

Sialic Acids on Tumor Cells Modulate IgA Therapy by Neutrophils via Inhibitory Receptors Siglec-7 and Siglec-9
 ,
,  , , ,
, , ,
Abstract
:Simple Summary
Abstract
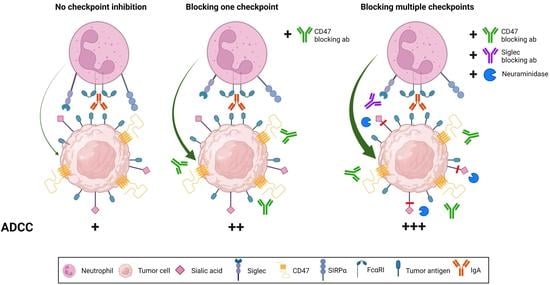
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Material and Methods
2.1. Cell Culture and Cell Lines
2.2. 51Cr Release ADCC Assay
2.3. Antibodies and Reagents
2.4. Flow Cytometry
2.5. CRISPR/Cas9 of GNE
2.6. Short i.p. In Vivo Model
2.7. Data Processing and Statistical Analyses
3. Results
3.1. Hypersialylation of Tumor Cells Inhibit Killing by Neutrophils
3.2. Knockout GNE Impairs Sialic Acid Biosynthesis
3.3. Siglec-7 and Siglec-9 as Key Receptors on Neutrophils Interacting with Hypersialylated Tumors
3.4. Enhancing IgA-Mediated ADCC in Resistant Tumor Cells through a Combination of Siglec-9 and CD47 Blockade
3.5. Desialylation Improved IgA Therapy In Vivo
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ADCC | Antibody-dependent cellular cytotoxicity |
| ADCP | Antibody-dependent cellular phagocytosis |
| CD47 | Cluster of differentiation 47 |
| CTLA-4 | Cytotoxic T lymphocyte-associated protein 4 |
| E:T | Effector-to-target cell ratio |
| FCS | Fetal calf serum |
| GNE | UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine |
| ITAM | Immunoreceptor tyrosine-based activation motif |
| ITIM | immunoreceptor tyrosine-based inhibitory motif |
| MAL II | Maackia amurensis Lectin II |
| NEU | Neuraminidase |
| Neu5Ac | N-Acetylneuraminic acid |
| PBMCs | Peripheral blood mononuclear cells |
| PD-1 | Programmed cell death protein 1 |
| PDAC | Pancreatic ductal adenocarcinoma |
| PD-L1 | Programmed death ligand 1 |
| PMNs | Polymorphonuclear leukocytes |
| RBC | Red blood cells |
| Sialoglycans | Sialic acid-containing carbohydrates |
| STinh | Sialyltransferase inhibitor |
| Siglec | Sialic acid-binding immunoglobulin-like lectins |
| SIRPα | Signal regulatory protein alpha |
| TAA | Tumor-associated antigen |
| TME | Tumor microenvironment |
References
- Evers, M.; Stip, M.; Keller, K.; Willemen, H.; Nederend, M.; Jansen, M.; Chan, C.; Budding, K.; Nierkens, S.; Valerius, T.; et al. Anti-GD2 IgA kills tumors by neutrophils without antibody-associated pain in the preclinical treatment of high-risk neuroblastoma. J. Immunother. Cancer 2021, 9, e003163. [Google Scholar] [CrossRef]
- Evers, M.; Ten Broeke, T.; Jansen, J.H.M.; Nederend, M.; Hamdan, F.; Reiding, K.R.; Meyer, S.; Moerer, P.; Brinkman, I.; Rösner, T.; et al. Novel chimerized IgA CD20 antibodies: Improving neutrophil activation against CD20-positive malignancies. mAbs 2020, 12, 1795505. [Google Scholar] [CrossRef]
- Boross, P.; Lohse, S.; Nederend, M.; Jansen, J.H.M.; van Tetering, G.; Dechant, M.; Peipp, M.; Royle, L.; Liew, L.P.; Boon, L.; et al. IgA EGFR antibodies mediate tumour killing in vivo. EMBO Mol. Med. 2013, 5, 1213–1226. [Google Scholar] [CrossRef]
- Brandsma, A.M.; Bondza, S.; Evers, M.; Koutstaal, R.; Nederend, M.; Jansen, J.H.M.; Rösner, T.; Valerius, T.; Leusen, J.H.W.; ten Broeke, T. Potent Fc Receptor Signaling by IgA Leads to Superior Killing of Cancer Cells by Neutrophils Compared to IgG. Front. Immunol. 2019, 10, 704. [Google Scholar] [CrossRef] [Green Version]
- Van Tetering, G.; Evers, M.; Chan, C.; Stip, M.; Leusen, J. Fc Engineering Strategies to Advance IgA Antibodies as Therapeutic Agents. Antibodies 2020, 9, 70. [Google Scholar] [CrossRef]
- Ten Broeke, T.; Honing, H.; Brandsma, A.M.; Jacobino, S.; Bakema, J.E.; Kanters, D.; Van Der Linden, J.A.M.M.; Bracke, M.; Koenderman, L.; Leusen, J.H.W. FcαRI dynamics are regulated by GSK-3 and PKCζ during cytokine mediated inside-out signaling. Front. Immunol. 2019, 9, 3191. [Google Scholar] [CrossRef] [Green Version]
- Matlung, H.L.; Babes, L.; Zhao, X.W.; van Houdt, M.; Treffers, L.W.; van Rees, D.J.; Franke, K.; Schornagel, K.; Verkuijlen, P.; Janssen, H.; et al. Neutrophils Kill Antibody-Opsonized Cancer Cells by Trogoptosis. Cell Rep. 2018, 23, 3946–3959.e6. [Google Scholar] [CrossRef]
- Chan, C.; Lustig, M.; Baumann, N.; Valerius, T.; van Tetering, G.; Leusen, J.H.W. Targeting Myeloid Checkpoint Molecules in Combination with Antibody Therapy: A Novel Anti-Cancer Strategy with IgA Antibodies? Front. Immunol. 2022, 13, 932155. [Google Scholar] [CrossRef] [PubMed]
- Vinay, D.S.; Ryan, E.P.; Pawelec, G.; Talib, W.H.; Stagg, J.; Elkord, E.; Lichtor, T.; Decker, W.K.; Whelan, R.L.; Kumara, H.M.C.S.; et al. Immune evasion in cancer: Mechanistic basis and therapeutic strategies. Semin. Cancer Biol. 2015, 35, S185–S198. [Google Scholar] [CrossRef] [PubMed]
- Majeti, R.; Chao, M.P.; Alizadeh, A.A.; Pang, W.W.; Jaiswal, S.; Gibbs, K.D.; van Rooijen, N.; Weissman, I.L. CD47 Is an Adverse Prognostic Factor and Therapeutic Antibody Target on Human Acute Myeloid Leukemia Stem Cells. Cell 2009, 138, 286–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willingham, S.B.; Volkmer, J.P.; Gentles, A.J.; Sahoo, D.; Dalerba, P.; Mitra, S.S.; Wang, J.; Contreras-Trujillo, H.; Martin, R.; Cohen, J.D.; et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 6662–6667. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Sanz, P.; Hoogendijk, A.J.; Verkuijlen, P.J.J.H.; Schornagel, K.; van Bruggen, R.; van den Berg, T.K.; Tytgat, G.A.M.; Franke, K.; Kuijpers, T.W.; Matlung, H.L. CD47-SIRPα Checkpoint Inhibition Enhances Neutrophil-Mediated Killing of Dinutuximab-Opsonized Neuroblastoma Cells. Cancers 2021, 13, 4261. [Google Scholar] [CrossRef] [PubMed]
- Logtenberg, M.E.W.; Jansen, J.H.M.; Raaben, M.; Toebes, M.; Franke, K.; Brandsma, A.M.; Matlung, H.L.; Fauster, A.; Gomez-Eerland, R.; Bakker, N.A.M.; et al. Glutaminyl cyclase is an enzymatic modifier of the CD47-SIRPα axis and a target for cancer immunotherapy. Nat. Med. 2019, 25, 612–619. [Google Scholar] [CrossRef] [PubMed]
- Treffers, L.W.; ten Broeke, T.; Rosner, T.; Marco Jansen, J.H.; van Houdt, M.; Kahle, S.; Schornagel, K.; Verkuijlen, P.J.J.H.; Prins, J.M.; Franke, K.; et al. IgA-mediated killing of tumor cells by neutrophils is enhanced by CD47–SIRPA checkpoint inhibition. Cancer Immunol. Res. 2020, 8, 120–130. [Google Scholar] [CrossRef] [Green Version]
- Mehta, K.A.; Patel, K.A.; Pandya, S.J.; Patel, P.S. Aberrant sialylation plays a significant role in oral squamous cell carcinoma progression. J. Oral Pathol. Med. 2020, 49, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Munkley, J. Aberrant Sialylation in Cancer: Therapeutic Opportunities. Cancers 2022, 14, 4248. [Google Scholar] [CrossRef]
- Kannagi, R.; Sakuma, K.; Miyazaki, K.; Lim, K.-T.; Yusa, A.; Yin, J.; Izawa, M. Altered expression of glycan genes in cancers induced by epigenetic silencing and tumor hypoxia: Clues in the ongoing search for new tumor markers. Cancer Sci. 2010, 101, 586–593. [Google Scholar] [CrossRef]
- Crocker, P.R.; Paulson, J.C.; Varki, A. Siglecs and their roles in the immune system. Nat. Rev. Immunol. 2007, 7, 255–266. [Google Scholar] [CrossRef]
- Ohta, M.; Ishida, A.; Toda, M.; Akita, K.; Inoue, M.; Yamashita, K.; Watanabe, M.; Murata, T.; Usui, T.; Nakada, H. Immunomodulation of monocyte-derived dendritic cells through ligation of tumor-produced mucins to Siglec-9. Biochem. Biophys. Res. Commun. 2010, 402, 663–669. [Google Scholar] [CrossRef]
- Kawasaki, Y.; Ito, A.; Withers, D.A.; Taima, T.; Kakoi, N.; Saito, S.; Arai, Y. Ganglioside DSGb5, preferred ligand for Siglec-7, inhibits NK cell cytotoxicity against renal cell carcinoma cells. Glycobiology 2010, 20, 1373–1379. [Google Scholar] [CrossRef] [Green Version]
- Miyazaki, K.; Sakuma, K.; Kawamura, Y.I.; Izawa, M.; Ohmori, K.; Mitsuki, M.; Yamaji, T.; Hashimoto, Y.; Suzuki, A.; Saito, Y.; et al. Colonic epithelial cells express specific ligands for mucosal macrophage immunosuppressive receptors siglec-7 and -9. J. Immunol. 2012, 188, 4690–4700. [Google Scholar] [CrossRef] [Green Version]
- Tyler, C.; Kapur, A.; Felder, M.; Belisle, J.A.; Trautman, C.; Gubbels, J.A.A.; Connor, J.P.; Patankar, M.S. The mucin MUC16 (CA125) binds to NK cells and monocytes from peripheral blood of women with healthy pregnancy and preeclampsia. Am. J. Reprod. Immunol. 2012, 68, 28–37. [Google Scholar] [CrossRef] [Green Version]
- Lustig, M.; Chan, C.; Jansen, J.H.M.; Brautigam, M.; Mester, S.; Foss, S.; Andersen, J.T.; Bastian, L.; Sondermann, P.; Peipp, M.; et al. Disruption of the Sialic Acid/Siglec-9 Axis Improves Antibody-Mediated Neutrophil Cytotoxicity towards Tumor Cells. Front. Immunol. 2023, 14, 1178817. [Google Scholar] [CrossRef] [PubMed]
- Alhallak, K.; Sun, J.; Muz, B.; Azab, A.K. CD24 signalling through macrophage Siglec-10 is a new target for cancer immunotherapy. Biomater. Cancer Ther. 2020, 572, 499–526. [Google Scholar]
- Nicoll, G.; Avril, T.; Lock, K.; Furukawa, K.; Bovin, N.; Crocker, P.R. Ganglioside GD3 expression on target cells can modulate NK cell cytotoxicity via siglec-7-dependent and -independent mechanisms. Eur. J. Immunol. 2003, 33, 1642–1648. [Google Scholar] [CrossRef]
- Beatson, R.; Tajadura-Ortega, V.; Achkova, D.; Picco, G.; Tsourouktsoglou, T.D.; Klausing, S.; Hillier, M.; Maher, J.; Noll, T.; Crocker, P.R.; et al. The mucin MUC1 modulates the tumor immunological microenvironment through engagement of the lectin Siglec-9. Nat. Immunol. 2016, 17, 1273–1281. [Google Scholar] [CrossRef] [Green Version]
- Brandsma, A.M.; ten Broeke, T.; Nederend, M.; Meulenbroek, L.A.P.M.; van Tetering, G.; Meyer, S.; Jansen, J.H.M.; Beltran Buitrago, M.A.; Nagelkerke, S.Q.; Nemeth, I.; et al. Simultaneous Targeting of Fc Rs and Fc RI Enhances Tumor Cell Killing. Cancer Immunol. Res. 2015, 3, 1316–1324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chernyavska, M.; Hermans, C.K.J.C.; Chan, C.; Baumann, N.; Rösner, T.; Leusen, J.H.W.; Valerius, T.; Verdurmen, W.P.R. Evaluation of immunotherapies improving macrophage anti-tumor response using a microfluidic model. Organs-on-a-Chip 2022, 4, 100019. [Google Scholar] [CrossRef]
- Hinderlich, S.; Weidemann, W.; Yardeni, T.; Horstkorte, R.; Huizing, M. UDP-GlcNAc 2-Epimerase/ManNAc Kinase (GNE): A Master Regulator of Sialic Acid Synthesis. Top. Curr. Chem. 2015, 366, 97–137. [Google Scholar]
- Zhang, J.Q.; Biedermann, B.; Nitschke, L.; Crocker, P.R. The Murine Inhibitory Receptor MSiglec-E Is Expressed Broadly on Cells of the Innate Immune System Whereas MSiglec-F Is Restricted to Eosinophils. Eur. J. Immunol. 2004, 34, 1175–1184. [Google Scholar] [CrossRef]
- Pai, S.; Bamodu, O.A.; Lin, Y.-K.; Lin, C.-S.; Chu, P.-Y.; Chien, M.-H.; Wang, L.-S.; Hsiao, M.; Yeh, C.-T.; Tsai, J.-T. CD47-SIRPα Signaling Induces Epithelial-Mesenchymal Transition and Cancer Stemness and Links to a Poor Prognosis in Patients with Oral Squamous Cell Carcinoma. Cells 2019, 8, 1658. [Google Scholar] [CrossRef] [Green Version]
- Hu, T.; Liu, H.; Liang, Z.; Wang, F.; Zhou, C.; Zheng, X.; Zhang, Y.; Song, Y.; Hu, J.; He, X.; et al. Tumor-intrinsic CD47 signal regulates glycolysis and promotes colorectal cancer cell growth and metastasis. Theranostics 2020, 10, 4056–4072. [Google Scholar] [CrossRef] [PubMed]
- Kamber, R.A.; Nishiga, Y.; Morton, B.; Banuelos, A.M.; Barkal, A.A.; Vences-Catalán, F.; Gu, M.; Fernandez, D.; Seoane, J.A.; Yao, D.; et al. Inter-cellular CRISPR screens reveal regulators of cancer cell phagocytosis. Nature 2021, 597, 549–554. [Google Scholar] [CrossRef] [PubMed]
- Barkal, A.A.; Brewer, R.E.; Markovic, M.; Kowarsky, M.; Barkal, S.A.; Zaro, B.W.; Krishnan, V.; Hatakeyama, J.; Dorigo, O.; Barkal, L.J.; et al. CD24 signalling through macrophage Siglec-10 is a target for cancer immunotherapy. Nature 2019, 572, 392–396. [Google Scholar] [CrossRef]
- Bärenwaldt, A.; Läubli, H. The sialoglycan-Siglec glyco-immune checkpoint—A target for improving innate and adaptive anti-cancer immunity. Expert Opin. Ther. Targets 2019, 23, 839–853. [Google Scholar] [CrossRef] [PubMed]
- Dobie, C.; Skropeta, D. Insights into the role of sialylation in cancer progression and metastasis. Br. J. Cancer 2021, 124, 76–90. [Google Scholar] [CrossRef]
- Szabo, R.; Skropeta, D. Advancement of Sialyltransferase Inhibitors: Therapeutic Challenges and Opportunities. Med. Res. Rev. 2017, 37, 219–270. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.Q.; Nicoll, G.; Jones, C.; Crocker, P.R. Siglec-9, a novel sialic acid binding member of the immunoglobulin superfamily expressed broadly on human blood leukocytes. J. Biol. Chem. 2000, 275, 22121–22126. [Google Scholar] [CrossRef] [Green Version]
- Ando, M.; Tu, W.; Nishijima, K.I.; Iijima, S. Siglec-9 enhances IL-10 production in macrophages via tyrosine-based motifs. Biochem. Biophys. Res. Commun. 2008, 369, 878–883. [Google Scholar] [CrossRef]
- Jandus, C.; Boligan, K.F.; Chijioke, O.; Liu, H.; Dahlhaus, M.; Démoulins, T.; Schneider, C.; Wehrli, M.; Hunger, R.E.; Baerlocher, G.M.; et al. Interactions between Siglec-7/9 receptors and ligands influence NK cell-dependent tumor immunosurveillance. J. Clin. Investig. 2014, 124, 1810–1820. [Google Scholar] [CrossRef] [Green Version]
- Xiao, H.; Woods, E.C.; Vukojicic, P.; Bertozzi, C.R. Precision glycocalyx editing as a strategy for cancer immunotherapy. Proc. Natl. Acad. Sci. USA 2016, 113, 10304–10309. [Google Scholar] [CrossRef] [Green Version]
- Von Gunten, S.; Yousefi, S.; Seitz, M.; Jakob, S.M.; Schaffner, T.; Seger, R.; Takala, J.; Villiger, P.M.; Simon, H.U. Siglec-9 transduces apoptotic and nonapoptotic death signals into neutrophils depending on the proinflammatory cytokine environment. Blood 2005, 106, 1423–1431. [Google Scholar] [CrossRef] [Green Version]
- McMillan, S.J.; Sharma, R.S.; McKenzie, E.J.; Richards, H.E.; Zhang, J.; Prescott, A.; Crocker, P.R. Siglec-E is a negative regulator of acute pulmonary neutrophil inflammation and suppresses CD11b β2-integrin-dependent signaling. Blood 2013, 121, 2084–2094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lizcano, A.; Secundino, I.; Dohrmann, S.; Corriden, R.; Rohena, C.; Diaz, S.; Ghosh, P.; Deng, L.; Nizet, V.; Varki, A. Erythrocyte sialoglycoproteins engage Siglec-9 on neutrophils to suppress activation. Blood 2017, 129, 3100–3110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, E.; Boelaars, K.; Brown, K.; Eveline Li, R.J.; Kruijssen, L.; Bruijns, S.C.M.; van Ee, T.; Schetters, S.T.T.; Crommentuijn, M.H.W.; van der Horst, J.C.; et al. Sialic acids in pancreatic cancer cells drive tumour-associated macrophage differentiation via the Siglec receptors Siglec-7 and Siglec-9. Nat. Commun. 2021, 12, 1270. [Google Scholar] [CrossRef]
- Belisle, J.A.; Horibata, S.; Jennifer, G.A.A.; Petrie, S.; Kapur, A.; André, S.; Gabius, H.J.; Rancourt, C.; Connor, J.; Paulson, J.C.; et al. Identification of Siglec-9 as the receptor for MUC16 on human NK cells, B cells, and monocytes. Mol. Cancer 2010, 9, 118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanida, S.; Akita, K.; Ishida, A.; Mori, Y.; Toda, M.; Inoue, M.; Ohta, M.; Yashiro, M.; Sawada, T.; Hirakawa, K.; et al. Binding of the sialic acid-binding lectin, siglec-9, to the membrane mucin, MUC1, induces recruitment of β-catenin and subsequent cell growth. J. Biol. Chem. 2013, 288, 31842–31852. [Google Scholar] [CrossRef] [Green Version]
- Gray, M.A.; Stanczak, M.A.; Mantuano, N.R.; Xiao, H.; Pijnenborg, J.F.A.; Malaker, S.A.; Miller, C.L.; Weidenbacher, P.A.; Tanzo, J.T.; Ahn, G.; et al. Targeted glycan degradation potentiates the anticancer immune response in vivo. Nat. Chem. Biol. 2020, 16, 1376–1384. [Google Scholar] [CrossRef]
- Picco, G.; Julien, S.; Brockhausen, I.; Beatson, R.; Antonopoulos, A.; Haslam, S.; Mandel, U.; Dell, A.; Pinder, S.; Taylor-Papadimitriou, J.; et al. Over-expression of ST3Gal-I promotes mammary tumorigenesis. Glycobiology 2010, 20, 1241–1250. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, A.S.; Harduin-Lepers, A.; Magalhães, A.; Machado, E.; Mendes, N.; Costa, L.T.; Matthiesen, R.; Almeida, R.; Costa, J.; Reis, C.A. Differential expression of α-2,3-sialyltransferases and α-1,3/4-fucosyltransferases regulates the levels of sialyl Lewis a and sialyl Lewis x in gastrointestinal carcinoma cells. Int. J. Biochem. Cell Biol. 2010, 42, 80–89. [Google Scholar] [CrossRef]
- Wu, X.; Zhao, J.; Ruan, Y.; Sun, L.; Xu, C.; Jiang, H. Sialyltransferase ST3GAL1 promotes cell migration, invasion, and TGF-β1-induced EMT and confers paclitaxel resistance in ovarian cancer. Cell Death Dis. 2018, 9, 1102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burchell, J.; Poulsom, R.; Hanby, A.; Whitehouse, C.; Cooper, L.; Clausen, H.; Miles, D.; Taylor-Papadimitriou, J. An alpha2,3 sialyltransferase (ST3Gal I) is elevated in primary breast carcinomas. Glycobiology 1999, 9, 1307–1311. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chan, C.; Lustig, M.; Jansen, J.H.M.; Garcia Villagrasa, L.; Raymakers, L.; Daamen, L.A.; Valerius, T.; van Tetering, G.; Leusen, J.H.W. Sialic Acids on Tumor Cells Modulate IgA Therapy by Neutrophils via Inhibitory Receptors Siglec-7 and Siglec-9. Cancers 2023, 15, 3405. https://doi.org/10.3390/cancers15133405
Chan C, Lustig M, Jansen JHM, Garcia Villagrasa L, Raymakers L, Daamen LA, Valerius T, van Tetering G, Leusen JHW. Sialic Acids on Tumor Cells Modulate IgA Therapy by Neutrophils via Inhibitory Receptors Siglec-7 and Siglec-9. Cancers. 2023; 15(13):3405. https://doi.org/10.3390/cancers15133405
Chicago/Turabian StyleChan, Chilam, Marta Lustig, J. H. Marco Jansen, Laura Garcia Villagrasa, Leon Raymakers, Lois A. Daamen, Thomas Valerius, Geert van Tetering, and Jeanette H. W. Leusen. 2023. "Sialic Acids on Tumor Cells Modulate IgA Therapy by Neutrophils via Inhibitory Receptors Siglec-7 and Siglec-9" Cancers 15, no. 13: 3405. https://doi.org/10.3390/cancers15133405