
A Local and Abscopal Effect Observed with Liposomal Encapsulation of Intratumorally Injected Oncolytic Adenoviral Therapy
 , , , , ,
, , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents and Cell Lines
2.2. Liposome-Encapsulated Ad Synthesis
2.3. Transduction
2.4. Immunohistochemistry of Anti-GFP Staining
2.5. Cell Viability Assay
2.6. Animal Studies
2.7. Multiplex Immunofluorescence Staining
2.8. RT-qPCR Analysis
2.9. Statistical Analysis
3. Results and Discussion
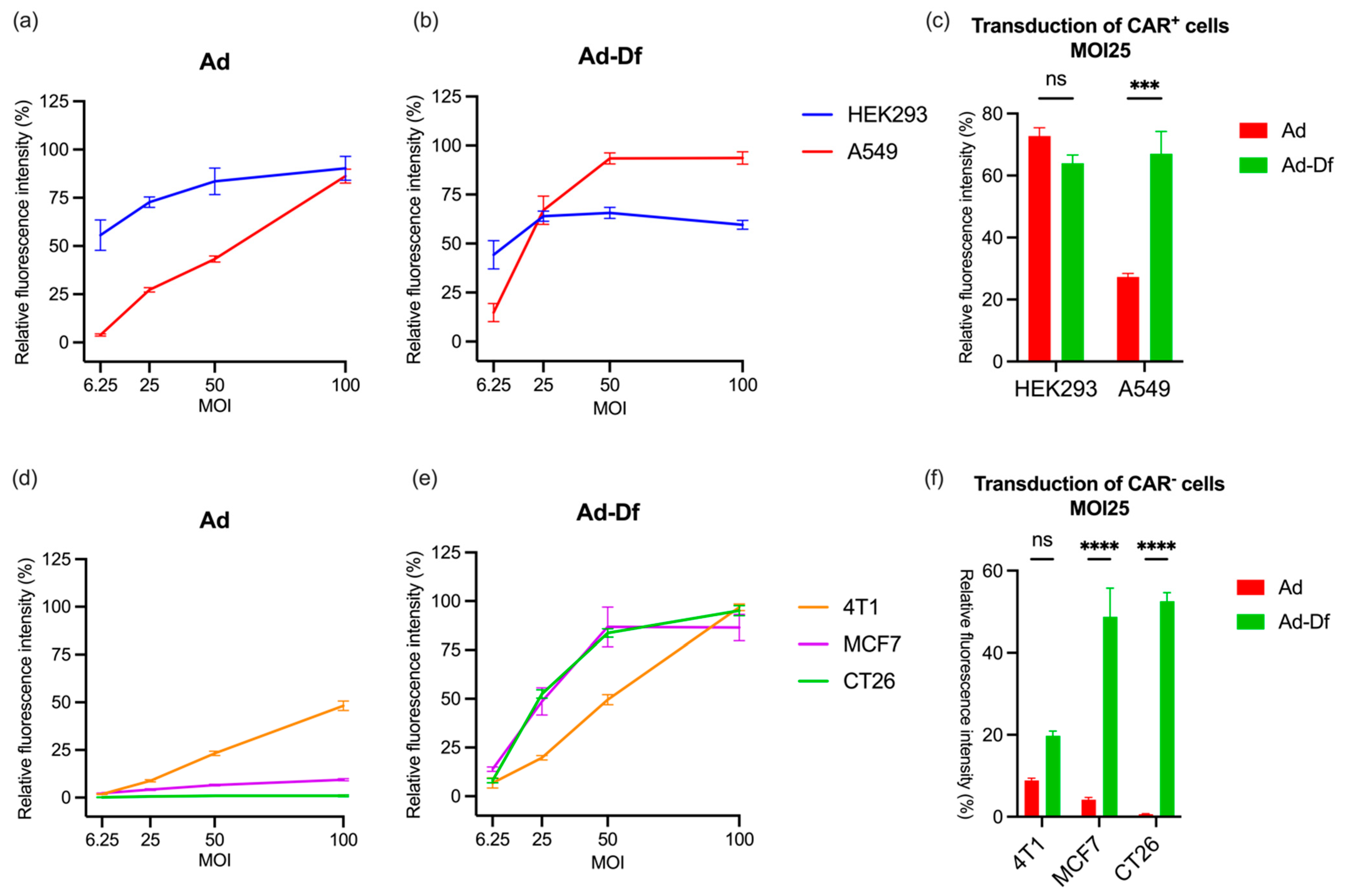
3.1. Transduction of CAR+ and CAR− Cells by Ad with or without Encapsulation In Vitro
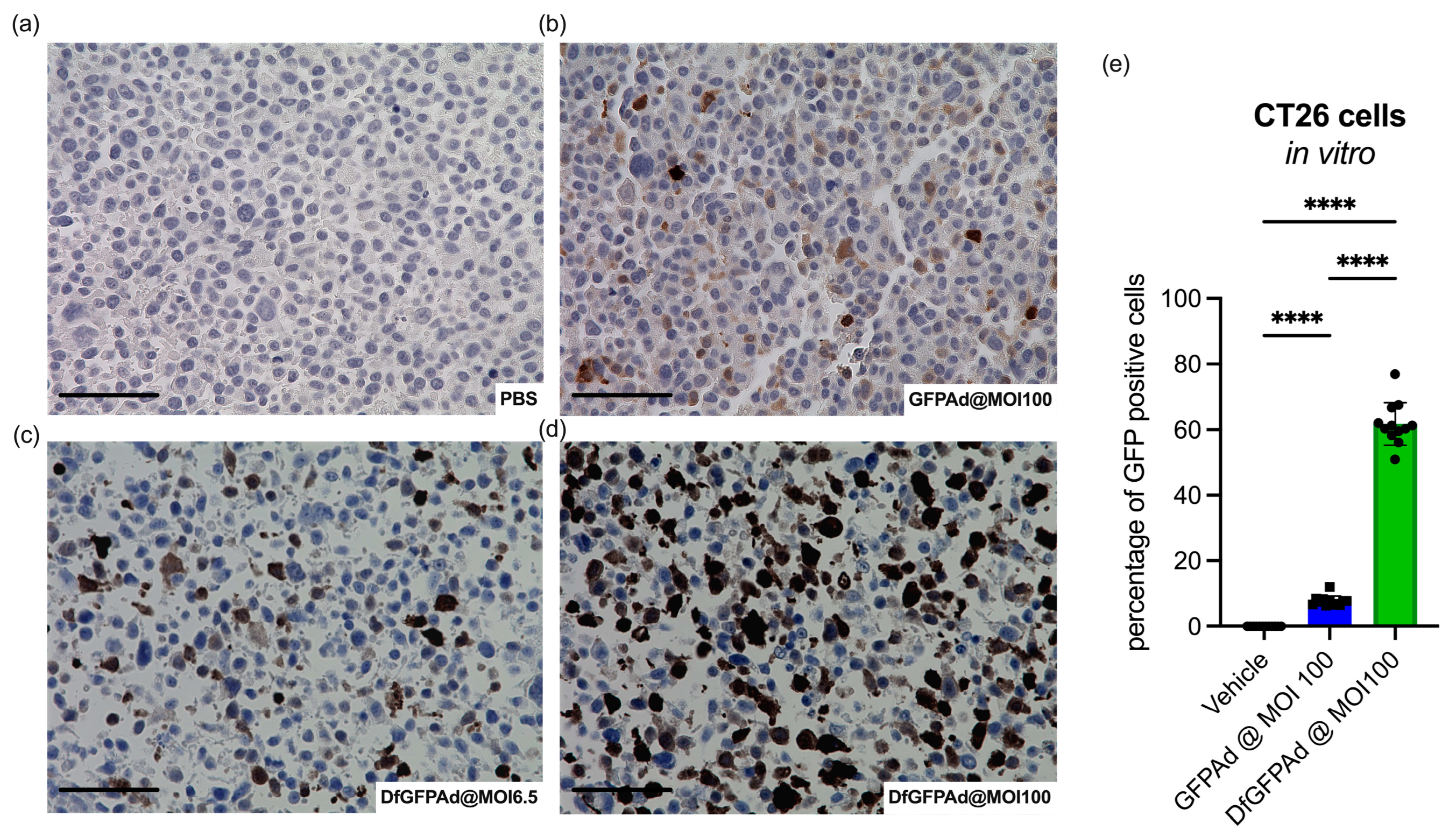
3.2. Ad Transduces CT26 Cells at High MOI as Confirmed by High Sensitivity Immunohistology Staining
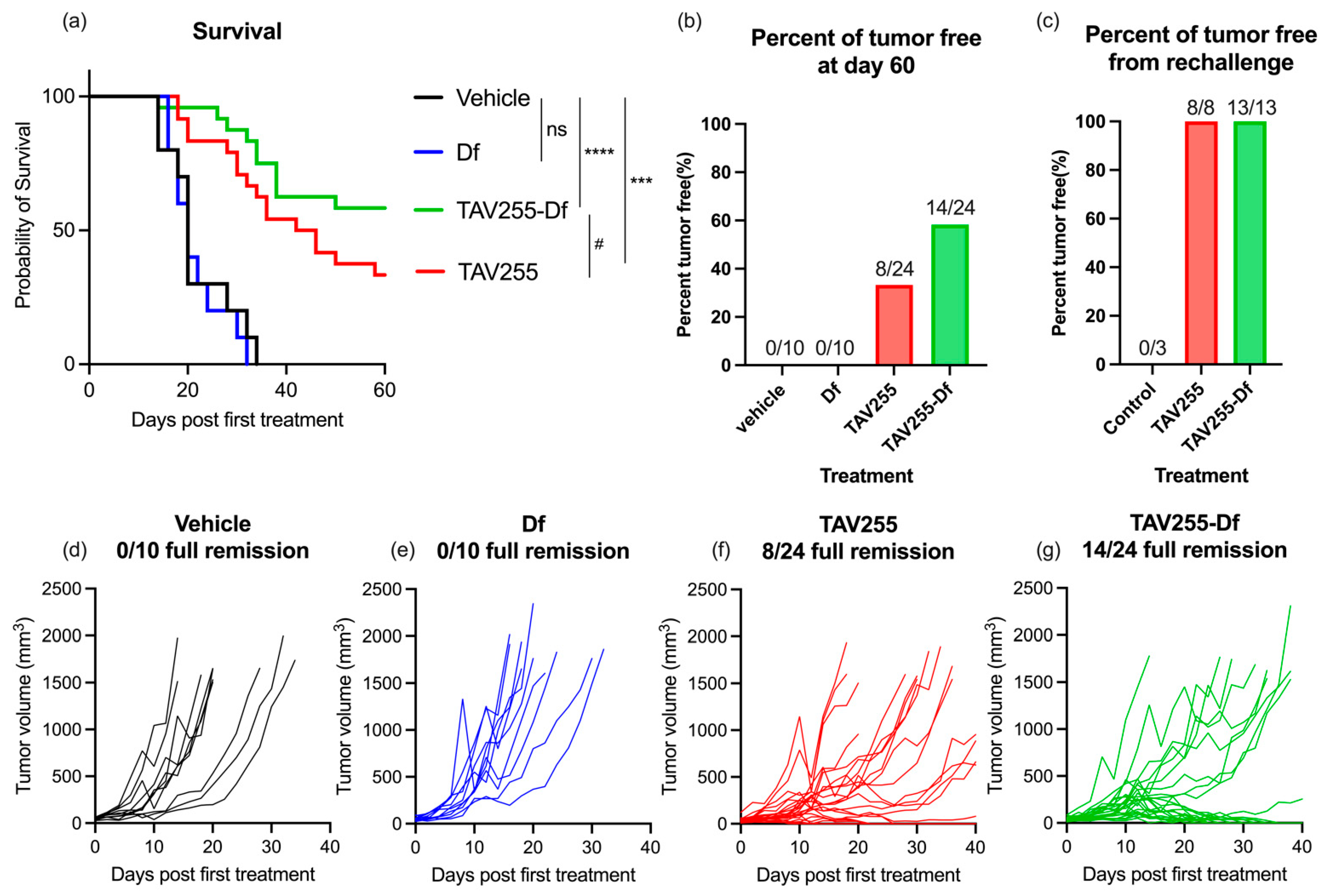
3.3. Anti-Tumor Effects of Unencapsulated and Encapsulated TAV-255 in a CT26 Subcutaneous Tumor Model
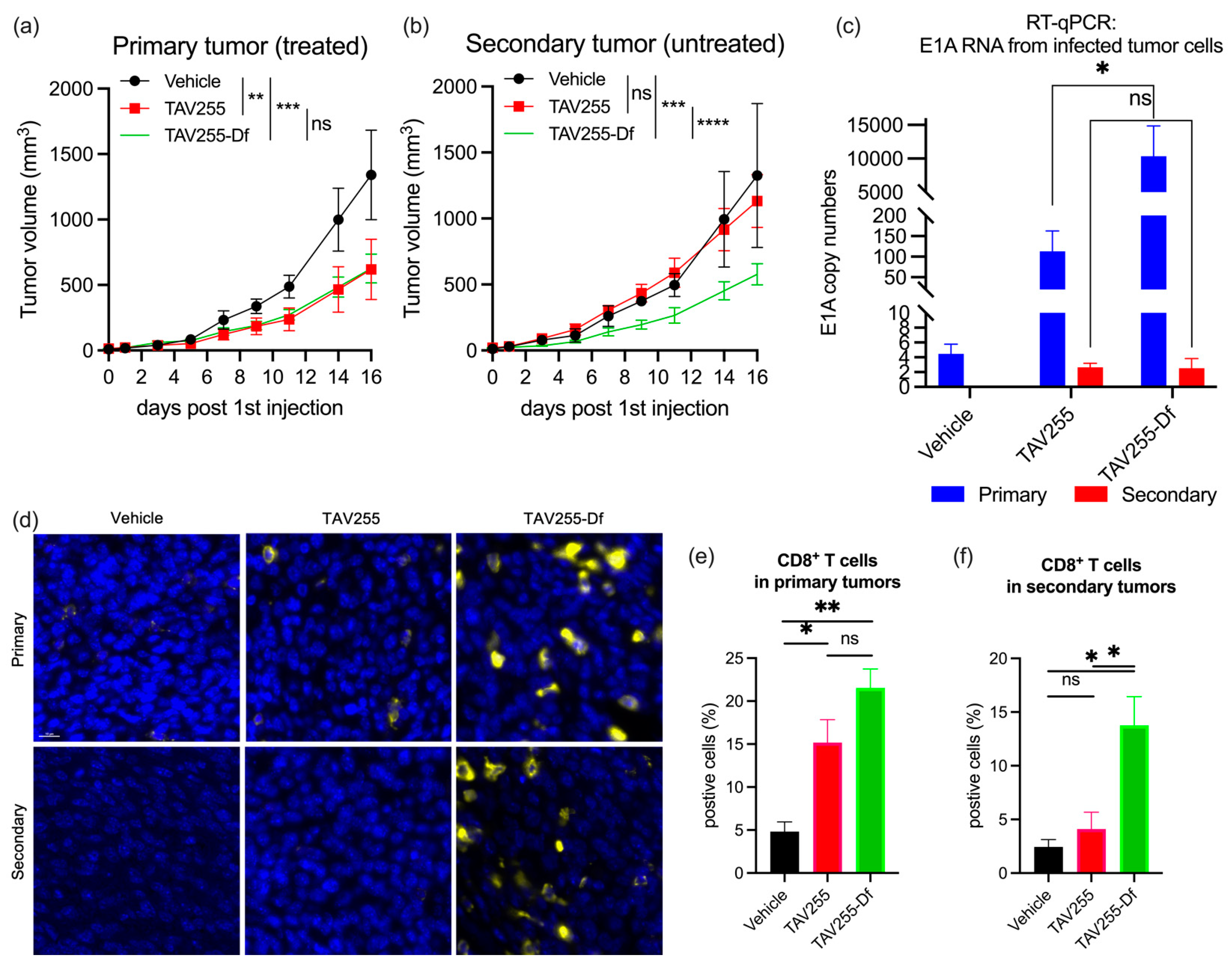
3.4. Unencapsulated and Encapsulated TAV255 Suppresses Tumor Growth in a CT26 Bilateral Tumor Model
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Robert, C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat. Commun. 2020, 11, 3801. [Google Scholar] [CrossRef]
- Xin Yu, J.; Hubbard-Lucey, V.M.; Tang, J. Immuno-oncology drug development goes global. Nat. Rev. Drug Discov. 2019, 18, 899–900. [Google Scholar] [CrossRef] [Green Version]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef] [Green Version]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [Green Version]
- Robert, C.; Schachter, J.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2015, 372, 2521–2532. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, E.V.; Chisamore, M.J.; Chaney, M.F.; Maradeo, M.E.; Anderson, J.; Baltus, G.A.; Pinheiro, E.M.; Uebele, V.N. Assessment of Clinical Activity of PD-1 Checkpoint Inhibitor Combination Therapies Reported in Clinical Trials. JAMA Netw. Open 2020, 3, e1920833. [Google Scholar] [CrossRef] [Green Version]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef] [Green Version]
- Larson, C.; Oronsky, B.; Carter, C.A.; Oronsky, A.; Knox, S.J.; Sher, D.; Reid, T.R. TGF-beta: A master immune regulator. Expert Opin. Ther. Targets 2020, 24, 427–438. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; He, F.; Li, D.; Xie, Y.J.; Jiang, Z.B.; Huang, J.M.; Zhao, X.P.; Nasim, A.A.; Chen, J.H.; Hou, J.C.; et al. PA-MSHA induces inflamed tumor microenvironment and sensitizes tumor to anti-PD-1 therapy. Cell Death Dis. 2022, 13, 931. [Google Scholar] [CrossRef] [PubMed]
- Harrington, K.; Freeman, D.J.; Kelly, B.; Harper, J.; Soria, J.C. Optimizing oncolytic virotherapy in cancer treatment. Nat. Rev. Drug Discov. 2019, 18, 689–706. [Google Scholar] [CrossRef]
- Peter, M.; Kühnel, F. Oncolytic Adenovirus in Cancer Immunotherapy. Cancers 2020, 12, 3354. [Google Scholar] [CrossRef]
- Russell, S.J.; Peng, K.W.; Bell, J.C. Oncolytic virotherapy. Nat. Biotechnol. 2012, 30, 658–670. [Google Scholar] [CrossRef] [Green Version]
- Sze, D.Y.; Reid, T.R.; Rose, S.C. Oncolytic virotherapy. J. Vasc. Interv. Radiol. 2013, 24, 1115–1122. [Google Scholar] [CrossRef]
- Bischoff, J.R.; Kirn, D.H.; Williams, A.; Heise, C.; Horn, S.; Muna, M.; Ng, L.; Nye, J.A.; Sampson-Johannes, A.; Fattaey, A.; et al. An adenovirus mutant that replicates selectively in p53-deficient human tumor cells. Science 1996, 274, 373–376. [Google Scholar] [CrossRef]
- Zhang, L.; Hedjran, F.; Larson, C.; Perez, G.L.; Reid, T. A novel immunocompetent murine model for replicating oncolytic adenoviral therapy. Cancer Gene Ther. 2015, 22, 17–22. [Google Scholar] [CrossRef] [Green Version]
- Chianese, A.; Santella, B.; Ambrosino, A.; Stelitano, D.; Rinaldi, L.; Galdiero, M.; Zannella, C.; Franci, G. Oncolytic Viruses in Combination Therapeutic Approaches with Epigenetic Modulators: Past, Present, and Future Perspectives. Cancers 2021, 13, 2761. [Google Scholar] [CrossRef]
- Hedjran, F.; Shantanu, K.; Tony, R. Deletion analysis of Ad5 E1a transcriptional control region: Impact on tumor-selective expression of E1a and E1b. Cancer Gene Ther. 2011, 18, 717–723. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Fueyo, J. Healing after death: Antitumor immunity induced by oncolytic adenoviral therapy. Oncoimmunology 2014, 3, e947872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kesari, S.; Bessudo, A.; Gastman, B.R.; Conley, A.P.; Villaflor, V.M.; Nabell, L.M.; Madere, D.; Chacon, E.; Spencer, C.; Li, L.; et al. BETA PRIME: Phase I study of AdAPT-001 as monotherapy and combined with a checkpoint inhibitor in superficially accessible, treatment-refractory solid tumors. Future Oncol. 2022, 18, 3245–3254. [Google Scholar] [CrossRef] [PubMed]
- Larson, C.; Oronsky, B.; Abrouk, N.E.; Oronsky, A.; Reid, T.R. Toxicology and biodistribution of AdAPT-001, a replication-competent type 5 adenovirus with a trap for the immunosuppressive cytokine, TGF-beta. Am. J. Cancer Res. 2021, 11, 5184–5189. [Google Scholar] [PubMed]
- Larson, C.; Oronsky, B.; Reid, T. AdAPT-001, an oncolytic adenovirus armed with a TGF-β trap, overcomes in vivo resistance to PD-L1-immunotherapy. Am. J. Cancer Res. 2022, 12, 3141–3147. [Google Scholar] [PubMed]
- Bergelson, J.M.; Cunningham, J.A.; Droguett, G.; Kurt-Jones, E.A.; Krithivas, A.; Hong, J.S.; Horwitz, M.S.; Crowell, R.L.; Finberg, R.W. Isolation of a common receptor for Coxsackie B viruses and adenoviruses 2 and 5. Science 1997, 275, 1320–1323. [Google Scholar] [CrossRef]
- Mach, N.; Gao, J.; Schaffarczyk, L.; Janz, S.; Ehrke-Schulz, E.; Dittmar, T.; Ehrhardt, A.; Zhang, W. Spectrum-Wide Exploration of Human Adenoviruses for Breast Cancer Therapy. Cancers 2020, 12, 1403. [Google Scholar] [CrossRef]
- Mendez, N.; Herrera, V.; Zhang, L.; Hedjran, F.; Feuer, R.; Blair, S.L.; Trogler, W.C.; Reid, T.R.; Kummel, A.C. Encapsulation of adenovirus serotype 5 in anionic lecithin liposomes using a bead-based immunoprecipitation technique enhances transfection efficiency. Biomaterials 2014, 35, 9554–9561. [Google Scholar] [CrossRef] [Green Version]
- Tomko, R.P.; Xu, R.; Philipson, L. HCAR and MCAR: The human and mouse cellular receptors for subgroup C adenoviruses and group B coxsackieviruses. Proc. Natl. Acad. Sci. USA 1997, 94, 3352–3356. [Google Scholar] [CrossRef] [Green Version]
- Law, L.K.; Davidson, B.L. What does it take to bind CAR? Mol. Ther. 2005, 12, 599–609. [Google Scholar] [CrossRef]
- Roelvink, P.W.; Mi Lee, G.; Einfeld, D.A.; Kovesdi, I.; Wickham, T.J. Identification of a conserved receptor-binding site on the fiber proteins of CAR-recognizing adenoviridae. Science 1999, 286, 1568–1571. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Bergelson, J.M. Adenovirus receptors. J. Virol. 2005, 79, 12125–12131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wunder, T.; Schmid, K.; Wicklein, D.; Groitl, P.; Dobner, T.; Lange, T.; Anders, M.; Schumacher, U. Expression of the coxsackie adenovirus receptor in neuroendocrine lung cancers and its implications for oncolytic adenoviral infection. Cancer Gene Ther. 2013, 20, 25–32. [Google Scholar] [CrossRef]
- Wunder, T.; Schumacher, U.; Friedrich, R.E. Coxsackie adenovirus receptor expression in carcinomas of the head and neck. Anticancer Res. 2012, 32, 1057–1062. [Google Scholar]
- Anders, M.; Vieth, M.; Röcken, C.; Ebert, M.; Pross, M.; Gretschel, S.; Schlag, P.M.; Wiedenmann, B.; Kemmner, W.; Höcker, M. Loss of the coxsackie and adenovirus receptor contributes to gastric cancer progression. Br. J. Cancer 2009, 100, 352–359. [Google Scholar] [CrossRef]
- Sato, Y.; Fu, Y.; Liu, H.; Lee, M.Y.; Shaw, M.H. Tumor-immune profiling of CT-26 and Colon 26 syngeneic mouse models reveals mechanism of anti-PD-1 response. BMC Cancer 2021, 21, 1222. [Google Scholar] [CrossRef] [PubMed]
- Vijayaraghavan, S.; Karakas, C.; Doostan, I.; Chen, X.; Bui, T.; Yi, M.; Raghavendra, A.S.; Zhao, Y.; Bashour, S.I.; Ibrahim, N.K.; et al. CDK4/6 and autophagy inhibitors synergistically induce senescence in Rb positive cytoplasmic cyclin E negative cancers. Nat. Commun. 2017, 8, 15916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.; Skora, A.D.; Li, Z.; Liu, Q.; Tam, A.J.; Blosser, R.L.; Diaz, L.A., Jr.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B.; et al. Eradication of metastatic mouse cancers resistant to immune checkpoint blockade by suppression of myeloid-derived cells. Proc. Natl. Acad. Sci. USA 2014, 111, 11774–11779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Napolitano, F.; Di Somma, S.; Castellano, G.; Amato, J.; Pagano, B.; Randazzo, A.; Portella, G.; Malfitano, A.M. Combination of dl922-947 Oncolytic Adenovirus and G-Quadruplex Binders Uncovers Improved Antitumor Activity in Breast Cancer. Cells 2022, 11, 2482. [Google Scholar] [CrossRef]
- Tanaka, T.; Uchida, H. Inhibition of Survivin by Adenovirus Vector Enhanced Paclitaxel-induced Apoptosis in Breast Cancer Cells. Anticancer. Res. 2018, 38, 4281–4288. [Google Scholar] [CrossRef] [PubMed]
- McKenna, M.K.; Rosewell-Shaw, A.; Suzuki, M. Modeling the Efficacy of Oncolytic Adenoviruses In Vitro and In Vivo: Current and Future Perspectives. Cancers 2020, 12, 619. [Google Scholar] [CrossRef] [Green Version]
- Han, S.Y.; Lee, Y.J.; Jung, H.I.; Lee, S.W.; Lim, S.J.; Hong, S.H.; Jeong, J.S. Gene transfer using liposome-complexed adenovirus seems to overcome limitations due to coxsackievirus and adenovirus receptor-deficiency of cancer cells, both in vitro and in vivo. Exp. Mol. Med. 2008, 40, 427–434. [Google Scholar] [CrossRef]
- Shah, J.R.; Dong, T.; Phung, A.T.; Reid, T.; Larson, C.; Sanchez, A.B.; Oronsky, B.; Blair, S.L.; Aisagbonhi, O.; Trogler, W.C.; et al. Development of Adenovirus Containing Liposomes Produced by Extrusion vs. Homogenization: A Comparison for Scale-Up Purposes. Bioengineering 2022, 9, 620. [Google Scholar] [CrossRef]
- Huang, C.H.; Dong, T.; Phung, A.T.; Shah, J.R.; Larson, C.; Sanchez, A.B.; Blair, S.L.; Oronsky, B.; Trogler, W.C.; Reid, T.; et al. Full Remission of CAR-Deficient Tumors by DOTAP-Folate Liposome Encapsulation of Adenovirus. ACS Biomater. Sci. Eng. 2022, 8, 5199–5209. [Google Scholar] [CrossRef]
- Yamano, T.; Kubo, S.; Fukumoto, M.; Yano, A.; Mawatari-Furukawa, Y.; Okamura, H.; Tomita, N. Whole cell vaccination using immunogenic cell death by an oncolytic adenovirus is effective against a colorectal cancer model. Mol. Ther. Oncolytics 2016, 3, 16031. [Google Scholar] [CrossRef] [PubMed]
- Bahrami, B.; Mohammadnia-Afrouzi, M.; Bakhshaei, P.; Yazdani, Y.; Ghalamfarsa, G.; Yousefi, M.; Sadreddini, S.; Jadidi-Niaragh, F.; Hojjat-Farsangi, M. Folate-conjugated nanoparticles as a potent therapeutic approach in targeted cancer therapy. Tumor Biol. 2015, 36, 5727–5742. [Google Scholar] [CrossRef] [PubMed]
- Handali, S.; Moghimipour, E.; Rezaei, M.; Ramezani, Z.; Kouchak, M.; Amini, M.; Angali, K.A.; Saremy, S.; Dorkoosh, F.A. A novel 5-Fluorouracil targeted delivery to colon cancer using folic acid conjugated liposomes. Biomed. Pharmacother. 2018, 108, 1259–1273. [Google Scholar] [CrossRef] [PubMed]
- Guzik, P.; Siwowska, K.; Fang, H.-Y.; Cohrs, S.; Bernhardt, P.; Schibli, R.; Müller, C. Promising potential of [177Lu]Lu-DOTA-folate to enhance tumor response to immunotherapy—A preclinical study using a syngeneic breast cancer model. Eur. J. Nucl. Med. Mol. Imaging 2021, 48, 984–994. [Google Scholar] [CrossRef]
- Toth, Z.E.; Shahar, T.; Leker, R.; Szalayova, I.; Bratincsák, A.; Key, S.; Lonyai, A.; Németh, K.; Mezey, E. Sensitive detection of GFP utilizing tyramide signal amplification to overcome gene silencing. Exp. Cell Res. 2007, 313, 1943–1950. [Google Scholar] [CrossRef] [Green Version]
- Shi, G.; Yang, Q.; Zhang, Y.; Jiang, Q.; Lin, Y.; Yang, S.; Wang, H.; Cheng, L.; Zhang, X.; Li, Y.; et al. Modulating the Tumor Microenvironment via Oncolytic Viruses and CSF-1R Inhibition Synergistically Enhances Anti-PD-1 Immunotherapy. Mol. Ther. 2019, 27, 244–260. [Google Scholar] [CrossRef] [Green Version]
- Preston, C.C.; Maurer, M.J.; Oberg, A.L.; Visscher, D.W.; Kalli, K.R.; Hartmann, L.C.; Goode, E.L.; Knutson, K.L. The ratios of CD8+ T cells to CD4+CD25+ FOXP3+ and FOXP3- T cells correlate with poor clinical outcome in human serous ovarian cancer. PLoS ONE 2013, 8, e80063. [Google Scholar] [CrossRef] [Green Version]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dong, T.; Shah, J.R.; Phung, A.T.; Larson, C.; Sanchez, A.B.; Aisagbonhi, O.; Blair, S.L.; Oronsky, B.; Trogler, W.C.; Reid, T.; et al. A Local and Abscopal Effect Observed with Liposomal Encapsulation of Intratumorally Injected Oncolytic Adenoviral Therapy. Cancers 2023, 15, 3157. https://doi.org/10.3390/cancers15123157
Dong T, Shah JR, Phung AT, Larson C, Sanchez AB, Aisagbonhi O, Blair SL, Oronsky B, Trogler WC, Reid T, et al. A Local and Abscopal Effect Observed with Liposomal Encapsulation of Intratumorally Injected Oncolytic Adenoviral Therapy. Cancers. 2023; 15(12):3157. https://doi.org/10.3390/cancers15123157
Chicago/Turabian StyleDong, Tao, Jaimin R. Shah, Abraham T. Phung, Christopher Larson, Ana B. Sanchez, Omonigho Aisagbonhi, Sarah L. Blair, Bryan Oronsky, William C. Trogler, Tony Reid, and et al. 2023. "A Local and Abscopal Effect Observed with Liposomal Encapsulation of Intratumorally Injected Oncolytic Adenoviral Therapy" Cancers 15, no. 12: 3157. https://doi.org/10.3390/cancers15123157