
In Vitro and In Vivo Drug-Response Profiling Using Patient-Derived High-Grade Glioma
 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Tumor Processing
2.2. FDA-Approved NCI Drug Library
2.3. 3D Culture and 3D Viability Assay
2.4. Data Processing
2.5. Concentration Response Assays
2.6. In Vivo Profiling
2.7. Intracranial Orthotopic Xenotransplantation
2.8. Subcutaneous Xenotransplantation and Pharmacology
2.8.1. Subcutaneous Model Build
2.8.2. Pharmacology
2.8.3. Flow Cytometry and Phenotypic Stemness Characterization
3. Results
3.1. In Vitro Cytotoxic Effects of Cobimetinib, Vemurafenib, and Their Synergistic Combination to 3D GBM Organoids
3.2. Patient-Derived 3D Organoids Show Differentially Enriched Pathways of Growth and Development
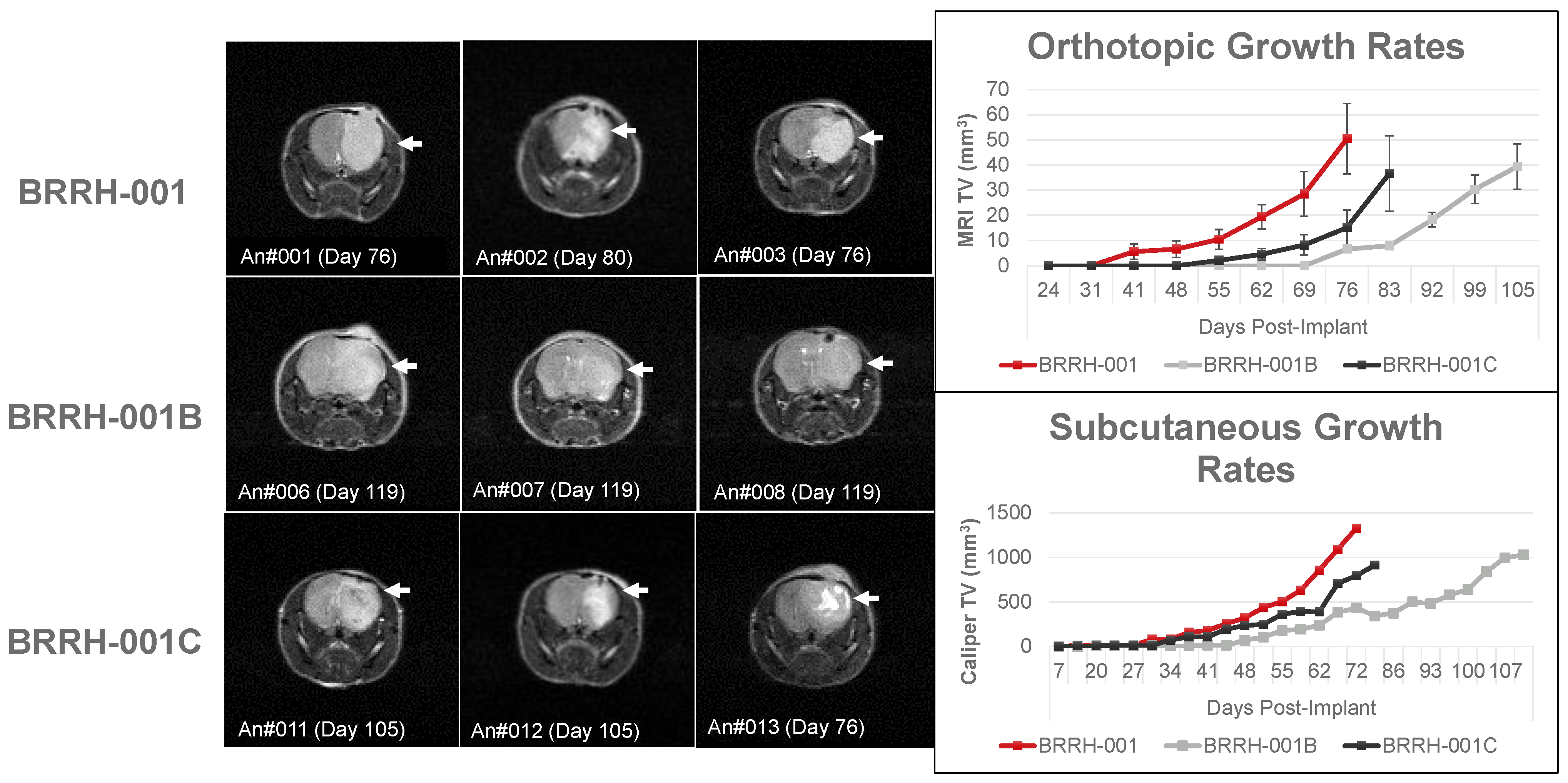
3.3. Orthotopic or Subcutaneously Implanted GBM Organoids Have Similar Growth Profile in Mice
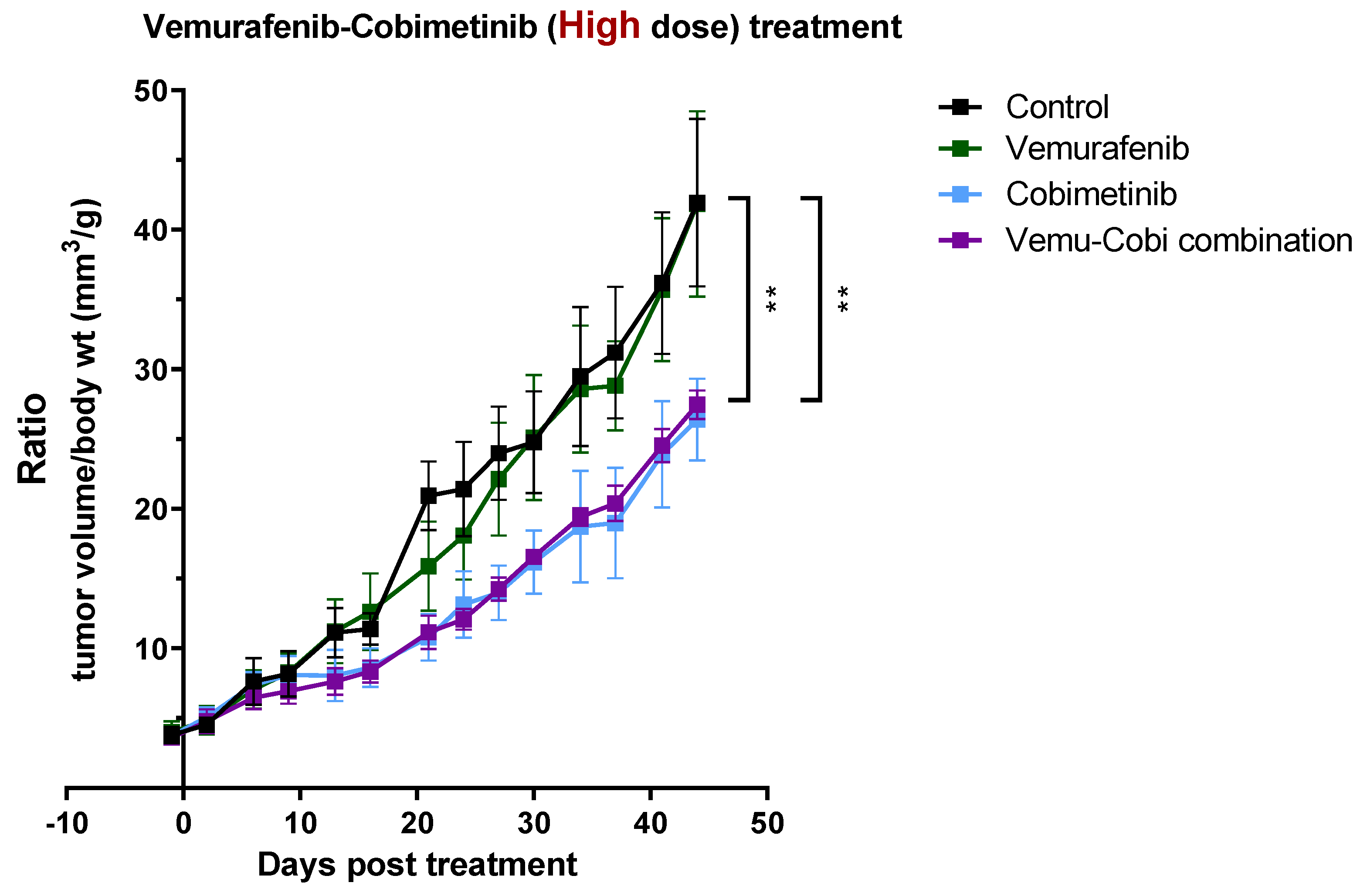
3.4. In Vivo Validation for Toxicity of Vemurafenib and Cobimetinib and Their Combination to Patient-Derived GBM Organoids
3.5. Patient-Derived 3D GBM Organoids from a Primary and Recurrent Tumor Show a Heterogeneous “Stemness” Signature
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Thakkar, J.P.; Dolecek, T.A.; Horbinski, C.; Ostrom, Q.T.; Lightner, D.D.; Barnholtz-Sloan, J.S.; Villano, J.L. Epidemiologic and molecular prognostic review of glioblastoma. Cancer Epidemiol. Biomarkers Prev. 2014, 23, 1985–1996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohgaki, H.; Kleihues, P. Epidemiology and etiology of gliomas. Acta Neuropathol. 2005, 109, 93–108. [Google Scholar] [CrossRef] [PubMed]
- Fazzari, F.G.T.; Rose, F.; Pauls, M.; Guay, E.; Ibrahim, M.F.K.; Basulaiman, B.; Tu, M.; Hutton, B.; Nicholas, G.; Ng, T.L. The current landscape of systemic therapy for recurrent glioblastoma: A systematic review of randomized-controlled trials. Crit. Rev. Oncol. Hematol. 2022, 169, 103540. [Google Scholar] [CrossRef] [PubMed]
- Vollmann-Zwerenz, A.; Leidgens, V.; Feliciello, G.; Klein, C.A.; Hau, P. Tumor Cell Invasion in Glioblastoma. Int. J. Mol. Sci. 2020, 21, 1932. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.Y. Temozolomide resistance in glioblastoma multiforme. Genes Dis. 2016, 3, 198–210. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Zhang, B.; Zhou, C.; Li, Y.; Li, B.; Yu, M.; Luo, Y.; Gao, L.; Zhang, D.; Xue, Q.; et al. The comparison genomics analysis with glioblastoma multiforme (GBM) cells under 3D and 2D cell culture conditions. Colloids Surf. B Biointerfaces 2018, 172, 665–673. [Google Scholar] [CrossRef]
- Darrigues, E.; Zhao, E.H.; De Loose, A.; Lee, M.P.; Borrelli, M.J.; Eoff, R.L.; Galileo, D.S.; Penthala, N.R.; Crooks, P.A.; Rodriguez, A. Biobanked Glioblastoma Patient-Derived Organoids as a Precision Medicine Model to Study Inhibition of Invasion. Int. J. Mol. Sci. 2021, 22, 10720. [Google Scholar] [CrossRef]
- Quereda, V.; Hou, S.; Madoux, F.; Scampavia, L.; Spicer, T.P.; Duckett, D. A Cytotoxic Three-Dimensional-Spheroid, High-Throughput Assay Using Patient-Derived Glioma Stem Cells. SLAS Discov. 2018, 23, 842–849. [Google Scholar] [CrossRef] [Green Version]
- Hubert, C.G.; Rivera, M.; Spangler, L.C.; Wu, Q.; Mack, S.C.; Prager, B.C.; Couce, M.; McLendon, R.E.; Sloan, A.E.; Rich, J.N. A Three-Dimensional Organoid Culture System Derived from Human Glioblastomas Recapitulates the Hypoxic Gradients and Cancer Stem Cell Heterogeneity of Tumors Found In Vivo. Cancer Res. 2016, 76, 2465–2477. [Google Scholar] [CrossRef] [Green Version]
- Gomez, G.A.; Oksdath, M.; Brown, M.P.; Ebert, L.M. New approaches to model glioblastoma in vitro using brain organoids: Implications for precision oncology. Transl. Cancer Res. 2019, 8, S606–S611. [Google Scholar] [CrossRef]
- Lenin, S.; Ponthier, E.; Scheer, K.G.; Yeo, E.C.F.; Tea, M.N.; Ebert, L.M.; Oksdath Mansilla, M.; Poonnoose, S.; Baumgartner, U.; Day, B.W.; et al. A Drug Screening Pipeline Using 2D and 3D Patient-Derived In Vitro Models for Pre-Clinical Analysis of Therapy Response in Glioblastoma. Int. J. Mol. Sci. 2021, 22, 4322. [Google Scholar] [CrossRef] [PubMed]
- Jacob, F.; Ming, G.L.; Song, H. Generation and biobanking of patient-derived glioblastoma organoids and their application in CAR T cell testing. Nat. Protoc. 2020, 15, 4000–4033. [Google Scholar] [CrossRef] [PubMed]
- Jacob, F.; Salinas, R.D.; Zhang, D.Y.; Nguyen, P.T.T.; Schnoll, J.G.; Wong, S.Z.H.; Thokala, R.; Sheikh, S.; Saxena, D.; Prokop, S.; et al. A Patient-Derived Glioblastoma Organoid Model and Biobank Recapitulates Inter- and Intra-tumoral Heterogeneity. Cell 2020, 180, 188–204.e122. [Google Scholar] [CrossRef]
- Julien, S.; Merino-Trigo, A.; Lacroix, L.; Pocard, M.; Goéré, D.; Mariani, P.; Landron, S.; Bigot, L.; Nemati, F.; Dartigues, P.; et al. Characterization of a large panel of patient-derived tumor xenografts representing the clinical heterogeneity of human colorectal cancer. Clin. Cancer Res. 2012, 18, 5314–5328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopetz, S.; Lemos, R.; Powis, G. The promise of patient-derived xenografts: The best laid plans of mice and men. Clin. Cancer Res. 2012, 18, 5160–5162. [Google Scholar] [CrossRef] [Green Version]
- Letai, A.; Bhola, P.; Welm, A.L. Functional precision oncology: Testing tumors with drugs to identify vulnerabilities and novel combinations. Cancer Cell 2022, 40, 26–35. [Google Scholar] [CrossRef]
- Ho, W.W.; Pittet, M.J.; Fukumura, D.; Jain, R.K. The local microenvironment matters in preclinical basic and translational studies of cancer immunology and immunotherapy. Cancer Cell 2022, 40, 701–702. [Google Scholar] [CrossRef]
- Ignatova, T.N.; Kukekov, V.G.; Laywell, E.D.; Suslov, O.N.; Vrionis, F.D.; Steindler, D.A. Human cortical glial tumors contain neural stem-like cells expressing astroglial and neuronal markers in vitro. Glia 2002, 39, 193–206. [Google Scholar] [CrossRef]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef]
- Galli, R.; Binda, E.; Orfanelli, U.; Cipelletti, B.; Gritti, A.; De Vitis, S.; Fiocco, R.; Foroni, C.; Dimeco, F.; Vescovi, A. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004, 64, 7011–7021. [Google Scholar] [CrossRef] [Green Version]
- Ignatova, T.N.; Chaitin, H.J.; Kukekov, N.V.; Suslov, O.N.; Dulatova, G.I.; Hanafy, K.A.; Vrionis, F.D. Gliomagenesis is orchestrated by the Oct3/4 regulatory network. J. Neurosurg. Sci. 2021. [Google Scholar] [CrossRef] [PubMed]
- Lathia, J.D.; Mack, S.C.; Mulkearns-Hubert, E.E.; Valentim, C.L.; Rich, J.N. Cancer stem cells in glioblastoma. Genes Dev. 2015, 29, 1203–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, C.; Shervington, A. Chemoresistance in gliomas. Mol. Cell. Biochem. 2008, 312, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Khan, M.A.; Weiler, M.; Blaes, J.; Jestaedt, L.; Geibert, M.; Zou, P.; Gronych, J.; Bernhardt, O.; Korshunov, A.; et al. Targeting self-renewal in high-grade brain tumors leads to loss of brain tumor stem cells and prolonged survival. Cell Stem Cell 2014, 15, 185–198. [Google Scholar] [CrossRef] [Green Version]
- Spencer, D.A.; Auffinger, B.M.; Murphy, J.P.; Muroski, M.E.; Qiao, J.; Gorind, Y.; Lesniak, M.S. Hitting a Moving Target: Glioma Stem Cells Demand New Approaches in Glioblastoma Therapy. Curr. Cancer Drug Targets 2017, 17, 236–254. [Google Scholar] [CrossRef]
- Reed, M.R.; Lyle, A.G.; De Loose, A.; Maddukuri, L.; Learned, K.; Beale, H.C.; Kephart, E.T.; Cheney, A.; van den Bout, A.; Lee, M.P.; et al. A Functional Precision Medicine Pipeline Combines Comparative Transcriptomics and Tumor Organoid Modeling to Identify Bespoke Treatment Strategies for Glioblastoma. Cells 2021, 10, 3400. [Google Scholar] [CrossRef]
- Hou, S.; Tiriac, H.; Sridharan, B.P.; Scampavia, L.; Madoux, F.; Seldin, J.; Souza, G.R.; Watson, D.; Tuveson, D.; Spicer, T.P. Advanced Development of Primary Pancreatic Organoid Tumor Models for High-Throughput Phenotypic Drug Screening. SLAS Discov. 2018, 23, 574–584. [Google Scholar] [CrossRef] [Green Version]
- Spicer, T.P.; Vega, V.F.; Scampavia, L.; Willetts, L.; Vessels, M. A Novel 3D Culture System for High-Throughput Hepatoxicity Screening. Bioprocess Int. 2018, 16, 1–5. [Google Scholar]
- Baillargeon, P.; Shumate, J.; Hou, S.; Fernandez-Vega, V.; Marques, N.; Souza, G.; Seldin, J.; Spicer, T.P.; Scampavia, L. Automating a Magnetic 3D Spheroid Model Technology for High-Throughput Screening. SLAS Technol. 2019, 24, 420–428. [Google Scholar] [CrossRef]
- Madoux, F.; Tanner, A.; Vessels, M.; Willetts, L.; Hou, S.; Scampavia, L.; Spicer, T.P. A 1536-Well 3D Viability Assay to Assess the Cytotoxic Effect of Drugs on Spheroids. SLAS Discov. 2017, 22, 516–524. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.H.; Chung, T.D.; Oldenburg, K.R. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J. Biomol. Screen. 1999, 4, 67–73. [Google Scholar] [CrossRef]
- Smith, E.; Chase, P.; Niswender, C.M.; Utley, T.J.; Sheffler, D.J.; Noetzel, M.J.; Lamsal, A.; Wood, M.R.; Conn, P.J.; Lindsley, C.W.; et al. Application of Parallel Multiparametric Cell-Based FLIPR Detection Assays for the Identification of Modulators of the Muscarinic Acetylcholine Receptor 4 (M4). J. Biomol. Screen. 2015, 20, 858–868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Sun, D.; Chen, Y.J.; Xie, X.; Shi, Y.; Tabar, V.; Brennan, C.W.; Bale, T.A.; Jayewickreme, C.D.; Laks, D.R.; et al. Cell Lineage-Based Stratification for Glioblastoma. Cancer Cell 2020, 38, 366–379.e368. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Yu, L.; Li, Z.; Li, W.; Huang, W. Patient-derived organoid (PDO) platforms to facilitate clinical decision making. J. Transl. Med. 2021, 19, 40. [Google Scholar] [CrossRef]
- Tuveson, D.; Clevers, H. Cancer modeling meets human organoid technology. Science 2019, 364, 952–955. [Google Scholar] [CrossRef] [PubMed]
- Veninga, V.; Voest, E.E. Tumor organoids: Opportunities and challenges to guide precision medicine. Cancer Cell 2021, 39, 1190–1201. [Google Scholar] [CrossRef]
- Wolff, R.A.; Wang-Gillam, A.; Alvarez, H.; Tiriac, H.; Engle, D.; Hou, S.; Groff, A.F.; San Lucas, A.; Bernard, V.; Allenson, K.; et al. Dynamic changes during the treatment of pancreatic cancer. Oncotarget 2018, 9, 14764–14790. [Google Scholar] [CrossRef] [Green Version]
- Kazda, T.; Dziacky, A.; Burkon, P.; Pospisil, P.; Slavik, M.; Rehak, Z.; Jancalek, R.; Slampa, P.; Slaby, O.; Lakomy, R. Radiotherapy of glioblastoma 15 years after the landmark Stupp’s trial: More controversies than standards? Radiol. Oncol. 2018, 52, 121–128. [Google Scholar] [CrossRef] [Green Version]
- Chen, A.P.; Kummar, S.; Moore, N.; Rubinstein, L.V.; Zhao, Y.; Williams, P.M.; Palmisano, A.; Sims, D.; O’Sullivan Coyne, G.; Rosenberger, C.L. Molecular profiling-based assignment of cancer therapy (NCI-MPACT): A randomized multicenter phase II trial. JCO Precis. Oncol. 2021, 5, 133–144. [Google Scholar] [CrossRef]
- Flaherty, K.T.; Gray, R.J.; Chen, A.P.; Li, S.; McShane, L.M.; Patton, D.; Hamilton, S.R.; Williams, P.M.; Iafrate, A.J.; Sklar, J.; et al. Molecular Landscape and Actionable Alterations in a Genomically Guided Cancer Clinical Trial: National Cancer Institute Molecular Analysis for Therapy Choice (NCI-MATCH). J. Clin. Oncol. 2020, 38, 3883–3894. [Google Scholar] [CrossRef]
- Sicklick, J.K.; Kato, S.; Okamura, R.; Schwaederle, M.; Hahn, M.E.; Williams, C.B.; De, P.; Krie, A.; Piccioni, D.E.; Miller, V.A. Molecular profiling of cancer patients enables personalized combination therapy: The I-PREDICT study. Nat. Med. 2019, 25, 744–750. [Google Scholar] [CrossRef] [PubMed]
- Massard, C.; Michiels, S.; Ferté, C.; Le Deley, M.-C.; Lacroix, L.; Hollebecque, A.; Verlingue, L.; Ileana, E.; Rosellini, S.; Ammari, S. High-Throughput Genomics and Clinical Outcome in Hard-to-Treat Advanced Cancers: Results of the MOSCATO 01 TrialGenomics to Improve Cancer Outcome. Cancer Discov. 2017, 7, 586–595. [Google Scholar] [CrossRef] [Green Version]
- Baylin, S.B.; Jones, P.A. Epigenetic Determinants of Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallette, F.M.; Olivier, C.; Lézot, F.; Oliver, L.; Cochonneau, D.; Lalier, L.; Cartron, P.F.; Heymann, D. Dormant, quiescent, tolerant and persister cells: Four synonyms for the same target in cancer. Biochem. Pharmacol. 2019, 162, 169–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boca, S.; Bhuvaneshwar, K.; Fernandez-Vega, V.; Kancherla, J.; Rao, S.; Madhavan, S.; Riggins, R.; Beckman, R.A.; Bravo, H.C.; Scampavia, L.; et al. Prioritizing targeted therapies in an evidence-based manner, integrating biological context and functional precision medicine. J. Clin. Oncol. 2020, 38, e14065. [Google Scholar] [CrossRef]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rajan, R.G.; Fernandez-Vega, V.; Sperry, J.; Nakashima, J.; Do, L.H.; Andrews, W.; Boca, S.; Islam, R.; Chowdhary, S.A.; Seldin, J.; et al. In Vitro and In Vivo Drug-Response Profiling Using Patient-Derived High-Grade Glioma. Cancers 2023, 15, 3289. https://doi.org/10.3390/cancers15133289
Rajan RG, Fernandez-Vega V, Sperry J, Nakashima J, Do LH, Andrews W, Boca S, Islam R, Chowdhary SA, Seldin J, et al. In Vitro and In Vivo Drug-Response Profiling Using Patient-Derived High-Grade Glioma. Cancers. 2023; 15(13):3289. https://doi.org/10.3390/cancers15133289
Chicago/Turabian StyleRajan, Robin G., Virneliz Fernandez-Vega, Jantzen Sperry, Jonathan Nakashima, Long H. Do, Warren Andrews, Simina Boca, Rezwanul Islam, Sajeel A. Chowdhary, Jan Seldin, and et al. 2023. "In Vitro and In Vivo Drug-Response Profiling Using Patient-Derived High-Grade Glioma" Cancers 15, no. 13: 3289. https://doi.org/10.3390/cancers15133289