
Unpacking the Complexity of Epithelial Plasticity: From Master Regulator Transcription Factors to Non-Coding RNAs
 ,
,  , , and
, , and 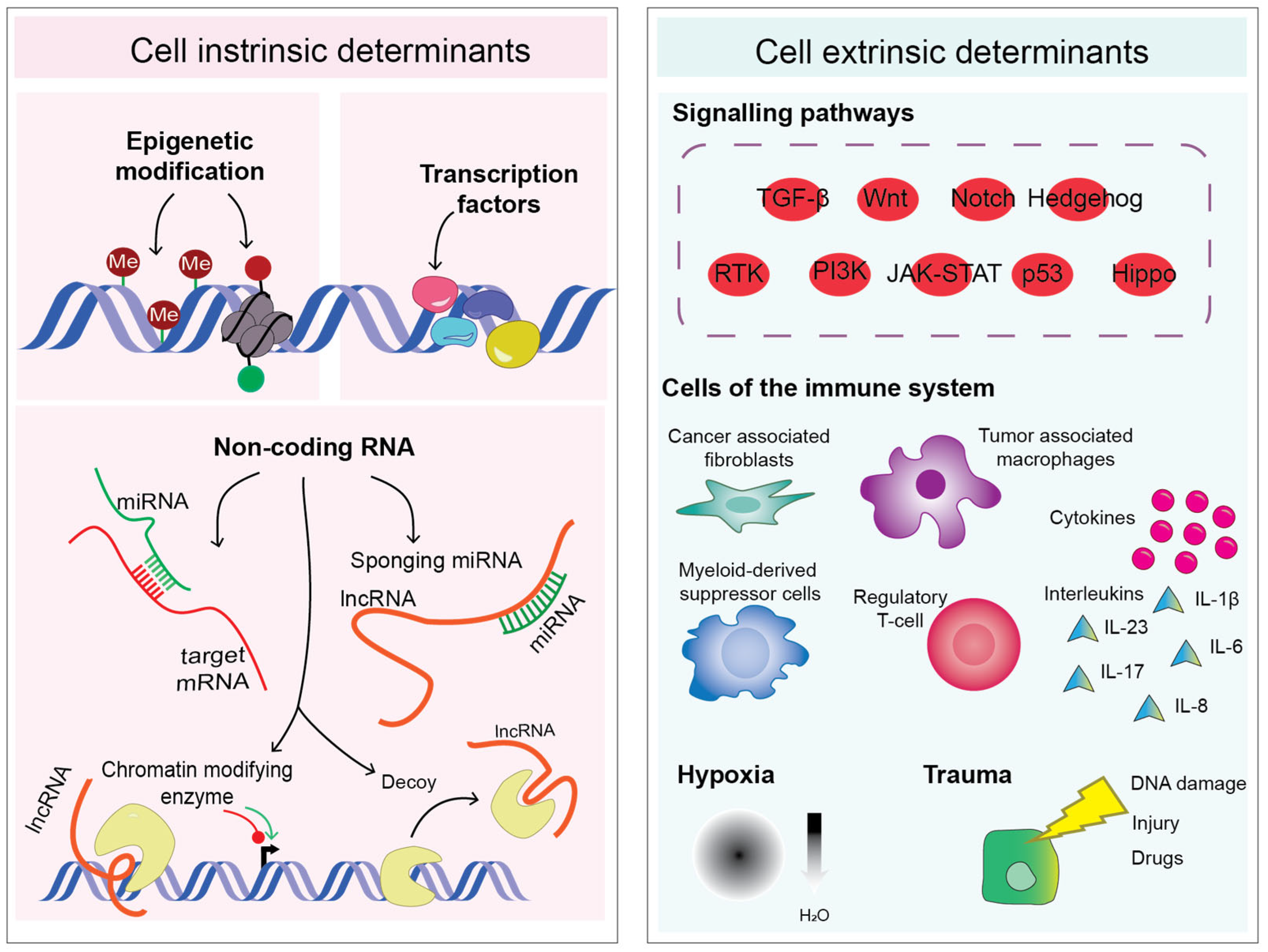{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
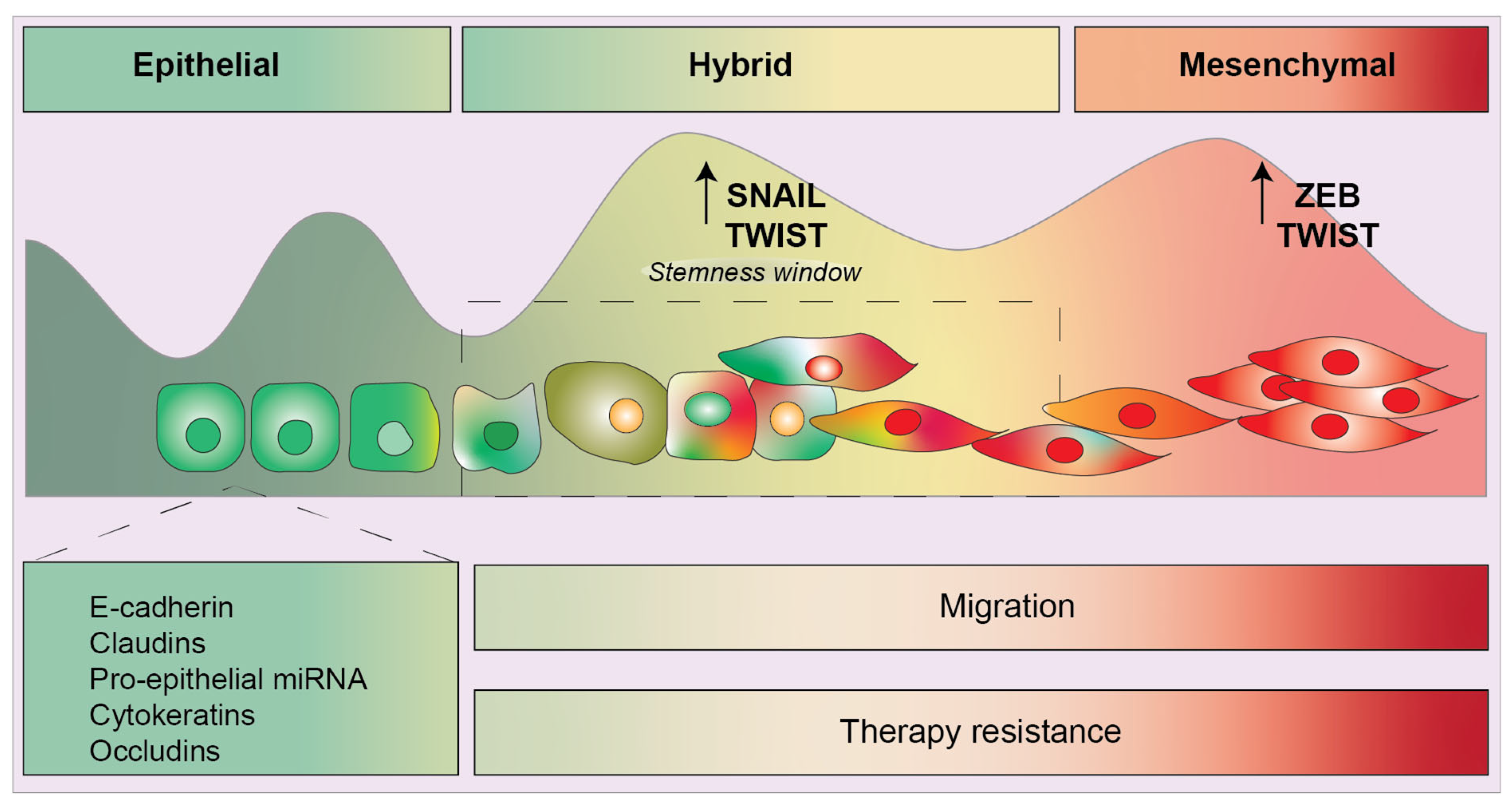
1. Introduction
2. Epigenetics and the Transcription Factor Network
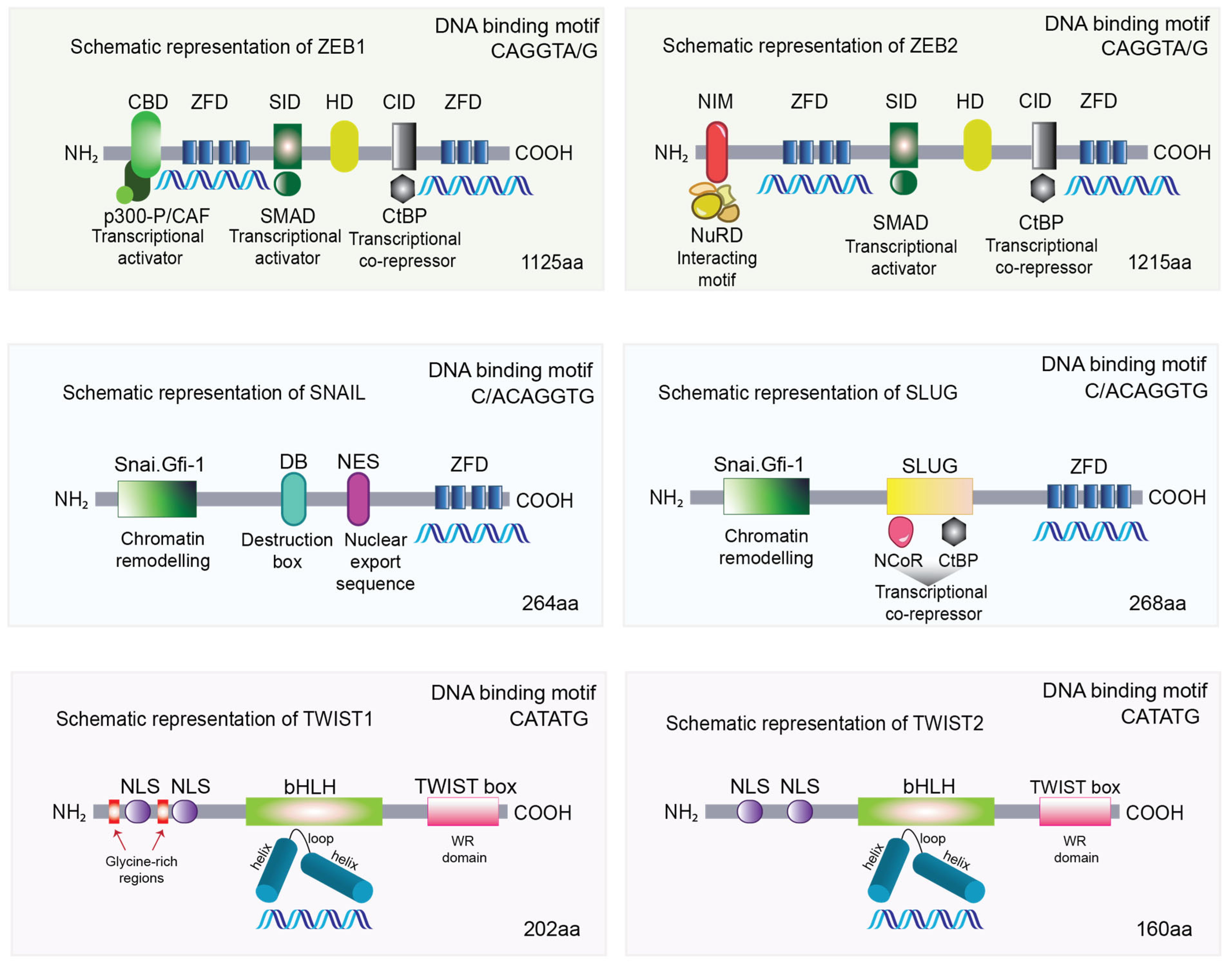
2.1. SNAIL and SLUG
2.2. TWIST1 and TWIST2
2.3. ZEB1 and ZEB2
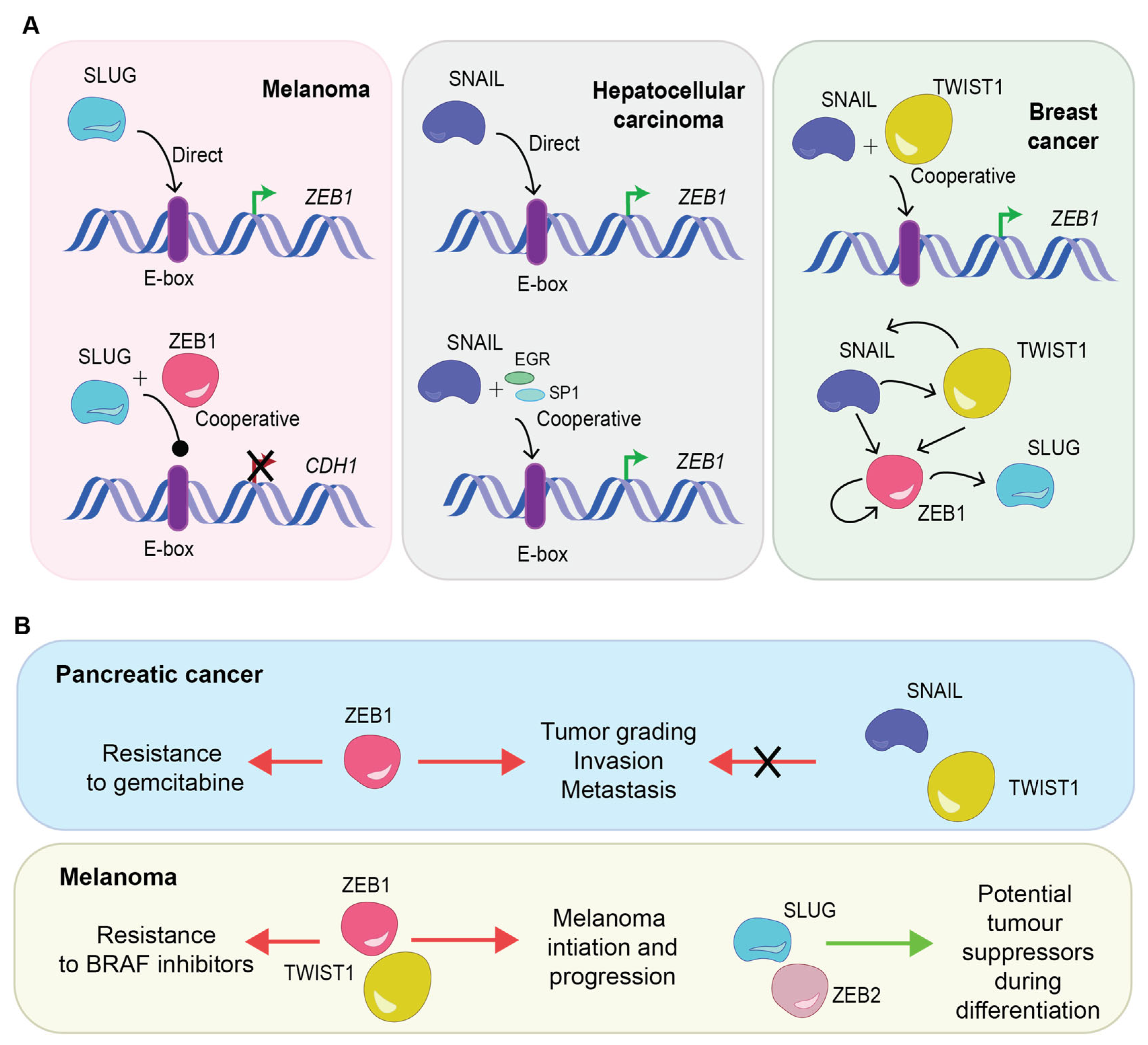
3. Transcription Factor Cooperative Regulation
4. EMT-TFs Are Induced by Proinflammatory Mediators
5. EMT-TFs Facilitate Immune Evasion
6. Non-Coding RNAs Add Another Layer of Intricacy
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Greenburg, G.; Hay, E.D. Epithelia Suspended in Collagen Gels Can Lose Polarity and Express Characteristics of Migrating Mesenchymal Cells. J. Cell Biol. 1982, 95, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef]
- Lachat, C.; Peixoto, P.; Hervouet, E. Epithelial to Mesenchymal Transition History: From Embryonic Development to Cancers. Biomolecules 2021, 11, 782. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and definitions for research on epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef]
- San Juan, B.P.; Garcia-Leon, M.J.; Rangel, L.; Goetz, J.G.; Chaffer, C.L. The Complexities of Metastasis. Cancers 2019, 11, 1575. [Google Scholar] [CrossRef] [PubMed]
- Jolly, M.K.; Boareto, M.; Huang, B.; Jia, D.; Lu, M.; Ben-Jacob, E.; Onuchic, J.N.; Levine, H. Implications of the Hybrid Epithelial/Mesenchymal Phenotype in Metastasis. Front. Oncol. 2015, 5, 155. [Google Scholar] [CrossRef] [PubMed]
- Jolly, M.K.; Murphy, R.J.; Bhatia, S.; Whitfield, H.J.; Redfern, A.; Davis, M.J.; Thompson, E.W. Measuring and Modelling the Epithelial- Mesenchymal Hybrid State in Cancer: Clinical Implications. Cells Tissues Organs 2022, 211, 110–133. [Google Scholar] [CrossRef]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef]
- Grosse-Wilde, A.; D’hérouël, A.F.; McIntosh, E.; Ertaylan, G.; Skupin, A.; Kuestner, R.E.; del Sol, A.; Walters, K.-A.; Huang, S. Stemness of the hybrid Epithelial/Mesenchymal State in Breast Cancer and Its Association with Poor Survival. PLoS ONE 2015, 10, e0126522. [Google Scholar] [CrossRef]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef]
- Cursons, J.; Pillman, K.A.; Scheer, K.G.; Gregory, P.A.; Foroutan, M.; Hediyeh-Zadeh, S.; Toubia, J.; Crampin, E.J.; Goodall, G.J.; Bracken, C.P.; et al. Combinatorial Targeting by MicroRNAs Co-ordinates Post-transcriptional Control of EMT. Cell Syst. 2018, 7, 77–91.e77. [Google Scholar] [CrossRef]
- Gregory, P.A.; Bert, A.G.; Paterson, E.L.; Barry, S.C.; Tsykin, A.; Farshid, G.; Vadas, M.A.; Khew-Goodall, Y.; Goodall, G.J. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat. Cell Biol. 2008, 10, 593–601. [Google Scholar] [CrossRef]
- Bracken, C.P.; Gregory, P.A.; Kolesnikoff, N.; Bert, A.G.; Wang, J.; Shannon, M.F.; Goodall, G.J. A double-negative feedback loop between ZEB1-SIP1 and the microRNA-200 family regulates epithelial-mesenchymal transition. Cancer Res. 2008, 68, 7846–7854. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.A.; Guo, W.; Liao, M.-J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Gregory, P.A.; Bracken, C.P.; Bert, A.G.; Goodall, G.J. MicroRNAs as regulators of epithelial-mesenchymal transition. Cell Cycle 2008, 7, 3112–3118. [Google Scholar] [CrossRef]
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt signaling in cancer. Oncogene 2017, 36, 1461–1473. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Sleeman, J.P. Complex networks orchestrate epithelial-mesenchymal transitions. Nat. Rev. Mol. Cell Biol. 2006, 7, 131–142. [Google Scholar] [CrossRef]
- Sahlgren, C.; Gustafsson, M.V.; Jin, S.; Poellinger, L.; Lendahl, U. Notch signaling mediates hypoxia-induced tumor cell migration and invasion. Proc. Natl. Acad. Sci. USA 2008, 105, 6392–6397. [Google Scholar] [CrossRef]
- Pan, G.; Liu, Y.; Shang, L.; Zhou, F.; Yang, S. EMT-associated microRNAs and their roles in cancer stemness and drug resistance. Cancer Commun. 2021, 41, 199–217. [Google Scholar] [CrossRef]
- Drak Alsibai, K.; Meseure, D. Tumor microenvironment and noncoding RNAs as co-drivers of epithelial-mesenchymal transition and cancer metastasis. Dev. Dyn. 2018, 247, 405–431. [Google Scholar] [CrossRef]
- Pastushenko, I.; Blanpain, C. EMT Transition States during Tumor Progression and Metastasis. Trends Cell Biol. 2019, 29, 212–226. [Google Scholar] [CrossRef] [PubMed]
- Pastushenko, I.; Brisebarre, A.; Sifrim, A.; Fioramonti, M.; Revenco, T.; Boumahdi, S.; Van Keymeulen, A.; Brown, D.; Moers, V.; Lemaire, S.; et al. Identification of the tumour transition states occurring during EMT. Nature 2018, 556, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Murakami, R.; Matsumura, N.; Mandai, M.; Yoshihara, K.; Tanabe, H.; Nakai, H.; Yamanoi, K.; Abiko, K.; Yoshioka, Y.; Hamanishi, J.; et al. Establishment of a Novel Histopathological Classification of High-Grade Serous Ovarian Carcinoma Correlated with Prognostically Distinct Gene Expression Subtypes. Am. J. Pathol. 2016, 186, 1103–1113. [Google Scholar] [CrossRef] [PubMed]
- Chae, Y.K.; Chang, S.; Ko, T.; Anker, J.; Agte, S.; Iams, W.; Choi, W.M.; Lee, K.; Cruz, M. Epithelial-mesenchymal transition (EMT) signature is inversely associated with T-cell infiltration in non-small cell lung cancer (NSCLC). Sci. Rep. 2018, 8, 2918. [Google Scholar] [CrossRef]
- Vasaikar, S.V.; Deshmukh, A.P.; Hollander, P.D.; Addanki, S.; Kuburich, N.A.; Kudaravalli, S.; Joseph, R.; Chang, J.T.; Soundararajan, R.; Mani, S.A. EMTome: A resource for pan-cancer analysis of epithelial-mesenchymal transition genes and signatures. Br. J. Cancer 2021, 124, 259–269. [Google Scholar] [CrossRef]
- Ray, I.; Michael, A.; Meira, L.B.; Ellis, P.E. The Role of Cytokines in Epithelial-Mesenchymal Transition in Gynaecological Cancers: A Systematic Review. Cells 2023, 12, 416. [Google Scholar] [CrossRef]
- Adekoya, T.O.; Richardson, R.M. Cytokines and Chemokines as Mediators of Prostate Cancer Metastasis. Int. J. Mol. Sci. 2021, 21, 4449. [Google Scholar] [CrossRef]
- Bhat, A.A.; Nisar, S.; Singh, M.; Ashraf, B.; Masoodi, T.; Prasad, C.P.; Sharma, A.; Maacha, S.; Karedath, T.; Hashem, S.; et al. Cytokine- and chemokine-induced inflammatory colorectal tumor microenvironment: Emerging avenue for targeted therapy. Cancer Commun. 2022, 42, 689–715. [Google Scholar] [CrossRef]
- Bhat, A.A.; Nisar, S.; Maacha, S.; Carneiro-Lobo, T.C.; Akhtar, S.; Siveen, K.S.; Wani, N.A.; Rizwan, A.; Bagga, P.; Singh, M.; et al. Cytokine-chemokine network driven metastasis in esophageal cancer; promising avenue for targeted therapy. Mol. Cancer 2021, 20, 2. [Google Scholar] [CrossRef]
- Sistigu, A.; Di Modugno, F.; Manic, G.; Nistico, P. Deciphering the loop of epithelial-mesenchymal transition, inflammatory cytokines and cancer immunoediting. Cytokine Growth Factor Rev. 2017, 36, 67–77. [Google Scholar] [CrossRef]
- Chaffer, C.L.; San Juan, B.P.; Lim, E.; Weinberg, R.A. EMT, cell plasticity and metastasis. Cancer Metastasis Rev. 2016, 35, 645–654. [Google Scholar] [CrossRef]
- Krebs, A.M.; Mitschke, J.; Lasierra Losada, M.; Schmalhofer, O.; Boerries, M.; Busch, H.; Boettcher, M.; Mougiakakos, D.; Reichardt, W.; Bronsert, P.; et al. The EMT-activator Zeb1 is a key factor for cell plasticity and promotes metastasis in pancreatic cancer. Nat. Cell Biol. 2017, 19, 518–529. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Tam, W.L.; Shibue, T.; Kaygusuz, Y.; Reinhardt, F.; Eaton, E.N.; Weinberg, R.A. Distinct EMT programs control normal mammary stem cells and tumour-initiating cells. Nature 2015, 525, 256–260. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A.; Bennett, M.F.; Sargent, M.G.; Wilkinson, D.G. Cloning and developmental expression of Sna, a murine homologue of the Drosophila snail gene. Development 1992, 116, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.E.; Franco del Amo, F.; Gridley, T. Isolation of Sna, a mouse gene homologous to the Drosophila genes snail and escargot: Its expression pattern suggests multiple roles during postimplantation development. Development 1992, 116, 1033–1039. [Google Scholar] [CrossRef] [PubMed]
- Prokop, J.W.; Liu, Y.; Milsted, A.; Peng, H.; Rauscher, F.J., 3rd. A method for in silico identification of SNAIL/SLUG DNA binding potentials to the E-box sequence using molecular dynamics and evolutionary conserved amino acids. J. Mol. Model. 2013, 19, 3463–3469. [Google Scholar] [CrossRef]
- Lin, Y.; Wu, Y.; Li, J.; Dong, C.; Ye, X.; Chi, Y.-I.; Evers, B.M.; Zhou, B.P. The SNAG domain of Snail1 functions as a molecular hook for recruiting lysine-specific demethylase 1. EMBO J. 2010, 29, 1803–1816. [Google Scholar] [CrossRef]
- Metzger, E.; Wissmann, M.; Yin, N.; Müller, J.M.; Schneider, R.; Peters, A.H.F.M.; Günther, T.; Buettner, R.; Schüle, R. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature 2005, 437, 436–439. [Google Scholar] [CrossRef]
- Peinado, H.; Ballestar, E.; Esteller, M.; Cano, A. Snail mediates E-cadherin repression by the recruitment of the Sin3A/histone deacetylase 1 (HDAC1)/HDAC2 complex. Mol. Cell Biol. 2004, 24, 306–319. [Google Scholar] [CrossRef]
- Dong, C.; Wu, Y.; Yao, J.; Wang, Y.; Yu, Y.; Rychahou, P.; Evers, B.M.; Zhou, B.P. G9a interacts with Snail and is critical for Snail-mediated E-cadherin repression in human breast cancer. J. Clin. Investig. 2012, 122, 1469–1486. [Google Scholar] [CrossRef]
- Zhang, J.; Fan, X.; Zhou, Y.; Chen, L.; Rao, H. The PRMT5-LSD1 axis confers Slug dual transcriptional activities and promotes breast cancer progression. J. Exp. Clin. Cancer Res. 2022, 41, 191. [Google Scholar] [CrossRef]
- Hou, Z.; Peng, H.; Ayyanathan, K.; Yan, K.-P.; Langer, E.M.; Longmore, G.D.; Rauscher, F.J. The LIM protein AJUBA recruits protein arginine methyltransferase 5 to mediate SNAIL-dependent transcriptional repression. Mol. Cell. Biol. 2008, 28, 3198–3207. [Google Scholar] [CrossRef] [PubMed]
- Molina-Ortiz, P.; Villarejo, A.; MacPherson, M.; Santos, V.; Montes, A.; Souchelnytskyi, S.; Portillo, F.; Cano, A. Characterization of the SNAG and SLUG domains of Snail2 in the repression of E-cadherin and EMT induction: Modulation by serine 4 phosphorylation. PLoS ONE 2012, 7, e36132. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Gross, K.M.; Kuperwasser, C. Molecular regulation of Snai2 in development and disease. J. Cell Sci. 2019, 132, jcs235127. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Estrada, O.M.; Cullerés, A.; Soriano, F.X.; Peinado, H.; Bolós, V.; Martínez, F.O.; Reina, M.; Cano, A.; Fabre, M.; Vilaró, S. The transcription factors Slug and Snail act as repressors of Claudin-1 expression in epithelial cells. Biochem. J. 2006, 394, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Guaita, S.; Puig, I.; Francí, C.; Garrido, M.; Domínguez, D.; Batlle, E.; Sancho, E.; Dedhar, S.; de Herreros, A.G.; Baulida, J. Snail induction of epithelial to mesenchymal transition in tumor cells is accompanied by MUC1 repression and ZEB1 expression. J. Biol. Chem. 2002, 277, 39209–39216. [Google Scholar] [CrossRef] [PubMed]
- Phillips, S.; Kuperwasser, C. SLUG: Critical regulator of epithelial cell identity in breast development and cancer. Cell Adh. Migr. 2014, 8, 578–587. [Google Scholar] [CrossRef]
- Mittal, M.K.; Myers, J.N.; Misra, S.; Bailey, C.K.; Chaudhuri, G. In vivo binding to and functional repression of the VDR gene promoter by SLUG in human breast cells. Biochem. Biophys. Res. Commun. 2008, 372, 30–34. [Google Scholar] [CrossRef]
- Turner, F.E.; Broad, S.; Khanim, F.L.; Jeanes, A.; Talma, S.; Hughes, S.; Tselepis, C.; Hotchin, N. Slug regulates integrin expression and cell proliferation in human epidermal keratinocytes. J. Biol. Chem. 2006, 281, 21321–21331. [Google Scholar] [CrossRef]
- Tripathi, M.K.; Misra, S.; Chaudhuri, G. Negative regulation of the expressions of cytokeratins 8 and 19 by SLUG repressor protein in human breast cells. Biochem. Biophys. Res. Commun. 2005, 329, 508–515. [Google Scholar] [CrossRef]
- Ikenouchi, J.; Matsuda, M.; Furuse, M.; Tsukita, S. Regulation of tight junctions during the epithelium-mesenchyme transition: Direct repression of the gene expression of claudins/occludin by Snail. J. Cell Sci. 2003, 116, 1959–1967. [Google Scholar] [CrossRef]
- Moses, C.; Nugent, F.; Waryah, C.B.; Garcia-Bloj, B.; Harvey, A.R.; Blancafort, P. Activating PTEN Tumor Suppressor Expression with the CRISPR/dCas9 System. Mol. Ther. Nucleic Acids 2019, 14, 287–300. [Google Scholar] [CrossRef]
- Escriva, M.; Peiró, S.; Herranz, N.; Villagrasa, P.; Dave, N.; Montserrat-Sentís, B.; Murray, S.A.; Francí, C.; Gridley, T.; Virtanen, I.; et al. Repression of PTEN phosphatase by Snail1 transcriptional factor during gamma radiation-induced apoptosis. Mol. Cell. Biol. 2008, 28, 1528–1540. [Google Scholar] [CrossRef]
- Uygur, B.; Abramo, K.; Leikina, E.; Vary, C.; Liaw, L.; Wu, W.-S. SLUG is a direct transcriptional repressor of PTEN tumor suppressor. Prostate 2015, 75, 907–916. [Google Scholar] [CrossRef]
- Costa, C.; Wang, Y.; Ly, A.; Hosono, Y.; Murchie, E.; Walmsley, C.S.; Huynh, T.; Healy, C.; Peterson, R.; Yanase, S.; et al. PTEN Loss Mediates Clinical Cross-Resistance to CDK4/6 and PI3Kalpha Inhibitors in Breast Cancer. Cancer Discov. 2020, 10, 72–85. [Google Scholar] [CrossRef] [PubMed]
- Nagata, Y.; Lan, K.-H.; Zhou, X.; Tan, M.; Esteva, F.J.; Sahin, A.A.; Klos, K.S.; Li, P.; Monia, B.P.; Nguyen, N.T.; et al. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell 2004, 6, 117–127. [Google Scholar] [CrossRef]
- Steelman, L.S.; Navolanic, P.M.; Sokolosky, M.L.; Taylor, J.R.; Lehmann, B.D.; Chappell, W.H.; Abrams, S.L.; Wong, E.W.T.; Stadelman, K.M.; Terrian, D.M.; et al. Suppression of PTEN function increases breast cancer chemotherapeutic drug resistance while conferring sensitivity to mTOR inhibitors. Oncogene 2008, 27, 4086–4095. [Google Scholar] [CrossRef]
- Tripathi, M.K.; Misra, S.; Khedkar, S.V.; Hamilton, N.; Irvin-Wilson, C.; Sharan, C.; Sealy, L.; Chaudhuri, G. Regulation of BRCA2 gene expression by the SLUG repressor protein in human breast cells. J. Biol. Chem. 2005, 280, 17163–17171. [Google Scholar] [CrossRef]
- Murre, C.; McCaw, P.S.; Vaessin, H.; Caudy, M.; Jan, L.; Jan, Y.; Cabrera, C.V.; Buskin, J.N.; Hauschka, S.D.; Lassar, A.B.; et al. Interactions between heterologous helix-loop-helix proteins generate complexes that bind specifically to a common DNA sequence. Cell 1989, 58, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Ledent, V.; Vervoort, M. The basic helix-loop-helix protein family: Comparative genomics and phylogenetic analysis. Genome Res. 2001, 11, 754–770. [Google Scholar] [CrossRef] [PubMed]
- Beck, K.; Peak, M.M.; Ota, T.; Nemazee, D.; Murre, C. Distinct roles for E12 and E47 in B cell specification and the sequential rearrangement of immunoglobulin light chain loci. J. Exp. Med. 2009, 206, 2271–2284. [Google Scholar] [CrossRef] [PubMed]
- Gajula, R.P.; Chettiar, S.T.; Williams, R.D.; Nugent, K.; Kato, Y.; Wang, H.; Malek, R.; Taparra, K.; Cades, J.; Annadanam, A.; et al. Structure-function studies of the bHLH phosphorylation domain of TWIST1 in prostate cancer cells. Neoplasia 2015, 17, 16–31. [Google Scholar] [CrossRef] [PubMed]
- Firulli, A.B.; Conway, S.J. Phosphoregulation of Twist1 provides a mechanism of cell fate control. Curr. Med. Chem. 2008, 15, 2641–2647. [Google Scholar] [CrossRef]
- Gajula, R.P.; Chettiar, S.T.; Williams, R.D.; Thiyagarajan, S.; Kato, Y.; Aziz, K.; Wang, R.; Gandhi, N.; Wild, A.T.; Vesuna, F.; et al. The twist box domain is required for Twist1-induced prostate cancer metastasis. Mol. Cancer Res. 2013, 11, 1387–1400. [Google Scholar] [CrossRef]
- Hamamori, Y.; Sartorelli, V.; Ogryzko, V.; Puri, P.L.; Wu, H.-Y.; Wang, J.Y.; Nakatani, Y.; Kedes, L. Regulation of histone acetyltransferases p300 and PCAF by the bHLH protein twist and adenoviral oncoprotein E1A. Cell 1999, 96, 405–413. [Google Scholar] [CrossRef]
- Fu, J.; Qin, L.; He, T.; Qin, J.; Hong, J.; Wong, J.; Liao, L.; Xu, J. The TWIST/Mi2/NuRD protein complex and its essential role in cancer metastasis. Cell Res. 2011, 21, 275–289. [Google Scholar] [CrossRef]
- Koh, H.S.; Lee, C.; Lee, K.S.; Park, E.J.; Seong, R.H.; Hong, S.; Jeon, S.H. Twist2 regulates CD7 expression and galectin-1-induced apoptosis in mature T-cells. Mol. Cells 2009, 28, 553–558. [Google Scholar] [CrossRef]
- Gong, X.Q.; Li, L. Dermo-1, a multifunctional basic helix-loop-helix protein, represses MyoD transactivation via the HLH domain, MEF2 interaction, and chromatin deacetylation. J. Biol. Chem. 2002, 277, 12310–12317. [Google Scholar] [CrossRef]
- Kim, J.Y.; Park, M.; Ohn, J.; Seong, R.H.; Chung, J.H.; Kim, K.H.; Jo, S.J.; Kwon, O. Twist2-driven chromatin remodeling governs the postnatal maturation of dermal fibroblasts. Cell Rep. 2022, 39, 110821. [Google Scholar] [CrossRef]
- Li, C.W.; Xia, W.; Huo, L.; Lim, S.-O.; Wu, Y.; Hsu, J.L.; Chao, C.-H.; Yamaguchi, H.; Yang, N.-K.; Ding, Q.; et al. Epithelial-mesenchymal transition induced by TNF-alpha requires NF-kappaB-mediated transcriptional upregulation of Twist1. Cancer Res. 2012, 72, 1290–1300. [Google Scholar] [CrossRef] [PubMed]
- Ioannou, M.; Kouvaras, E.; Papamichali, R.; Samara, M.; Chiotoglou, I.; Koukoulis, G. Smad4 and epithelial-mesenchymal transition proteins in colorectal carcinoma: An immunohistochemical study. J. Mol. Histol. 2018, 49, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Chen, K.; Zhang, Y.; Barnes, S.D.; Jaichander, P.; Zheng, Y.; Hassan, M.; Malladi, V.S.; Skapek, S.X.; Xu, L.; et al. Twist2 amplification in rhabdomyosarcoma represses myogenesis and promotes oncogenesis by redirecting MyoD DNA binding. Genes Dev. 2019, 33, 626–640. [Google Scholar] [CrossRef]
- Cripps, R.M.; Black, B.L.; Zhao, B.; Lien, C.-L.; Schulz, R.A.; Olson, E.N. The myogenic regulatory gene Mef2 is a direct target for transcriptional activation by Twist during Drosophila myogenesis. Genes Dev. 1998, 12, 422–434. [Google Scholar] [CrossRef] [PubMed]
- Sharabi, A.B.; Aldrich, M.; Šošić, A.; Olson, E.N.; Friedman, A.D.; Lee, S.-H.; Chen, S.-Y. Twist-2 controls myeloid lineage development and function. PLoS Biol. 2008, 6, e316. [Google Scholar] [CrossRef] [PubMed]
- Pham, C.G.; Bubici, C.; Zazzeroni, F.; Knabb, J.R.; Papa, S.; Kuntzen, C.; Franzoso, G. Upregulation of Twist-1 by NF-kappaB blocks cytotoxicity induced by chemotherapeutic drugs. Mol. Cell Biol. 2007, 27, 3920–3935. [Google Scholar] [CrossRef]
- Chaffer, C.L.; Marjanovic, N.D.; Lee, T.; Bell, G.; Kleer, C.G.; Reinhardt, F.; D’Alessio, A.C.; Young, R.A.; Weinberg, R.A. Poised chromatin at the ZEB1 promoter enables breast cancer cell plasticity and enhances tumorigenicity. Cell 2013, 154, 61–74. [Google Scholar] [CrossRef]
- Drapela, S.; Bouchal, J.; Jolly, M.K.; Culig, Z.; Soucek, K. ZEB1: A Critical Regulator of Cell Plasticity, DNA Damage Response, and Therapy Resistance. Front. Mol. Biosci. 2020, 7, 36. [Google Scholar] [CrossRef]
- Lehmann, W.; Mossmann, D.; Kleemann, J.; Mock, K.; Meisinger, C.; Brummer, T.; Herr, R.; Brabletz, S.; Stemmler, M.P.; Brabletz, T. ZEB1 turns into a transcriptional activator by interacting with YAP1 in aggressive cancer types. Nat. Commun. 2016, 7, 10498. [Google Scholar] [CrossRef]
- Brabletz, S.; Bajdak-Rusinek, K.; Meidhof, S.; Burk, U.; Niedermann, G.; Firat, E.; Wellner, U.; Dimmler, A.; Faller, G.; Schubert, J.; et al. The ZEB1/miR-200 feedback loop controls Notch signalling in cancer cells. EMBO J. 2011, 30, 770–782. [Google Scholar] [CrossRef]
- Gu, K.; Li, M.M.; Shen, J.; Liu, F.; Cao, J.Y.; Jin, S.; Yu, Y. Interleukin-17-induced EMT promotes lung cancer cell migration and invasion via NF-kappaB/ZEB1 signal pathway. Am. J. Cancer Res. 2015, 5, 1169–1179. [Google Scholar]
- Vandewalle, C.; Van Roy, F.; Berx, G. The role of the ZEB family of transcription factors in development and disease. Cell Mol. Life Sci. 2009, 66, 773–787. [Google Scholar] [CrossRef] [PubMed]
- Postigo, A.A.; Dean, D.C. ZEB represses transcription through interaction with the corepressor CtBP. Proc. Natl. Acad. Sci. USA 1999, 96, 6683–6688. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Sawada, J.-I.; Sui, G.; Affar, E.B.; Whetstine, J.R.; Lan, F.; Ogawa, H.; Luke, M.P.-S.; Nakatani, Y.; Shi, Y. Coordinated histone modifications mediated by a CtBP co-repressor complex. Nature 2003, 422, 735–738. [Google Scholar] [CrossRef]
- Wang, W.; Chi, T.; Xue, Y.; Zhou, S.; Kuo, A.; Crabtree, G.R. Architectural DNA binding by a high-mobility-group/kinesin-like subunit in mammalian SWI/SNF-related complexes. Proc. Natl. Acad. Sci. USA 1998, 95, 492–498. [Google Scholar] [CrossRef]
- Feldker, N.; Ferrazzi, F.; Schuhwerk, H.; Widholz, S.A.; Guenther, K.; Frisch, I.; Jakob, K.; Kleemann, J.; Riegel, D.; Bönisch, U.; et al. Genome-wide cooperation of EMT transcription factor ZEB1 with YAP and AP-1 in breast cancer. EMBO J. 2020, 39, e103209. [Google Scholar] [CrossRef] [PubMed]
- Hugo, H.J.; Pereira, L.; Suryadinata, R.; Drabsch, Y.; Gonda, T.J.; Gunasinghe, N.P.A.D.; Pinto, C.; Soo, E.T.L.; Van Denderen, B.J.W.; Hill, P.; et al. Direct repression of MYB by ZEB1 suppresses proliferation and epithelial gene expression during epithelial-to-mesenchymal transition of breast cancer cells. Breast Cancer Res. 2013, 15, R113. [Google Scholar] [CrossRef]
- Postigo, A.A. Opposing functions of ZEB proteins in the regulation of the TGFbeta/BMP signaling pathway. EMBO J. 2003, 22, 2443–2452. [Google Scholar] [CrossRef]
- Burks, H.E.; Matossian, M.D.; Rhodes, L.V.; Phamduy, T.; Elliott, S.; Buechlein, A.; Rusch, D.B.; Miller, D.F.B.; Nephew, K.P.; Chrisey, D.; et al. ZEB2 regulates endocrine therapy sensitivity and metastasis in luminal a breast cancer cells through a non-canonical mechanism. Breast Cancer Res. Treat. 2021, 189, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Sun, Y.; Ma, L. ZEB1: At the crossroads of epithelial-mesenchymal transition, metastasis and therapy resistance. Cell Cycle 2015, 14, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Waryah, C.; Cursons, J.; Foroutan, M.; Pflueger, C.; Wang, E.; Molania, R.; Woodward, E.; Sorolla, A.; Wallis, C.; Moses, C.; et al. Synthetic Epigenetic Reprogramming of Mesenchymal to Epithelial States Using the CRISPR/dCas9 Platform in Triple Negative Breast Cancer. Adv. Sci. 2023, 2301802. [Google Scholar] [CrossRef]
- Perez-Oquendo, M.; Gibbons, D.L. Regulation of ZEB1 Function and Molecular Associations in Tumor Progression and Metastasis. Cancers 2022, 14, 1864. [Google Scholar] [CrossRef] [PubMed]
- Katsura, A.; Tamura, Y.; Hokari, S.; Harada, M.; Morikawa, M.; Sakurai, T.; Takahashi, K.; Mizutani, A.; Nishida, J.; Yokoyama, Y.; et al. ZEB1-regulated inflammatory phenotype in breast cancer cells. Mol. Oncol. 2017, 11, 1241–1262. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Kang, Y. Epithelial-Mesenchymal Plasticity in Cancer Progression and Metastasis. Dev. Cell 2019, 49, 361–374. [Google Scholar] [CrossRef] [PubMed]
- Wels, C.; Joshi, S.; Koefinger, P.; Bergler, H.; Schaider, H. Transcriptional activation of ZEB1 by Slug leads to cooperative regulation of the epithelial-mesenchymal transition-like phenotype in melanoma. J. Investig. Dermatol. 2011, 131, 1877–1885. [Google Scholar] [CrossRef]
- Wu, W.S.; You, R.-I.; Cheng, C.-C.; Lee, M.-C.; Lin, T.-Y.; Hu, C.-T. Snail collaborates with EGR-1 and SP-1 to directly activate transcription of MMP 9 and ZEB1. Sci. Rep. 2017, 7, 17753. [Google Scholar] [CrossRef] [PubMed]
- Dave, N.; Guaita-Esteruelas, S.; Gutarra, S.; Frias, Á.; Beltran, M.; Peiró, S.; de Herreros, A.G. Functional cooperation between Snail1 and twist in the regulation of ZEB1 expression during epithelial to mesenchymal transition. J. Biol. Chem. 2011, 286, 12024–12032. [Google Scholar] [CrossRef]
- Stemmler, M.P.; Eccles, R.L.; Brabletz, S.; Brabletz, T. Non-redundant functions of EMT transcription factors. Nat. Cell Biol. 2019, 21, 102–112. [Google Scholar] [CrossRef]
- Kroger, C.; Afeyan, A.; Mraz, J.; Eaton, E.N.; Reinhardt, F.; Khodor, Y.L.; Thiru, P.; Bierie, B.; Ye, X.; Burge, C.B.; et al. Acquisition of a hybrid E/M state is essential for tumorigenicity of basal breast cancer cells. Proc. Natl. Acad. Sci. USA 2019, 116, 7353–7362. [Google Scholar] [CrossRef]
- Vazquez-Martin, A.; Anatskaya, O.V.; Giuliani, A.; Erenpreisa, J.; Huang, S.; Salmina, K.; Inashkina, I.; Huna, A.; Nikolsky, N.N.; Vinogradov, A.E. Somatic polyploidy is associated with the upregulation of c-MYC interacting genes and EMT-like signature. Oncotarget 2016, 7, 75235–75260. [Google Scholar] [CrossRef]
- Huang, Y.; Hong, W.; Wei, X. The molecular mechanisms and therapeutic strategies of EMT in tumor progression and metastasis. J. Hematol. Oncol. 2022, 15, 129. [Google Scholar] [CrossRef]
- Haslehurst, A.M.; Koti, M.; Dharsee, M.; Nuin, P.; Evans, K.; Geraci, J.; Childs, T.; Chen, J.; Li, J.; Weberpals, J.; et al. EMT transcription factors snail and slug directly contribute to cisplatin resistance in ovarian cancer. BMC Cancer 2012, 12, 91. [Google Scholar] [CrossRef]
- Kurrey, N.K.; Jalgaonkar, S.P.; Joglekar, A.V.; Ghanate, A.D.; Chaskar, P.D.; Doiphode, R.Y.; Bapat, S.A. Snail and slug mediate radioresistance and chemoresistance by antagonizing p53-mediated apoptosis and acquiring a stem-like phenotype in ovarian cancer cells. Stem Cells 2009, 27, 2059–2068. [Google Scholar] [CrossRef]
- Lee, A.F.; Chen, M.-C.; Chen, C.-J.; Yang, C.-J.; Huang, M.-S.; Liu, Y.-P. Reverse epithelial-mesenchymal transition contributes to the regain of drug sensitivity in tyrosine kinase inhibitor-resistant non-small cell lung cancer cells. PLoS ONE 2017, 12, e0180383. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, Z.; Zhang, Q.; Zhang, Q.; Sun, P.; Xiang, R.; Ren, G.; Yang, S. ZEB1 confers chemotherapeutic resistance to breast cancer by activating ATM. Cell Death Dis. 2018, 9, 57. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Liao, W.; Wu, Q.; Huang, X.; Pan, Z.; Chen, W.; Gu, S.; Huang, Z.; Wang, Y.; Tang, X.; et al. LINC00152 upregulates ZEB1 expression and enhances epithelial-mesenchymal transition and oxaliplatin resistance in esophageal cancer by interacting with EZH2. Cancer Cell Int. 2020, 20, 569. [Google Scholar] [CrossRef]
- Hanrahan, K.; O’Neill, A.; Prencipe, M.; Bugler, J.; Murphy, L.; Fabre, A.; Puhr, M.; Culig, Z.; Murphy, K.; Watson, R.W. The role of epithelial-mesenchymal transition drivers ZEB1 and ZEB2 in mediating docetaxel-resistant prostate cancer. Mol. Oncol. 2017, 11, 251–265. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Chen, Y.; Lin, Y.; Wang, X.; Cui, X.; Zhang, Z.; Xian, G.; Qin, C. Novel crosstalk between KLF4 and ZEB1 regulates gemcitabine resistance in pancreatic ductal adenocarcinoma. Int. J. Oncol. 2017, 51, 1239–1248. [Google Scholar] [CrossRef]
- Redfern, A.D.; Spalding, L.J.; Thompson, E.W. The Kraken Wakes: Induced EMT as a driver of tumour aggression and poor outcome. Clin. Exp. Metastasis 2018, 35, 285–308. [Google Scholar] [CrossRef]
- Tran, H.D.; Luitel, K.; Kim, M.; Zhang, K.; Longmore, G.D.; Tran, D.D. Transient SNAIL1 expression is necessary for metastatic competence in breast cancer. Cancer Res. 2014, 74, 6330–6340. [Google Scholar] [CrossRef]
- Caramel, J.; Papadogeorgakis, E.; Hill, L.; Browne, G.J.; Richard, G.; Wierinckx, A.; Saldanha, G.; Osborne, J.; Hutchinson, P.; Tse, G.; et al. A switch in the expression of embryonic EMT-inducers drives the development of malignant melanoma. Cancer Cell 2013, 24, 466–480. [Google Scholar] [CrossRef]
- Asiedu, M.K.; Ingle, J.N.; Behrens, M.D.; Radisky, D.C.; Knutson, K.L. TGFbeta/TNF(alpha)-mediated epithelial-mesenchymal transition generates breast cancer stem cells with a claudin-low phenotype. Cancer Res. 2011, 71, 4707–4719. [Google Scholar] [CrossRef] [PubMed]
- Kudo-Saito, C.; Shirako, H.; Takeuchi, T.; Kawakami, Y. Cancer metastasis is accelerated through immunosuppression during Snail-induced EMT of cancer cells. Cancer Cell 2009, 15, 195–206. [Google Scholar] [CrossRef]
- Yang, J.; Wang, X.; Huang, B.; Liu, R.; Xiong, H.; Ye, F.; Zeng, C.; Fu, X.; Li, L. An IFNgamma/STAT1/JMJD3 Axis Induces ZEB1 Expression and Promotes Aggressiveness in Lung Adenocarcinoma. Mol. Cancer Res. 2021, 19, 1234–1246. [Google Scholar] [CrossRef]
- Lo, U.G.; Pong, R.-C.; Yang, D.; Gandee, L.; Hernandez, E.; Dang, A.; Lin, C.-J.; Santoyo, J.; Ma, S.; Sonavane, R.; et al. IFNgamma-Induced IFIT5 Promotes Epithelial-to-Mesenchymal Transition in Prostate Cancer via miRNA Processing. Cancer Res. 2019, 79, 1098–1112. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, L.; Pappan, L.; Galliher-Beckley, A.; Shi, J. IL-1beta promotes stemness and invasiveness of colon cancer cells through Zeb1 activation. Mol. Cancer 2012, 11, 87. [Google Scholar] [CrossRef]
- Lee, C.H.; Chang, J.S.; Syu, S.; Wong, T.; Chan, J.Y.; Tang, Y.; Yang, Z.; Yang, W.; Chen, C.; Lu, S.; et al. IL-1beta promotes malignant transformation and tumor aggressiveness in oral cancer. J. Cell Physiol. 2015, 230, 875–884. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, N.J.; Sasser, A.K.; Axel, A.; Vesuna, F.; Raman, V.; Ramirez, N.; Oberyszyn, T.M.; Hall, B.M. Interleukin-6 induces an epithelial-mesenchymal transition phenotype in human breast cancer cells. Oncogene 2009, 28, 2940–2947. [Google Scholar] [CrossRef]
- Yadav, A.; Kumar, B.; Datta, J.; Teknos, T.N.; Kumar, P. IL-6 promotes head and neck tumor metastasis by inducing epithelial-mesenchymal transition via the JAK-STAT3-SNAIL signaling pathway. Mol. Cancer Res. 2011, 9, 1658–1667. [Google Scholar] [CrossRef]
- Hwang, W.L.; Yang, M.; Tsai, M.; Lan, H.; Su, S.; Chang, S.; Teng, H.; Yang, S.; Lan, Y.; Chiou, S.; et al. SNAIL regulates interleukin-8 expression, stem cell-like activity, and tumorigenicity of human colorectal carcinoma cells. Gastroenterology 2011, 141, 279–291.e5. [Google Scholar] [CrossRef]
- Visciano, C.; Liotti, F.; Prevete, N.; Cali’, G.; Franco, R.; Collina, F.; De Paulis, A.; Marone, G.; Santoro, M.; Melillo, R.M. Mast cells induce epithelial-to-mesenchymal transition and stem cell features in human thyroid cancer cells through an IL-8-Akt-Slug pathway. Oncogene 2015, 34, 5175–5186. [Google Scholar] [CrossRef]
- Zhang, Q.; Liu, S.; Parajuli, K.R.; Zhang, W.; Zhang, K.; Mo, Z.; Liu, J.; Chen, Z.; Yang, S.; Wang, A.R.; et al. Interleukin-17 promotes prostate cancer via MMP7-induced epithelial-to-mesenchymal transition. Oncogene 2017, 36, 687–699. [Google Scholar] [CrossRef]
- Chen, D.; Li, W.; Liu, S.; Su, Y.; Han, G.; Xu, C.; Liu, H.; Zheng, T.; Zhou, Y.; Mao, C. Interleukin-23 promotes the epithelial-mesenchymal transition of oesophageal carcinoma cells via the Wnt/beta-catenin pathway. Sci. Rep. 2015, 5, 8604. [Google Scholar] [CrossRef]
- Izumi, K.; Fang, L.Y.; Mizokami, A.; Namiki, M.; Li, L.; Lin, W.J.; Chang, C. Targeting the androgen receptor with siRNA promotes prostate cancer metastasis through enhanced macrophage recruitment via CCL2/CCR2-induced STAT3 activation. EMBO Mol. Med. 2013, 5, 1383–1401. [Google Scholar] [CrossRef]
- Rao, Q.; Chen, Y.; Yeh, C.-R.; Ding, J.; Li, L.; Chang, C.; Yeh, S. Recruited mast cells in the tumor microenvironment enhance bladder cancer metastasis via modulation of ERbeta/CCL2/CCR2 EMT/MMP9 signals. Oncotarget 2016, 7, 7842–7855. [Google Scholar] [CrossRef] [PubMed]
- Long, H.; Xiang, T.; Qi, W.; Huang, J.; Chen, J.; He, L.; Liang, Z.; Guo, B.; Li, Y.; Xie, R.; et al. CD133+ ovarian cancer stem-like cells promote non-stem cancer cell metastasis via CCL5 induced epithelial-mesenchymal transition. Oncotarget 2015, 6, 5846–5859. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Zhao, J.; Li, J.; Zhu, Z.; Cui, Z.; Liu, R.; Lu, R.; Yao, Z.; Xu, Q. Cancer associated fibroblast-derived CCL5 promotes hepatocellular carcinoma metastasis through activating HIF1alpha/ZEB1 axis. Cell Death Dis. 2022, 13, 478. [Google Scholar] [CrossRef]
- Zhang, B.; Yin, C.; Li, H.; Shi, L.; Liu, N.; Sun, Y.; Lu, S.; Liu, Y.; Sun, L.; Li, X.; et al. Nir1 promotes invasion of breast cancer cells by binding to chemokine (C-C motif) ligand 18 through the PI3K/Akt/GSK3beta/Snail signalling pathway. Eur. J. Cancer 2013, 49, 3900–3913. [Google Scholar] [CrossRef]
- Bonde, A.K.; Tischler, V.; Kumar, S.; Soltermann, A.; Schwendener, R.A. Intratumoral macrophages contribute to epithelial-mesenchymal transition in solid tumors. BMC Cancer 2012, 12, 35. [Google Scholar] [CrossRef] [PubMed]
- Toh, B.; Wang, X.; Keeble, J.; Sim, W.J.; Khoo, K.; Wong, W.-C.; Kato, M.; Prevost-Blondel, A.; Thiery, J.-P.; Abastado, J.-P. Mesenchymal transition and dissemination of cancer cells is driven by myeloid-derived suppressor cells infiltrating the primary tumor. PLoS Biol. 2011, 9, e1001162. [Google Scholar] [CrossRef]
- Camisaschi, C.; Casati, C.; Rini, F.; Perego, M.; De Filippo, A.; Triebel, F.; Parmiani, G.; Belli, F.; Rivoltini, L.; Castelli, C. LAG-3 expression defines a subset of CD4(+)CD25(high)Foxp3(+) regulatory T cells that are expanded at tumor sites. J. Immunol. 2010, 184, 6545–6551. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Liu, Q.; Chen, J.; Chen, J.; Chen, F.; He, C.; Huang, D.; Wu, W.; Lin, L.; Huang, W.; et al. A positive feedback loop between mesenchymal-like cancer cells and macrophages is essential to breast cancer metastasis. Cancer Cell 2014, 25, 605–620. [Google Scholar] [CrossRef]
- Bronte, V.; Chappell, D.B.; Apolloni, E.; Cabrelle, A.; Wang, M.; Hwu, P.; Restifo, N.P. Unopposed production of granulocyte-macrophage colony-stimulating factor by tumors inhibits CD8+ T cell responses by dysregulating antigen-presenting cell maturation. J. Immunol. 1999, 162, 5728–5737. [Google Scholar] [CrossRef] [PubMed]
- Nagaraj, S.; Gupta, K.; Pisarev, V.; Kinarsky, L.; Sherman, S.; Kang, L.; Herber, D.L.; Schneck, J.; Gabrilovich, D.I. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat. Med. 2007, 13, 828–835. [Google Scholar] [CrossRef]
- Chen, L.; Gibbons, D.L.; Goswami, S.; Cortez, M.A.; Ahn, Y.-H.; Byers, L.A.; Zhang, X.; Yi, X.; Dwyer, D.; Lin, W.; et al. Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nat. Commun. 2014, 5, 5241. [Google Scholar] [CrossRef] [PubMed]
- Ock, C.Y.; Kim, S.; Keam, B.; Kim, M.; Kim, T.M.; Kim, J.-H.; Jeon, Y.K.; Lee, J.-S.; Kwon, S.K.; Hah, J.H.; et al. PD-L1 expression is associated with epithelial-mesenchymal transition in head and neck squamous cell carcinoma. Oncotarget 2016, 7, 15901–15914. [Google Scholar] [CrossRef]
- Alsuliman, A.; Colak, D.; Al-Harazi, O.; Fitwi, H.; Tulbah, A.; Al-Tweigeri, T.; Al-Alwan, M.; Ghebeh, H. Bidirectional crosstalk between PD-L1 expression and epithelial to mesenchymal transition: Significance in claudin-low breast cancer cells. Mol. Cancer 2015, 14, 149. [Google Scholar] [CrossRef] [PubMed]
- Dongre, A.; Rashidian, M.; Eaton, E.N.; Reinhardt, F.; Thiru, P.; Zagorulya, M.; Nepal, S.; Banaz, T.; Martner, A.; Spranger, S.; et al. Direct and Indirect Regulators of Epithelial-Mesenchymal Transition-Mediated Immunosuppression in Breast Carcinomas. Cancer Discov. 2021, 11, 1286–1305. [Google Scholar] [CrossRef]
- Zheng, Q.; Gao, J.; Yin, P.; Wang, W.; Wang, B.; Li, Y.; Zhao, C. CD155 contributes to the mesenchymal phenotype of triple-negative breast cancer. Cancer Sci. 2020, 111, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Mak, M.P.; Tong, P.; Diao, L.; Cardnell, R.J.; Gibbons, D.L.; William, W.N.; Skoulidis, F.; Parra, E.R.; Rodriguez-Canales, J.; Wistuba, I.I.; et al. A Patient-Derived, Pan-Cancer EMT Signature Identifies Global Molecular Alterations and Immune Target Enrichment Following Epithelial-to-Mesenchymal Transition. Clin. Cancer Res. 2016, 22, 609–620. [Google Scholar] [CrossRef]
- Lou, Y.; Diao, L.; Cuentas, E.R.P.; Denning, W.L.; Chen, L.; Fan, Y.H.; Byers, L.A.; Wang, J.; Papadimitrakopoulou, V.A.; Behrens, C.; et al. Epithelial-Mesenchymal Transition Is Associated with a Distinct Tumor Microenvironment Including Elevation of Inflammatory Signals and Multiple Immune Checkpoints in Lung Adenocarcinoma. Clin. Cancer Res. 2016, 22, 3630–3642. [Google Scholar] [CrossRef]
- Lee, J.H.; Shklovskaya, E.; Lim, S.Y.; Carlino, M.S.; Menzies, A.M.; Stewart, A.; Pedersen, B.; Irvine, M.; Alavi, S.; Yang, J.Y.H.; et al. Transcriptional downregulation of MHC class I and melanoma de- differentiation in resistance to PD-1 inhibition. Nat. Commun. 2020, 11, 1897. [Google Scholar] [CrossRef]
- Chen, X.H.; Liu, Z.-C.; Zhang, G.; Wei, W.; Wang, X.-X.; Wang, H.; Ke, H.-P.; Zhang, F.; Wang, H.-S.; Cai, S.-H.; et al. TGF-beta and EGF induced HLA-I downregulation is associated with epithelial-mesenchymal transition (EMT) through upregulation of snail in prostate cancer cells. Mol. Immunol. 2015, 65, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Alves, E.; Taifour, S.; Dolcetti, R.; Chee, J.; Nowak, A.K.; Gaudieri, S.; Blancafort, P. Reprogramming the anti-tumor immune response via CRISPR genetic and epigenetic editing. Mol. Ther. Methods Clin. Dev. 2021, 21, 592–606. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, S.C.; Peters, H.L.; Taguchi, A.; Katayama, H.; Wang, H.; Momin, A.; Jolly, M.K.; Celiktas, M.; Rodriguez-Canales, J.; Liu, H.; et al. Immunoproteasome deficiency is a feature of non-small cell lung cancer with a mesenchymal phenotype and is associated with a poor outcome. Proc. Natl. Acad. Sci. USA 2016, 113, E1555–E1564. [Google Scholar] [CrossRef]
- He, Y.; Bunn, P.A.; Zhou, C.; Chan, D. KIR 2D (L1, L3, L4, S4) and KIR 3DL1 protein expression in non-small cell lung cancer. Oncotarget 2016, 7, 82104–82111. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Tian, X.; Li, Y.; Liu, Y.; Yang, T.; Han, Z.; An, J.; Kong, L.; Li, Y. Epithelial-mesenchymal transition may be involved in the immune evasion of circulating gastric tumor cells via downregulation of ULBP1. Cancer Med. 2020, 9, 2686–2697. [Google Scholar] [CrossRef]
- Lopez-Soto, A.; Huergo-Zapico, L.; Galvan, J.A.; Rodrigo, L.; de Herreros, A.G.; Astudillo, A.; Gonzalez, S. Epithelial-mesenchymal transition induces an antitumor immune response mediated by NKG2D receptor. J. Immunol. 2013, 190, 4408–4419. [Google Scholar] [CrossRef]
- Alves, E.; McLeish, E.; Blancafort, P.; Coudert, J.D.; Gaudieri, S. Manipulating the NKG2D Receptor-Ligand Axis Using CRISPR: Novel Technologies for Improved Host Immunity. Front. Immunol. 2021, 12, 712722. [Google Scholar] [CrossRef]
- Anastasiadou, E.; Jacob, L.S.; Slack, F.J. Non-coding RNA networks in cancer. Nat. Rev. Cancer 2018, 18, 5–18. [Google Scholar] [CrossRef]
- Lin, C.C.; Liu, L.-Z.; Addison, J.B.; Wonderlin, W.F.; Ivanov, A.V.; Ruppert, J.M. A KLF4-miRNA-206 autoregulatory feedback loop can promote or inhibit protein translation depending upon cell context. Mol. Cell Biol. 2011, 31, 2513–2527. [Google Scholar] [CrossRef]
- Ma, F.; Liu, X.; Li, D.; Wang, P.; Li, N.; Lu, L.; Cao, X. MicroRNA-466l upregulates IL-10 expression in TLR-triggered macrophages by antagonizing RNA-binding protein tristetraprolin-mediated IL-10 mRNA degradation. J. Immunol. 2010, 184, 6053–6059. [Google Scholar] [CrossRef]
- Vasudevan, S.; Tong, Y.; Steitz, J.A. Switching from repression to activation: microRNAs can up-regulate translation. Science 2007, 318, 1931–1934. [Google Scholar] [CrossRef]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. miRBase: From microRNA sequences to function. Nucleic Acids Res. 2019, 47, D155–D162. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ma, L. MicroRNA control of epithelial-mesenchymal transition and metastasis. Cancer Metastasis Rev. 2012, 31, 653–662. [Google Scholar] [CrossRef]
- Burk, U.; Schubert, J.; Wellner, U.; Schmalhofer, O.; Vincan, E.; Spaderna, S.; Brabletz, T. A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep. 2008, 9, 582–589. [Google Scholar] [CrossRef]
- Siemens, H.; Jackstadt, R.; Hünten, S.; Kaller, M.; Menssen, A.; Götz, U.; Hermeking, H. miR-34 and SNAIL form a double-negative feedback loop to regulate epithelial-mesenchymal transitions. Cell Cycle 2011, 10, 4256–4271. [Google Scholar] [CrossRef]
- Zaravinos, A. The Regulatory Role of MicroRNAs in EMT and Cancer. J. Oncol. 2015, 2015, 865816. [Google Scholar] [CrossRef]
- Davis-Dusenbery, B.N.; Hata, A. MicroRNA in Cancer: The Involvement of Aberrant MicroRNA Biogenesis Regulatory Pathways. Genes Cancer 2010, 1, 1100–1114. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Xi, Q.; Wang, H.; Zhang, Z.; Liu, H.; Cheng, Y.; Guo, X.; Zhang, J.; Zhang, Q.; Zhang, L.; et al. miR-142-5p regulates tumor cell PD-L1 expression and enhances anti-tumor immunity. Biochem. Biophys. Res. Commun. 2017, 488, 425–431. [Google Scholar] [CrossRef]
- Zhao, L.; Yu, H.; Yi, S.; Peng, X.; Su, P.; Xiao, Z.; Liu, R.; Tang, A.; Li, X.; Liu, F.; et al. The tumor suppressor miR-138-5p targets PD-L1 in colorectal cancer. Oncotarget 2016, 7, 45370–45384. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Tan, W.; Liu, S.; Huang, X.; Lin, J.; Liang, R.; Su, L.; Su, Q.; Wang, C. MiR-570 inhibited the cell proliferation and invasion through directly targeting B7-H1 in hepatocellular carcinoma. Tumour Biol. 2015, 36, 9049–9057. [Google Scholar] [CrossRef] [PubMed]
- Omar, H.A.; El-Serafi, A.T.; Hersi, F.; Arafa, E.A.; Zaher, D.M.; Madkour, M.; Arab, H.H.; Tolba, M.F. Immunomodulatory MicroRNAs in cancer: Targeting immune checkpoints and the tumor microenvironment. FEBS J. 2019, 286, 3540–3557. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, J.; Dong, K.; Lin, F.; Long, M.; Ouyang, Y.; Wei, J.; Chen, X.; Weng, Y.; He, T.; et al. Tumor suppressor miR-34a targets PD-L1 and functions as a potential immunotherapeutic target in acute myeloid leukemia. Cell Signal. 2015, 27, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Pavlic, A.; Hauptman, N.; Bostjancic, E.; Zidar, N. Long Non-Coding RNAs as Potential Regulators of EMT-Related Transcription Factors in Colorectal Cancer-A Systematic Review and Bioinformatics Analysis. Cancers 2022, 14, 2280. [Google Scholar] [CrossRef] [PubMed]
- Xiong, H.G.; Li, H.; Xiao, Y.; Yang, Q.-C.; Yang, L.-L.; Chen, L.; Bu, L.-L.; Zhang, W.F.; Zhang, J.L.; Sun, Z.-J. Long noncoding RNA MYOSLID promotes invasion and metastasis by modulating the partial epithelial-mesenchymal transition program in head and neck squamous cell carcinoma. J. Exp. Clin. Cancer Res. 2019, 38, 278. [Google Scholar] [CrossRef]
- Xu, Q.; Deng, F.; Qin, Y.; Zhao, Z.; Wu, Z.; Xing, Z.; Ji, A.; Wang, Q.J. Long non-coding RNA regulation of epithelial-mesenchymal transition in cancer metastasis. Cell Death Dis. 2016, 7, e2254. [Google Scholar] [CrossRef]
- Xue, M.; Pang, H.; Li, X.; Li, H.; Pan, J.; Chen, W. Long non-coding RNA urothelial cancer-associated 1 promotes bladder cancer cell migration and invasion by way of the hsa-miR-145-ZEB1/2-FSCN1 pathway. Cancer Sci. 2016, 107, 18–27. [Google Scholar] [CrossRef]
- Li, H.; Zhu, L.; Xu, L.; Qin, K.; Liu, C.; Yu, Y.; Su, D.; Wu, K.; Sheng, Y. Long noncoding RNA linc00617 exhibits oncogenic activity in breast cancer. Mol. Carcinog. 2017, 56, 3–17. [Google Scholar] [CrossRef]
- Wu, J.; Zhang, J.; Shen, B.; Yin, K.; Xu, J.; Gao, W.; Zhang, L. Long noncoding RNA lncTCF7, induced by IL-6/STAT3 transactivation, promotes hepatocellular carcinoma aggressiveness through epithelial-mesenchymal transition. J. Exp. Clin. Cancer Res. 2015, 34, 116. [Google Scholar] [CrossRef]
- Su, W.; Xu, M.; Chen, X.; Ni Chen, N.; Gong, J.; Nie, L.; Li, L.; Li, X.; Zhang, M.; Zhou, Q. Long noncoding RNA ZEB1-AS1 epigenetically regulates the expressions of ZEB1 and downstream molecules in prostate cancer. Mol. Cancer 2017, 16, 142. [Google Scholar] [CrossRef]
- Beltran, M.; Puig, I.; Peña, C.; García, J.M.; Álvarez, A.B.; Peña, R.; Bonilla, F.; de Herreros, A.G. A natural antisense transcript regulates Zeb2/Sip1 gene expression during Snail1-induced epithelial-mesenchymal transition. Genes Dev. 2008, 22, 756–769. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.H.; Li, B.; Liu, D.-G.; Zhang, B.; Yang, X.; Tu, Y.-L. LncRNA KCNQ1OT1 sponges miR-15a to promote immune evasion and malignant progression of prostate cancer via up-regulating PD-L1. Cancer Cell Int. 2020, 20, 394. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.H.; Yang, F.; Wang, F.; Ma, J.-Z.; Guo, Y.-J.; Tao, Q.-F.; Liu, F.; Pan, W.; Wang, T.-T.; Zhou, C.-C.; et al. A long noncoding RNA activated by TGF-beta promotes the invasion-metastasis cascade in hepatocellular carcinoma. Cancer Cell 2014, 25, 666–681. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Lee, D.W.; Yim, G.W.; Nam, E.J.; Kim, S.; Kim, S.W.; Kim, Y.T. Long non-coding RNA HOTAIR is associated with human cervical cancer progression. Int. J. Oncol. 2015, 46, 521–530. [Google Scholar] [CrossRef]
- Liu, Y.W.; Sun, M.; Xia, R.; Zhang, E.-B.; Liu, X.-H.; Zhang, Z.-H.; Xu, T.-P.; De, W.; Liu, B.-R.; Wang, Z.-X. LincHOTAIR epigenetically silences miR34a by binding to PRC2 to promote the epithelial-to-mesenchymal transition in human gastric cancer. Cell Death Dis. 2015, 6, e1802. [Google Scholar] [CrossRef]
- Liu, M.; Zhang, H.; Li, Y.; Wang, R.; Li, Y.; Zhang, H.; Ren, D.; Liu, H.; Kang, C.; Chen, J. HOTAIR, a long noncoding RNA, is a marker of abnormal cell cycle regulation in lung cancer. Cancer Sci. 2018, 109, 2717–2733. [Google Scholar] [CrossRef]
- Zhang, H.; Cai, K.; Wang, J.; Wang, X.; Cheng, K.; Shi, F.; Jiang, L.; Zhang, Y.; Dou, J. MiR-7, inhibited indirectly by lincRNA HOTAIR, directly inhibits SETDB1 and reverses the EMT of breast cancer stem cells by downregulating the STAT3 pathway. Stem Cells 2014, 32, 2858–2868. [Google Scholar] [CrossRef]
- Jin, L.; Pan, Y.L.; Zhang, J.; Cao, P.G. LncRNA HOTAIR recruits SNAIL to inhibit the transcription of HNF4alpha and promote the viability, migration, invasion and EMT of colorectal cancer. Transl Oncol. 2021, 14, 101036. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Waryah, C.; Alves, E.; Mazzieri, R.; Dolcetti, R.; Thompson, E.W.; Redfern, A.; Blancafort, P. Unpacking the Complexity of Epithelial Plasticity: From Master Regulator Transcription Factors to Non-Coding RNAs. Cancers 2023, 15, 3152. https://doi.org/10.3390/cancers15123152
Waryah C, Alves E, Mazzieri R, Dolcetti R, Thompson EW, Redfern A, Blancafort P. Unpacking the Complexity of Epithelial Plasticity: From Master Regulator Transcription Factors to Non-Coding RNAs. Cancers. 2023; 15(12):3152. https://doi.org/10.3390/cancers15123152
Chicago/Turabian StyleWaryah, Charlene, Eric Alves, Roberta Mazzieri, Riccardo Dolcetti, Erik W. Thompson, Andrew Redfern, and Pilar Blancafort. 2023. "Unpacking the Complexity of Epithelial Plasticity: From Master Regulator Transcription Factors to Non-Coding RNAs" Cancers 15, no. 12: 3152. https://doi.org/10.3390/cancers15123152