
Identification, and Experimental and Bioinformatics Validation of an Immune-Related Prognosis Gene Signature for Low-Grade Glioma Based on mRNAsi

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. LGG and Perineural Tissue Acquisition
2.2. Collecting Immune-Related Genes and Datasets
2.3. Calculation of the mRNA Stemness Index
2.4. WGCNA to Filter Key Module
2.5. Determination of Immune-Related Genes with Differential Expression
2.6. Hub Gene Identification
2.7. Identification of Hub Genes
2.8. Chemotherapy Sensitivity Response Predictions
2.9. Translation Level of Hub Gene Expression Identification
2.10. Prognostic Risk System Establishment
2.11. Multivariate Cox Regression Analysis
2.12. Nomogram Construction and Verification
2.13. Prognostic Risk Signature Functional Exploration
2.14. Relevance between Hub Genes’ Expression and Immune Cells
2.15. qPCR
2.16. Statistical Analysis
3. Results
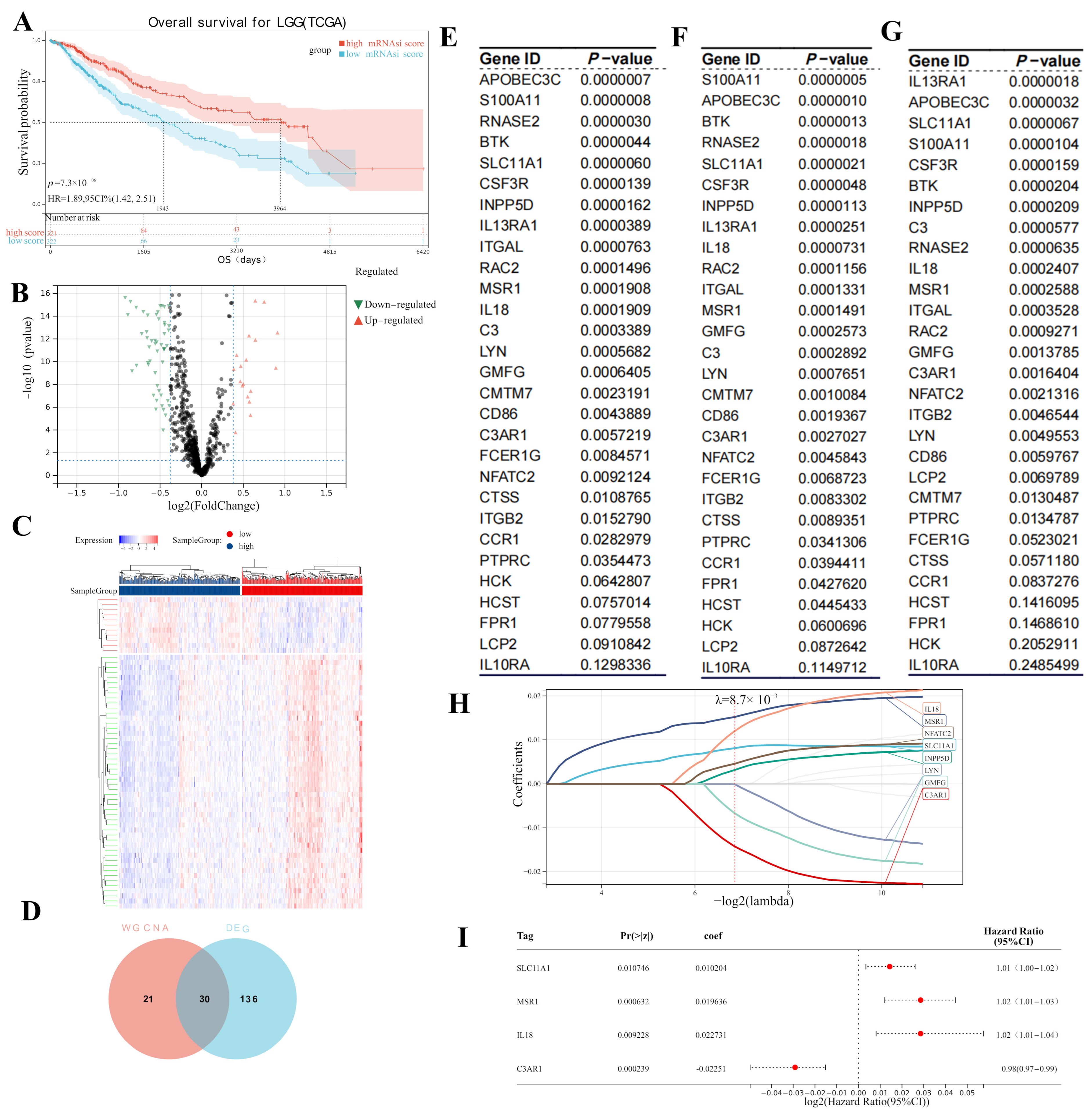
3.1. Key Module Identification
3.2. Hub Gene Screening
3.3. Screening for Potential Prognostic Genes
3.4. Potential Function of DEGs
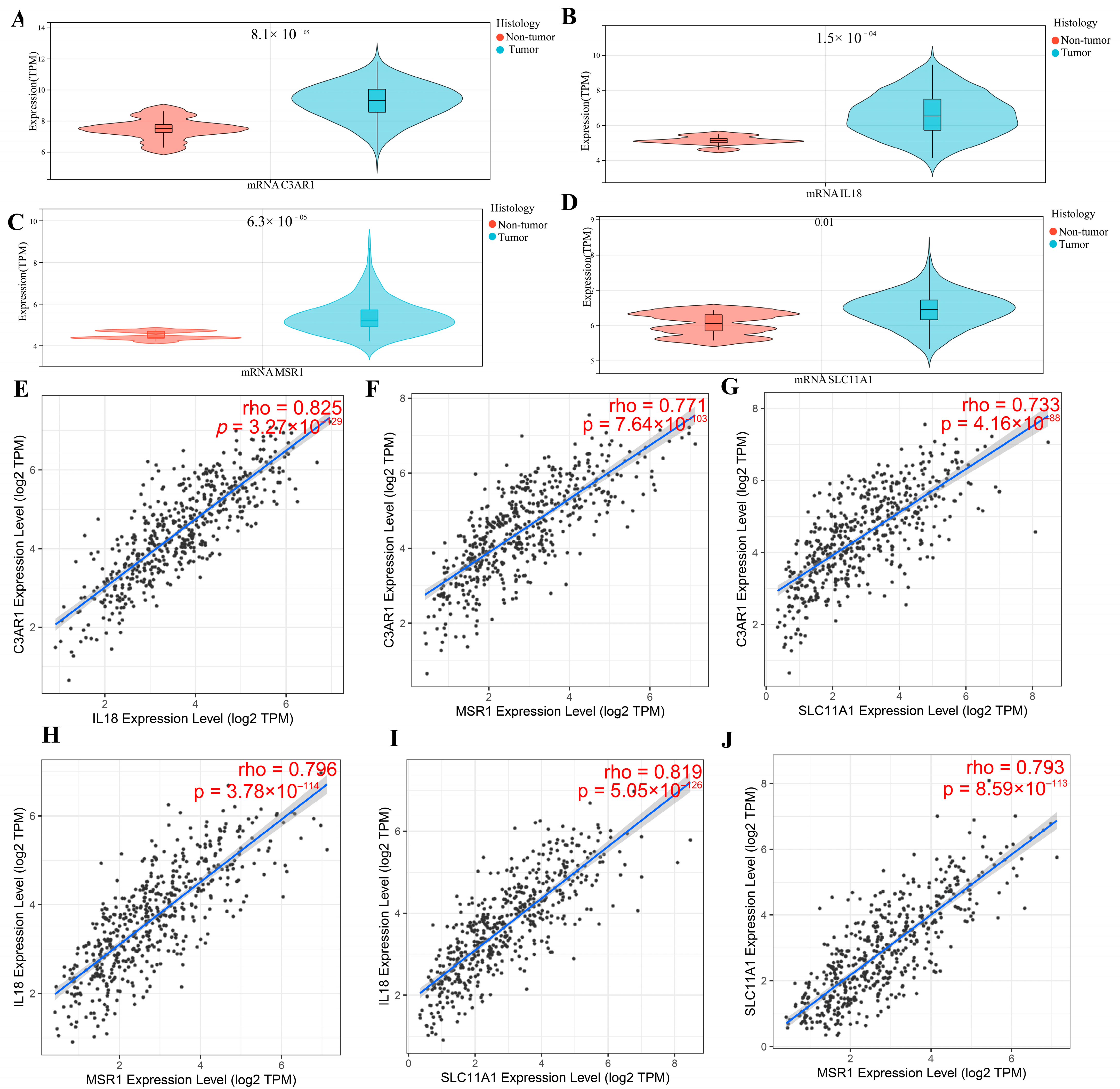
3.5. Verification of Hub Gene Expression in Normal Tissues and LGG Tissues
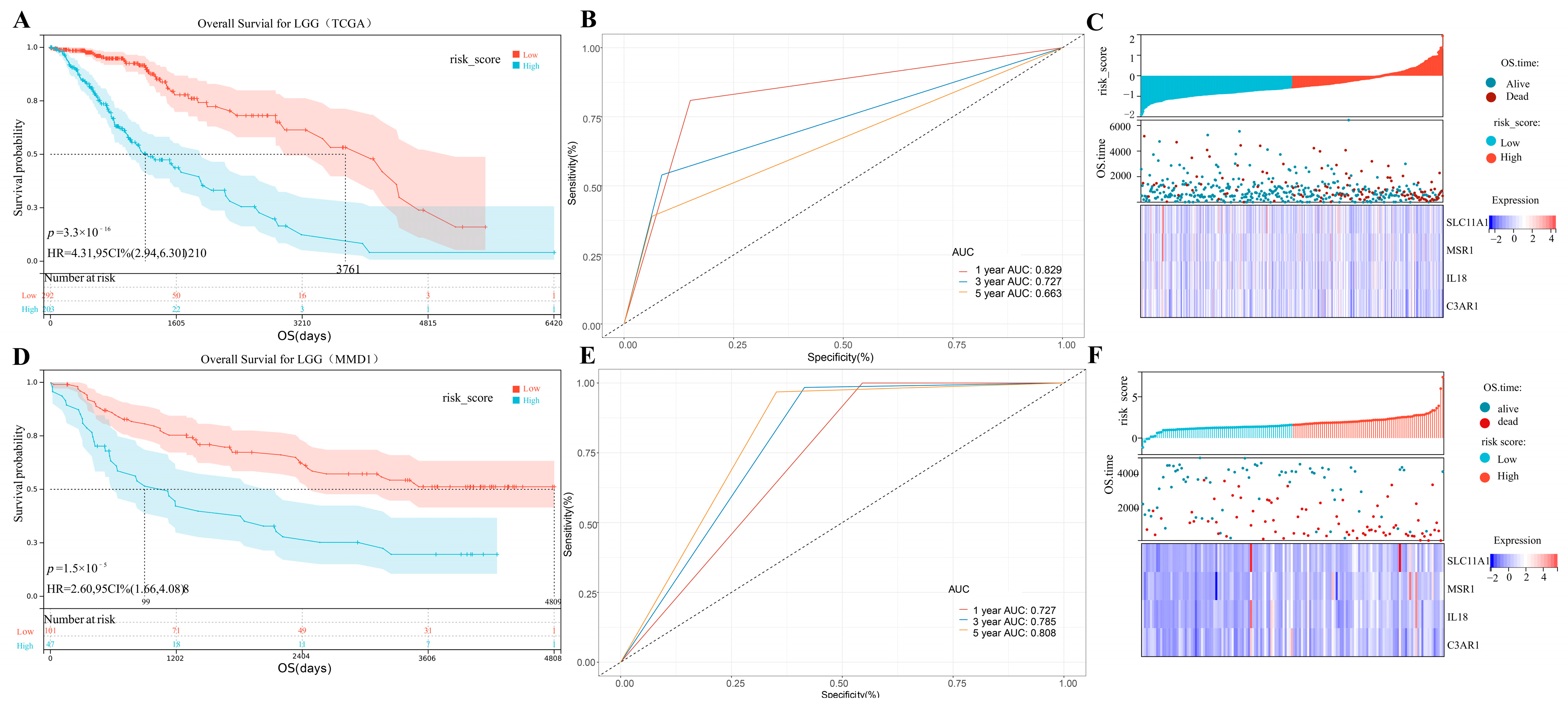
3.6. IPS Building
3.7. Clinical Nomogram Based on Created Immune-Related Prognostic Signature
3.8. GSEA Analysis
3.9. Correlation between Immune Infiltration and IPS in LGG
3.10. Predicting Immunotherapy Response
3.11. Validation of Hub Genes’ Expression
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Davis, M.E. Glioblastoma: Overview of Disease and Treatment. Clin. J. Oncol. Nurs. 2016, 20, S2–S8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sprooten, J.; Ceusters, J.; Coosemans, A.; Agostinis, P.; De Vleeschouwer, S.; Zitvogel, L.; Kroemer, G.; Galluzzi, L.; Garg, A.D. Trial watch: Dendritic cell vaccination for cancer immunotherapy. Oncoimmunology 2019, 8, e1638212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumthekar, P.; Raizer, J.; Singh, S. Low-Grade Glioma. In Current Understanding and Treatment of Gliomas; Cancer Treatment and Research; Raizer, J., Parsa, A., Eds.; Springer: Cham, Switzerland, 2015; Volume 163. [Google Scholar] [CrossRef]
- Zhang, X.; Li, X.; Xie, J.; Zhu, Q.; Yuan, Y. A Novel Immune-Related Prognostic Signature Predicting Survival in Patients with Pancreatic Adenocarcinoma. J. Oncol. 2022, 2022, 8909631. [Google Scholar] [CrossRef]
- Andersen, B.M.; Reardon, D.A. Immunotherapy approaches for adult glioma: Knowledge gained from recent clinical trials. Curr. Opin. Neurol. 2022, 35, 803–813. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Quan, W.; Luo, Q.; Pan, Y.; Peng, D.; Zhang, G. Identification of an Immune-Related Prognostic Predictor in Hepatocellular Carcinoma. Front. Mol. Biosci. 2020, 7, 567950. [Google Scholar] [CrossRef]
- Fu, M.; Wang, Q.; Wang, H.; Dai, Y.; Wang, J.; Kang, W.; Cui, Z.; Jin, X. Immune-Related Genes Are Prognostic Markers for Prostate Cancer Recurrence. Front. Genet. 2021, 12, 639642. [Google Scholar] [CrossRef]
- Tao, H.; Li, Z.; Mei, Y.; Li, X.; Lou, H.; Dong, L.; Zhou, L. Integrative bioinformatics analysis of a prognostic index and immunotherapeutic targets in renal cell carcinoma. Int. Immunopharmacol. 2020, 87, 106832. [Google Scholar] [CrossRef]
- Ye, S.; Yang, B.; Zhang, T.; Wei, W.; Li, Z.; Chen, J.; Li, X. Identification of an Immune-Related Prognostic Signature for Glioblastoma by Comprehensive Bioinformatics and Experimental Analyses. Cells 2022, 11, 3000. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Grzmil, M.; Morin, P.J.; Lino, M.M.; Merlo, A.; Frank, S.; Wang, Y.; Moncayo, G.; Hemmings, B.A. MAP kinase-interacting kinase 1 regulates SMAD2-dependent TGF-β signaling pathway in human glioblastoma. Cancer Res. 2011, 71, 2392–2402. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Hui, A.M.; Su, Q.; Vortmeyer, A.; Kotliarov, Y.; Pastorino, S.; Passaniti, A.; Menon, J.; Walling, J.; Bailey, R.; et al. Neuronal and glioma-derived stem cell factor induces angiogenesis within the brain. Cancer Cell 2006, 9, 287–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, A.M.; Kapoor, G.S.; Buonato, J.M.; Mathew, L.K.; Bi, Y.; Davuluri, R.V.; Martinez-Lage, M.; Simon, M.C.; O’Rourke, D.M.; Lazzara, M.J. Sprouty2 Drives Drug Resistance and Proliferation in Glioblastoma. Mol. Cancer Res. 2015, 13, 1227–1237. [Google Scholar] [CrossRef] [Green Version]
- Malta, T.M.; Sokolov, A.; Gentles, A.J.; Burzykowski, T.; Poisson, L.; Weinstein, J.N.; Kaminska, B.; Huelsken, J.; Omberg, L.; Gevaert, O.; et al. Machine learning identifies stemness features associated with oncogenic dedifferentiation. Cell 2018, 173, 338–354.e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, G.; Zhang, K.; Zhao, Y.; Yang, Q.; Lv, X. A novel stemness-hypoxia-related signature for prognostic stratification and immunotherapy response in hepatocellular carcinoma. BMC Cancer 2022, 22, 1103. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Z.; Cha, G.; Wang, Y.; Gao, J.; Jia, J. Characterizing the Crosstalk of NCAPG with Tumor Microenvironment and Tumor Stemness in Stomach Adenocarcinoma. Stem Cells Int. 2022, 2022, 1888358. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivliev, A.E.; Pa, T.H.; Sergeeva, M.G. Coexpression network analysis identifies transcriptional modules related to proastrocytic differentiation and sprouty signaling in glioma. Cancer Res. 2010, 70, 10060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [Green Version]
- Therneau, T.M. survival: Survival Analysis. Technometrics 2015, 46, 111–112. [Google Scholar]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef] [Green Version]
- Heagerty, P.J.; Lumley, T.; Pepe, M.S. Time-dependent ROC curves for censored survival data and a diagnostic marker. Biometrics 2000, 56, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Aut, M.G.; Aut, T.L. Forestplot: Advanced Forest Plot Using ‘Grid’ Graphics. 2016. Available online: http://CRAN.R-project.org/package=forestplot (accessed on 16 March 2023).
- Vickers, A.; Elkin, E. Decision curve analysis: A novel method for evaluating prediction models. Med. Decis. Mak. Int. J. Soc. Med. Decis. Mak. 2006, 26, 565. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Fan, J.; Wang, B.; Traugh, N.; Chen, Q.; Liu, J.S.; Li, B.; Liu, X.S. TIMER: A Web Server for Comprehensive Analysis of Tumor-Infiltrating Immune Cells. Cancer Res. 2017, 77, e108–e110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, A.M.; Steen, C.B.; Liu, C.L.; Gentles, A.J.; Chaudhuri, A.A.; Scherer, F.; Khodadoust, M.S.; Esfahani, M.S.; Luca, B.A.; Steiner, D.; et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol. 2019, 37, 773–782. [Google Scholar] [CrossRef]
- Carlsson, S.K.; Brothers, S.P.; Wahlestedt, C. Emerging treatment strategies for glioblastoma multiforme. Embo Mol. Med. 2014, 6, 1359–1370. [Google Scholar] [CrossRef]
- Hou, X.; Chen, J.; Zhang, Q.; Fan, Y.; Xiang, C.; Zhou, G.; Cao, F.; Yao, S. Interaction network of immune-associated genes affecting the prognosis of patients with glioblastoma. Exp. Ther. Med. 2021, 21, 61. [Google Scholar] [CrossRef]
- Klemm, F.; Maas, R.R.; Bowman, R.L.; Kornete, M.; Soukup, K.; Nassiri, S.; Brouland, J.P.; Iacobuzio-Donahue, C.A.; Brennan, C.; Tabar, V.; et al. Interrogation of the Microenvironmental Landscape in Brain Tumors Reveals Disease-Specific Alterations of Immune Cells. Cell 2020, 181, 1643–1660.e17. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.Q.; Li, Y.T.; Yan, T.F.; Xu, Y.; Liu, B.H.; Yang, J.A.; Yang, X.; Chen, Q.X.; Zhang, H.B. Six Immune Associated Genes Construct Prognostic Model Evaluate Low-Grade Glioma. Front. Immunol. 2020, 11, 606164. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Wang, X.; Chen, X.; Guo, F.; Hong, J. Prognostic Value of a Stemness Index-Associated Signature in Primary Lower-Grade Glioma. Front. Genet. 2020, 11, 441. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Tian, S.; Pan, Y.; Li, W.; Wang, Q.; Tang, Y.; Yu, T.; Wu, X.; Shi, Y.; Ma, P.; et al. Pyroptosis: A new frontier in cancer. Biomed. Pharmacother. 2020, 121, 109595. [Google Scholar] [CrossRef] [PubMed]
- Padala, C.; Puranam, K.; Shyamala, N.; Kupsal, K.; Kummari, R.; Galimudi, R.K.; Gundapaneni, K.K.; Tupurani, M.A.; Suryadevera, A.; Chinta, S.K.; et al. Genotypic and haplotype analysis of Interleukin-6 and -18 gene polymorphisms in association with clinicopathological factors in breast cancer. Cytokine 2022, 160, 156024. [Google Scholar] [CrossRef]
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef] [Green Version]
- Ji, Q.; Huang, K.; Jiang, Y.; Lei, K.; Tu, Z.; Luo, H.; Zhu, X. Comprehensive analysis of the prognostic and role in immune cell infiltration of MSR1 expression in lower-grade gliomas. Cancer Med. 2022, 11, 2020–2035. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.; Li, Z.; Guan, X.; Cai, K.; Wang, L.; Liu, J.; Tong, Y. The Research Progress of Host Genes and Tuberculosis Susceptibility. Oxid. Med. Cell. Longev. 2019, 2019, 9273056. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Q.; Meng, Y.; Li, S.; Xin, J.; Du, M.; Wang, M.; Cheng, G. Association of genetic variants in autophagy-lysosome pathway genes with susceptibility and survival to prostate cancer. Gene 2022, 808, 145953. [Google Scholar] [CrossRef]
- Qu, J.; Zhao, Q.; Yang, L.; Ping, Y.; Zhang, K.; Lei, Q.; Liu, F.; Zhang, Y. Identification and characterization of prognosis-related genes in the tumor microenvironment of esophageal squamous cell carcinoma. Int. Immunopharmacol. 2021, 96, 107616. [Google Scholar] [CrossRef]
- Yang, H.; Li, L.; Liu, X.; Zhao, Y. High Expression of the Component 3a Receptor 1 (C3AR1) Gene in Stomach Adenocarcinomas Infers a Poor Prognosis and High Immune-Infiltration Levels. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2021, 27, e927977. [Google Scholar] [CrossRef]












Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Ye, S.; Wu, D.; Xu, Z.; Wei, W.; Duan, F.; Luo, M. Identification, and Experimental and Bioinformatics Validation of an Immune-Related Prognosis Gene Signature for Low-Grade Glioma Based on mRNAsi. Cancers 2023, 15, 3238. https://doi.org/10.3390/cancers15123238
Wang Y, Ye S, Wu D, Xu Z, Wei W, Duan F, Luo M. Identification, and Experimental and Bioinformatics Validation of an Immune-Related Prognosis Gene Signature for Low-Grade Glioma Based on mRNAsi. Cancers. 2023; 15(12):3238. https://doi.org/10.3390/cancers15123238
Chicago/Turabian StyleWang, Yuan, Shengda Ye, Du Wu, Ziyue Xu, Wei Wei, Faliang Duan, and Ming Luo. 2023. "Identification, and Experimental and Bioinformatics Validation of an Immune-Related Prognosis Gene Signature for Low-Grade Glioma Based on mRNAsi" Cancers 15, no. 12: 3238. https://doi.org/10.3390/cancers15123238