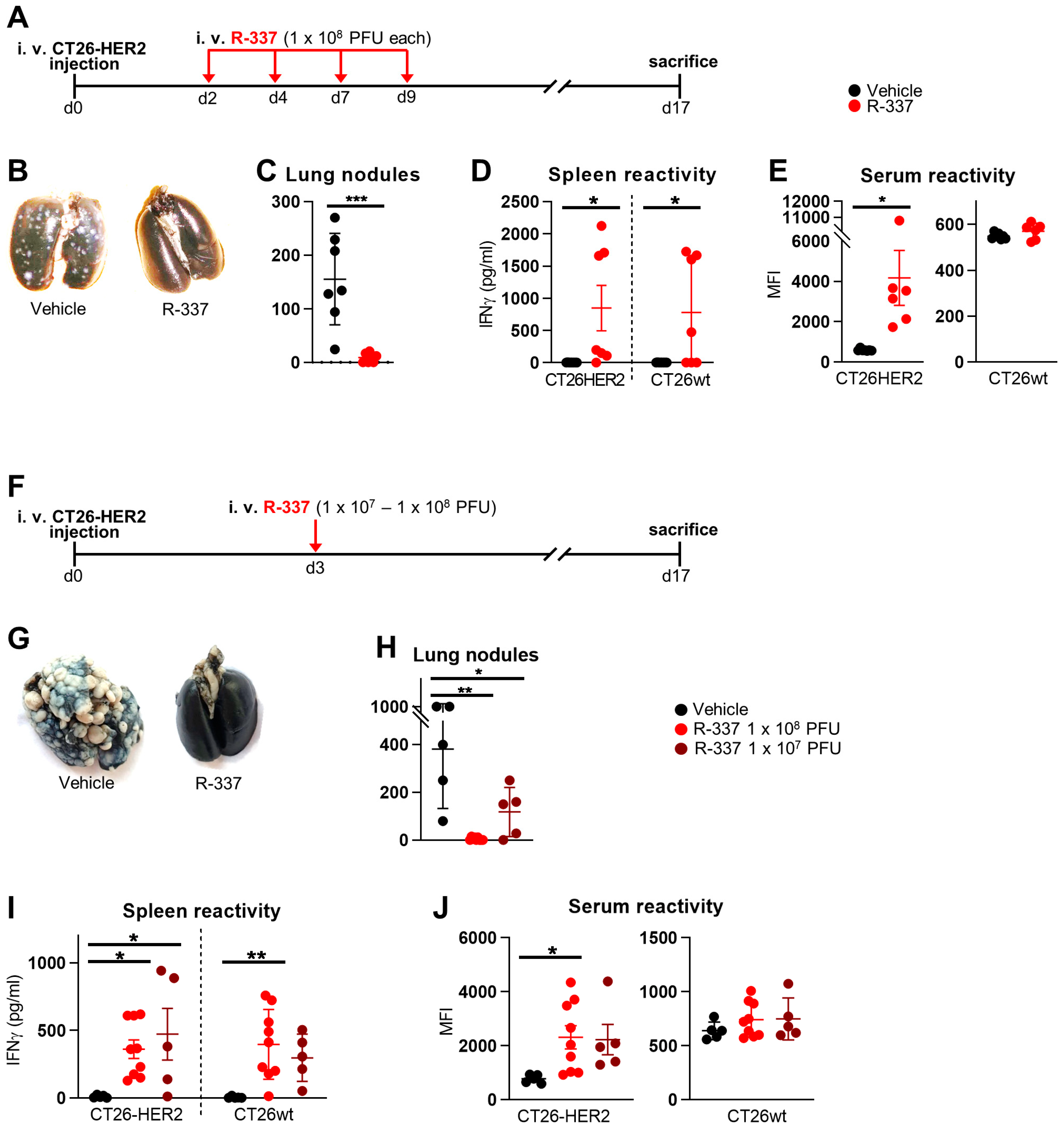
3.1. Therapeutic Efficacy of Systemically Administered R-337 Monotherapy against CT26-HER2 Lung Tumors, a Model of Metastatic Disease. Early Administration
3.2. Combination with α-CTLA4 Antibodies Increases the Therapeutic Efficacy of Systemically Administered R-337
3.3. Systemically Administered ReHV R-405 Inhibits the Growth of a Subcutaneous Tumor
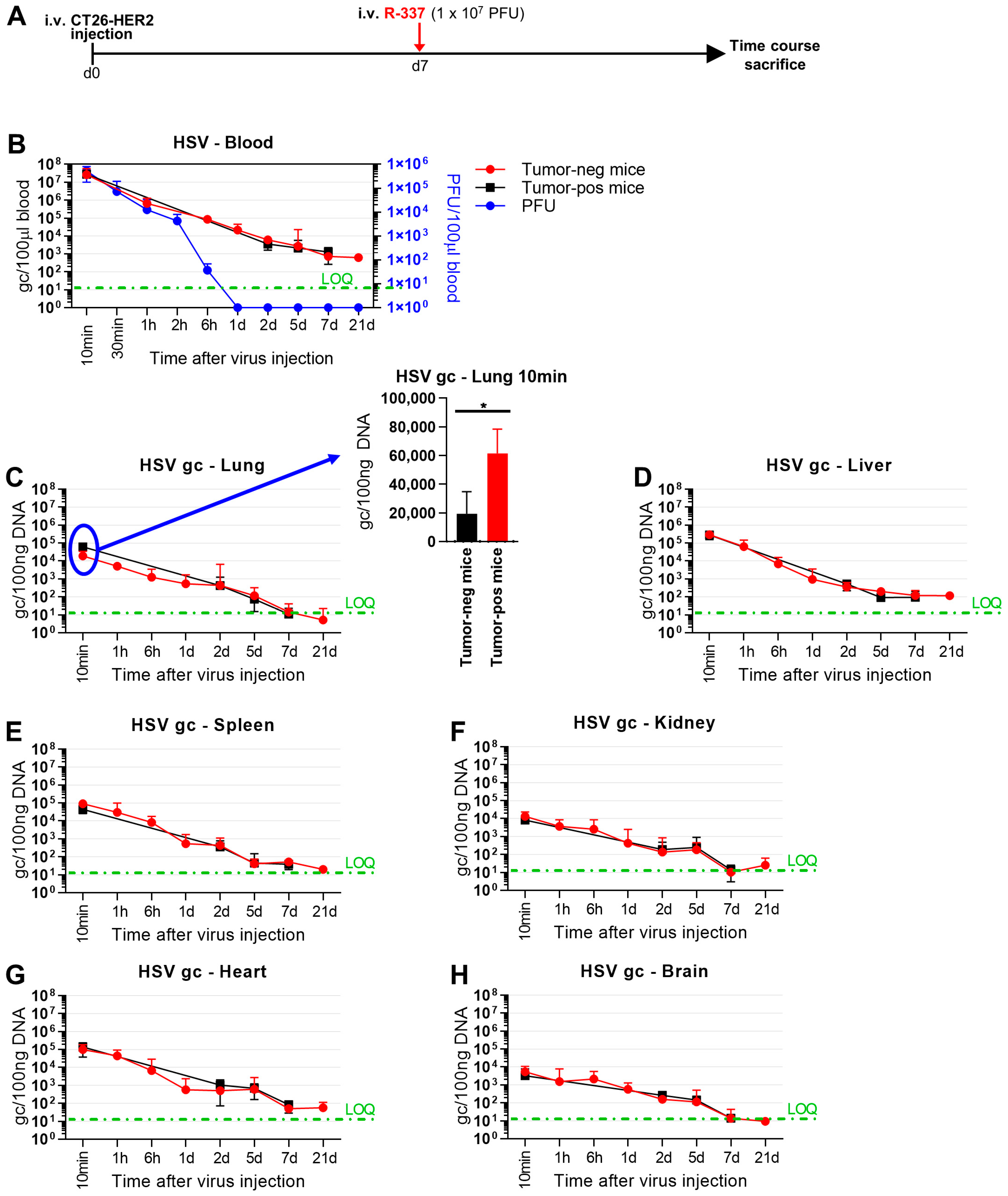
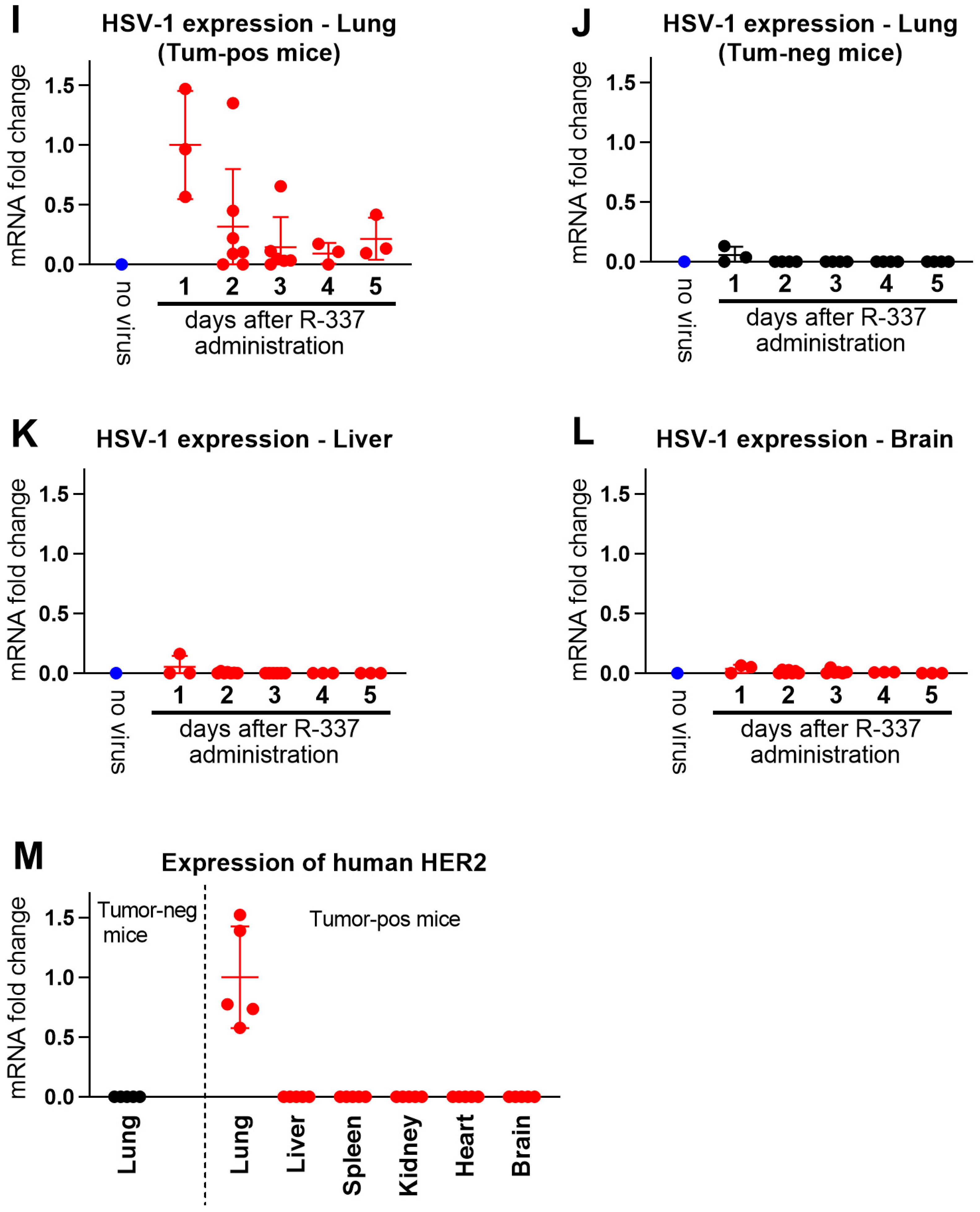
3.4. Clearance and Biodistribution of Systemically Administered R-337
3.5. Lack of Efficacy of Systemically Administered R-337 in HSV-Preimmune Mice
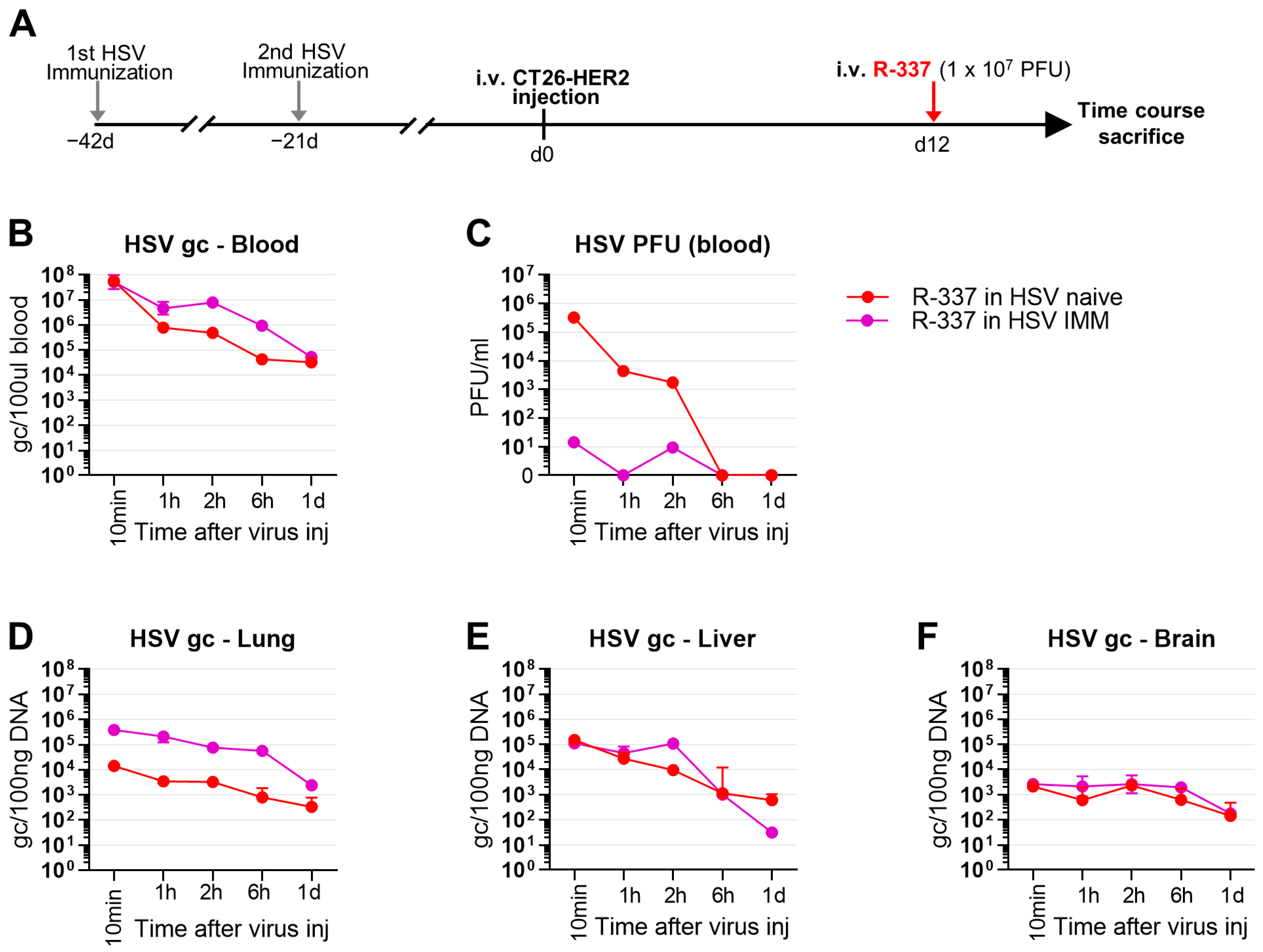
3.6. Clearance and Biodistribution of Systemically Administered R-337 in HSV-IMM Mice
3.7. Factors Affecting the Stability of R-337 in Serum and in Whole Blood In Vivo
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mineta, T.; Rabkin, S.D.; Yazaki, T.; Hunter, W.D.; Martuza, R.L. Attenuated multi-mutated herpes simplex virus-1 for the treatment of malignant gliomas. Nat. Med. 1995, 1, 938–943. [Google Scholar] [CrossRef]
- Chambers, R.; Gillespie, G.Y.; Soroceanu, L.; Andreansky, S.; Chatterjee, S.; Chou, J.; Roizman, B.; Whitley, R.J. Comparison of genetically engineered herpes simplex viruses for the treatment of brain tumors in a scid mouse model of human malignant glioma. Proc. Natl. Acad. Sci. USA 1995, 92, 1411–1415. [Google Scholar] [CrossRef]
- Nakamura, T.; Russell, S.J. Oncolytic measles viruses for cancer therapy. Expert. Opin. Biol. Ther. 2004, 4, 1685–1692. [Google Scholar] [CrossRef] [PubMed]
- Prestwich, R.J.; Harrington, K.J.; Vile, R.G.; Melcher, A.A. Immunotherapeutic potential of oncolytic virotherapy. Lancet Oncol. 2008, 9, 610–612. [Google Scholar] [CrossRef] [PubMed]
- Cattaneo, R.; Russell, S.J. How to develop viruses into anticancer weapons. PLoS Pathog. 2017, 13, e1006190. [Google Scholar] [CrossRef] [PubMed]
- Russell, S.J.; Peng, K.W.; Bell, J.C. Oncolytic virotherapy. Nat. Biotechnol. 2012, 30, 658–670. [Google Scholar] [CrossRef] [Green Version]
- Alberts, P.; Tilgase, A.; Rasa, A.; Bandere, K.; Venskus, D. The advent of oncolytic virotherapy in oncology: The Rigvir® story. Eur. J. Pharmacol. 2018, 837, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Fang, H. Clinical trials with oncolytic adenovirus in China. Curr. Cancer Drug Targets 2007, 7, 141–148. [Google Scholar] [CrossRef]
- Andtbacka, R.H.; Kaufman, H.L.; Collichio, F.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; Puzanov, I.; Agarwala, S.S.; et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients With Advanced Melanoma. J. Clin. Oncol. 2015, 33, 2780–2788. [Google Scholar] [CrossRef]
- Todo, T.; Ito, H.; Ino, Y.; Ohtsu, H.; Ota, Y.; Shibahara, J.; Tanaka, M. Intratumoral oncolytic herpes virus G47∆ for residual or recurrent glioblastoma: A phase 2 trial. Nat. Med. 2022, 28, 1630–1639. [Google Scholar] [CrossRef]
- Veerapong, J.; Bickenbach, K.A.; Shao, M.Y.; Smith, K.D.; Posner, M.C.; Roizman, B.; Weichselbaum, R.R. Systemic delivery of (gamma1)34.5-deleted herpes simplex virus-1 selectively targets and treats distant human xenograft tumors that express high MEK activity. Cancer Res. 2007, 67, 8301–8306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrington, K.; Freeman, D.J.; Kelly, B.; Harper, J.; Soria, J.-C. Optimizing oncolytic virotherapy in cancer treatment. Nat. Rev. Drug Discov. 2019, 18, 689–706. [Google Scholar] [CrossRef]
- Hill, C.; Carlisle, R. Achieving systemic delivery of oncolytic viruses. Expert Opin. Drug Deliv. 2019, 16, 607–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cripe, T.P.; Chen, C.Y.; Denton, N.L.; Haworth, K.B.; Hutzen, B.; Leddon, J.L.; Streby, K.A.; Wang, P.Y.; Markert, J.M.; Waters, A.M.; et al. Pediatric cancer gone viral. Part I: Strategies for utilizing oncolytic herpes simplex virus-1 in children. Mol. Ther. Oncolytics 2015, 2, 15015. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Martuza, R.; Rabkin, S. Intracarotid delivery of oncolytic HSV vector G47Δ to metastatic breast cancer in the brain. Gene Ther. 2005, 12, 647–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, R.J.; Chan, M.K.; Yu, Z.; Kim, T.H.; Bhargava, A.; Stiles, B.M.; Horsburgh, B.C.; Shah, J.P.; Ghossein, R.A.; Singh, B.; et al. Effective intravenous therapy of murine pulmonary metastases with an oncolytic herpes virus expressing interleukin 12. Clin. Cancer Res. 2004, 10, 251–259. [Google Scholar] [CrossRef] [Green Version]
- Ferguson, M.S.; Lemoine, N.R.; Wang, Y. Systemic delivery of oncolytic viruses: Hopes and hurdles. Adv. Virol. 2012, 2012, 805629. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Nakashima, H.; Decklever, T.; Nace, R.; Russell, S. HSV-NIS, an oncolytic herpes simplex virus type 1 encoding human sodium iodide symporter for preclinical prostate cancer radiovirotherapy. Cancer Gene Ther. 2013, 20, 478–485. [Google Scholar] [CrossRef] [Green Version]
- Nanni, P.; Gatta, V.; Menotti, L.; De Giovanni, C.; Ianzano, M.; Palladini, A.; Grosso, V.; Dall’ora, M.; Croci, S.; Nicoletti, G.; et al. Preclinical Therapy of Disseminated HER-2(+) Ovarian and Breast Carcinomas with a HER-2-Retargeted Oncolytic Herpesvirus. PLoS Pathog. 2013, 9, e1003155. [Google Scholar] [CrossRef] [Green Version]
- Shikano, T.; Kasuya, H.; T Sahin, T.; Nomura, N.; Kanzaki, A.; Misawa, M.; Nishikawa, Y.; Shirota, T.; Yamada, S.; Fujii, T. High therapeutic potential for systemic delivery of a liposomeconjugated herpes simplex virus. Curr. Cancer Drug Targets 2011, 11, 111–122. [Google Scholar] [CrossRef]
- Delman, K.A.; Bennett, J.J.; Zager, J.S.; Burt, B.M.; McAuliffe, P.F.; Petrowsky, H.; Kooby, D.A.; Hawkins, W.G.; Horsburgh, B.C.; Johnson, P.; et al. Effects of preexisting immunity on the response to herpes simplex-based oncolytic viral therapy. Hum. Gene Ther. 2000, 11, 2465–2472. [Google Scholar] [CrossRef]
- De Lucia, M.; Cotugno, G.; Bignone, V.; Garzia, I.; Nocchi, L.; Langone, F.; Petrovic, B.; Sasso, E.; Pepe, S.; Froechlich, G. Retargeted and multi-cytokine-armed herpes virus is a potent cancer endovaccine for local and systemic anti-tumor treatment. Mol. Ther. Oncolytics 2020, 19, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Hotte, S.J.; Lorence, R.M.; Hirte, H.W.; Polawski, S.R.; Bamat, M.K.; O’Neil, J.D.; Roberts, M.S.; Groene, W.S.; Major, P.P. An optimized clinical regimen for the oncolytic virus PV701. Clin. Cancer Res. 2007, 13, 977–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leoni, V.; Gatta, V.; Palladini, A.; Nicoletti, G.; Ranieri, D.; Dall’Ora, M.; Grosso, V.; Rossi, M.; Alviano, F.; Bonsi, L.; et al. Systemic delivery of HER2-retargeted oncolytic-HSV by mesenchymal stromal cells protects from lung and brain metastases. Oncotarget 2015, 6, 34774–34787. [Google Scholar] [CrossRef] [Green Version]
- Hadryś, A.; Sochanik, A.; McFadden, G.; Jazowiecka-Rakus, J. Mesenchymal stem cells as carriers for systemic delivery of oncolytic viruses. Eur. J. Pharmacol. 2020, 874, 172991. [Google Scholar] [CrossRef]
- Ilett, E.; Kottke, T.; Donnelly, O.; Thompson, J.; Willmon, C.; Diaz, R.; Zaidi, S.; Coffey, M.; Selby, P.; Harrington, K. Cytokine conditioning enhances systemic delivery and therapy of an oncolytic virus. Mol. Ther. 2014, 22, 1851–1863. [Google Scholar] [CrossRef] [Green Version]
- Carpenter, S.G.; Carson, J.; Fong, Y. Regional liver therapy using oncolytic virus to target hepatic colorectal metastases. In Seminars in Oncology; Elsevier: Amsterdam, The Netherlands, 2010. [Google Scholar]
- Cook, M.; Chauhan, A. Clinical application of oncolytic viruses: A systematic review. Int. J. Mol. Sci. 2020, 21, 7505. [Google Scholar] [CrossRef] [PubMed]
- Campadelli-Fiume, G.; De Giovanni, C.; Gatta, V.; Nanni, P.; Lollini, P.L.; Menotti, L. Rethinking herpes simplex virus: The way to oncolytic agents. Rev. Med. Virol. 2011, 21, 213–226. [Google Scholar] [CrossRef]
- Vannini, A.; Leoni, V.; Sanapo, M.; Gianni, T.; Giordani, G.; Gatta, V.; Barboni, C.; Zaghini, A.; Campadelli-Fiume, G. Immunotherapeutic Efficacy of Retargeted oHSVs Designed for Propagation in an Ad Hoc Cell Line. Cancers 2021, 13, 266. [Google Scholar] [CrossRef]
- Campadelli-Fiume, G.; Petrovic, B.; Leoni, V.; Gianni, T.; Avitabile, E.; Casiraghi, C.; Gatta, V. Retargeting Strategies for Oncolytic Herpes Simplex Viruses. Viruses 2016, 8, 63. [Google Scholar] [CrossRef] [Green Version]
- Leoni, V.; Petrovic, B.; Gianni, T.; Gatta, V.; Campadelli-Fiume, G. Simultaneous Insertion of Two Ligands in gD for Cultivation of Oncolytic Herpes Simplex Viruses in Noncancer Cells and Retargeting to Cancer Receptors. J. Virol. 2018, 92, e02132-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menotti, L.; Cerretani, A.; Hengel, H.; Campadelli-Fiume, G. Construction of a fully retargeted herpes simplex virus 1 recombinant capable of entering cells solely via human epidermal growth factor receptor 2. J. Virol. 2008, 20, 10153–10161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gatta, V.; Petrovic, B.; Campadelli-Fiume, G. The Engineering of a Novel Ligand in gH Confers to HSV an Expanded Tropism Independent of gD Activation by Its Receptors. PLoS Pathog. 2015, 11, e1004907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrovic, B.; Gianni, T.; Gatta, V.; Campadelli-Fiume, G. Insertion of a ligand to HER2 in gB retargets HSV tropism and obviates the need for activation of the other entry glycoproteins. PLoS Pathog. 2017, 13, e1006352. [Google Scholar] [CrossRef] [Green Version]
- Menotti, L.; Avitabile, E.; Gatta, V.; Petrovic, B.; Campadelli-Fiume, G. HSV as a platform for the generation of retargeted, armed, and reporter-expressing oncolytic viruses. Viruses 2018, 10, 352. [Google Scholar] [CrossRef] [Green Version]
- Vannini, A.; Parenti, F.; Bressanin, D.; Barboni, C.; Zaghini, A.; Campadelli-Fiume, G.; Gianni, T. Towards a Precision Medicine Approach and In Situ Vaccination against Prostate Cancer by PSMA-Retargeted oHSV. Viruses 2021, 13, 2085. [Google Scholar] [CrossRef]
- Vannini, A.; Parenti, F.; Forghieri, C.; Barboni, C.; Zaghini, A.; Campadelli-Fiume, G.; Gianni, T. Innovative retargeted oncolytic herpesvirus against nectin4-positive cancers. Front. Mol. Biosci. 2023, 10, 1149973. [Google Scholar] [CrossRef]
- Leoni, V.; Vannini, A.; Gatta, V.; Rambaldi, J.; Sanapo, M.; Barboni, C.; Zaghini, A.; Nanni, P.; Lollini, P.L.; Casiraghi, C.; et al. A fully-virulent retargeted oncolytic HSV armed with IL-12 elicits local immunity and vaccine therapy towards distant tumors. PLoS Pathog. 2018, 14, e1007209. [Google Scholar] [CrossRef] [Green Version]
- Gianni, T.; Leoni, V.; Sanapo, M.; Parenti, F.; Bressanin, D.; Barboni, C.; Zaghini, A.; Campadelli-Fiume, G.; Vannini, A. Genotype of Immunologically Hot or Cold Tumors Determines the Antitumor Immune Response and Efficacy by Fully Virulent Retargeted oHSV. Viruses 2021, 13, 1747. [Google Scholar] [CrossRef]
- Menotti, L.; Nicoletti, G.; Gatta, V.; Croci, S.; Landuzzi, L.; De Giovanni, C.; Nanni, P.; Lollini, P.L.; Campadelli-Fiume, G. Inhibition of human tumor growth in mice by an oncolytic herpes simplex virus designed to target solely HER-2-positive cells. Proc. Natl. Acad. Sci. USA 2009, 106, 9039–9044. [Google Scholar] [CrossRef]
- Cocchi, F.; Menotti, L.; Mirandola, P.; Lopez, M.; Campadelli-Fiume, G. The ectodomain of a novel member of the immunoglobulin subfamily related to the poliovirus receptor has the attributes of a bona fide receptor for herpes simplex virus types 1 and 2 in human cells. J. Virol. 1998, 72, 9992–10002. [Google Scholar] [CrossRef] [PubMed]
- Campadelli-Fiume, G.; Cocchi, F.; Menotti, L.; Lopez, M. The novel receptors that mediate the entry of herpes simplex viruses and animal alphaherpesviruses into cells. Rev. Med. Virol. 2000, 10, 305–319. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Campadelli Fiume, G. Anti-idiotypic antibodies mimicking glycoprotein D of herpes simplex virus identify a cellular protein required for virus spread from cell to cell and virus-induced polykaryocytosis. Proc. Natl. Acad. Sci. USA 1996, 93, 1836–1840. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, R.I.; Warner, M.S.; Lum, B.J.; Spear, P.G. Herpes simplex virus-1 entry into cells mediated by a novel member of the TNF/NGF receptor family. Cell 1996, 87, 427–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fulci, G.; Breymann, L.; Gianni, D.; Kurozomi, K.; Rhee, S.S.; Yu, J.; Kaur, B.; Louis, D.N.; Weissleder, R.; Caligiuri, M.A.; et al. Cyclophosphamide enhances glioma virotherapy by inhibiting innate immune responses. Proc. Natl. Acad. Sci. USA 2006, 103, 12873–12878. [Google Scholar] [CrossRef] [PubMed]
- Currier, M.A.; Gillespie, R.A.; Sawtell, N.M.; Mahller, Y.Y.; Stroup, G.; Collins, M.H.; Kambara, H.; Chiocca, E.A.; Cripe, T.P. Efficacy and safety of the oncolytic herpes simplex virus rRp450 alone and combined with cyclophosphamide. Mol. Ther. 2008, 16, 879–885. [Google Scholar] [CrossRef] [Green Version]
- Gambini, E.; Reisoli, E.; Appolloni, I.; Gatta, V.; Campadelli-Fiume, G.; Menotti, L.; Malatesta, P. Replication-competent herpes simplex virus retargeted to HER2 as therapy for high-grade glioma. Mol. Ther. 2012, 20, 994–1001. [Google Scholar] [CrossRef] [Green Version]
- Reisoli, E.; Gambini, E.; Appolloni, I.; Gatta, V.; Barilari, M.; Menotti, L.; Malatesta, P. Efficacy of HER2 retargeted herpes simplex virus as therapy for high-grade glioma in immunocompetent mice. Cancer Gene Ther. 2012, 19, 788–795. [Google Scholar] [CrossRef]
- Li, H.; Peng, K.; Dingli, D.; Kratzke, R.; Russell, S. Oncolytic measles viruses encoding interferon β and the thyroidal sodium iodide symporter gene for mesothelioma virotherapy. Cancer Gene Ther. 2010, 17, 550–558. [Google Scholar] [CrossRef]
- Roos, F.C.; Roberts, A.M.; Hwang, I.I.; Moriyama, E.H.; Evans, A.J.; Sybingco, S.; Watson, I.R.; Carneiro, L.A.; Gedye, C.; Girardin, S.E. Oncolytic targeting of renal cell carcinoma via encephalomyocarditis virus. EMBO Mol. Med. 2010, 2, 275–288. [Google Scholar] [CrossRef]
- Farrell, C.J.; Zaupa, C.; Barnard, Z.; Maley, J.; Martuza, R.L.; Rabkin, S.D.; Curry Jr, W.T. Combination immunotherapy for tumors via sequential intratumoral injections of oncolytic herpes simplex virus 1 and immature dendritic cells. Clin. Cancer Res. 2008, 14, 7711–7716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreansky, S.; Soroceanu, L.; Flotte, E.R.; Chou, J.; Markert, J.M.; Gillespie, G.Y.; Roizman, B.; Whitley, R.J. Evaluation of genetically engineered herpes simplex viruses as oncolytic agents for human malignant brain tumors. Cancer Res. 1997, 57, 1502–1509. [Google Scholar] [PubMed]
- Oseledchyk, A.; Ricca, J.M.; Gigoux, M.; Ko, B.; Redelman-Sidi, G.; Walther, T.; Liu, C.; Iyer, G.; Merghoub, T.; Wolchok, J.D. Lysis-independent potentiation of immune checkpoint blockade by oncolytic virus. Oncotarget 2018, 9, 28702. [Google Scholar] [CrossRef] [Green Version]
- Ricca, J.M.; Oseledchyk, A.; Walther, T.; Liu, C.; Mangarin, L.; Merghoub, T.; Wolchok, J.D.; Zamarin, D. Pre-existing Immunity to Oncolytic Virus Potentiates Its Immunotherapeutic Efficacy. Mol. Ther. 2018, 26, 1008–1019. [Google Scholar] [CrossRef] [Green Version]
- Veinalde, R.; Pidelaserra-Martí, G.; Moulin, C.; Tan, C.L.; Schäfer, T.E.; Kang, N.; Ball, C.R.; Leichsenring, J.; Stenzinger, A.; Kaderali, L. Virotherapy combined with anti-PD-1 transiently reshapes the tumor immune environment and induces anti-tumor immunity in a preclinical PDAC model. Front. Immunol. 2023, 13, 1096162. [Google Scholar] [CrossRef]
- Engeland, C.E.; Grossardt, C.; Veinalde, R.; Bossow, S.; Lutz, D.; Kaufmann, J.K.; Shevchenko, I.; Umansky, V.; Nettelbeck, D.M.; Weichert, W.; et al. CTLA-4 and PD-L1 checkpoint blockade enhances oncolytic measles virus therapy. Mol. Ther. 2014, 22, 1949–1959. [Google Scholar] [CrossRef] [Green Version]
- Saha, D.; Martuza, R.L.; Rabkin, S.D. Oncolytic herpes simplex virus immunovirotherapy in combination with immune checkpoint blockade to treat glioblastoma. Immunotherapy 2018, 10, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Rahman, A.; Tsai, V.; Goudreau, A.; Shinoda, J.Y.; Wen, S.F.; Ramachandra, M.; Ralston, R.; Maneval, D.; LaFace, D.; Shabram, P. Specific depletion of human anti-adenovirus antibodies facilitates transduction in an in vivo model for systemic gene therapy. Mol. Ther. 2001, 3, 768–778. [Google Scholar] [CrossRef] [PubMed]
- Howard, F.; Muthana, M. Designer nanocarriers for navigating the systemic delivery of oncolytic viruses. Nanomedicine 2020, 15, 93–110. [Google Scholar] [CrossRef]
- Tse, L.V.; Moller-Tank, S.; Asokan, A. Strategies to circumvent humoral immunity to adeno-associated viral vectors. Expert Opin. Biol. Ther. 2015, 15, 845–855. [Google Scholar] [CrossRef]
- Orlowski, A.; Katz, M.G.; Gubara, S.M.; Fargnoli, A.S.; Fish, K.M.; Weber, T. Successful transduction with AAV vectors after selective depletion of anti-AAV antibodies by immunoadsorption. Mol. Ther. Methods Clin. Dev. 2020, 16, 192–203. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Haller, E.; Horak, J.; Brandstetter, M.; Heuser, T.; Lämmerhofer, M. Protein A-and Protein G-gold nanoparticle bioconjugates as nano-immunoaffinity platform for human IgG depletion in plasma and antibody extraction from cell culture supernatant. Talanta 2019, 194, 664–672. [Google Scholar] [CrossRef] [PubMed]
- Ettorre, A.; Rösli, C.; Silacci, M.; Brack, S.; McCombie, G.; Knochenmuss, R.; Elia, G.; Neri, D. Recombinant antibodies for the depletion of abundant proteins from human serum. Proteomics 2006, 6, 4496–4505. [Google Scholar] [CrossRef]
- Cambridge, G.; Stohl, W.; Leandro, M.J.; Migone, T.S.; Hilbert, D.M.; Edwards, J.C. Circulating levels of B lymphocyte stimulator in patients with rheumatoid arthritis following rituximab treatment: Relationships with B cell depletion, circulating antibodies, and clinical relapse. Arthritis Rheum. 2006, 54, 723–732. [Google Scholar] [CrossRef]
- Afzali, B.; Noris, M.; Lambrecht, B.N.; Kemper, C. The state of complement in COVID-19. Nat. Rev. Immunol. 2022, 22, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Leber, M.F.; Neault, S.; Jirovec, E.; Barkley, R.; Said, A.; Bell, J.C.; Ungerechts, G. Engineering and combining oncolytic measles virus for cancer therapy. Cytokine Growth Factor Rev. 2020, 56, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Cerullo, V.; Diaconu, I.; Kangasniemi, L.; Rajecki, M.; Escutenaire, S.; Koski, A.; Romano, V.; Rouvinen, N.; Tuuminen, T.; Laasonen, L. Immunological effects of low-dose cyclophosphamide in cancer patients treated with oncolytic adenovirus. Mol. Ther. 2011, 19, 1737–1746. [Google Scholar] [CrossRef] [Green Version]
- Peng, K.; Myers, R.; Greenslade, A.; Mader, E.; Greiner, S.; Federspiel, M.; Dispenzieri, A.; Russell, S. Using clinically approved cyclophosphamide regimens to control the humoral immune response to oncolytic viruses. Gene Ther. 2013, 20, 255–261. [Google Scholar] [CrossRef] [Green Version]
- Ban, W.; Guan, J.; Huang, H.; He, Z.; Sun, M.; Liu, F.; Sun, J. Emerging systemic delivery strategies of oncolytic viruses: A key step toward cancer immunotherapy. Nano Res. 2022, 15, 4137–4153. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vannini, A.; Parenti, F.; Barboni, C.; Forghieri, C.; Leoni, V.; Sanapo, M.; Bressanin, D.; Zaghini, A.; Campadelli-Fiume, G.; Gianni, T. Efficacy of Systemically Administered Retargeted Oncolytic Herpes Simplex Viruses—Clearance and Biodistribution in Naïve and HSV-Preimmune Mice. Cancers 2023, 15, 4042. https://doi.org/10.3390/cancers15164042
Vannini A, Parenti F, Barboni C, Forghieri C, Leoni V, Sanapo M, Bressanin D, Zaghini A, Campadelli-Fiume G, Gianni T. Efficacy of Systemically Administered Retargeted Oncolytic Herpes Simplex Viruses—Clearance and Biodistribution in Naïve and HSV-Preimmune Mice. Cancers. 2023; 15(16):4042. https://doi.org/10.3390/cancers15164042
Chicago/Turabian StyleVannini, Andrea, Federico Parenti, Catia Barboni, Cristina Forghieri, Valerio Leoni, Mara Sanapo, Daniela Bressanin, Anna Zaghini, Gabriella Campadelli-Fiume, and Tatiana Gianni. 2023. "Efficacy of Systemically Administered Retargeted Oncolytic Herpes Simplex Viruses—Clearance and Biodistribution in Naïve and HSV-Preimmune Mice" Cancers 15, no. 16: 4042. https://doi.org/10.3390/cancers15164042