
Imidazole-4-N-acetamide Derivatives as a Novel Scaffold for Selective Targeting of Cyclin Dependent Kinases

 , , ,
, , , 
Abstract
:Simple Summary
Abstract
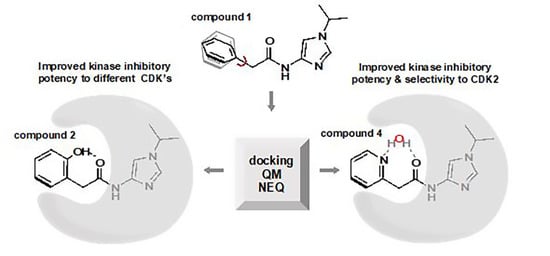
1. Introduction
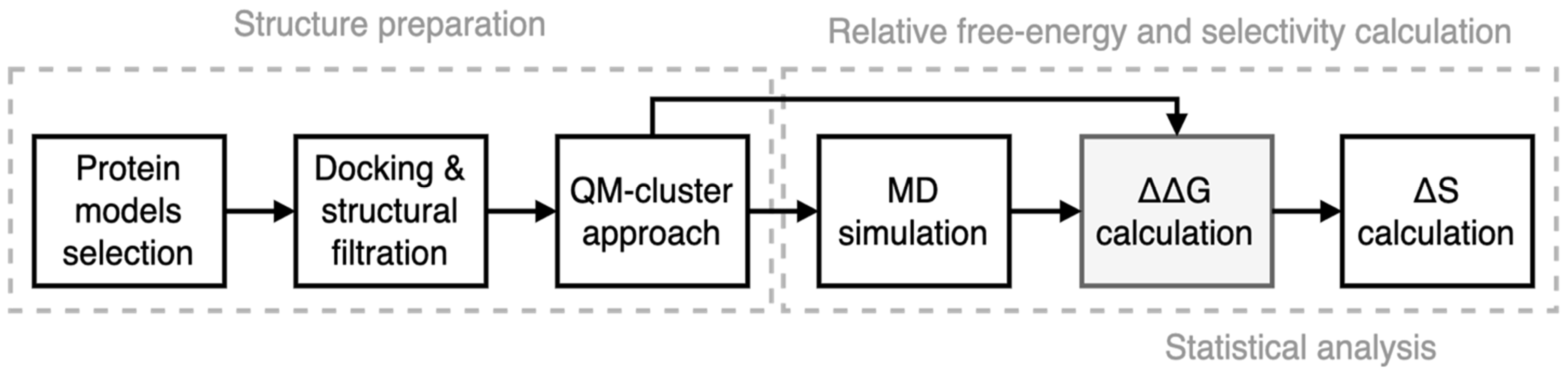
2. Materials and Methods
2.1. Chemicals
2.2. In Silico Prediction of the Binding Affinity of Imidazole-4-N-acetamide Derivatives
2.2.1. Preparation of CDK–Ligand Complexes
2.2.2. NEQ Thermodynamics and Free Energy Calculations
2.3. In Vitro Kinase Assays
2.4. Relative Selectivity Calculation
2.5. Anti-Proliferative Activity of New Imidazole-4-N-acetamide Derivatives
2.6. Statistical Analysis
3. Results
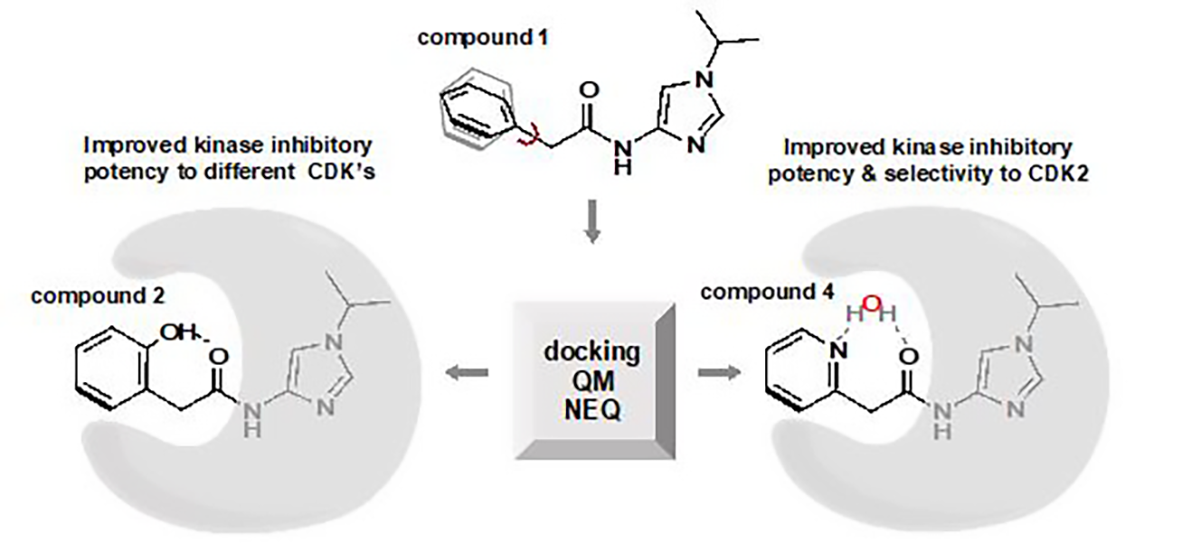
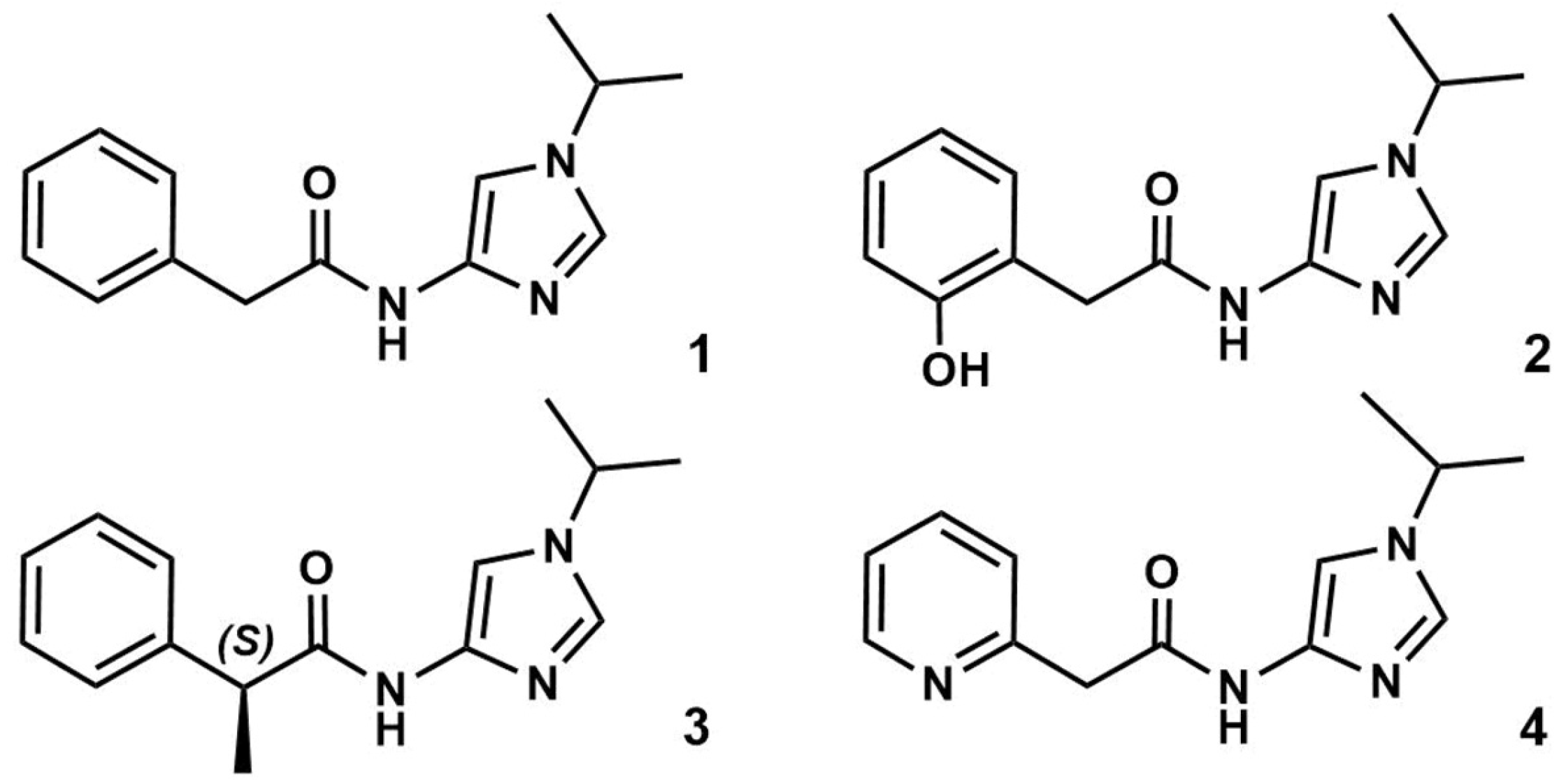
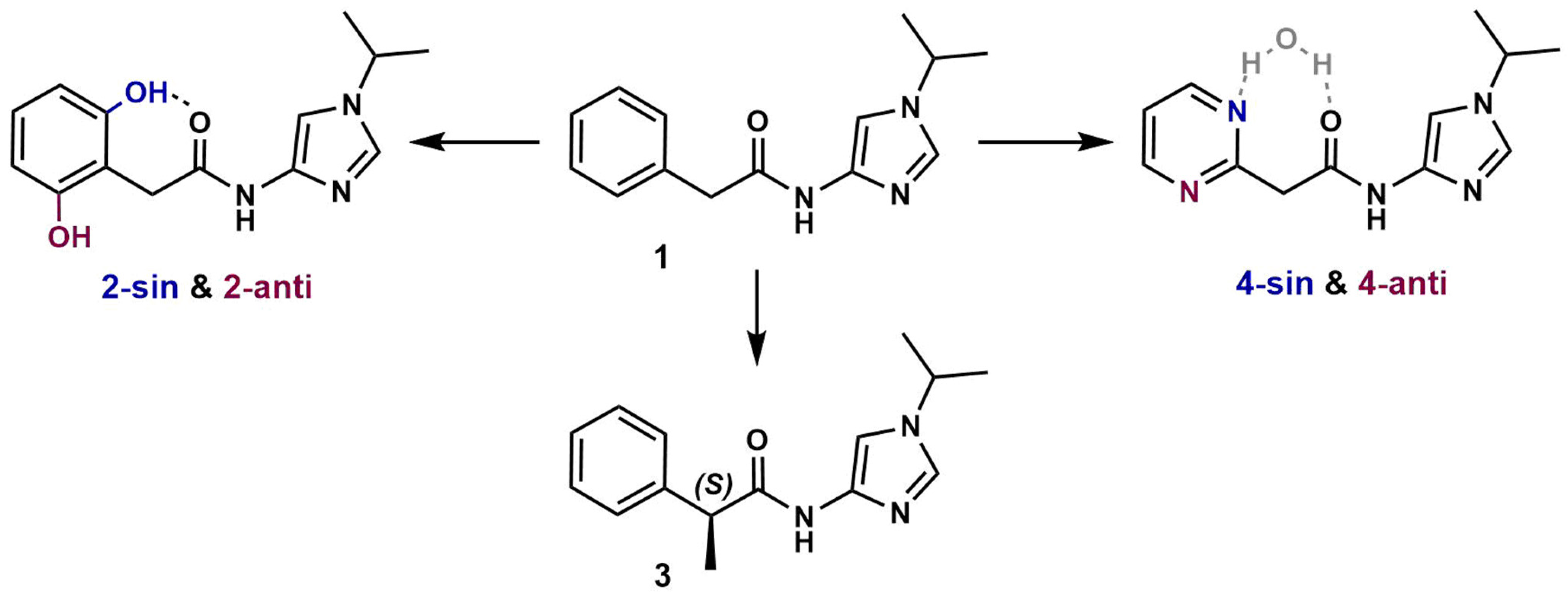
3.1. Preparation of Starting Target–Ligand Complexes and Calculations of Binding Energies
3.2. CDK Inhibitory Potency of New Imidazole-4-N-acetamide Derivatives
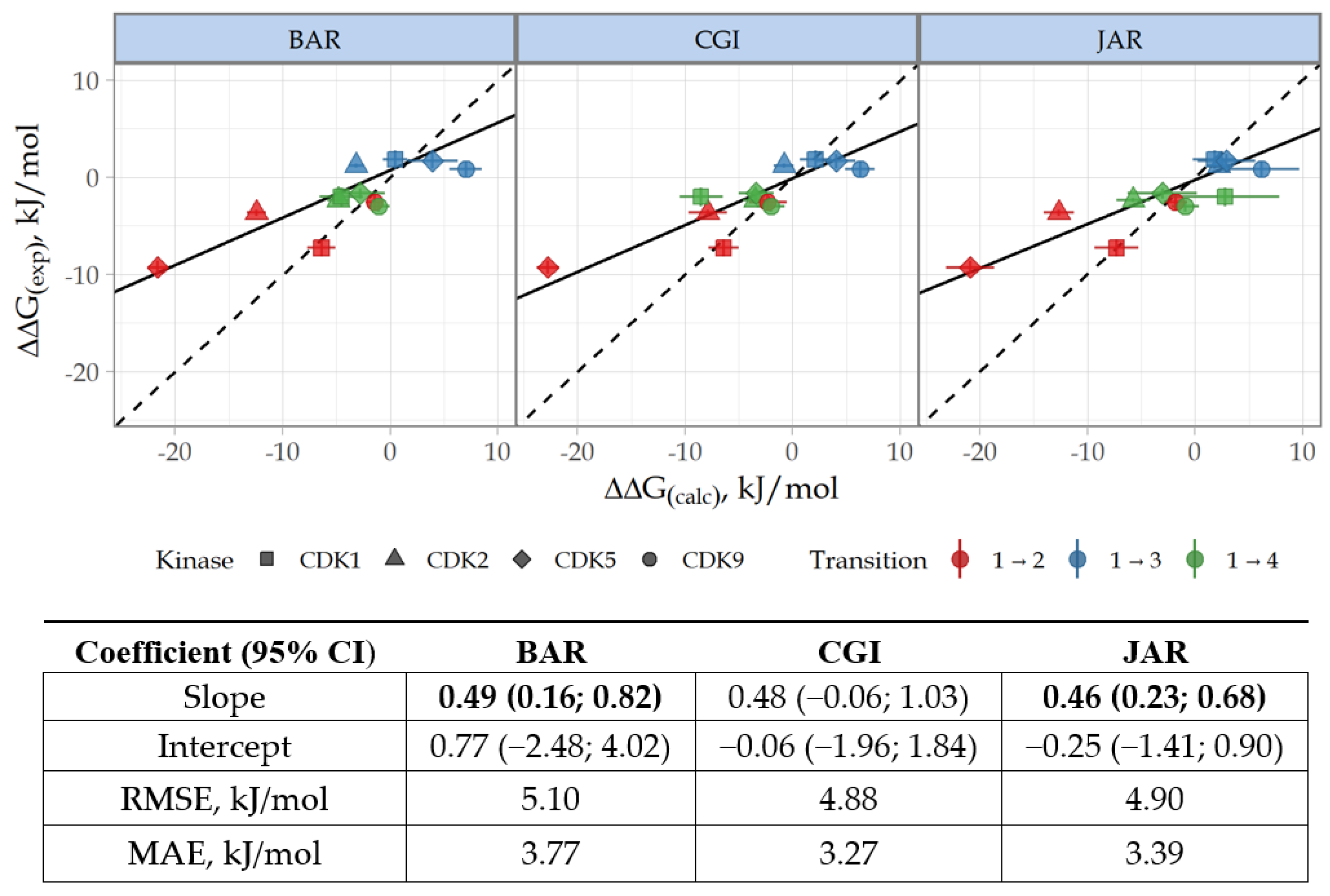
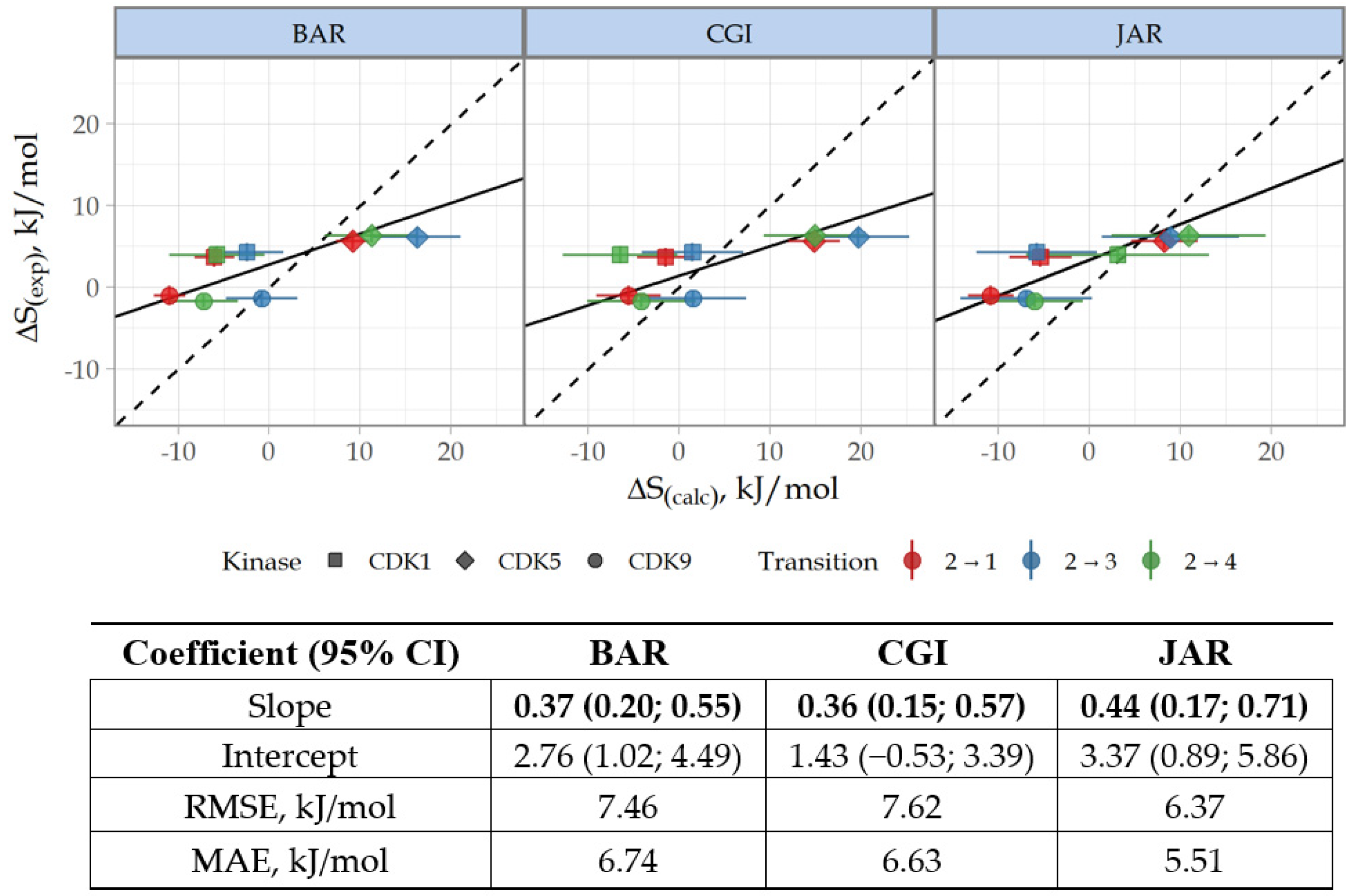
3.3. Prediction of CDK Inhibitory Potency and Selectivity of Imidazole-4-N-acetamide Derivatives
3.4. Cytotoxicity of Novel Imidazole-4-N-acetamide Derivatives
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Malumbres, M.; Barbacid, M. Cell cycle, CDKs and cancer: A changing paradigm. Nat. Rev. Cancer 2009, 9, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Greenwood, J.; Jones, A.W.; Nurse, P. Core control principles of the eukaryotic cell cycle. Nature 2022, 607, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Echalier, A.; Hole, A.J.; Lolli, G.; Endicott, J.A.; Noble, M.E.M. An inhibitor’s-eye view of the ATP-binding site of CDKs in different regulatory states. ACS Chem. Biol. 2014, 9, 1251–1256. [Google Scholar] [CrossRef] [PubMed]
- Whittaker, S.R.; Mallinger, A.; Workman, P.; Clarke, P.A. Inhibitors of cyclin-dependent kinases as cancer therapeutics. Pharmacol. Ther. 2017, 173, 83–105. [Google Scholar] [CrossRef]
- Rusina, P.V.; Lisov, A.A.; Denisova, A.A.; Gandalipov, E.R.; Novikov, F.N.; Shtil, A.A. Clinical CDK2 Inhibitors: Trends To Selectivity and Efficacy. Recent Pat. Anticancer Drug Discov. 2022, 18, 102–107. [Google Scholar] [CrossRef]
- Koirala, N.; Dey, N.; Aske, J.; De, P. Targeting Cell Cycle Progression in HER2+ Breast Cancer: An Emerging Treatment Opportunity. Int. J. Mol. Sci. 2022, 23, 6547. [Google Scholar] [CrossRef]
- Hassan, M.A.-K.; Ates-Alagoz, Z. Cyclin-Dependent Kinase 4/6 Inhibitors Against Breast Cancer. Mini Rev. Med. Chem. 2022, 166, 41–54. [Google Scholar] [CrossRef]
- Brasca, M.G.; Albanese, C.; Alzani, R.; Amici, R.; Avanzi, N.; Ballinari, D.; Bischoff, J.; Borghi, D.; Casale, E.; Croci, V.; et al. Optimization of 6,6-dimethyl pyrrolo[3,4-c]pyrazoles: Identification of PHA-793887, a potent CDK inhibitor suitable for intravenous dosing. Bioorg. Med. Chem. 2010, 18, 1844–1853. [Google Scholar] [CrossRef]
- Massard, C.; Soria, J.-C.; Anthoney, D.A.; Proctor, A.; Scaburri, A.; Pacciarini, M.A.; Laffranchi, B.; Pellizzoni, C.; Kroemer, G.; Armand, J.-P.; et al. A first in man, phase I dose-escalation study of PHA-793887, an inhibitor of multiple cyclin-dependent kinases (CDK2, 1 and 4) reveals unexpected hepatotoxicity in patients with solid tumors. Cell Cycle 2011, 10, 963–970. [Google Scholar] [CrossRef] [Green Version]
- Rusina, P.V.; Titov, I.Y.; Panova, M.V.; Stroylov, V.S.; Abdyusheva, Y.R.; Murlatova, E.Y.; Svitanko, I.V.; Novikov, F.N. Modeling of novel CDK7 inhibitors activity by molecular dynamics and free energy perturbation methods. Mendeleev Commun. 2020, 30, 430–432. [Google Scholar] [CrossRef]
- Albrecht, W. Highlight report: Hepatotoxicity of triazole fungicides. Arch. Toxicol. 2019, 93, 3037–3038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kharb, R.; Sharma, P.C.; Yar, M.S. Pharmacological significance of triazole scaffold. J. Enzym. Inhib. Med. Chem. 2011, 26, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Gong, P.; Cederbaum, A.I. Pyrazole induced oxidative liver injury independent of CYP2E1/2A5 induction due to Nrf2 deficiency. Toxicology 2008, 252, 9–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Wu, D.; Yang, L.; Cederbaum, A.I. Hepatotoxicity mediated by pyrazole (cytochrome P450 2E1) plus tumor necrosis factor alpha treatment occurs in c-Jun N-terminal kinase 2−/− but not in c-Jun N-terminal kinase 1−/− mice. Hepatology 2011, 54, 1753–1766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, P.; Jia, X.; Wang, M.; Mei, Y. Comparison of Accuracy and Convergence Rate between Equilibrium and Nonequilibrium Alchemical Transformations for Calculation of Relative Binding Free Energy. Available online: http://cjcp.ustc.edu.cn/html/hxwlxb_cn/2017/6/cjcp1711204.htm (accessed on 3 May 2022).
- Breznik, M.; Ge, Y.; Bluck, J.P.; Briem, H.; Hahn, D.F.; Christ, C.D.; Mortier, J.; Mobley, D.L.; Meier, K. Prioritizing small sets of molecules for synthesis through in-silico tools: A comparison of common ranking methods. ChemMedChem 2023, 18, e202200425. [Google Scholar] [CrossRef]
- Procacci, P. Methodological uncertainties in drug-receptor binding free energy predictions based on classical molecular dynamics. Curr. Opin. Struct. Biol. 2021, 67, 127–134. [Google Scholar] [CrossRef]
- Gapsys, V.; Pérez-Benito, L.; Aldeghi, M.; Seeliger, D.; van Vlijmen, H.; Tresadern, G.; de Groot, B.L. Large scale relative protein ligand binding affinities using non-equilibrium alchemy. Chem. Sci. 2020, 11, 1140–1152. [Google Scholar] [CrossRef] [Green Version]
- Gapsys, V.; Yildirim, A.; Aldeghi, M.; Khalak, Y.; van der Spoel, D.; de Groot, B.L. Accurate absolute free energies for ligand–protein binding based on non-equilibrium approaches. Commun. Chem. 2021, 4, 61. [Google Scholar] [CrossRef]
- Albanese, S.K.; Chodera, J.D.; Volkamer, A.; Keng, S.; Abel, R.; Wang, L. Is Structure-Based Drug Design Ready for Selectivity Optimization? J. Chem. Inf. Model. 2020, 60, 6211–6227. [Google Scholar] [CrossRef]
- Chacón Simon, S.; Wang, F.; Thomas, L.R.; Phan, J.; Zhao, B.; Olejniczak, E.T.; Macdonald, J.D.; Shaw, J.G.; Schlund, C.; Payne, W.; et al. Discovery of WD Repeat-Containing Protein 5 (WDR5)–MYC Inhibitors Using Fragment-Based Methods and Structure-Based Design. J. Med. Chem. 2020, 63, 4315–4333. [Google Scholar] [CrossRef]
- Pevarello, P.; Brasca, M.G.; Orsini, P.; Traquandi, G.; Longo, A.; Nesi, M.; Orzi, F.; Piutti, C.; Sansonna, P.; Varasi, M.; et al. 3-Aminopyrazole Inhibitors of CDK2/Cyclin A as Antitumor Agents. 2. Lead Optimization. J. Med. Chem. 2005, 48, 2944–2956. [Google Scholar] [CrossRef]
- Gapsys, V.; Michielssens, S.; Seeliger, D.; de Groot, B.L. pmx: Automated protein structure and topology generation for alchemical perturbations. J. Comput. Chem. 2015, 36, 348–354. [Google Scholar] [CrossRef] [Green Version]
- PyMOL | pymol.org. Available online: https://pymol.org/2/ (accessed on 1 August 2022).
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Stroganov, O.V.; Novikov, F.N.; Stroylov, V.S.; Kulkov, V.; Chilov, G.G. Lead finder: An approach to improve accuracy of protein-ligand docking, binding energy estimation, and virtual screening. J. Chem. Inf. Model. 2008, 48, 2371–2385. [Google Scholar] [CrossRef] [PubMed]
- Stroganov, O.V.; Novikov, F.N.; Zeifman, A.A.; Stroylov, V.S.; Chilov, G.G. TSAR, a new graph–theoretical approach to computational modeling of protein side-chain flexibility: Modeling of ionization properties of proteins. Proteins Struct. Funct. Bioinform. 2011, 79, 2693–2710. [Google Scholar] [CrossRef]
- Novikov, F.N.; Stroylov, V.S.; Stroganov, O.V.; Chilov, G.G. Improving performance of docking-based virtual screening by structural filtration. J. Mol. Model. 2010, 16, 1223–1230. [Google Scholar] [CrossRef]
- Losev, T.V.; Gerasimov, I.S.; Panova, M.V.; Lisov, A.A.; Abdyusheva, Y.R.; Rusina, P.V.; Zaletskaya, E.; Stroganov, O.V.; Medvedev, M.G.; Novikov, F.N. Quantum Mechanical-Cluster Approach to Solve the Bioisosteric Replacement Problem in Drug Design. J. Chem. Inf. Model. 2023, 63, 1239–1248. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef]
- Sousa da Silva, A.W.; Vranken, W.F. ACPYPE-AnteChamber PYthon Parser interfacE. BMC Res. Notes 2012, 5, 367. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Gapsys, V.; de Groot, B.L. pmx Webserver: A User Friendly Interface for Alchemistry. J. Chem. Inf. Model. 2017, 57, 109–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarzynski, C. Nonequilibrium Equality for Free Energy Differences. Phys. Rev. Lett. 1997, 78, 2690–2693. [Google Scholar] [CrossRef] [Green Version]
- Crooks, G.E. Nonequilibrium Measurements of Free Energy Differences for Microscopically Reversible Markovian Systems. J. Stat. Phys. 1998, 90, 1481–1487. [Google Scholar] [CrossRef]
- Bennett, C.H. Efficient estimation of free energy differences from Monte Carlo data. J. Comput. Phys. 1976, 22, 245–268. [Google Scholar] [CrossRef]
- Home-GraphPad. Available online: https://www.graphpad.com/ (accessed on 9 August 2022).
- Cheng, Y.; Prusoff, W.H. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar] [CrossRef]
- Kõivomägi, M.; Valk, E.; Venta, R.; Iofik, A.; Lepiku, M.; Morgan, D.O.; Loog, M. Dynamics of Cdk1 Substrate Specificity during the Cell Cycle. Mol. Cell 2011, 42, 610–623. [Google Scholar] [CrossRef]
- Sitcheran, R.; Gupta, P.; Fisher, P.B.; Baldwin, A.S. Positive and negative regulation of EAAT2 by NF-κB: A role for N-myc in TNFα-controlled repression. EMBO J. 2005, 24, 510–520. [Google Scholar] [CrossRef] [Green Version]
- Hashiguchi, M.; Saito, T.; Hisanaga, S.; Hashiguchi, T. Truncation of CDK5 Activator p35 Induces Intensive Phosphorylation of Ser202/Thr205 of Human Tau. J. Biol. Chem. 2002, 277, 44525–44530. [Google Scholar] [CrossRef] [Green Version]
- Baumli, S.; Hole, A.J.; Wang, L.-Z.; Noble, M.E.M.; Endicott, J.A. The CDK9 Tail Determines the Reaction Pathway of Positive Transcription Elongation Factor b. Structure 2012, 20, 1788–1795. [Google Scholar] [CrossRef] [Green Version]
- Package Deming-CRAN. Available online: https://CRAN.R-project.org/package=deming (accessed on 9 August 2022).
- R: The R Project for Statistical Computing. Available online: https://www.r-project.org/ (accessed on 19 February 2023).
- Martin, R.F. General Deming Regression for Estimating Systematic Bias and Its Confidence Interval in Method-Comparison Studies. Clin. Chem. 2000, 46, 100–104. [Google Scholar] [CrossRef]
- Zeifman, A.A.; Stroylov, V.V.; Novikov, F.N.; Stroganov, O.V.; Kulkov, V.; Chilov, G.G. Alchemical FEP Calculations of Ligand Conformer Focusing in Explicit Solvent. J. Chem. Theory Comput. 2013, 9, 1093–1102. [Google Scholar] [CrossRef]
- Łukasik, P.; Załuski, M.; Gutowska, I. Cyclin-Dependent Kinases (CDK) and Their Role in Diseases Development—Review. Int. J. Mol. Sci. 2021, 22, 2935. [Google Scholar] [CrossRef]
- Martin, M.P.; Endicott, J.A.; Noble, M.E.M. Structure-based discovery and development of cyclin-dependent protein kinase inhibitors. Essays Biochem 2017, 61, 439–452. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Choi, H.; Kim, K.-H.; Jeong, S.; Park, J.-W.; Baek, C.-S.; Lee, S.-H. Synthesis and biological evaluation of 3,5-diaminoindazoles as cyclin-dependent kinase inhibitors. Bioorganic Med. Chem. Lett. 2008, 18, 2292–2295. [Google Scholar] [CrossRef]
- Helal, C.J.; Kang, Z.; Lucas, J.C.; Gant, T.; Ahlijanian, M.K.; Schachter, J.B.; Richter, K.E.G.; Cook, J.M.; Menniti, F.S.; Kelly, K.; et al. Potent and cellularly active 4-aminoimidazole inhibitors of cyclin-dependent kinase 5/p25 for the treatment of Alzheimer’s disease. Bioorganic Med. Chem. Lett. 2009, 19, 5703–5707. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Revision A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Mennucci, B. Polarizable continuum model. WIREs Comput. Mol. Sci. 2012, 2, 386–404. [Google Scholar] [CrossRef]
- Luchini, G.; Alegre-Requena, J.V.; Guan, Y.; Funes-Ardoiz, I.; Paton, R.S. GoodVibes. 2019. Available online: https://anaconda.org/patonlab/goodvibes (accessed on 30 May 2023).
- Zhang, J.; Lu, T. Efficient evaluation of electrostatic potential with computerized optimized code. Phys. Chem. Chem. Phys. 2021, 23, 20323–20328. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Compound 1 | Compound 2 | Compound 3 | Compound 4 | PHA-793887 |
|---|---|---|---|---|---|
| CDK1/cyclin E | 14 (8–27) | 0.72 (0.54–0.96) | 30 (19–49) | 6.2 (4.0–9.9) | 0.060 (cyclin B) |
| CDK2/cyclin E | 0.71 (0.64–0.80) | 0.16 (0.13–0.19) | 1.2 (1.0–1.3) | 0.27 (0.24–0.31) | 0.008 |
| CDK5/p35 | 40 (27–59) | 0.88 (0.65–1.19) | 79 (49–126) | 20 (16–25) | 0.006 (p25) |
| CDK9/cyclin K | 3.0 (1.7–5.3) | 1.0 (0.7–1.5) | 4.3 (3.0–6.1) | 0.88 (0.83–0.94) | 0.138 (cyclin T1) |
| Cell Line | Compound 2 | Compound 4 |
|---|---|---|
| SKOV-3 | 4.4 ± 1.2 | 3.3 ± 0.2 |
| OVCAR-3 | 6.9 ± 0.5 | 5.8 ± 0.4 |
| OV-90 | 3.9 ± 0.6 | 2.4 ± 0.3 |
| UWB1.289 | 2.1 ± 0.2 | 1.7 ± 0.1 |
| IMR-32 | 4.2 ± 0.1 | 3.2 ± 0.1 |
| Kelly | 8.6 ± 2.1 | 9.2 ± 2.4 |
| SH-SY5Y | 55.9 ± 3.7 | 57.8 ± 8.4 |
| HELF | >100 | >100 |
| MSC | >100 | >100 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rusina, P.; Gandalipov, E.; Abdusheva, Y.; Panova, M.; Burdenkova, A.; Chaliy, V.; Brachs, M.; Stroganov, O.; Guzeeva, K.; Svitanko, I.; et al. Imidazole-4-N-acetamide Derivatives as a Novel Scaffold for Selective Targeting of Cyclin Dependent Kinases. Cancers 2023, 15, 3766. https://doi.org/10.3390/cancers15153766
Rusina P, Gandalipov E, Abdusheva Y, Panova M, Burdenkova A, Chaliy V, Brachs M, Stroganov O, Guzeeva K, Svitanko I, et al. Imidazole-4-N-acetamide Derivatives as a Novel Scaffold for Selective Targeting of Cyclin Dependent Kinases. Cancers. 2023; 15(15):3766. https://doi.org/10.3390/cancers15153766
Chicago/Turabian StyleRusina, Polina, Erik Gandalipov, Yana Abdusheva, Maria Panova, Alexandra Burdenkova, Vasiliy Chaliy, Maria Brachs, Oleg Stroganov, Ksenia Guzeeva, Igor Svitanko, and et al. 2023. "Imidazole-4-N-acetamide Derivatives as a Novel Scaffold for Selective Targeting of Cyclin Dependent Kinases" Cancers 15, no. 15: 3766. https://doi.org/10.3390/cancers15153766