

Vitamin D Receptor Antagonist MeTC7 Inhibits PD-L1
 , , , , , , , , and add
Show full author list
, , , , , , , , and add
Show full author list
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
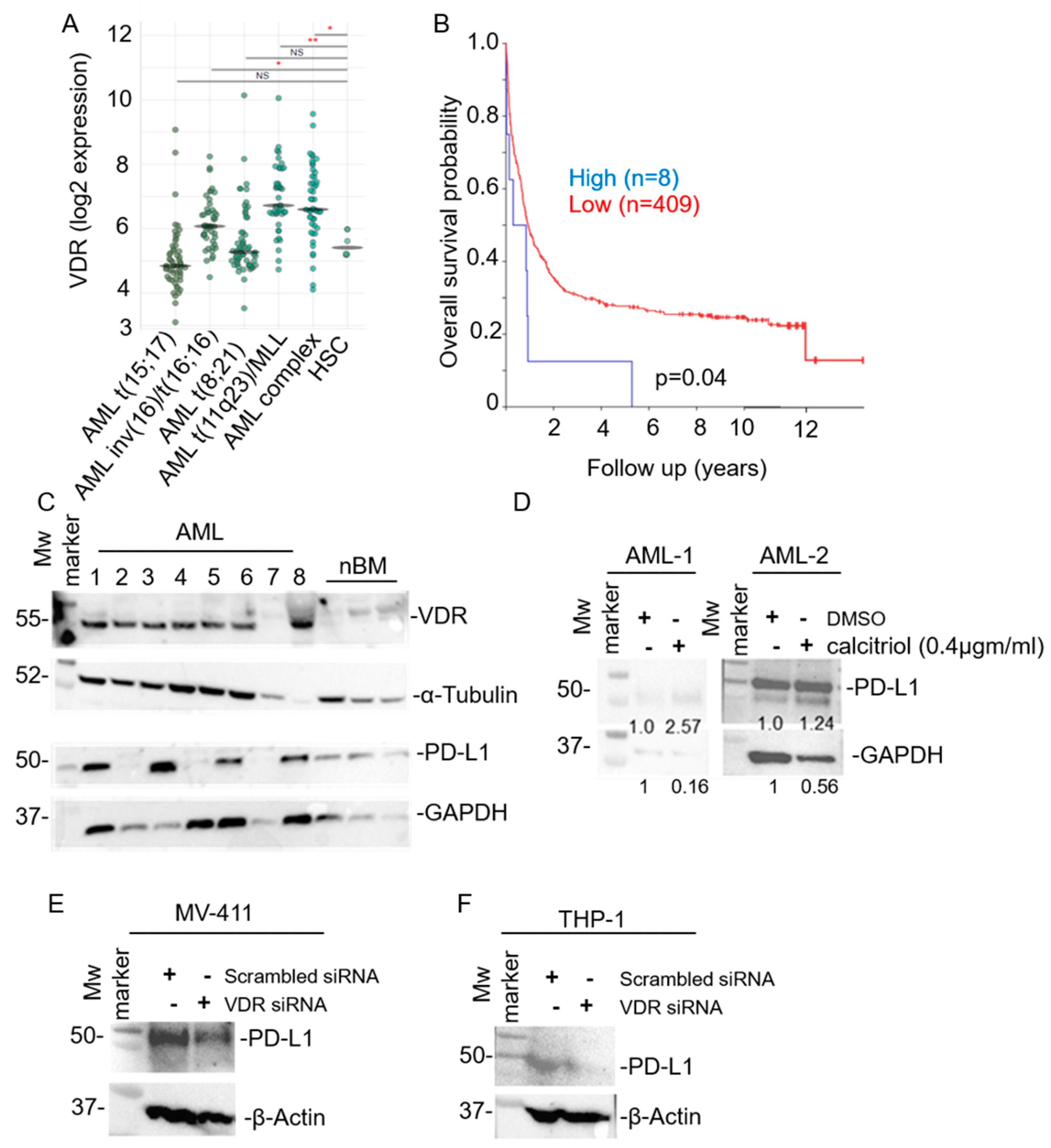
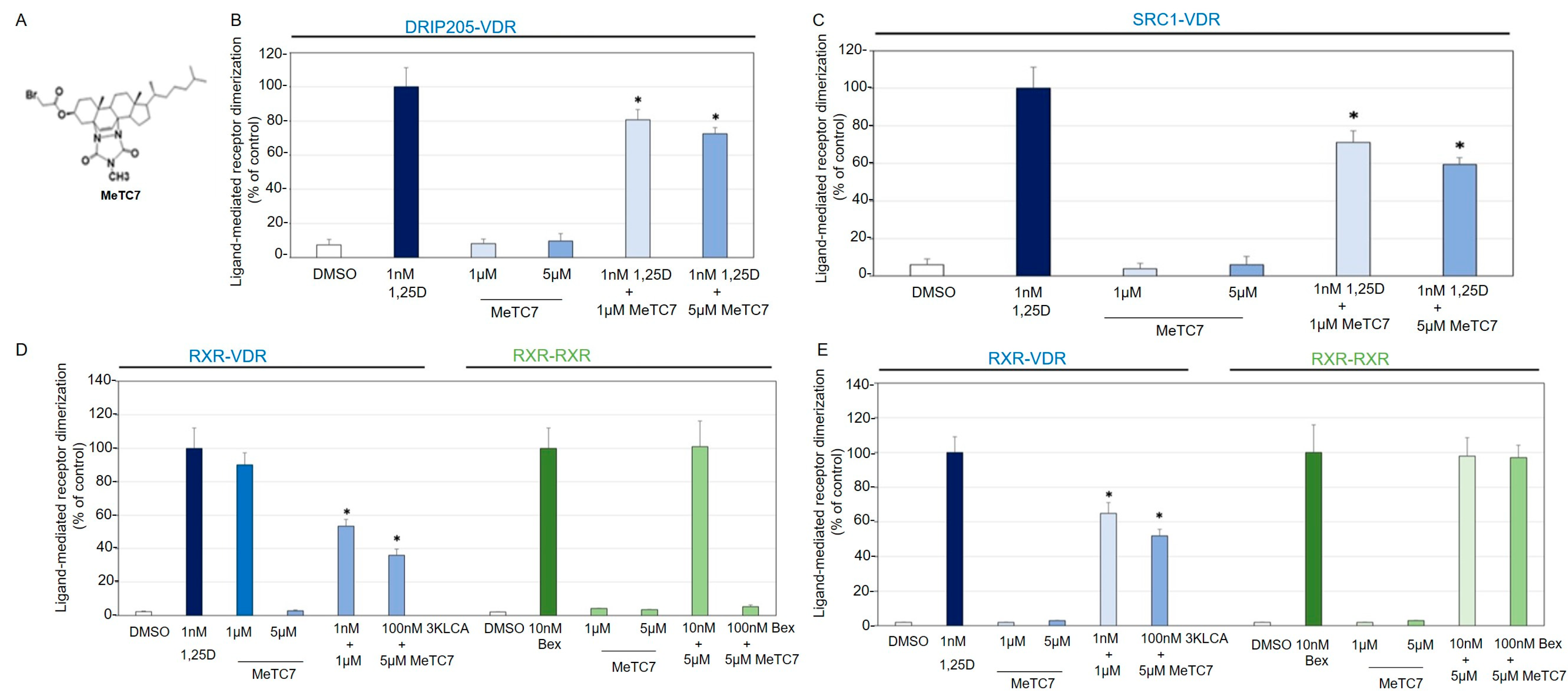
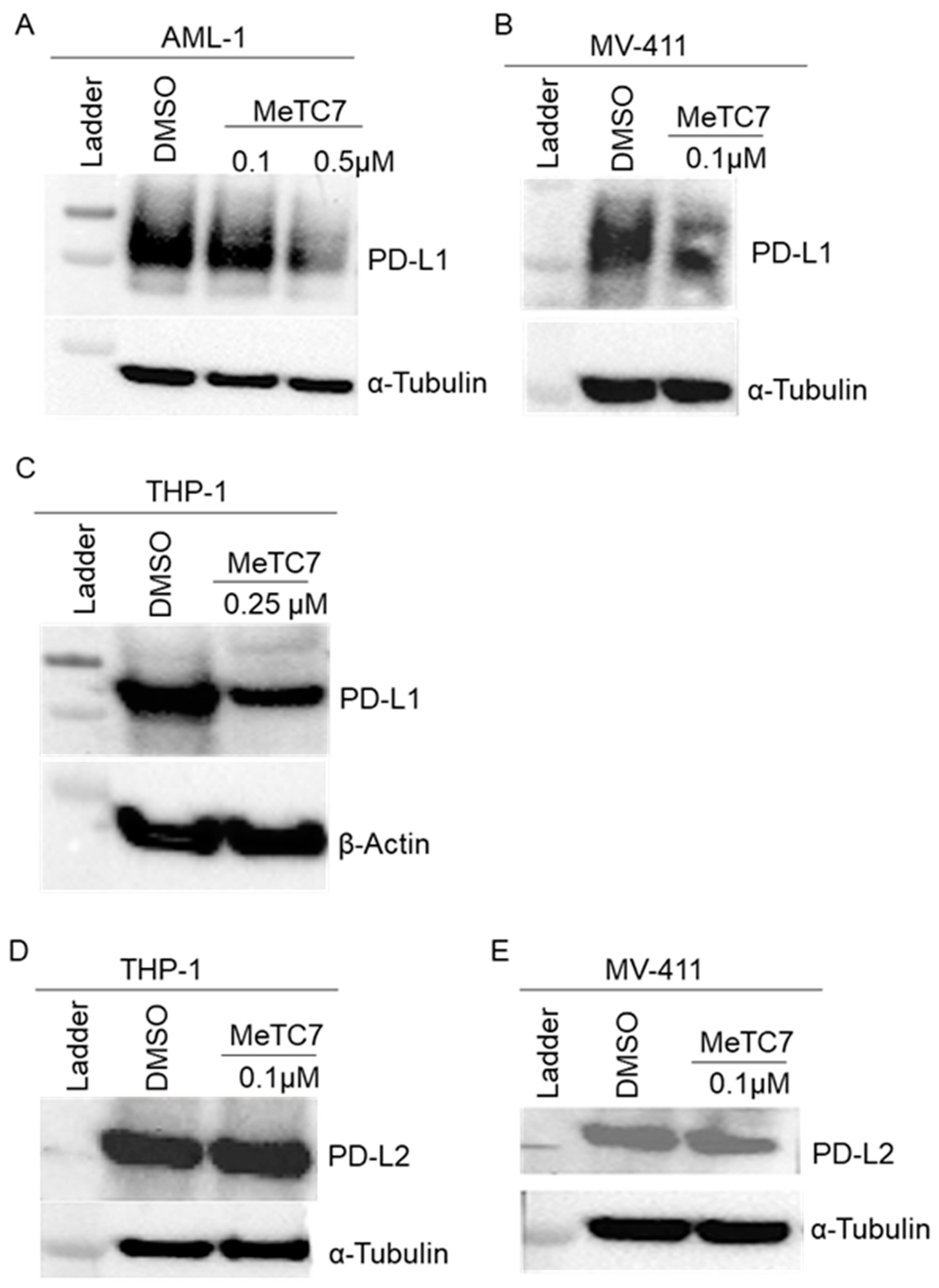
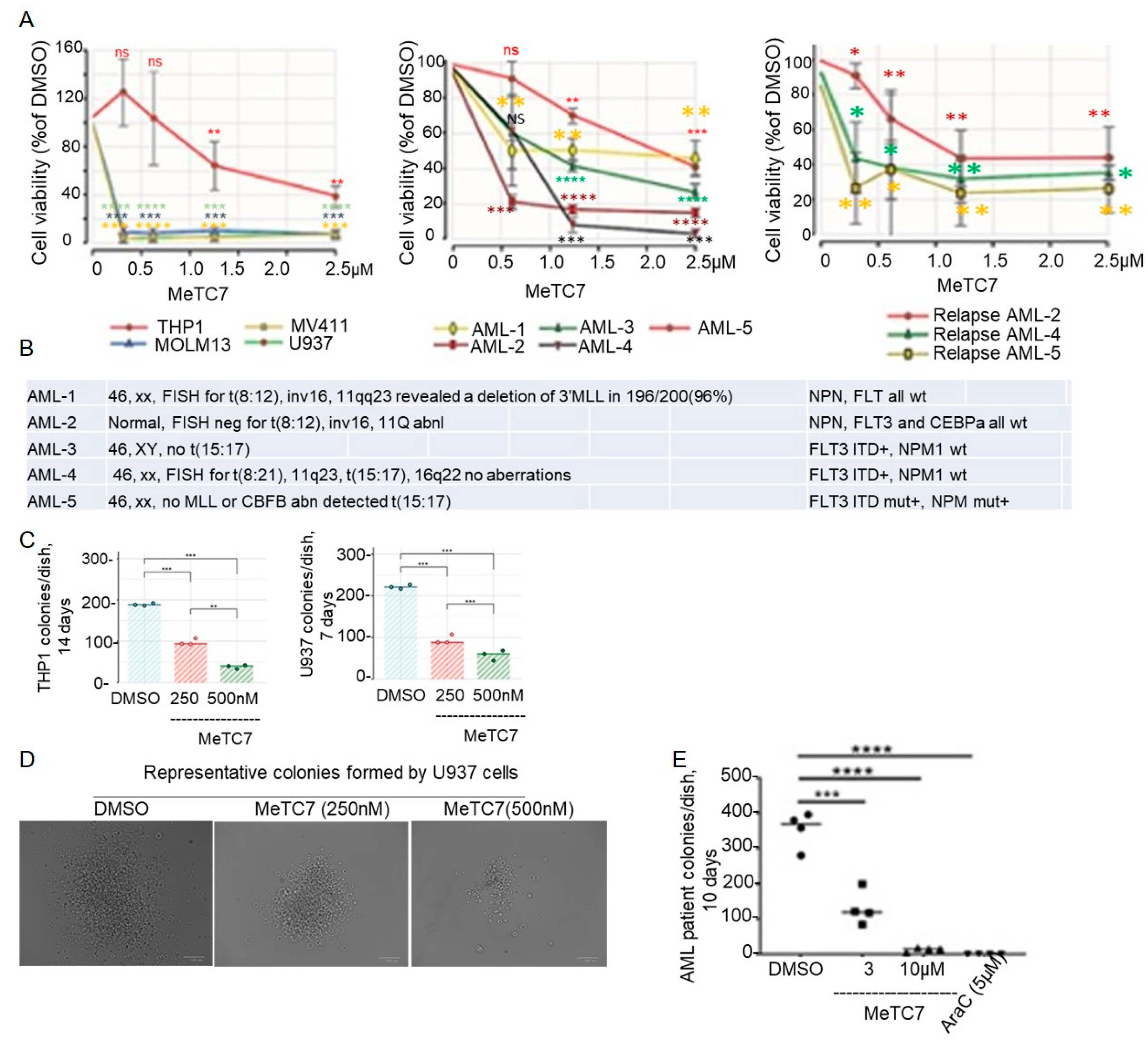
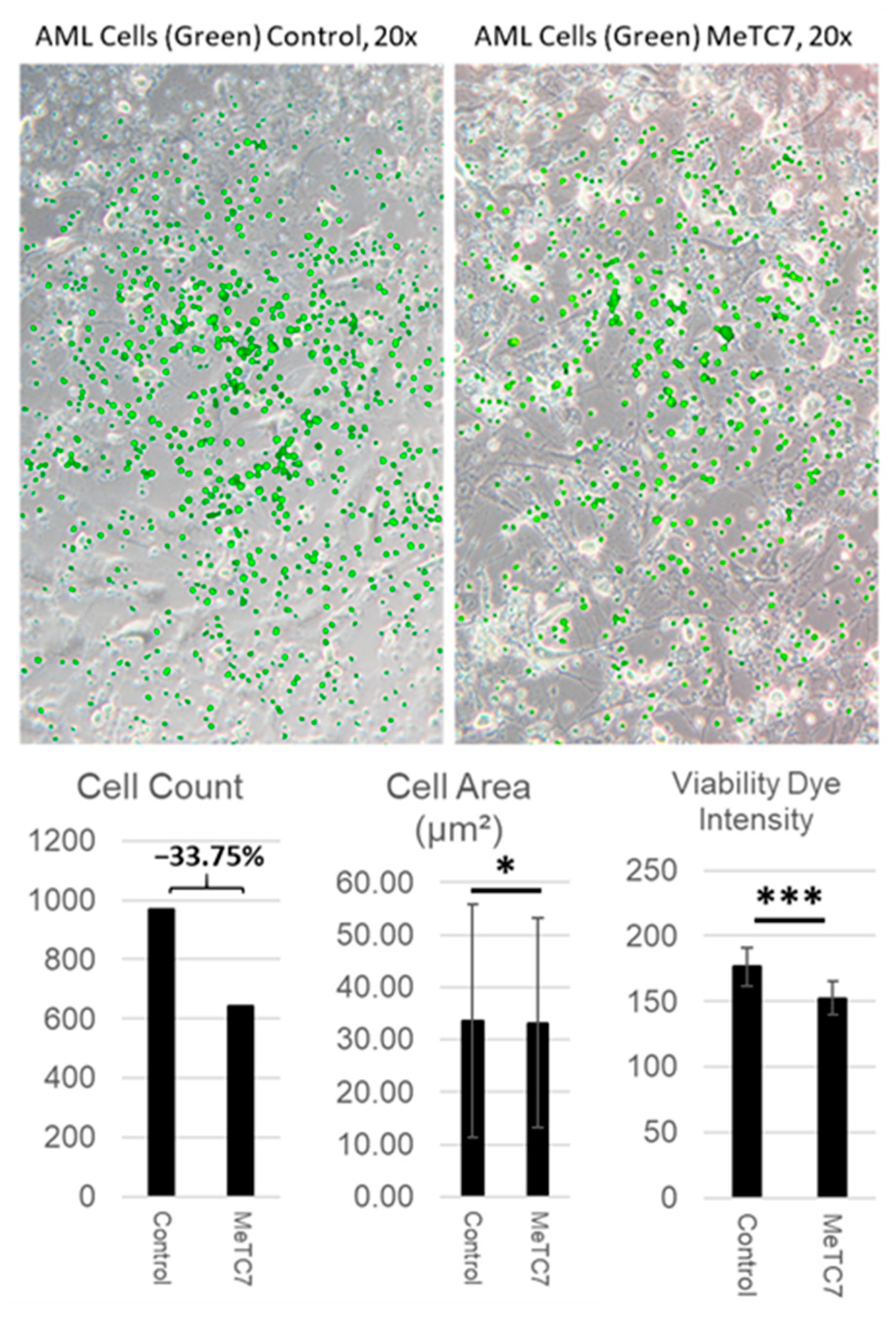
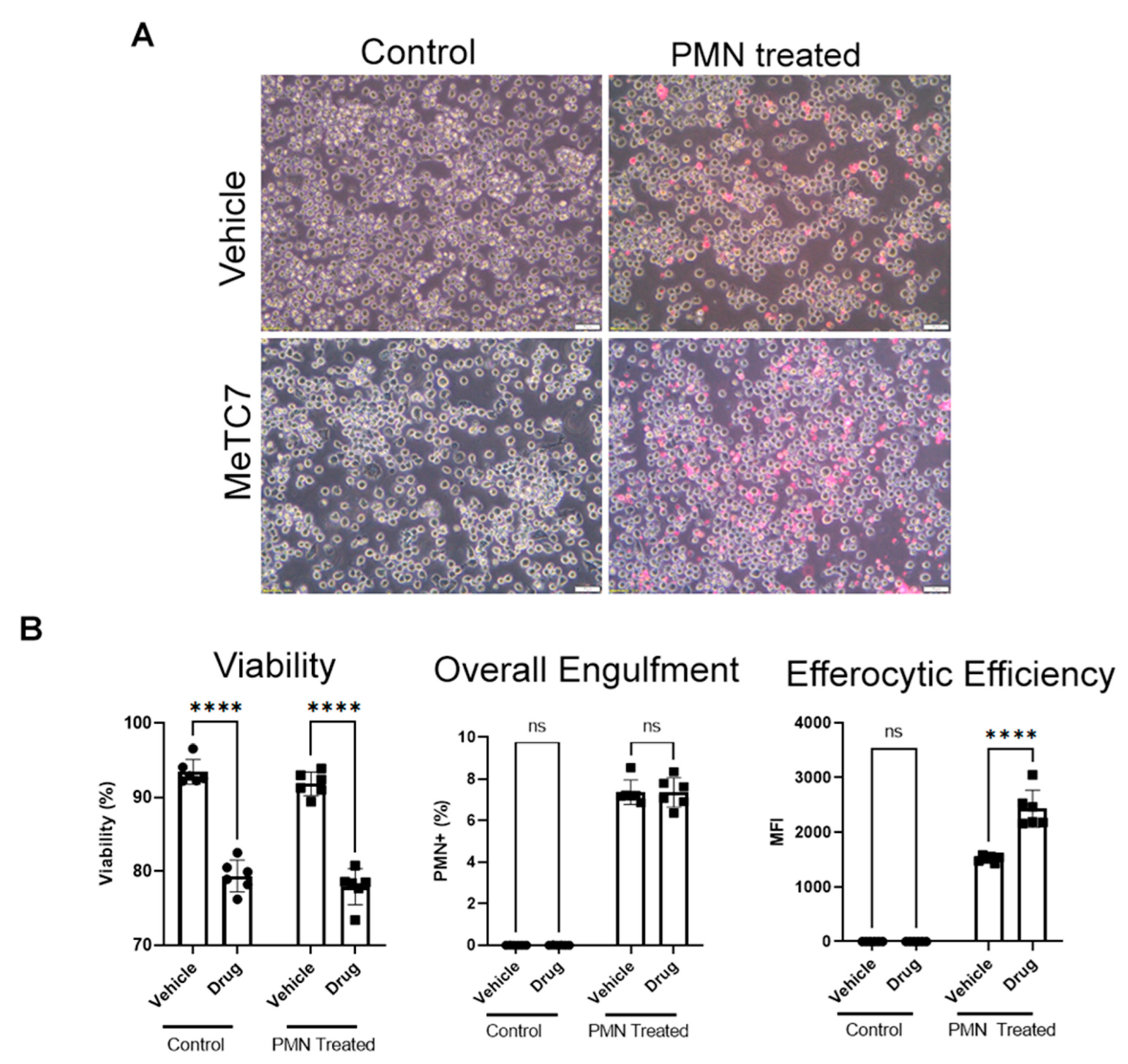
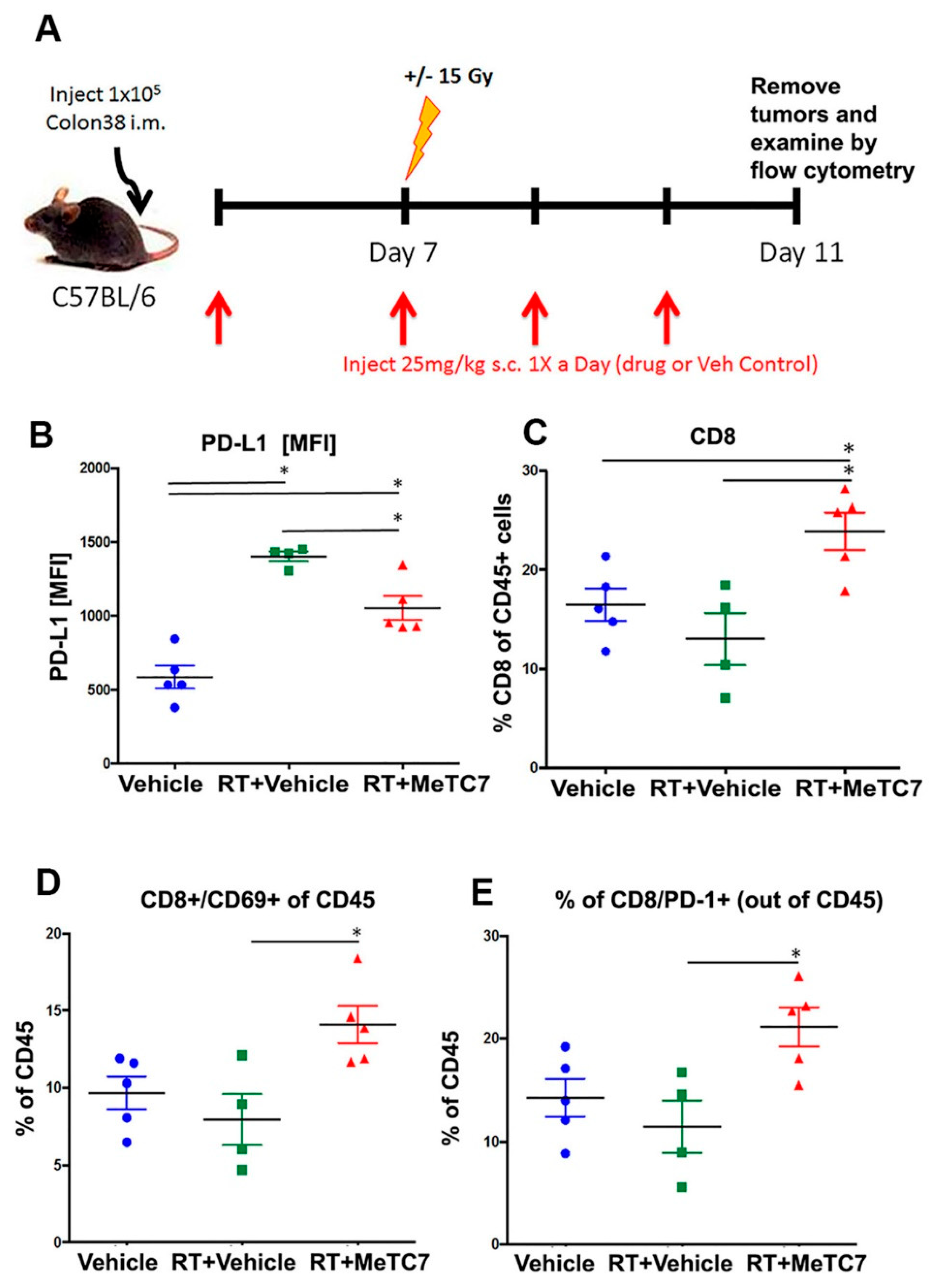
3. Results
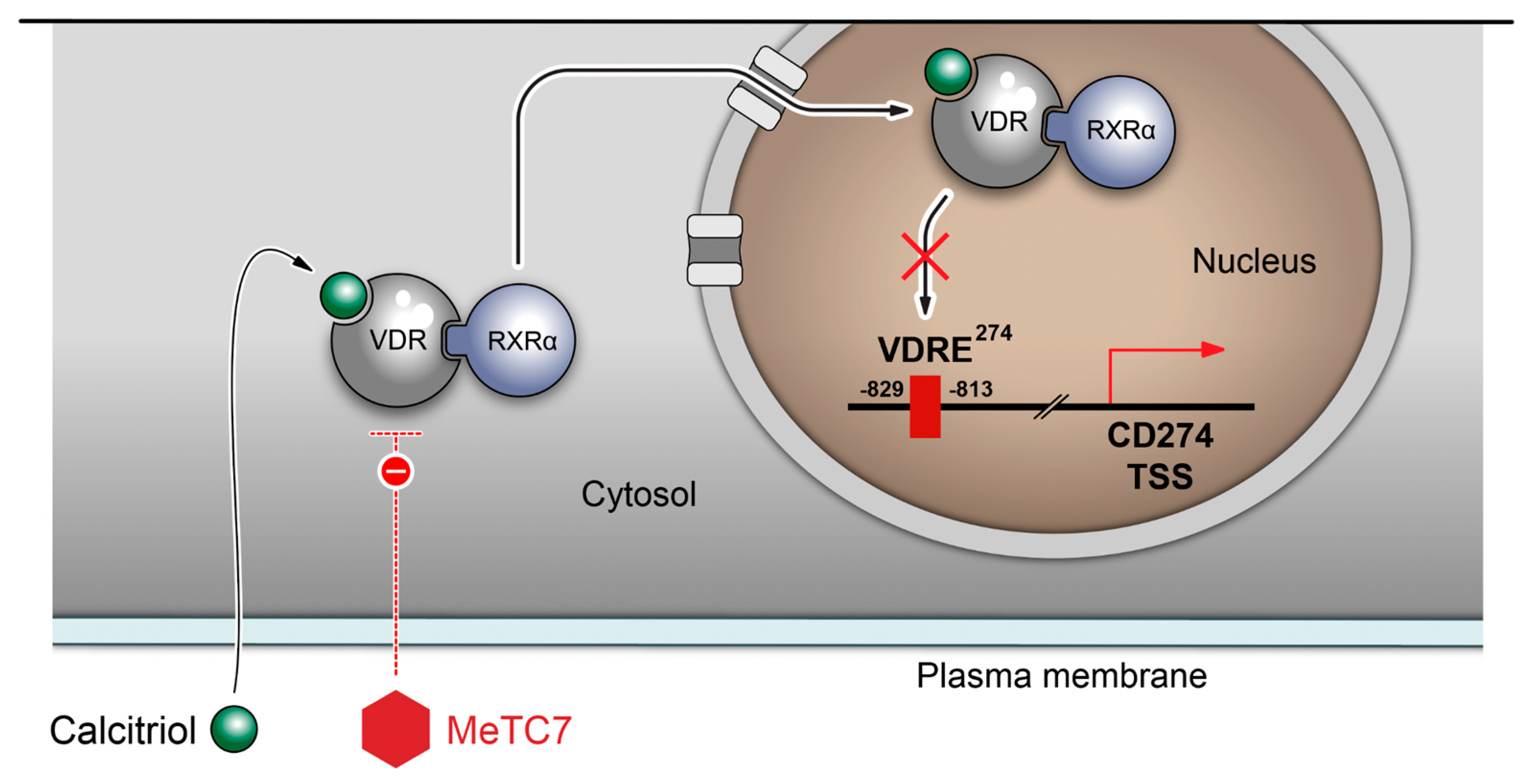
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dong, H.; Zhu, G.; Tamada, K.; Chen, L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat. Med. 1999, 5, 1365–1369. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Strome, S.E.; Salomao, D.R.; Tamura, H.; Hirano, F.; Flies, D.B.; Roche, P.C.; Lu, J.; Zhu, G.; Tamada, K.; et al. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat. Med. 2002, 8, 793–800. [Google Scholar] [CrossRef] [PubMed]
- Vandenborre, K.; Van Gool, S.W.; Kasran, A.; Ceuppens, J.L.; Boogaerts, M.A.; Vandenberghe, P. Interaction of CTLA-4 (CD152) with CD80 or CD86 inhibits human T-cell activation. Immunology 1999, 98, 413–421. [Google Scholar] [CrossRef] [PubMed]
- ElTanbouly, M.A.; Schaafsma, E.; Noelle, R.J.; Lines, J.L. VISTA: Coming of age as a multi-lineage immune checkpoint. Clin. Exp. Immunol. 2020, 200, 120–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamberti, G.; Sisi, M.; Andrini, E.; Palladini, A.; Giunchi, F.; Lollini, P.L.; Ardizzoni, A.; Gelsomino, F. The mechanisms of PD-L1 regulation in non-small-cell lung cancer (NSCLC): Which are the involved players? Cancers 2020, 12, 3129. [Google Scholar] [CrossRef]
- Zhang, W.; Pang, Q.; Yan, C.; Wang, Q.; Yang, J.; Yu, S.; Liu, X.; Yuan, Z.; Wang, P.; Xiao, Z. Induction of PD-L1 expression by epidermal growth factor receptor-mediated signaling in esophageal squamous cell carcinoma. OncoTargets Ther. 2017, 10, 763–771. [Google Scholar] [CrossRef] [Green Version]
- Dimitrov, V.; Bouttier, M.; Boukhaled, G.; Salehi-Tabar, R.; Avramescu, R.G.; Memari, B.; Hasaj, B.; Lukacs, G.L.; Krawczyk, C.M.; White, J.H. Hormonal vitamin D up-regulates tissue-specific PD-L1 and PD-L2 surface glycoprotein expression in humans but not mice. J. Biol. Chem. 2017, 292, 20657–20668. [Google Scholar] [CrossRef] [Green Version]
- Cao, D.; Qi, Z.; Pang, Y.; Li, H.; Xie, H.; Wu, J.; Huang, Y.; Zhu, Y.; Shen, Y.; Zhu, Y.; et al. Retinoic acid-related orphan receptor C regulates proliferation, glycolysis, and chemoresistance via the PD-L1/ITGB6/STAT3 signaling axis in bladder cancer. Cancer Res. 2019, 79, 2604–2618. [Google Scholar] [CrossRef] [Green Version]
- Antonangeli, F.; Natalini, A.; Garassino, M.C.; Sica, A.; Santoni, A.; Di Rosa, F. Regulation of PD-L1 Expression by NF-κB in Cancer. Front. Immunol. 2020, 11, 584626. [Google Scholar] [CrossRef]
- Garcia-Diaz, A.; Shin, D.S.; Moreno, B.H.; Saco, J.; Escuin-Ordinas, H.; Rodriguez, G.A.; Zaretsky, J.M.; Sun, L.; Hugo, W.; Wang, X.; et al. Interferon receptor signaling pathways regulating PD-L1 and PD-L2 expression. Cell Rep. 2017, 19, 1189–1201. [Google Scholar] [CrossRef] [Green Version]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Wang, J.; Deng, X.; Xiong, F.; Ge, J.; Xiang, B.; Wu, X.; Ma, J.; Zhou, M.; Li, X.; et al. Role of the tumor microenvironment in PD-L1/PD-1-mediated tumor immune escape. Mol. Cancer 2019, 18, 10. [Google Scholar] [CrossRef] [Green Version]
- Koustas, E.; Sarantis, P.; Papavassiliou, A.G.; Karamouzis, M.V. The resistance mechanisms of checkpoint inhibitors in solid tumors. Biomolecules 2020, 10, 666. [Google Scholar] [CrossRef] [PubMed]
- Escors, D.; Gato-Cañas, M.; Zuazo, M.; Arasanz, H.; García-Granda, M.J.; Vera, R.; Kochan, G. The intracellular signalosome of PD-L1 in cancer cells. Signal Transduct. Target. Ther. 2018, 3, 26. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Huang, A.C.; Zhang, W.; Zhang, G.; Wu, M.; Xu, W.; Yu, Z.; Yang, J.; Wang, B.; Sun, H.; et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature 2018, 560, 382–386. [Google Scholar] [CrossRef] [PubMed]
- Poggio, M.; Hu, T.; Pai, C.C.; Chu, B.; Belair, C.D.; Chang, A.; Montabana, E.; Lang, U.E.; Fu, Q.; Fong, L.; et al. Suppression of exosomal PD-L1 induces systemic anti-tumor immunity and memory. Cell 2019, 177, 414–427.e13. [Google Scholar] [CrossRef] [Green Version]
- Gong, B.; Kiyotani, K.; Sakata, S.; Nagano, S.; Kumehara, S.; Baba, S.; Besse, B.; Yanagitani, N.; Friboulet, L.; Nishio, M.; et al. Secreted PD-L1 variants mediate resistance to PD-L1 blockade therapy in non-small cell lung cancer. J. Exp. Med. 2019, 216, 982–1000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaguchi, H.; Hsu, J.M.; Yang, W.H.; Hung, M.C. Mechanisms regulating PD-L1 expression in cancers and associated opportunities for novel small-molecule therapeutics. Nat. Rev. Clin. Oncol. 2022, 19, 287–305. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Casals, M.; Brahmer, J.R.; Callahan, M.K.; Flores-Chávez, A.; Keegan, N.; Khamashta, M.A.; Lambotte, O.; Mariette, X.; Prat, A.; Suárez-Almazor, M.E. Immune-related adverse events of checkpoint inhibitors. Nat. Rev. Dis. Primers. 2020, 6, 38. [Google Scholar] [CrossRef] [PubMed]
- Koblish, H.K.; Wu, L.; Wang, L.S.; Liu, P.C.C.; Wynn, R.; Rios-Doria, J.; Spitz, S.; Liu, H.; Volgina, A.; Zolotarjova, N.; et al. Characterization of INCB086550: A potent and novel small-molecule PD-L1 inhibitor. Cancer. Discov. 2022, 12, 1482–1499. [Google Scholar] [CrossRef]
- Skalniak, L.; Zak, K.M.; Guzik, K.; Magiera, K.; Musielak, B.; Pachota, M.; Szelazek, B.; Kocik, J.; Grudnik, P.; Tomala, M.; et al. Small-molecule inhibitors of PD-1/PD-L1 immune checkpoint alleviate the PD-L1-induced exhaustion of T-cells. Oncotarget 2017, 8, 72167–72181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzik, K.; Zak, K.M.; Grudnik, P.; Magiera, K.; Musielak, B.; Törner, R.; Skalniak, L.; Dömling, A.; Dubin, G.; Holak, T.A. Small-molecule inhibitors of the programmed cell death-1/programmed death-ligand 1 (PD-1/PD-L1) Interaction via transiently induced protein states and dimerization of PD-L1. J. Med. Chem. 2017, 60, 5857–5867. [Google Scholar] [CrossRef] [PubMed]
- Musielak, B.; Kocik, J.; Skalniak, L.; Magiera-Mularz, K.; Sala, D.; Czub, M.; Stec, M.; Siedlar, M.; Holak, T.A.; Plewka, J. CA-170—A potent small-molecule PD-L1 inhibitor or not? Molecules 2019, 24, 2804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khazan, N.; Kim, K.K.; Hansen, J.N.; Singh, N.A.; Moore, T.; Snyder, C.W.A.; Pandita, R.; Strawderman, M.; Fujihara, M.; Takamura, Y.; et al. Identification of a Vitamin-D Receptor antagonist, MeTC7, which inhibits the growth of xenograft and transgenic tumors in vivo. J. Med. Chem. 2022, 65, 6039–6055. [Google Scholar] [CrossRef]
- Azadniv, M.; Myers, J.R.; McMurray, H.R.; Guo, N.; Rock, P.; Coppage, M.L.; Ashton, J.; Becker, M.W.; Calvi, L.M.; Liesveld, J.L. Bone marrow mesenchymal stromal cells from acute myelogenous leukemia patients demonstrate adipogenic differentiation propensity with implications for leukemia cell support. Leukemia 2020, 34, 391–403. [Google Scholar] [CrossRef]
- Connolly, K.A.; Belt, B.A.; Figueroa, N.M.; Murthy, A.; Patel, A.; Kim, M.; Lord, E.M.; Linehan, D.C.; Gerber, S.A. Increasing the efficacy of radiotherapy by modulating the CCR2/CCR5 chemokine axes. Oncotarget 2016, 7, 86522–86535. [Google Scholar] [CrossRef] [Green Version]
- Benekli, M.; Xia, Z.; Donohue, K.A.; Ford, L.A.; Pixley, L.A.; Baer, M.R.; Baumann, H.; Wetzler, M. Constitutive activity of signal transducer and activator of transcription 3 protein in acute myeloid leukemia blasts is associated with short disease-free survival. Blood 2002, 99, 252–257. [Google Scholar] [CrossRef] [Green Version]
- Brenner, A.K.; Nepstad, I.; Bruserud, Ø. Mesenchymal stem cells support survival and proliferation of primary human acute myeloid leukemia cells through heterogeneous molecular mechanisms. Front. Immunol. 2017, 8, 106. [Google Scholar] [CrossRef] [Green Version]
- Arandjelovic, S.; Ravichandran, K.S. Phagocytosis of apoptotic cells in homeostasis. Nat. Immunol. 2015, 16, 907–917. [Google Scholar] [CrossRef] [Green Version]
- Kawano, M.; Nagata, S. Efferocytosis and autoimmune disease. Int. Immunol. 2018, 30, 551–558. [Google Scholar] [CrossRef] [Green Version]
- Gordon, S.R.; Maute, R.L.; Dulken, B.W.; Hutter, G.; George, B.M.; McCracken, M.N.; Gupta, R.; Tsai, J.M.; Sinha, R.; Corey, D.; et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature 2017, 545, 495–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, S.A.; Rahimzadeh, M.; Brierley, C.; Gration, B.; Doree, C.; Kimber, C.E.; Cajide, A.P.; Lamikanra, A.A.; Roberts, D.J. The role of vitamin D in increasing circulating T regulatory cell numbers and modulating T regulatory cell phenotypes in patients with inflammatory disease or in healthy volunteers: A systematic review. PLoS ONE. 2019, 14, e0222313. [Google Scholar] [CrossRef] [Green Version]
- Cantorna, M.T. Why do T cells express the vitamin D receptor? Ann. N. Y. Acad. Sci. 2011, 1217, 77–82. [Google Scholar] [CrossRef] [Green Version]
- Cantorna, M.T.; Snyder, L.; Lin, Y.D.; Yang, L. Vitamin D and 1,25(OH)2D regulation of T cells. Nutrients 2015, 7, 3011–3021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von Essen, M.R.; Kongsbak, M.; Schjerling, P.; Olgaard, K.; Odum, N.; Geisler, C. Vitamin D controls T cell antigen receptor signaling and activation of human T cells. Nat. Immunol. 2010, 11, 344–349. [Google Scholar] [CrossRef] [PubMed]
- Brodská, B.; Otevřelová, P.; Šálek, C.; Fuchs, O.; Gašová, Z.; Kuželová, K. High PD-L1 expression predicts for worse outcome of leukemia patients with concomitant NPM1 and FLT3 Mutations. Int. J. Mol. Sci. 2019, 20, 2823. [Google Scholar] [CrossRef] [Green Version]
- Dong, Y.; Han, Y.; Huang, Y.; Jiang, S.; Huang, Z.; Chen, R.; Yu, Z.; Yu, K.; Zhang, S. PD-L1 Is expressed and promotes the expansion of regulatory T cells in acute myeloid leukemia. Front. Immunol. 2020, 11, 1710. [Google Scholar] [CrossRef]
- Toffalori, C.; Zito, L.; Gambacorta, V.; Riba, M.; Oliveira, G.; Bucci, G.; Barcella, M.; Spinelli, O.; Greco, R.; Crucitti, L.; et al. Immune signature drives leukemia escape and relapse after hematopoietic cell transplantation. Nat. Med. 2019, 25, 603–611. [Google Scholar] [CrossRef]
- Daver, N.; Garcia-Manero, G.; Basu, S.; Boddu, P.C.; Alfayez, M.; Cortes, J.E.; Konopleva, M.; Ravandi-Kashani, F.; Jabbour, E.; Kadia, T.; et al. Efficacy, safety, and biomarkers of response to azacitidine and nivolumab in relapsed/refractory acute myeloid leukemia: A nonrandomized, open-label, phase II study. Cancer Discov. 2019, 9, 370–383. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, A.; Barba, P.; Perales, M.A. Checkpoint inhibitors in AML: Are we there yet? Br. J. Haematol. 2020, 188, 159–167. [Google Scholar] [CrossRef]
- Waclawiczek, A.; Hamilton, A.; Rouault-Pierre, K.; Abarrategi, A.; Albornoz, M.G.; Miraki-Moud, F.; Bah, N.; Gribben, J.; Fitzgibbon, J.; Taussig, D.; et al. Mesenchymal niche remodeling impairs hematopoiesis via stanniocalcin 1 in acute myeloid leukemia. J. Clin. Investig. 2020, 130, 3038–3050. [Google Scholar] [CrossRef] [PubMed]
- von der Heide, E.K.; Neumann, M.; Vosberg, S.; James, A.R.; Schroeder, M.P.; Ortiz-Tanchez, J.; Isaakidis, K.; Schlee, C.; Luther, M.; Jöhrens, K.; et al. Molecular alterations in bone marrow mesenchymal stromal cells derived from acute myeloid leukemia patients. Leukemia 2016, 31, 1069–1078. [Google Scholar] [CrossRef]
- Querfeld, U. Vitamin D and inflammation. Pediatr. Nephrol. 2013, 28, 605–610. [Google Scholar] [CrossRef] [PubMed]
- Doran, A.C.; Yurdagul, A.; Tabas, I. Efferocytosis in health and disease. Nat. Rev. Immunol. 2020, 20, 254–267. [Google Scholar] [CrossRef]
- Mehrotra, P.; Ravichandran, K.S. Drugging the efferocytosis process: Concepts and opportunities. Nat. Rev. Drug Discov. 2022, 21, 601–620. [Google Scholar] [CrossRef] [PubMed]
- Greenlee-Wacker, M.C. Clearance of apoptotic neutrophils and resolution of inflammation. Immunol. Rev. 2016, 273, 357–370. [Google Scholar] [CrossRef] [Green Version]
- Padmanabhan, S.; Zou, Y.; Vancurova, I. Immunoblotting analysis of intracellular PD-L1 levels in interferon-γ-treated ovarian cancer cells stably transfected with Bcl3 shRNA. Methods Mol. Biol. 2020, 2108, 211–220. [Google Scholar] [CrossRef]
- Ghebeh, H.; Lehe, C.; Barhoush, E.; Al-Romaih, K.; Tulbah, A.; Al-Alwan, M.; Hendrayani, S.F.; Manogaran, P.; Alaiya, A.; Al-Tweigeri, T.; et al. Doxorubicin downregulates cell surface B7-H1 expression and upregulates its nuclear expression in breast cancer cells: Role of B7-H1 as an anti-apoptotic molecule. Breast Cancer Res. 2010, 12, R48. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Chen, L.; Feng, Y.; Shen, J.; Gao, Y.; Cote, G.; Choy, E.; Harmon, D.; Mankin, H.; Hornicek, F.; et al. Targeting programmed cell death ligand 1 by CRISPR/Cas9 in osteosarcoma cells. Oncotarget 2017, 8, 30276–30287. [Google Scholar] [CrossRef] [Green Version]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khazan, N.; Quarato, E.R.; Singh, N.A.; Snyder, C.W.A.; Moore, T.; Miller, J.P.; Yasui, M.; Teramoto, Y.; Goto, T.; Reshi, S.; et al. Vitamin D Receptor Antagonist MeTC7 Inhibits PD-L1. Cancers 2023, 15, 3432. https://doi.org/10.3390/cancers15133432
Khazan N, Quarato ER, Singh NA, Snyder CWA, Moore T, Miller JP, Yasui M, Teramoto Y, Goto T, Reshi S, et al. Vitamin D Receptor Antagonist MeTC7 Inhibits PD-L1. Cancers. 2023; 15(13):3432. https://doi.org/10.3390/cancers15133432
Chicago/Turabian StyleKhazan, Negar, Emily R. Quarato, Niloy A. Singh, Cameron W. A. Snyder, Taylor Moore, John P. Miller, Masato Yasui, Yuki Teramoto, Takuro Goto, Sabeeha Reshi, and et al. 2023. "Vitamin D Receptor Antagonist MeTC7 Inhibits PD-L1" Cancers 15, no. 13: 3432. https://doi.org/10.3390/cancers15133432