
Tumor Microenvironment as a Therapeutic Target in Melanoma Treatment

 , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
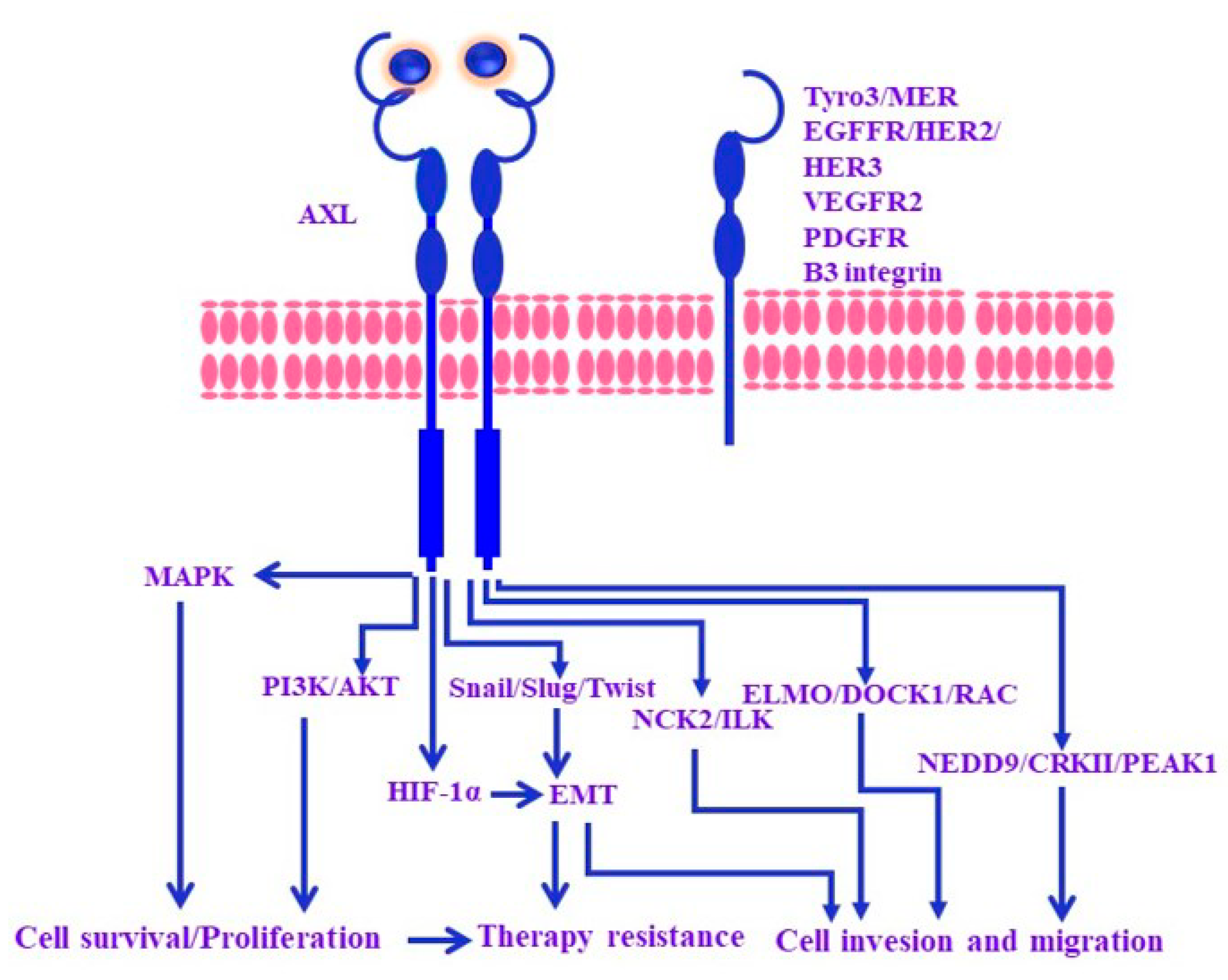
2. Mechanisms of Melanoma Resistance
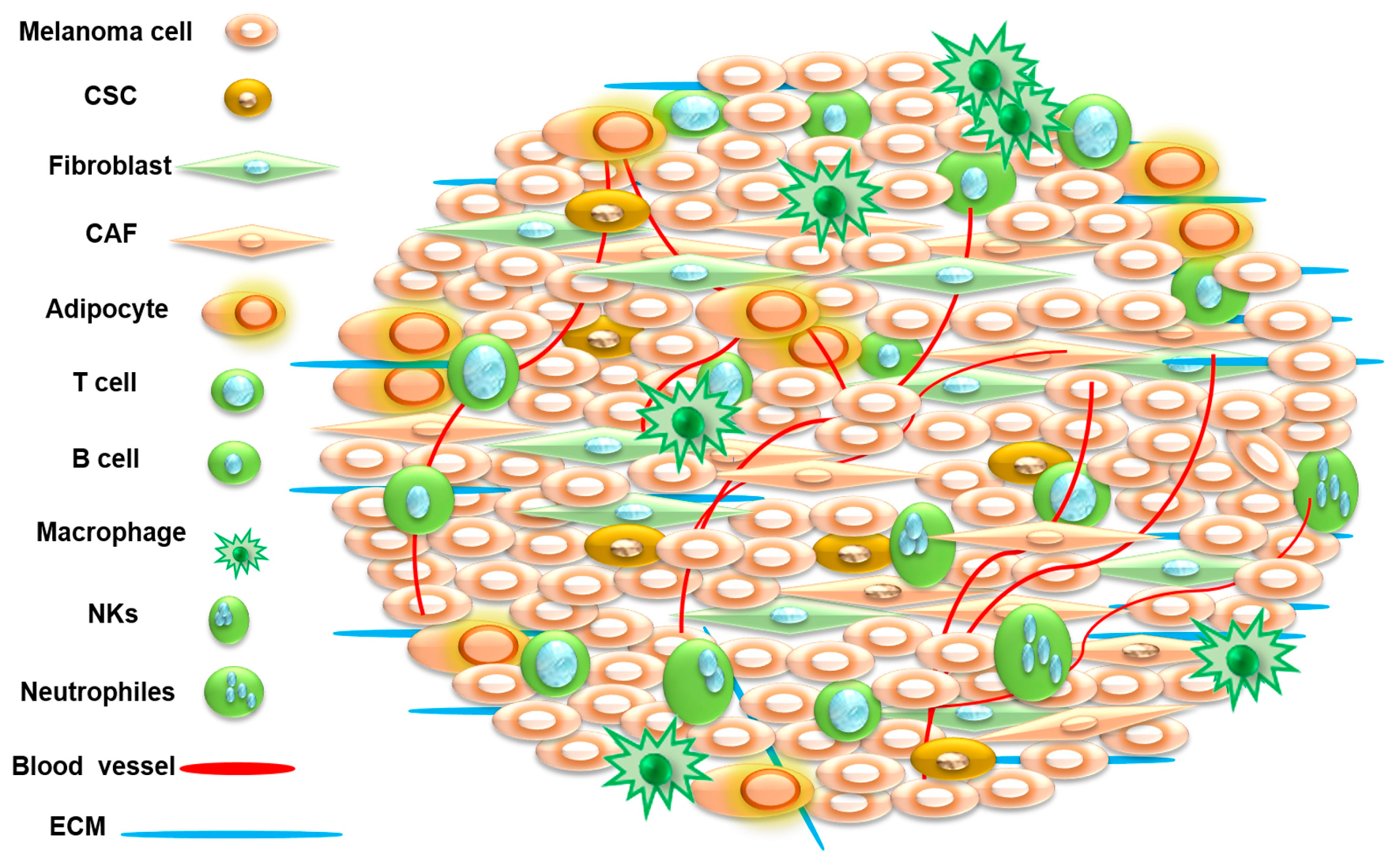
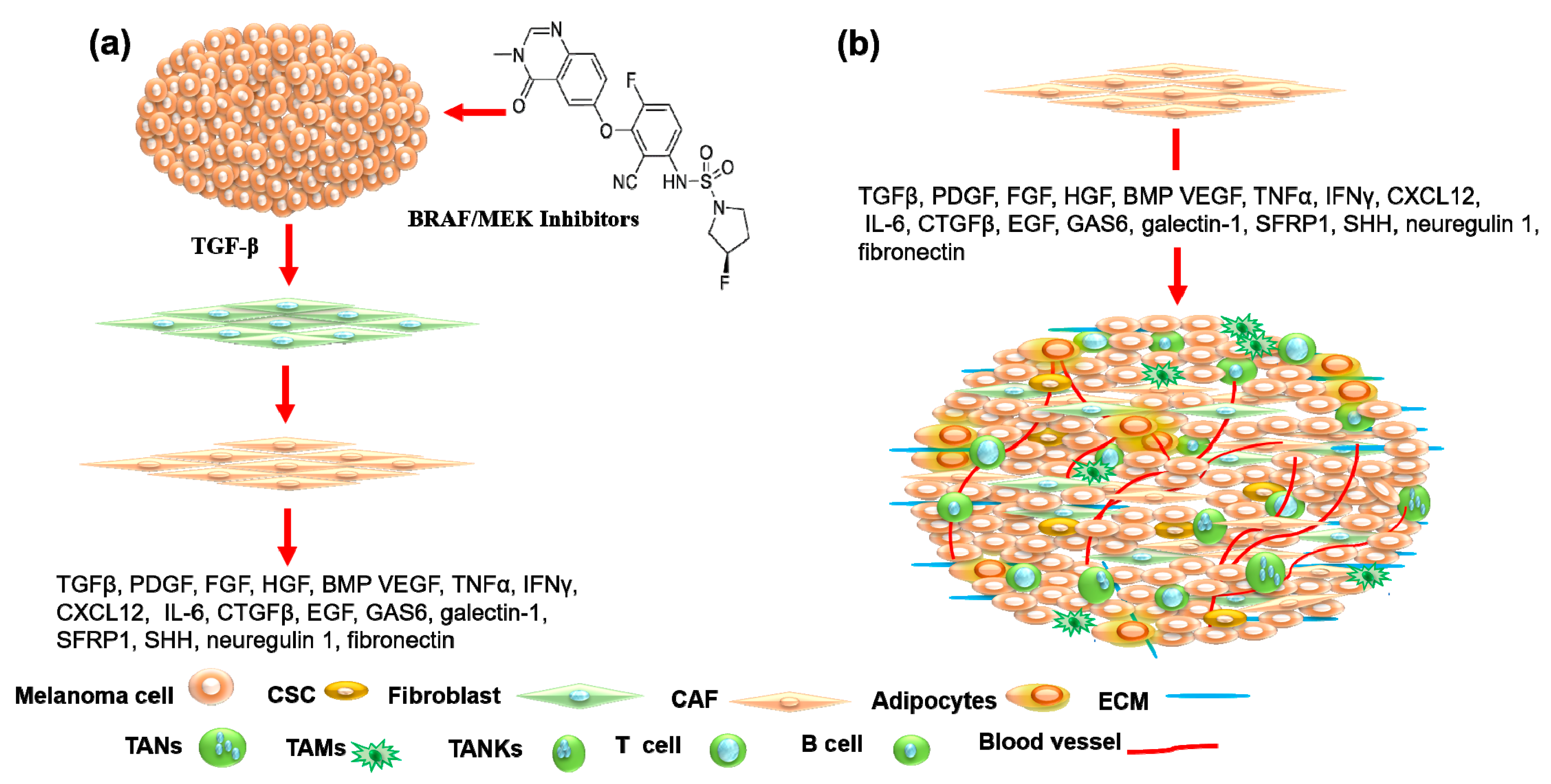
3. Tumor Microenvironment
4. Tumor Microenvironment as Therapeutic Target in Melanoma Treatment
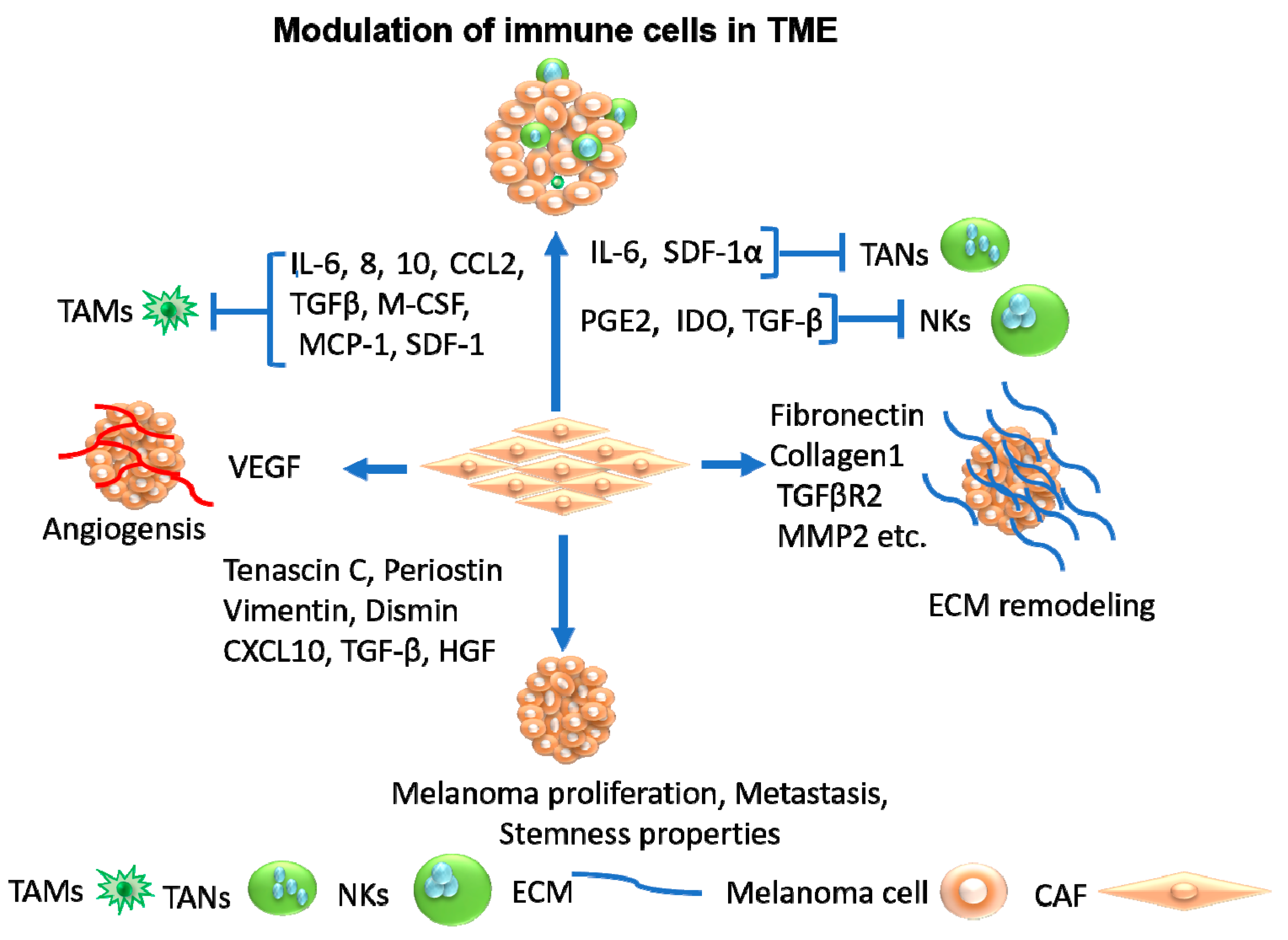
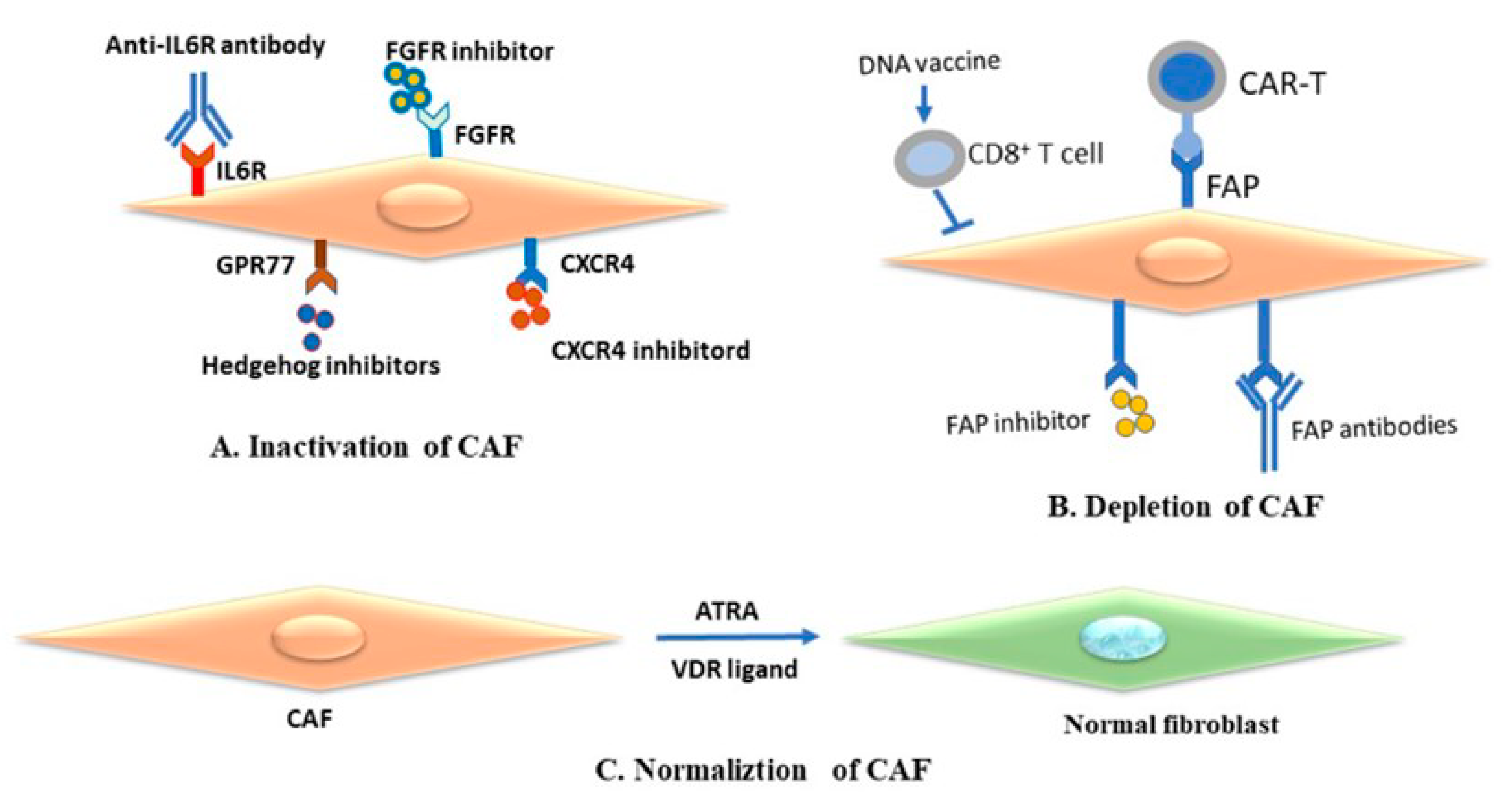
4.1. Cancer-Associated Fibroblasts as Therapeutic Target
4.2. Tumor-Associated Macrophages as Therapeutic Target
4.3. Tumor-Associated Neutrophils
5. Tumor-Promoting Chronic Inflammation as Therapeutic Target for Melanoma Treatment
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Karras, P.; Bordeu, I.; Pozniak, J.; Nowosad, A.; Pazzi, C.; Van Raemdonck, N.; Landeloos, E.; Van Herck, Y.; Pedri, D.; Bervoets, G.; et al. A cellular hierarchy in melanoma uncouples growth and metastasis. Nature 2022, 610, 190–198. [Google Scholar] [CrossRef]
- Motwani, J.; Eccles, M.R. Genetic and Genomic Pathways of Melanoma Development, Invasion and Metastasis. Genes 2021, 12, 1543. [Google Scholar] [CrossRef]
- Weiss, S.A.; Hanniford, D.; Hernando, E.; Osman, I. Revisiting determinants of prognosis in cutaneous melanoma. Cancer 2015, 121, 4108–4123. [Google Scholar] [CrossRef] [Green Version]
- Teixido, C.; Castillo, P.; Martinez-Vila, C.; Arance, A.; Alos, L. Molecular Markers and Targets in Melanoma. Cells 2021, 10, 2320. [Google Scholar] [CrossRef] [PubMed]
- Ascierto, P.A.; Kirkwood, J.M.; Grob, J.J.; Simeone, E.; Grimaldi, A.M.; Maio, M.; Palmieri, G.; Testori, A.; Marincola, F.M.; Mozzillo, N. The role of BRAF V600 mutation in melanoma. J. Transl. Med. 2012, 10, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czarnecka, A.M.; Bartnik, E.; Fiedorowicz, M.; Rutkowski, P. Targeted Therapy in Melanoma and Mechanisms of Resistance. Int. J. Mol. Sci. 2020, 21, 4576. [Google Scholar] [CrossRef] [PubMed]
- Dankner, M.; Rose, A.A.N.; Rajkumar, S.; Siegel, P.M.; Watson, I.R. Classifying BRAF alterations in cancer: New rational therapeutic strategies for actionable mutations. Oncogene 2018, 37, 3183–3199. [Google Scholar] [CrossRef]
- Faghfuri, E.; Nikfar, S.; Niaz, K.; Faramarzi, M.A.; Abdollahi, M. Mitogen-activated protein kinase (MEK) inhibitors to treat melanoma alone or in combination with other kinase inhibitors. Expert. Opin. Drug. Metab. Toxicol. 2018, 14, 317–330. [Google Scholar] [CrossRef]
- Goldinger, S.M.; Buder-Bakhaya, K.; Lo, S.N.; Forschner, A.; McKean, M.; Zimmer, L.; Khoo, C.; Dummer, R.; Eroglu, Z.; Buchbinder, E.I.; et al. Chemotherapy after immune checkpoint inhibitor failure in metastatic melanoma: A retrospective multicentre analysis. Eur. J. Cancer. 2022, 162, 22–33. [Google Scholar] [CrossRef]
- Han, J.; Jung, Y.; Jun, Y.; Park, S.; Lee, S. Elucidating molecular mechanisms of acquired resistance to BRAF inhibitors in melanoma using a microfluidic device and deep sequencing. Genom. Inform. 2021, 19, e2. [Google Scholar] [CrossRef]
- Bucheit, A.D.; Davies, M.A. Emerging insights into resistance to BRAF inhibitors in melanoma. Biochem. Pharmacol. 2014, 87, 381–389. [Google Scholar] [CrossRef]
- Li, B.; Shen, W.; Peng, H.; Li, Y.; Chen, F.; Zheng, L.; Xu, J.; Jia, L. Fibronectin 1 promotes melanoma proliferation and metastasis by inhibiting apoptosis and regulating EMT. Onco. Targets Ther. 2019, 12, 3207–3221. [Google Scholar] [CrossRef] [Green Version]
- Pasqual-Melo, G.; Sagwal, S.K.; Freund, E.; Gandhirajan, R.K.; Frey, B.; von Woedtke, T.; Gaipl, U.; Bekeschus, S. Combination of Gas Plasma and Radiotherapy Has Immunostimulatory Potential and Additive Toxicity in Murine Melanoma Cells in Vitro. Int. J. Mol. Sci. 2020, 21, 1379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boumahdi, S.; de Sauvage, F.J. The great escape: Tumour cell plasticity in resistance to targeted therapy. Nat. Rev. Drug Discov. 2020, 19, 39–56. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Yang, R.; Wei, R.; Du, Y.; He, P.; Liu, X. Pan-cancer analysis reveals RIPK2 predicts prognosis and promotes immune therapy resistance via triggering cytotoxic T lymphocytes dysfunction. Mol. Med. 2022, 28, 47. [Google Scholar] [CrossRef]
- Dulgar, O.; Kutuk, T.; Eroglu, Z. Mechanisms of Resistance to BRAF-Targeted Melanoma Therapies. Am. J. Clin. Dermatol. 2021, 22, 1–10. [Google Scholar] [CrossRef]
- Wessely, A.; Steeb, T.; Berking, C.; Heppt, M.V. How Neural Crest Transcription Factors Contribute to Melanoma Heterogeneity, Cellular Plasticity, and Treatment Resistance. Int. J. Mol. Sci. 2021, 22, 5761. [Google Scholar] [CrossRef]
- Arozarena, I.; Wellbrock, C. Phenotype plasticity as enabler of melanoma progression and therapy resistance. Nat. Rev. Cancer 2019, 19, 377–391. [Google Scholar] [CrossRef] [Green Version]
- Luís, R.; Brito, C.; Pojo, M. Melanoma Metabolism: Cell Survival and Resistance to Therapy. Adv. Exp. Med. Biol. 2020, 1219, 203–223. [Google Scholar] [CrossRef] [PubMed]
- Santourlidis, S.; Schulz, W.A.; Araúzo-Bravo, M.J.; Gerovska, D.; Ott, P.; Bendhack, M.L.; Hassan, M.; Erichsen, L. Epigenetics in the Diagnosis and Therapy of Malignant Melanoma. Int. J. Mol. Sci. 2022, 23, 1531. [Google Scholar] [CrossRef]
- Lee, J.; Molley, T.G.; Seward, C.H.; Abdeen, A.A.; Zhang, H.; Wang, X.; Gandhi, H.; Yang, J.L.; Gaus, K.; Kilian, K.A. Geometric regulation of histone state directs melanoma reprogramming. Commun. Biol. 2020, 3, 341. [Google Scholar] [CrossRef] [PubMed]
- Wellbrock, C.; Arozarena, I. Microphthalmia-associated transcription factor in melanoma development and MAP-kinase pathway targeted therapy. Pigment Cell Melanoma Res. 2015, 28, 390–406. [Google Scholar] [CrossRef] [Green Version]
- Seftor, E.A.; Meltzer, P.S.; Kirschmann, D.A.; Margaryan, N.V.; Seftor, R.E.; Hendrix, M.J. The epigenetic reprogramming of poorly aggressive melanoma cells by a metastatic microenvironment. J. Cell Mol. Med. 2006, 10, 174–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Najem, A.; Soumoy, L.; Sabbah, M.; Krayem, M.; Awada, A.; Journe, F.; Ghanem, G.E. Understanding Molecular Mechanisms of Phenotype Switching and Crosstalk with TME to Reveal New Vulnerabilities of Melanoma. Cells 2022, 11, 1157. [Google Scholar] [CrossRef] [PubMed]
- Pillai, M.; Rajaram, G.; Thakur, P.; Agarwal, N.; Muralidharan, S.; Ray, A.; Barbhaya, D.; Somarelli, J.A.; Jolly, M.K. Mapping phenotypic heterogeneity in melanoma onto the epithelial-hybrid-mesenchymal axis. Front. Oncol. 2022, 12, 913803. [Google Scholar] [CrossRef]
- Corrales, E.; Levit-Zerdoun, E.; Metzger, P.; Mertes, R.; Lehmann, A.; Münch, J.; Lemke, S.; Kowar, S.; Boerries, M. PI3K/AKT signaling allows for MAPK/ERK pathway independency mediating dedifferentiation-driven treatment resistance in melanoma. Cell Commun. Signal 2022, 20, 187. [Google Scholar] [CrossRef] [PubMed]
- Jamal, S.M.E.; Alamodi, A.; Wahl, R.U.; Grada, Z.; Shareef, M.A.; Hassan, S.Y.; Murad, F.; Hassan, S.L.; Santourlidis, S.; Gomez, C.R.; et al. Melanoma stem cell maintenance and chemo-resistance are mediated by CD133 signal to PI3K-dependent pathways. Oncogene 2020, 39, 5468–5478. [Google Scholar] [CrossRef] [PubMed]
- Takata, M.; Morita, R.; Takehara, K. Clonal heterogeneity in sporadic melanomas as revealed by loss-of-heterozygosity analysis. Int. J. Cancer 2000, 85, 492–497. [Google Scholar] [CrossRef]
- Gelmi, M.C.; Houtzagers, L.E.; Strub, T.; Krossa, I.; Jager, M.J. MITF in Normal Melanocytes, Cutaneous and Uveal Melanoma: A Delicate Balance. Int. J. Mol. Sci. 2022, 23, 6001. [Google Scholar] [CrossRef]
- Ballesteros-Álvarez, J.; Dilshat, R.; Fock, V.; Möller, K.; Karl, L.; Larue, L.; Ögmundsdóttir, M.H.; Steingrímsson, E. MITF and TFEB cross-regulation in melanoma cells. PLoS ONE 2020, 15, e0238546. [Google Scholar] [CrossRef]
- Travnickova, J.; Wojciechowska, S.; Khamseh, A.; Gautier, P.; Brown, D.V.; Lefevre, T.; Brombin, A.; Ewing, A.; Capper, A.; Spitzer, M.; et al. Zebrafish MITF-Low Melanoma Subtype Models Reveal Transcriptional Subclusters and MITF-Independent Residual Disease. Cancer Res. 2019, 79, 5769–5784. [Google Scholar] [CrossRef] [Green Version]
- Almeida, F.V.; Douglass, S.M.; Fane, M.E.; Weeraratna, A.T. Bad company: Microenvironmentally mediated resistance to targeted therapy in melanoma. Pigment Cell Melanoma Res. 2019, 32, 237–247. [Google Scholar] [CrossRef]
- Matsuo, T.; Daishaku, S.; Sadzuka, Y. Lactic Acid Promotes Cell Survival by Blocking Autophagy of B16F10 Mouse Melanoma Cells under Glucose Deprivation and Hypoxic Conditions. Biol. Pharm. Bull. 2019, 42, 837–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimm, E.A.; Sikora, A.G.; Ekmekcioglu, S. Molecular pathways: Inflammation-associated nitric-oxide production as a cancer-supporting redox mechanism and a potential therapeutic target. Clin. Cancer Res. 2013, 19, 5557–5563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Möller, K.; Sigurbjornsdottir, S.; Arnthorsson, A.O.; Pogenberg, V.; Dilshat, R.; Fock, V.; Brynjolfsdottir, S.H.; Bindesboll, C.; Bessadottir, M.; Ogmundsdottir, H.M.; et al. MITF has a central role in regulating starvation-induced autophagy in melanoma. Sci. Rep. 2019, 9, 1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Khattouti, A.; Selimovic, D.; Haikel, Y.; Hassan, M. Crosstalk between apoptosis and autophagy: Molecular mechanisms and therapeutic strategies in cancer. J. Cell Death 2013, 6, 37–55. [Google Scholar] [CrossRef] [Green Version]
- Catalani, E.; Giovarelli, M.; Zecchini, S.; Perrotta, C.; Cervia, D. Oxidative Stress and Autophagy as Key Targets in Melanoma Cell Fate. Cancers 2021, 13, 5791. [Google Scholar] [CrossRef] [PubMed]
- Puertollano, R.; Ferguson, S.M.; Brugarolas, J.; Ballabio, A. The complex relationship between TFEB transcription factor phosphorylation and subcellular localization. EMBO J. 2018, 37, e98804. [Google Scholar] [CrossRef]
- Paquette, M.; El-Houjeiri, L.; Zirden, L.C.; Puustinen, P.; Blanchette, P.; Jeong, H.; Dejgaard, K.; Siegel, P.M.; Pause, A. AMPK-dependent phosphorylation is required for transcriptional activation of TFEB and TFE3. Autophagy 2021, 17, 3957–3975. [Google Scholar] [CrossRef]
- Sakamaki, J.I.; Wilkinson, S.; Hahn, M.; Tasdemir, N.; O’Prey, J.; Clark, W.; Hedley, A.; Nixon, C.; Long, J.S.; New, M.; et al. Bromodomain Protein BRD4 Is a Transcriptional Repressor of Autophagy and Lysosomal Function. Mol. Cell 2017, 66, 517–532.e519. [Google Scholar] [CrossRef]
- Davis, I.J.; Hsi, B.L.; Arroyo, J.D.; Vargas, S.O.; Yeh, Y.A.; Motyckova, G.; Valencia, P.; Perez-Atayde, A.R.; Argani, P.; Ladanyi, M.; et al. Cloning of an Alpha-TFEB fusion in renal tumors harboring the t(6;11)(p21;q13) chromosome translocation. Proc. Natl. Acad. Sci. USA 2003, 100, 6051–6056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marathe, H.G.; Watkins-Chow, D.E.; Weider, M.; Hoffmann, A.; Mehta, G.; Trivedi, A.; Aras, S.; Basuroy, T.; Mehrotra, A.; Bennett, D.C.; et al. BRG1 interacts with SOX10 to establish the melanocyte lineage and to promote differentiation. Nucleic Acids Res. 2017, 45, 6442–6458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, L.; Arnheiter, H.; Pavan, W.J. Interspecies difference in the regulation of melanocyte development by SOX10 and MITF. Proc. Natl. Acad. Sci. USA 2006, 103, 9081–9085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Čermák, V.; Škarková, A.; Merta, L.; Kolomazníková, V.; Palušová, V.; Uldrijan, S.; Rösel, D.; Brábek, J. RNA-seq Characterization of Melanoma Phenotype Switch in 3D Collagen after p38 MAPK Inhibitor Treatment. Biomolecules 2021, 11, 449. [Google Scholar] [CrossRef] [PubMed]
- Miskolczi, Z.; Smith, M.P.; Rowling, E.J.; Ferguson, J.; Barriuso, J.; Wellbrock, C. Collagen abundance controls melanoma phenotypes through lineage-specific microenvironment sensing. Oncogene 2018, 37, 3166–3182. [Google Scholar] [CrossRef] [Green Version]
- Granados, K.; Hüser, L.; Federico, A.; Sachindra, S.; Wolff, G.; Hielscher, T.; Novak, D.; Madrigal-Gamboa, V.; Sun, Q.; Vierthaler, M.; et al. T-type calcium channel inhibition restores sensitivity to MAPK inhibitors in de-differentiated and adaptive melanoma cells. Br. J. Cancer 2020, 122, 1023–1036. [Google Scholar] [CrossRef] [Green Version]
- Schieven, S.M.; Traets, J.J.H.; Vliet, A.V.; Baalen, M.V.; Song, J.Y.; Guimaraes, M.D.S.; Kuilman, T.; Peeper, D.S. The Elongin BC complex negatively regulates AXL and marks a differentiated phenotype in melanoma. Mol. Cancer Res. 2023, 21, 428–443. [Google Scholar] [CrossRef]
- Paz, H.; Tsoi, J.; Kalbasi, A.; Grasso, C.S.; McBride, W.H.; Schaue, D.; Butterfield, L.H.; Maurer, D.M.; Ribas, A.; Graeber, T.G.; et al. Interleukin 32 expression in human melanoma. J. Transl. Med. 2019, 17, 113. [Google Scholar] [CrossRef] [Green Version]
- Tsoi, J.; Robert, L.; Paraiso, K.; Galvan, C.; Sheu, K.M.; Lay, J.; Wong, D.J.L.; Atefi, M.; Shirazi, R.; Wang, X.; et al. Multi-stage Differentiation Defines Melanoma Subtypes with Differential Vulnerability to Drug-Induced Iron-Dependent Oxidative Stress. Cancer Cell 2018, 33, 890–904. [Google Scholar] [CrossRef] [Green Version]
- Varum, S.; Baggiolini, A.; Zurkirchen, L.; Atak, Z.K.; Cantù, C.; Marzorati, E.; Bossart, R.; Wouters, J.; Häusel, J.; Tuncer, E.; et al. Yin Yang 1 Orchestrates a Metabolic Program Required for Both Neural Crest Development and Melanoma Formation. Cell Stem Cell 2019, 24, 637–653. [Google Scholar] [CrossRef] [Green Version]
- Sauka-Spengler, T.; Bronner-Fraser, M. A gene regulatory network orchestrates neural crest formation. Nat. Rev. Mol. Cell Biol. 2008, 9, 557–568. [Google Scholar] [CrossRef] [PubMed]
- Konieczkowski, D.J.; Johannessen, C.M.; Abudayyeh, O.; Kim, J.W.; Cooper, Z.A.; Piris, A.; Frederick, D.T.; Barzily-Rokni, M.; Straussman, R.; Haq, R.; et al. A melanoma cell state distinction influences sensitivity to MAPK pathway inhibitors. Cancer Discov. 2014, 4, 816–827. [Google Scholar] [CrossRef] [Green Version]
- Natarajan, V.T.; Ganju, P.; Singh, A.; Vijayan, V.; Kirty, K.; Yadav, S.; Puntambekar, S.; Bajaj, S.; Dani, P.P.; Kar, H.K.; et al. IFN-γ signaling maintains skin pigmentation homeostasis through regulation of melanosome maturation. Proc. Natl. Acad. Sci. USA 2014, 111, 2301–2306. [Google Scholar] [CrossRef] [Green Version]
- Müller, J.; Krijgsman, O.; Tsoi, J.; Robert, L.; Hugo, W.; Song, C.; Kong, X.; Possik, P.A.; Cornelissen-Steijger, P.D.; Geukes Foppen, M.H.; et al. Low MITF/AXL ratio predicts early resistance to multiple targeted drugs in melanoma. Nat. Commun. 2014, 5, 5712. [Google Scholar] [CrossRef] [Green Version]
- Mikolajczyk, A.; Mitula, F.; Popiel, D.; Kaminska, B.; Wieczorek, M.; Pieczykolan, J. Two-Front War on Cancer-Targeting TAM Receptors in Solid Tumour Therapy. Cancers 2022, 14, 2488. [Google Scholar] [CrossRef]
- Grit, J.L.; Pridgeon, M.G.; Essenburg, C.J.; Wolfrum, E.; Madaj, Z.B.; Turner, L.; Wulfkuhle, J.; Petricoin, E.F.; Graveel, C.R.; Steensma, M.R. Kinome Profiling of NF1-Related MPNSTs in Response to Kinase Inhibition and Doxorubicin Reveals Therapeutic Vulnerabilities. Genes 2020, 11, 331. [Google Scholar] [CrossRef] [Green Version]
- Karachaliou, N.; Rosell, R.; Molina, M.A.; Viteri, S. Predicting resistance by selection of signaling pathways. Transl. Lung Cancer Res. 2014, 3, 107–115. [Google Scholar] [CrossRef]
- Fane, M.E.; Chhabra, Y.; Alicea, G.M.; Maranto, D.A.; Douglass, S.M.; Webster, M.R.; Rebecca, V.W.; Marino, G.E.; Almeida, F.; Ecker, B.L.; et al. Stromal changes in the aged lung induce an emergence from melanoma dormancy. Nature 2022, 606, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Balaji, K.; Vijayaraghavan, S.; Diao, L.; Tong, P.; Fan, Y.; Carey, J.P.; Bui, T.N.; Warner, S.; Heymach, J.V.; Hunt, K.K.; et al. AXL Inhibition Suppresses the DNA Damage Response and Sensitizes Cells to PARP Inhibition in Multiple Cancers. Mol. Cancer Res. 2017, 15, 45–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Polo, A.; Luo, Z.; Gerarduzzi, C.; Chen, X.; Little, J.B.; Yuan, Z.M. AXL receptor signalling suppresses p53 in melanoma through stabilization of the MDMX-MDM2 complex. J. Mol. Cell Biol. 2017, 9, 154–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sensi, M.; Catani, M.; Castellano, G.; Nicolini, G.; Alciato, F.; Tragni, G.; De Santis, G.; Bersani, I.; Avanzi, G.; Tomassetti, A.; et al. Human cutaneous melanomas lacking MITF and melanocyte differentiation antigens express a functional Axl receptor kinase. J. Investig. Dermatol. 2011, 131, 2448–2457. [Google Scholar] [CrossRef] [Green Version]
- Hedrich, V.; Breitenecker, K.; Ortmayr, G.; Pupp, F.; Huber, H.; Chen, D.; Sahoo, S.; Jolly, M.K.; Mikulits, W. PRAME Is a Novel Target of Tumor-Intrinsic Gas6/Axl Activation and Promotes Cancer Cell Invasion in Hepatocellular Carcinoma. Cancers 2023, 15, 2415. [Google Scholar] [CrossRef]
- Sala, M.; Allain, N.; Moreau, M.; Jabouille, A.; Henriet, E.; Abou-Hammoud, A.; Uguen, A.; Di-Tommaso, S.; Dourthe, C.; Raymond, A.A.; et al. Discoidin Domain Receptor 2 orchestrates melanoma resistance combining phenotype switching and proliferation. Oncogene 2022, 41, 2571–2586. [Google Scholar] [CrossRef]
- Tworkoski, K.; Singhal, G.; Szpakowski, S.; Zito, C.I.; Bacchiocchi, A.; Muthusamy, V.; Bosenberg, M.; Krauthammer, M.; Halaban, R.; Stern, D.F. Phosphoproteomic screen identifies potential therapeutic targets in melanoma. Mol. Cancer Res. 2011, 9, 801–812. [Google Scholar] [CrossRef] [Green Version]
- Nyakas, M.; Fleten, K.G.; Haugen, M.H.; Engedal, N.; Sveen, C.; Farstad, I.N.; Flørenes, V.A.; Prasmickaite, L.; Mælandsmo, G.M.; Seip, K. AXL inhibition improves BRAF-targeted treatment in melanoma. Sci. Rep. 2022, 12, 5076. [Google Scholar] [CrossRef]
- Dieter, S.M.; Lovecchio, D.; Pataskar, A.; Zowada, M.K.; Körner, P.R.; Khalizieva, A.; van Tellingen, O.; Jäger, D.; Glimm, H.; Agami, R. Suppression of heparan sulfation re-sensitizes YAP1-driven melanoma to MAPK pathway inhibitors. Oncogene 2022, 41, 3953–3968. [Google Scholar] [CrossRef]
- Boshuizen, J.; Koopman, L.A.; Krijgsman, O.; Shahrabi, A.; van den Heuvel, E.G.; Ligtenberg, M.A.; Vredevoogd, D.W.; Kemper, K.; Kuilman, T.; Song, J.Y.; et al. Cooperative targeting of melanoma heterogeneity with an AXL antibody-drug conjugate and BRAF/MEK inhibitors. Nat. Med. 2018, 24, 203–212. [Google Scholar] [CrossRef]
- Brand, T.M.; Iida, M.; Stein, A.P.; Corrigan, K.L.; Braverman, C.M.; Coan, J.P.; Pearson, H.E.; Bahrar, H.; Fowler, T.L.; Bednarz, B.P.; et al. AXL Is a Logical Molecular Target in Head and Neck Squamous Cell Carcinoma. Clin Cancer Res. 2015, 21, 2601–2612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laberge, R.M.; Sun, Y.; Orjalo, A.V.; Patil, C.K.; Freund, A.; Zhou, L.; Curran, S.C.; Davalos, A.R.; Wilson-Edell, K.A.; Liu, S.; et al. Author Correction: MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat. Cell Biol. 2021, 23, 564–565. [Google Scholar] [CrossRef] [PubMed]
- Macleod, K.; Mullen, P.; Sewell, J.; Rabiasz, G.; Lawrie, S.; Miller, E.; Smyth, J.F.; Langdon, S.P. Altered ErbB receptor signaling and gene expression in cisplatin-resistant ovarian cancer. Cancer Res. 2005, 65, 6789–6800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.; Zhou, L.; Xiao, Y.; Andl, T.; Zhang, Y. BRAF Inhibitors Reprogram Cancer-Associated Fibroblasts to Drive Matrix Remodeling and Therapeutic Escape in Melanoma. Cancer Res. 2022, 82, 419–432. [Google Scholar] [CrossRef] [PubMed]
- Loria, R.; Laquintana, V.; Scalera, S.; Fraioli, R.; Caprara, V.; Falcone, I.; Bazzichetto, C.; Di Martile, M.; Rosanò, L.; Del Bufalo, D.; et al. SEMA6A/RhoA/YAP axis mediates tumor-stroma interactions and prevents response to dual BRAF/MEK inhibition in BRAF-mutant melanoma. J. Exp. Clin. Cancer Res. 2022, 41, 148. [Google Scholar] [CrossRef]
- Fedorenko, I.V.; Wargo, J.A.; Flaherty, K.T.; Messina, J.L.; Smalley, K.S.M. BRAF Inhibition Generates a Host-Tumor Niche that Mediates Therapeutic Escape. J. Investig. Dermatol. 2015, 135, 3115–3124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capparelli, C.; Rosenbaum, S.; Berger, A.C.; Aplin, A.E. Fibroblast-derived neuregulin 1 promotes compensatory ErbB3 receptor signaling in mutant BRAF melanoma. J. Biol. Chem. 2015, 290, 24267–24277. [Google Scholar] [CrossRef] [Green Version]
- Gaggioli, C.; Robert, G.; Bertolotto, C.; Bailet, O.; Abbe, P.; Spadafora, A.; Bahadoran, P.; Ortonne, J.P.; Baron, V.; Ballotti, R.; et al. Tumor-derived fibronectin is involved in melanoma cell invasion and regulated by V600E B-Raf signaling pathway. J. Investig. Dermatol. 2007, 127, 400–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.; Gan, G.; Wang, X.; Xu, T.; Xie, W. The HGF-MET axis coordinates liver cancer metabolism and autophagy for chemotherapeutic resistance. Autophagy 2019, 15, 1258–1279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czyz, M. HGF/c-MET Signaling in Melanocytes and Melanoma. Int. J. Mol. Sci. 2018, 19, 3844. [Google Scholar] [CrossRef] [Green Version]
- Asnaghi, L.; Gezgin, G.; Tripathy, A.; Handa, J.T.; Merbs, S.L.; van der Velden, P.A.; Jager, M.J.; Harbour, J.W.; Eberhart, C.G. EMT-associated factors promote invasive properties of uveal melanoma cells. Mol. Vis. 2015, 21, 919–929. [Google Scholar]
- Yoon, Y.S.; Lee, Y.J.; Choi, Y.H.; Park, Y.M.; Kang, J.L. Macrophages programmed by apoptotic cells inhibit epithelial-mesenchymal transition in lung alveolar epithelial cells via PGE2, PGD2, and HGF. Sci. Rep. 2016, 6, 20992. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Sui, X.; Weng, L.; Liu, Y. SNAIL1: Linking Tumor Metastasis to Immune Evasion. Front. Immunol. 2021, 12, 724200. [Google Scholar] [CrossRef]
- Kaufhold, S.; Bonavida, B. Central role of Snail1 in the regulation of EMT and resistance in cancer: A target for therapeutic intervention. J. Exp. Clin. Cancer Res. 2014, 33, 62. [Google Scholar] [CrossRef]
- Sil, H.; Sen, T.; Chatterjee, A. Fibronectin-integrin (alpha5beta1) modulates migration and invasion of murine melanoma cell line B16F10 by involving MMP-9. Oncol. Res. 2011, 19, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Natali, P.G.; Nicotra, M.R.; Di Filippo, F.; Bigotti, A. Expression of fibronectin, fibronectin isoforms and integrin receptors in melanocytic lesions. Br. J. Cancer. 1995, 71, 1243–1247. [Google Scholar] [CrossRef] [Green Version]
- Iida, J.; Skubitz, A.P.; Furcht, L.T.; Wayner, E.A.; McCarthy, J.B. Coordinate role for cell surface chondroitin sulfate proteoglycan and alpha 4 beta 1 integrin in mediating melanoma cell adhesion to fibronectin. J. Cell Biol. 1992, 118, 431–444. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.J.; Park, I.; Hong, I.K.; Byun, H.J.; Choi, J.; Kim, Y.M.; Lee, H. Fibronectin and vitronectin induce AP-1-mediated matrix metalloproteinase-9 expression through integrin α(5)β(1)/α(v)β(3)-dependent Akt, ERK and JNK signaling pathways in human umbilical vein endothelial cells. Cell Signal 2011, 23, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Yang, Q.; Shi, K.; Xiao, Y.; Wei, X.; Qian, Z. Intratumoral fate of functional nanoparticles in response to microenvironment factor: Implications on cancer diagnosis and therapy. Adv. Drug. Deliv. Rev. 2019, 143, 37–67. [Google Scholar] [CrossRef]
- Saha, S.; Lo, P.K.; Duan, X.; Chen, H.; Wang, Q. Breast tumour initiating cell fate is regulated by microenvironmental cues from an extracellular matrix. Integr. Biol. 2012, 4, 897–904. [Google Scholar] [CrossRef] [Green Version]
- Gargalionis, A.N.; Papavassiliou, K.A.; Papavassiliou, A.G. Mechanobiology of solid tumors. Biochim. Biophys. Acta. Mol. Basis Dis. 2022, 1868, 166555. [Google Scholar] [CrossRef]
- Tan, Q.; Saggar, J.K.; Yu, M.; Wang, M.; Tannock, I.F. Mechanisms of Drug Resistance Related to the Microenvironment of Solid Tumors and Possible Strategies to Inhibit Them. Cancer J. 2015, 21, 254–262. [Google Scholar] [CrossRef]
- Biffi, G.; Tuveson, D.A. Diversity and Biology of Cancer-Associated Fibroblasts. Physiol. Rev. 2021, 101, 147–176. [Google Scholar] [CrossRef]
- Chen, D.; Zhang, X.; Li, Z.; Zhu, B. Metabolic regulatory crosstalk between tumor microenvironment and tumor-associated macrophages. Theranostics 2021, 11, 1016–1030. [Google Scholar] [CrossRef]
- Barrett, R.L.; Puré, E. Cancer-associated fibroblasts and their influence on tumor immunity and immunotherapy. Elife 2020, 9. [Google Scholar] [CrossRef]
- Fiori, M.E.; Di Franco, S.; Villanova, L.; Bianca, P.; Stassi, G.; De Maria, R. Cancer-associated fibroblasts as abettors of tumor progression at the crossroads of EMT and therapy resistance. Mol. Cancer 2019, 18, 70. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Tang, L.L.; Ma, J. Survival-related indicators ALOX12B and SPRR1A are associated with DNA damage repair and tumor microenvironment status in HPV 16-negative head and neck squamous cell carcinoma patients. BMC Cancer 2022, 22, 714. [Google Scholar] [CrossRef]
- Heichler, C.; Scheibe, K.; Schmied, A.; Geppert, C.I.; Schmid, B.; Wirtz, S.; Thoma, O.M.; Kramer, V.; Waldner, M.J.; Büttner, C.; et al. STAT3 activation through IL-6/IL-11 in cancer-associated fibroblasts promotes colorectal tumour development and correlates with poor prognosis. Gut 2020, 69, 1269–1282. [Google Scholar] [CrossRef]
- Xiao, Y.; Yu, D. Tumor microenvironment as a therapeutic target in cancer. Pharmacol. Ther. 2021, 221, 107753. [Google Scholar] [CrossRef]
- Mao, X.; Xu, J.; Wang, W.; Liang, C.; Hua, J.; Liu, J.; Zhang, B.; Meng, Q.; Yu, X.; Shi, S. Crosstalk between cancer-associated fibroblasts and immune cells in the tumor microenvironment: New findings and future perspectives. Mol. Cancer 2021, 20, 131. [Google Scholar] [CrossRef]
- Hinshaw, D.C.; Shevde, L.A. The Tumor Microenvironment Innately Modulates Cancer Progression. Cancer Res. 2019, 79, 4557–4566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Contreras, O.; Rossi, F.M.; Brandan, E. Adherent muscle connective tissue fibroblasts are phenotypically and biochemically equivalent to stromal fibro/adipogenic progenitors. Matrix Biol. Plus 2019, 2, 100006. [Google Scholar] [CrossRef]
- Serban, A.I.; Stanca, L.; Geicu, O.I.; Munteanu, M.C.; Dinischiotu, A. RAGE and TGF-β1 Cross-Talk Regulate Extracellular Matrix Turnover and Cytokine Synthesis in AGEs Exposed Fibroblast Cells. PLoS ONE 2016, 11, e0152376. [Google Scholar] [CrossRef] [PubMed]
- Tschumperlin, D.J. Matrix, mesenchyme, and mechanotransduction. Ann. Am. Thorac. Soc. 2015, 12 (Suppl. 1), S24–S29. [Google Scholar] [CrossRef] [Green Version]
- Rippa, A.L.; Kalabusheva, E.P.; Vorotelyak, E.A. Regeneration of Dermis: Scarring and Cells Involved. Cells 2019, 8, 607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thulabandu, V.; Chen, D.; Atit, R.P. Dermal fibroblast in cutaneous development and healing. Wiley Interdiscip. Rev. Dev. Biol. 2018, 7, 10. [Google Scholar] [CrossRef] [PubMed]
- Monteran, L.; Erez, N. The Dark Side of Fibroblasts: Cancer-Associated Fibroblasts as Mediators of Immunosuppression in the Tumor Microenvironment. Front. Immunol. 2019, 10, 1835. [Google Scholar] [CrossRef] [Green Version]
- Lavie, D.; Ben-Shmuel, A.; Erez, N.; Scherz-Shouval, R. Cancer-associated fibroblasts in the single-cell era. Nat. Cancer 2022, 3, 793–807. [Google Scholar] [CrossRef]
- Papaccio, F.; Kovacs, D.; Bellei, B.; Caputo, S.; Migliano, E.; Cota, C.; Picardo, M. Profiling Cancer-Associated Fibroblasts in Melanoma. Int. J. Mol. Sci. 2021, 22, 7255. [Google Scholar] [CrossRef]
- Valkenburg, K.C.; de Groot, A.E.; Pienta, K.J. Targeting the tumour stroma to improve cancer therapy. Nat. Rev. Clin. Oncol. 2018, 15, 366–381. [Google Scholar] [CrossRef]
- Yu, B.; Wu, K.; Wang, X.; Zhang, J.; Wang, L.; Jiang, Y.; Zhu, X.; Chen, W.; Yan, M. Periostin secreted by cancer-associated fibroblasts promotes cancer stemness in head and neck cancer by activating protein tyrosine kinase 7. Cell Death Dis. 2018, 9, 1082. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.S.; Tang, X.T.; Lin, M.; Yuan, J.; Peng, Y.J.; Yin, X.; Shang, G.; Ge, G.; Ren, Z.; Zhou, B.O. Perivenous Stellate Cells Are the Main Source of Myofibroblasts and Cancer-Associated Fibroblasts Formed After Chronic Liver Injuries. Hepatology 2021, 74, 1578–1594. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.Y.; Chan, M.K.; Li, J.S.; Chan, A.S.; Tang, P.C.; Leung, K.T.; To, K.F.; Lan, H.Y.; Tang, P.M. TGF-β Signaling: From Tissue Fibrosis to Tumor Microenvironment. Int. J. Mol. Sci. 2021, 22, 7575. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.T.; Huang, Y.C.; Chen, W.C.; Chen, M.F. Effect of 1α,25-Dihydroxyvitamin D3 on the Radiation Response in Prostate Cancer: Association With IL-6 Signaling. Front. Oncol. 2021, 11, 619365. [Google Scholar] [CrossRef]
- Bremnes, R.M.; Dønnem, T.; Al-Saad, S.; Al-Shibli, K.; Andersen, S.; Sirera, R.; Camps, C.; Marinez, I.; Busund, L.T. The role of tumor stroma in cancer progression and prognosis: Emphasis on carcinoma-associated fibroblasts and non-small cell lung cancer. J. Thorac. Oncol. 2011, 6, 209–217. [Google Scholar] [CrossRef] [Green Version]
- Moubarak, R.S.; de Pablos-Aragoneses, A.; Ortiz-Barahona, V.; Gong, Y.; Gowen, M.; Dolgalev, I.; Shadaloey, S.A.A.; Argibay, D.; Karz, A.; Von Itter, R.; et al. The histone demethylase PHF8 regulates TGFβ signaling and promotes melanoma metastasis. Sci. Adv. 2022, 8, eabi7127. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Yamamoto, S.; Ishii, Y.; Sang, Y.; Hamashima, T.; Van De, N.; Nishizono, H.; Inoue, R.; Mori, H.; Sasahara, M. Glioma-Derived Platelet-Derived Growth Factor-BB Recruits Oligodendrocyte Progenitor Cells via Platelet-Derived Growth Factor Receptor-α and Remodels Cancer Stroma. Am. J. Pathol. 2016, 186, 1081–1091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.Y.; Hu, S.Q.; Xiao, L. The cancer-associated fibroblasts and drug resistance. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 2112–2119. [Google Scholar]
- Mazurkiewicz, J.; Simiczyjew, A.; Dratkiewicz, E.; Ziętek, M.; Matkowski, R.; Nowak, D. Stromal Cells Present in the Melanoma Niche Affect Tumor Invasiveness and Its Resistance to Therapy. Int. J. Mol. Sci. 2021, 22, 529. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Evans, K.; Xiao, C.; DeVito, N.; Theivanthiran, B.; Holtzhausen, A.; Siska, P.J.; Blobe, G.C.; Hanks, B.A. Stromal Fibroblasts Mediate Anti-PD-1 Resistance via MMP-9 and Dictate TGFβ Inhibitor Sequencing in Melanoma. Cancer Immunol. Res. 2018, 6, 1459–1471. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Sun, Z.; Peng, G.; Xiao, Y.; Guo, J.; Wu, B.; Zhou, W.; Li, J.; Li, Z.; Bai, C.; et al. Single-cell RNA sequencing reveals a pro-invasive cancer-associated fibroblast subgroup associated with poor clinical outcomes in patients with gastric cancer. Theranostics 2022, 12, 620–638. [Google Scholar] [CrossRef]
- Strnadová, K.; Pfeiferová, L.; Přikryl, P.; Dvořánková, B.; Vlčák, E.; Frýdlová, J.; Vokurka, M.; Novotný, J.; Šáchová, J.; Hradilová, M.; et al. Exosomes produced by melanoma cells significantly influence the biological properties of normal and cancer-associated fibroblasts. Histochem. Cell Biol. 2022, 157, 153–172. [Google Scholar] [CrossRef]
- Pessotti, D.S.; Andrade-Silva, D.; Serrano, S.M.T.; Zelanis, A. Heterotypic signaling between dermal fibroblasts and melanoma cells induces phenotypic plasticity and proteome rearrangement in malignant cells. Biochim. Biophys. Acta Proteins Proteom. 2020, 1868, 140525. [Google Scholar] [CrossRef]
- Ren, X.; Li, L.; Wu, J.; Lin, K.; He, Y.; Bian, L. PDGF-BB regulates the transformation of fibroblasts into cancer-associated fibroblasts via the lncRNA LURAP1L-AS1/LURAP1L/IKK/IκB/NF-κB signaling pathway. Oncol. Lett. 2021, 22, 537. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Yu, X.; Yang, F.; Zhang, Z.; Shen, J.; Sun, J.; Choksi, S.; Jitkaew, S.; Shu, Y. Reprogramming of Normal Fibroblasts into Cancer-Associated Fibroblasts by miRNAs-Mediated CCL2/VEGFA Signaling. PLoS Genet. 2016, 12, e1006244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaziri, N.; Shariati, L.; Zarrabi, A.; Farazmand, A.; Haghjooy Javanmard, S. Cancer-Associated Fibroblasts Regulate the Plasticity of Breast Cancer Stemness through the Production of Leukemia Inhibitory Factor. Life 2021, 11, 1298. [Google Scholar] [CrossRef]
- Hu, J.L.; Wang, W.; Lan, X.L.; Zeng, Z.C.; Liang, Y.S.; Yan, Y.R.; Song, F.Y.; Wang, F.F.; Zhu, X.H.; Liao, W.J.; et al. CAFs secreted exosomes promote metastasis and chemotherapy resistance by enhancing cell stemness and epithelial-mesenchymal transition in colorectal cancer. Mol. Cancer 2019, 18, 91. [Google Scholar] [CrossRef] [Green Version]
- Ren, J.; Ding, L.; Zhang, D.; Shi, G.; Xu, Q.; Shen, S.; Wang, Y.; Wang, T.; Hou, Y. Carcinoma-associated fibroblasts promote the stemness and chemoresistance of colorectal cancer by transferring exosomal lncRNA H19. Theranostics 2018, 8, 3932–3948. [Google Scholar] [CrossRef]
- Huang, K.; Zhang, X.; Hao, Y.; Feng, R.; Wang, H.; Shu, Z.; Li, A.; Du, M. Hypoxia Tumor Microenvironment Activates GLI2 through HIF-1. Comput. Math. Methods Med. 2022, 2022, 2032895. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.A.; Chen, Y.F.; Bao, Y.; Mahara, S.; Yatim, S.M.J.M.; Oguz, G.; Lee, P.L.; Feng, M.; Cai, Y.; Tan, E.Y.; et al. Hypoxic tumor microenvironment activates GLI2 via HIF-1α and TGF-β2 to promote chemoresistance in colorectal cancer. Proc. Natl. Acad. Sci. USA 2018, 115, E5990–E5999. [Google Scholar] [CrossRef] [Green Version]
- Mimeault, M.; Batra, S.K. Hypoxia-inducing factors as master regulators of stemness properties and altered metabolism of cancer- and metastasis-initiating cells. J. Cell Mol. Med. 2013, 17, 30–54. [Google Scholar] [CrossRef]
- Shin, J.; Kim, J.H.; Kim, E.K. Repeated exposure of human fibroblasts to UVR induces secretion of stem cell factor and senescence. J. Eur. Acad. Dermatol. Venereol. 2012, 26, 1577–1580. [Google Scholar] [CrossRef]
- Li, D.; Qu, C.; Ning, Z.; Wang, H.; Zang, K.; Zhuang, L.; Chen, L.; Wang, P.; Meng, Z. Radiation promotes epithelial-to-mesenchymal transition and invasion of pancreatic cancer cell by activating carcinoma-associated fibroblasts. Am. J. Cancer Res. 2016, 6, 2192–2206. [Google Scholar]
- Loo, T.M.; Miyata, K.; Tanaka, Y.; Takahashi, A. Cellular senescence and senescence-associated secretory phenotype via the cGAS-STING signaling pathway in cancer. Cancer Sci. 2020, 111, 304–311. [Google Scholar] [CrossRef] [Green Version]
- Strub, T.; Giuliano, S.; Ye, T.; Bonet, C.; Keime, C.; Kobi, D.; Le Gras, S.; Cormont, M.; Ballotti, R.; Bertolotto, C.; et al. Essential role of microphthalmia transcription factor for DNA replication, mitosis and genomic stability in melanoma. Oncogene 2011, 30, 2319–2332. [Google Scholar] [CrossRef] [Green Version]
- Paskaš, S.; Krajnović, T.; Basile, M.S.; Dunđerović, D.; Cavalli, E.; Mangano, K.; Mammana, S.; Al-Abed, Y.; Nicoletti, F.; Mijatović, S.; et al. Senescence as a main mechanism of Ritonavir and Ritonavir-NO action against melanoma. Mol. Carcinog. 2019, 58, 1362–1375. [Google Scholar] [CrossRef]
- Kawaguchi, K.; Komoda, K.; Mikawa, R.; Asai, A.; Sugimoto, M. Cellular senescence promotes cancer metastasis by enhancing soluble E-cadherin production. iScience 2021, 24, 103022. [Google Scholar] [CrossRef]
- Meierjohann, S. Oxidative stress in melanocyte senescence and melanoma transformation. Eur. J. Cell Biol. 2014, 93, 36–41. [Google Scholar] [CrossRef]
- Ou, H.L.; Hoffmann, R.; González-López, C.; Doherty, G.J.; Korkola, J.E.; Muñoz-Espín, D. Cellular senescence in cancer: From mechanisms to detection. Mol. Oncol. 2021, 15, 2634–2671. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, T.; Koiwa, M.; Yonemura, A.; Miyake, K.; Kariya, R.; Kubota, S.; Yokomizo-Nakano, T.; Yasuda-Yoshihara, N.; Uchihara, T.; Itoyama, R.; et al. Inflammation-driven senescence-associated secretory phenotype in cancer-associated fibroblasts enhances peritoneal dissemination. Cell Rep. 2021, 34, 108779. [Google Scholar] [CrossRef] [PubMed]
- Novotný, J.; Strnadová, K.; Dvořánková, B.; Kocourková, Š.; Jakša, R.; Dundr, P.; Pačes, V.; Smetana, K.; Kolář, M.; Lacina, L. Single-Cell RNA Sequencing Unravels Heterogeneity of the Stromal Niche in Cutaneous Melanoma Heterogeneous Spheroids. Cancers 2020, 12, 3324. [Google Scholar] [CrossRef] [PubMed]
- Ohtani, N. The roles and mechanisms of senescence-associated secretory phenotype (SASP): Can it be controlled by senolysis? Inflamm. Regen. 2022, 42, 11. [Google Scholar] [CrossRef]
- Roger, L.; Tomas, F.; Gire, V. Mechanisms and Regulation of Cellular Senescence. Int. J. Mol. Sci. 2021, 22, 13173. [Google Scholar] [CrossRef]
- Jochems, F.; Thijssen, B.; De Conti, G.; Jansen, R.; Pogacar, Z.; Groot, K.; Wang, L.; Schepers, A.; Wang, C.; Jin, H.; et al. The Cancer SENESCopedia: A delineation of cancer cell senescence. Cell Rep. 2021, 36, 109441. [Google Scholar] [CrossRef] [PubMed]
- Lopes-Paciencia, S.; Saint-Germain, E.; Rowell, M.C.; Ruiz, A.F.; Kalegari, P.; Ferbeyre, G. The senescence-associated secretory phenotype and its regulation. Cytokine 2019, 117, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Herranz, N.; Gallage, S.; Gil, J. TORn about SASP regulation. Cell Cycle 2015, 14, 3771–3772. [Google Scholar] [CrossRef] [Green Version]
- Ito, Y.; Hoare, M.; Narita, M. Spatial and Temporal Control of Senescence. Trends Cell Biol. 2017, 27, 820–832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miwa, S.; Kashyap, S.; Chini, E.; von Zglinicki, T. Mitochondrial dysfunction in cell senescence and aging. J Clin Invest 2022, 132, e158447. [Google Scholar] [CrossRef]
- Aguayo-Mazzucato, C.; Andle, J.; Lee, T.B.; Midha, A.; Talemal, L.; Chipashvili, V.; Hollister-Lock, J.; van Deursen, J.; Weir, G.; Bonner-Weir, S. Acceleration of β Cell Aging Determines Diabetes and Senolysis Improves Disease Outcomes. Cell Metab. 2019, 30, 129–142.e124. [Google Scholar] [CrossRef]
- Di Micco, R.; Krizhanovsky, V.; Baker, D.; d’Adda di Fagagna, F. Cellular senescence in ageing: From mechanisms to therapeutic opportunities. Nat. Rev. Mol. Cell Biol. 2021, 22, 75–95. [Google Scholar] [CrossRef]
- Homann, L.; Rentschler, M.; Brenner, E.; Böhm, K.; Röcken, M.; Wieder, T. IFN-γ and TNF Induce Senescence and a Distinct Senescence-Associated Secretory Phenotype in Melanoma. Cells 2022, 11, 1514. [Google Scholar] [CrossRef] [PubMed]
- Peeper, D.S. Oncogene-induced senescence and melanoma: Where do we stand? Pigment Cell Melanoma Res. 2011, 24, 1107–1111. [Google Scholar] [CrossRef]
- Shain, A.H.; Yeh, I.; Kovalyshyn, I.; Sriharan, A.; Talevich, E.; Gagnon, A.; Dummer, R.; North, J.; Pincus, L.; Ruben, B.; et al. The Genetic Evolution of Melanoma from Precursor Lesions. N. Engl. J. Med. 2015, 373, 1926–1936. [Google Scholar] [CrossRef]
- Farghaly, S.A. Combination therapy of cytotoxic t-lymphocyte-associated antigen 4 (ctla-4) and programmed death 1 (pd 1) blocker, check point inhibitors for treatment of patients with advanced and recurrent epithelial ovarian cancer. Eur. J. Gynaecol. Oncol. 2017, 38, 7–9. [Google Scholar]
- Khan, H.; Gucalp, R.; Shapira, I. Evolving Concepts: Immunity in Oncology from Targets to Treatments. J. Oncol. 2015, 2015, 847383. [Google Scholar] [CrossRef]
- Vokurka, M.; Lacina, L.; Brábek, J.; Kolář, M.; Ng, Y.Z.; Smetana, K. Cancer-Associated Fibroblasts Influence the Biological Properties of Malignant Tumours via Paracrine Secretion and Exosome Production. Int. J. Mol. Sci. 2022, 23, 964. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Sakakura, K.; Kawabata-Iwakawa, R.; Rokudai, S.; Toyoda, M.; Nishiyama, M.; Chikamatsu, K. Immunosuppressive activity of cancer-associated fibroblasts in head and neck squamous cell carcinoma. Cancer Immunol. Immunother. 2015, 64, 1407–1417. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Bosch, N.; Vinaixa, J.; Navarro, P. Immune Evasion in Pancreatic Cancer: From Mechanisms to Therapy. Cancers 2018, 10, 6. [Google Scholar] [CrossRef] [Green Version]
- van der Valk, M.J.M.; Marijnen, C.A.M.; van Etten, B.; Dijkstra, E.A.; Hilling, D.E.; Kranenbarg, E.M.; Putter, H.; Roodvoets, A.G.H.; Bahadoer, R.R.; Fokstuen, T.; et al. Compliance and tolerability of short-course radiotherapy followed by preoperative chemotherapy and surgery for high-risk rectal cancer - Results of the international randomized RAPIDO-trial. Radiother. Oncol. 2020, 147, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Wang, Q.; Zhang, B.; Yuan, Y.; Xie, W.; Huang, X.; Zhou, C.; Zhang, S.; Niu, S.; Chang, H.; et al. Neoadjuvant chemotherapy and radiotherapy followed by resection/ablation in stage IV rectal cancer patients with potentially resectable metastases. BMC Cancer 2021, 21, 1333. [Google Scholar] [CrossRef]
- Yamada, T.; Matsuda, A.; Takahashi, G.; Iwai, T.; Takeda, K.; Ueda, K.; Kuriyama, S.; Koizumi, M.; Shinji, S.; Yokoyama, Y.; et al. Emerging RAS, BRAF, and EGFR mutations in cell-free DNA of metastatic colorectal patients are associated with both primary and secondary resistance to first-line anti-EGFR therapy. Int. J. Clin. Oncol. 2020, 25, 1523–1532. [Google Scholar] [CrossRef]
- Fattore, L.; Marra, E.; Pisanu, M.E.; Noto, A.; de Vitis, C.; Belleudi, F.; Aurisicchio, L.; Mancini, R.; Torrisi, M.R.; Ascierto, P.A.; et al. Activation of an early feedback survival loop involving phospho-ErbB3 is a general response of melanoma cells to RAF/MEK inhibition and is abrogated by anti-ErbB3 antibodies. J. Transl. Med. 2013, 11, 180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Liu, M.; Huang, Y.; Wang, Z.; Wang, Y.; He, K.; Bai, R.; Ying, T.; Zheng, Y. Differential Gene Expression and Methylation Analysis of Melanoma in TCGA Database to Further Study the Expression Pattern of KYNU in Melanoma. J. Pers. Med. 2022, 12, 1209. [Google Scholar] [CrossRef]
- Delyon, J.; Lebbe, C.; Dumaz, N. Targeted therapies in melanoma beyond BRAF: Targeting NRAS-mutated and KIT-mutated melanoma. Curr. Opin. Oncol. 2020, 32, 79–84. [Google Scholar] [CrossRef]
- Ponti, G.; Manfredini, M.; Greco, S.; Pellacani, G.; Depenni, R.; Tomasi, A.; Maccaferri, M.; Cascinu, S. BRAF, NRAS and C-KIT Advanced Melanoma: Clinico-pathological Features, Targeted-Therapy Strategies and Survival. Anticancer Res. 2017, 37, 7043–7048. [Google Scholar] [CrossRef]
- Turner, E.; Chen, L.; Foulke, J.G.; Gu, Z.; Tian, F. CRISPR/Cas9 Edited RAS & MEK Mutant Cells Acquire BRAF and MEK Inhibitor Resistance with MEK1 Q56P Restoring Sensitivity to MEK/BRAF Inhibitor Combo and KRAS G13D Gaining Sensitivity to Immunotherapy. Cancers 2022, 14, 5449. [Google Scholar] [CrossRef]
- Sun, C.; Wang, L.; Huang, S.; Heynen, G.J.; Prahallad, A.; Robert, C.; Haanen, J.; Blank, C.; Wesseling, J.; Willems, S.M.; et al. Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature 2014, 508, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Walbrecq, G.; Lecha, O.; Gaigneaux, A.; Fougeras, M.R.; Philippidou, D.; Margue, C.; Tetsi Nomigni, M.; Bernardin, F.; Dittmar, G.; Behrmann, I.; et al. Hypoxia-Induced Adaptations of miRNomes and Proteomes in Melanoma Cells and Their Secreted Extracellular Vesicles. Cancers 2020, 12, 692. [Google Scholar] [CrossRef] [Green Version]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Marzagalli, M.; Ebelt, N.D.; Manuel, E.R. Unraveling the crosstalk between melanoma and immune cells in the tumor microenvironment. Semin. Cancer Biol. 2019, 59, 236–250. [Google Scholar] [CrossRef]
- Avagliano, A.; Fiume, G.; Pelagalli, A.; Sanità, G.; Ruocco, M.R.; Montagnani, S.; Arcucci, A. Metabolic Plasticity of Melanoma Cells and Their Crosstalk With Tumor Microenvironment. Front. Oncol. 2020, 10, 722. [Google Scholar] [CrossRef] [PubMed]
- Elia, A.R.; Caputo, S.; Bellone, M. Immune Checkpoint-Mediated Interactions Between Cancer and Immune Cells in Prostate Adenocarcinoma and Melanoma. Front. Immunol. 2018, 9, 1786. [Google Scholar] [CrossRef] [Green Version]
- Bagaev, A.; Kotlov, N.; Nomie, K.; Svekolkin, V.; Gafurov, A.; Isaeva, O.; Osokin, N.; Kozlov, I.; Frenkel, F.; Gancharova, O.; et al. Conserved pan-cancer microenvironment subtypes predict response to immunotherapy. Cancer Cell 2021, 39, 845–865.e847. [Google Scholar] [CrossRef]
- Dharanipragada, P.; Zhang, X.; Liu, S.; Lomeli, S.H.; Hong, A.; Wang, Y.; Yang, Z.; Lo, K.Z.; Vega-Crespo, A.; Ribas, A.; et al. Blocking Genomic Instability Prevents Acquired Resistance to MAPK Inhibitor Therapy in Melanoma. Cancer Discov. 2023, 13, 880–909. [Google Scholar] [CrossRef]
- Sensebé, L. Beyond genetic stability of mesenchymal stromal cells. Cytotherapy 2013, 15, 1307–1308. [Google Scholar] [CrossRef]
- Thompson, B.J. YAP/TAZ: Drivers of Tumor Growth, Metastasis, and Resistance to Therapy. Bioessays 2020, 42, e1900162. [Google Scholar] [CrossRef] [Green Version]
- Fujimura, T. Stromal Factors as a Target for Immunotherapy in Melanoma and Non-Melanoma Skin Cancers. Int. J. Mol. Sci. 2022, 23, 4044. [Google Scholar] [CrossRef]
- Bejarano, L.; Jordāo, M.J.C.; Joyce, J.A. Therapeutic Targeting of the Tumor Microenvironment. Cancer Discov. 2021, 11, 933–959. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Yang, K.; Andl, T.; Wickett, R.R.; Zhang, Y. Perspective of Targeting Cancer-Associated Fibroblasts in Melanoma. J. Cancer 2015, 6, 717–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamson, E.J.; Keane, F.M.; Tholen, S.; Schilling, O.; Gorrell, M.D. Understanding fibroblast activation protein (FAP): Substrates, activities, expression and targeting for cancer therapy. Proteomics Clin. Appl. 2014, 8, 454–463. [Google Scholar] [CrossRef] [PubMed]
- Zboralski, D.; Hoehne, A.; Bredenbeck, A.; Schumann, A.; Nguyen, M.; Schneider, E.; Ungewiss, J.; Paschke, M.; Haase, C.; von Hacht, J.L.; et al. Preclinical evaluation of FAP-2286 for fibroblast activation protein targeted radionuclide imaging and therapy. Eur. J. Nucl. Med. Mol. Imaging 2022, 49, 3651–3667. [Google Scholar] [CrossRef]
- Zhang, Y.; Ertl, H.C. Depletion of FAP+ cells reduces immunosuppressive cells and improves metabolism and functions CD8+T cells within tumors. Oncotarget 2016, 7, 23282–23299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brożyna, A.A.; Hoffman, R.M.; Slominski, A.T. Relevance of Vitamin D in Melanoma Development, Progression and Therapy. Anticancer Res. 2020, 40, 473–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nemazannikova, N.; Antonas, K.; Dass, C.R. Role of vitamin D metabolism in cutaneous tumour formation and progression. J. Pharm. Pharmacol. 2013, 65, 2–10. [Google Scholar] [CrossRef]
- Lederle, W.; Stark, H.J.; Skobe, M.; Fusenig, N.E.; Mueller, M.M. Platelet-derived growth factor-BB controls epithelial tumor phenotype by differential growth factor regulation in stromal cells. Am. J. Pathol. 2006, 169, 1767–1783. [Google Scholar] [CrossRef] [Green Version]
- Onimaru, M.; Yonemitsu, Y. Angiogenic and lymphangiogenic cascades in the tumor microenvironment. Front. Biosci. 2011, 3, 216–225. [Google Scholar] [CrossRef] [Green Version]
- Hawryluk, G.W.; Mothe, A.; Wang, J.; Wang, S.; Tator, C.; Fehlings, M.G. An in vivo characterization of trophic factor production following neural precursor cell or bone marrow stromal cell transplantation for spinal cord injury. Stem Cells Dev. 2012, 21, 2222–2238. [Google Scholar] [CrossRef] [Green Version]
- Di Desidero, T.; Orlandi, P.; Fioravanti, A.; Alì, G.; Cremolini, C.; Loupakis, F.; Gentile, D.; Banchi, M.; Cucchiara, F.; Antoniotti, C.; et al. Chemotherapeutic and antiangiogenic drugs beyond tumor progression in colon cancer: Evaluation of the effects of switched schedules and related pharmacodynamics. Biochem. Pharmacol. 2019, 164, 94–105. [Google Scholar] [CrossRef]
- Benjamin, R.S.; Schöffski, P.; Hartmann, J.T.; Van Oosterom, A.; Bui, B.N.; Duyster, J.; Schuetze, S.; Blay, J.Y.; Reichardt, P.; Rosen, L.S.; et al. Efficacy and safety of motesanib, an oral inhibitor of VEGF, PDGF, and Kit receptors, in patients with imatinib-resistant gastrointestinal stromal tumors. Cancer Chemother. Pharmacol. 2011, 68, 69–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; McAndrews, K.M.; Kalluri, R. Clinical and therapeutic relevance of cancer-associated fibroblasts. Nat. Rev. Clin. Oncol. 2021, 18, 792–804. [Google Scholar] [CrossRef]
- Pan, Y.; Yu, Y.; Wang, X.; Zhang, T. Tumor-Associated Macrophages in Tumor Immunity. Front. Immunol. 2020, 11, 583084. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Li, R.; Cao, Y.; Gu, Y.; Lin, C.; Liu, X.; Lv, K.; He, X.; Fang, H.; Jin, K.; et al. Poor Clinical Outcomes and Immunoevasive Contexture in Intratumoral IL-10-Producing Macrophages Enriched Gastric Cancer Patients. Ann. Surg. 2022, 275, e626–e635. [Google Scholar] [CrossRef]
- Hwang, I.; Kim, J.W.; Ylaya, K.; Chung, E.J.; Kitano, H.; Perry, C.; Hanaoka, J.; Fukuoka, J.; Chung, J.Y.; Hewitt, S.M. Tumor-associated macrophage, angiogenesis and lymphangiogenesis markers predict prognosis of non-small cell lung cancer patients. J. Transl. Med. 2020, 18, 443. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, T.; Yakoub, M.A.; Chandler, A.; Christ, A.B.; Yang, G.; Ouerfelli, O.; Rajasekhar, V.K.; Yoshida, A.; Kondo, H.; Hata, T.; et al. CSF1/CSF1R Signaling Inhibitor Pexidartinib (PLX3397) Reprograms Tumor-Associated Macrophages and Stimulates T-cell Infiltration in the Sarcoma Microenvironment. Mol. Cancer Ther. 2021, 20, 1388–1399. [Google Scholar] [CrossRef] [PubMed]
- Pridans, C.; Sauter, K.A.; Irvine, K.M.; Davis, G.M.; Lefevre, L.; Raper, A.; Rojo, R.; Nirmal, A.J.; Beard, P.; Cheeseman, M.; et al. Macrophage colony-stimulating factor increases hepatic macrophage content, liver growth, and lipid accumulation in neonatal rats. Am. J. Physiol. Gastrointest. Liver Physiol. 2018, 314, G388–G398. [Google Scholar] [CrossRef] [Green Version]
- Kumar, V.; Donthireddy, L.; Marvel, D.; Condamine, T.; Wang, F.; Lavilla-Alonso, S.; Hashimoto, A.; Vonteddu, P.; Behera, R.; Goins, M.A.; et al. Cancer-Associated Fibroblasts Neutralize the Anti-tumor Effect of CSF1 Receptor Blockade by Inducing PMN-MDSC Infiltration of Tumors. Cancer Cell 2017, 32, 654–668.e655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pyonteck, S.M.; Akkari, L.; Schuhmacher, A.J.; Bowman, R.L.; Sevenich, L.; Quail, D.F.; Olson, O.C.; Quick, M.L.; Huse, J.T.; Teijeiro, V.; et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 2013, 19, 1264–1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pylaeva, E.; Harati, M.D.; Spyra, I.; Bordbari, S.; Strachan, S.; Thakur, B.K.; Höing, B.; Franklin, C.; Skokowa, J.; Welte, K.; et al. NAMPT signaling is critical for the proangiogenic activity of tumor-associated neutrophils. Int. J. Cancer 2019, 144, 136–149. [Google Scholar] [CrossRef]
- Zhou, S.L.; Zhou, Z.J.; Hu, Z.Q.; Huang, X.W.; Wang, Z.; Chen, E.B.; Fan, J.; Cao, Y.; Dai, Z.; Zhou, J. Tumor-Associated Neutrophils Recruit Macrophages and T-Regulatory Cells to Promote Progression of Hepatocellular Carcinoma and Resistance to Sorafenib. Gastroenterology 2016, 150, 1646–1658. [Google Scholar] [CrossRef] [Green Version]
- Jaillon, S.; Ponzetta, A.; Di Mitri, D.; Santoni, A.; Bonecchi, R.; Mantovani, A. Neutrophil diversity and plasticity in tumour progression and therapy. Nat. Rev. Cancer 2020, 20, 485–503. [Google Scholar] [CrossRef]
- Galdiero, M.R.; Bonavita, E.; Barajon, I.; Garlanda, C.; Mantovani, A.; Jaillon, S. Tumor associated macrophages and neutrophils in cancer. Immunobiology 2013, 218, 1402–1410. [Google Scholar] [CrossRef]
- Vannitamby, A.; Seow, H.J.; Anderson, G.; Vlahos, R.; Thompson, M.; Steinfort, D.; Irving, L.B.; Bozinovski, S. Tumour-associated neutrophils and loss of epithelial PTEN can promote corticosteroid-insensitive MMP-9 expression in the chronically inflamed lung microenvironment. Thorax 2017, 72, 1140–1143. [Google Scholar] [CrossRef] [Green Version]
- Bekes, E.M.; Schweighofer, B.; Kupriyanova, T.A.; Zajac, E.; Ardi, V.C.; Quigley, J.P.; Deryugina, E.I. Tumor-recruited neutrophils and neutrophil TIMP-free MMP-9 regulate coordinately the levels of tumor angiogenesis and efficiency of malignant cell intravasation. Am. J. Pathol. 2011, 179, 1455–1470. [Google Scholar] [CrossRef]
- Gong, L.; Cumpian, A.M.; Caetano, M.S.; Ochoa, C.E.; De la Garza, M.M.; Lapid, D.J.; Mirabolfathinejad, S.G.; Dickey, B.F.; Zhou, Q.; Moghaddam, S.J. Promoting effect of neutrophils on lung tumorigenesis is mediated by CXCR2 and neutrophil elastase. Mol. Cancer 2013, 12, 154. [Google Scholar] [CrossRef] [Green Version]
- Mittendorf, E.A.; Alatrash, G.; Qiao, N.; Wu, Y.; Sukhumalchandra, P.; St John, L.S.; Philips, A.V.; Xiao, H.; Zhang, M.; Ruisaard, K.; et al. Breast cancer cell uptake of the inflammatory mediator neutrophil elastase triggers an anticancer adaptive immune response. Cancer Res. 2012, 72, 3153–3162. [Google Scholar] [CrossRef] [Green Version]
- Hurt, B.; Schulick, R.; Edil, B.; El Kasmi, K.C.; Barnett, C. Cancer-promoting mechanisms of tumor-associated neutrophils. Am. J. Surg. 2017, 214, 938–944. [Google Scholar] [CrossRef]
- Lim, C.L.; Lin, V.C. Estrogen markedly reduces circulating low-density neutrophils and enhances pro-tumoral gene expression in neutrophil of tumour-bearing mice. BMC Cancer 2021, 21, 1017. [Google Scholar] [CrossRef]
- Mishalian, I.; Bayuh, R.; Eruslanov, E.; Michaeli, J.; Levy, L.; Zolotarov, L.; Singhal, S.; Albelda, S.M.; Granot, Z.; Fridlender, Z.G. Neutrophils recruit regulatory T-cells into tumors via secretion of CCL17--a new mechanism of impaired antitumor immunity. Int. J. Cancer 2014, 135, 1178–1186. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, R.; Kawada, K.; Itatani, Y.; Ogawa, R.; Kiyasu, Y.; Sakai, Y. The Role of Tumor-Associated Neutrophils in Colorectal Cancer. Int. J. Mol. Sci. 2019, 20, 529. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Rahmy, S.; Liu, Z.; Zhang, C.; Lu, X. Rational targeting of immunosuppressive neutrophils in cancer. Pharmacol. Ther. 2020, 212, 107556. [Google Scholar] [CrossRef]
- Wang, Y.; Dembowsky, K.; Chevalier, E.; Stüve, P.; Korf-Klingebiel, M.; Lochner, M.; Napp, L.C.; Frank, H.; Brinkmann, E.; Kanwischer, A.; et al. C-X-C Motif Chemokine Receptor 4 Blockade Promotes Tissue Repair After Myocardial Infarction by Enhancing Regulatory T Cell Mobilization and Immune-Regulatory Function. Circulation 2019, 139, 1798–1812. [Google Scholar] [CrossRef] [PubMed]
- Shibata, F.; Konishi, K.; Nakagawa, H. Chemokine receptor CXCR2 activates distinct pathways for chemotaxis and calcium mobilization. Biol. Pharm. Bull. 2002, 25, 1217–1219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greene, S.; Robbins, Y.; Mydlarz, W.K.; Huynh, A.P.; Schmitt, N.C.; Friedman, J.; Horn, L.A.; Palena, C.; Schlom, J.; Maeda, D.Y.; et al. Inhibition of MDSC Trafficking with SX-682, a CXCR1/2 Inhibitor, Enhances NK-Cell Immunotherapy in Head and Neck Cancer Models. Clin. Cancer Res. 2020, 26, 1420–1431. [Google Scholar] [CrossRef] [Green Version]
- Tengesdal, I.W.; Menon, D.R.; Osborne, D.G.; Neff, C.P.; Powers, N.E.; Gamboni, F.; Mauro, A.G.; D’Alessandro, A.; Stefanoni, D.; Henen, M.A.; et al. Targeting tumor-derived NLRP3 reduces melanoma progression by limiting MDSCs expansion. Proc. Natl. Acad. Sci. USA 2021, 118, e2000915118. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Li, A.; Tian, Y.; Wu, J.D.; Liu, Y.; Li, T.; Chen, Y.; Han, X.; Wu, K. The CXCL8-CXCR1/2 pathways in cancer. Cytokine Growth Factor Rev. 2016, 31, 61–71. [Google Scholar] [CrossRef] [Green Version]
- Liebow, M.; Larson, M.C.; Thompson, C.A.; Nowakowski, G.S.; Call, T.G.; Macon, W.R.; Kay, N.E.; Habermann, T.M.; Slager, S.L.; Cerhan, J.R. Aspirin and other nonsteroidal anti-inflammatory drugs, statins and risk of non-Hodgkin lymphoma. Int. J. Cancer 2021, 149, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Neill, A.S.; Nagle, C.M.; Protani, M.M.; Obermair, A.; Spurdle, A.B.; Webb, P.M.; Group, A.N.E.C.S. Aspirin, nonsteroidal anti-inflammatory drugs, paracetamol and risk of endometrial cancer: A case-control study, systematic review and meta-analysis. Int. J. Cancer 2013, 132, 1146–1155. [Google Scholar] [CrossRef]
- Hao, N.B.; Lü, M.H.; Fan, Y.H.; Cao, Y.L.; Zhang, Z.R.; Yang, S.M. Macrophages in tumor microenvironments and the progression of tumors. Clin. Dev. Immunol. 2012, 2012, 948098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klemke, L.; De Oliveira, T.; Witt, D.; Winkler, N.; Bohnenberger, H.; Bucala, R.; Conradi, L.C.; Schulz-Heddergott, R. Hsp90-stabilized MIF supports tumor progression via macrophage recruitment and angiogenesis in colorectal cancer. Cell Death Dis. 2021, 12, 155. [Google Scholar] [CrossRef]
- Dong, H.; Strome, S.E.; Salomao, D.R.; Tamura, H.; Hirano, F.; Flies, D.B.; Roche, P.C.; Lu, J.; Zhu, G.; Tamada, K.; et al. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat. Med. 2002, 8, 793–800. [Google Scholar] [CrossRef]
- Dallavalasa, S.; Beeraka, N.M.; Basavaraju, C.G.; Tulimilli, S.V.; Sadhu, S.P.; Rajesh, K.; Aliev, G.; Madhunapantula, S.V. The Role of Tumor Associated Macrophages (TAMs) in Cancer Progression, Chemoresistance, Angiogenesis and Metastasis - Current Status. Curr. Med. Chem. 2021, 28, 8203–8236. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Velotti, F.; Barchetta, I.; Cimini, F.A.; Cavallo, M.G. Granzyme B in Inflammatory Diseases: Apoptosis, Inflammation, Extracellular Matrix Remodeling, Epithelial-to-Mesenchymal Transition and Fibrosis. Front. Immunol. 2020, 11, 587581. [Google Scholar] [CrossRef]
- Chen, Q.; Jin, M.; Yang, F.; Zhu, J.; Xiao, Q.; Zhang, L. Matrix metalloproteinases: Inflammatory regulators of cell behaviors in vascular formation and remodeling. Mediators Inflamm. 2013, 2013, 928315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maimon, A.; Levi-Yahid, V.; Ben-Meir, K.; Halpern, A.; Talmi, Z.; Priya, S.; Mizraji, G.; Mistriel-Zerbib, S.; Berger, M.; Baniyash, M.; et al. Myeloid cell-derived PROS1 inhibits tumor metastasis by regulating inflammatory and immune responses via IL-10. J. Clin. Investig. 2021, 131, e126089. [Google Scholar] [CrossRef]
- Savant, S.S.; Sriramkumar, S.; O’Hagan, H.M. The Role of Inflammation and Inflammatory Mediators in the Development, Progression, Metastasis, and Chemoresistance of Epithelial Ovarian Cancer. Cancers 2018, 10, 251. [Google Scholar] [CrossRef] [Green Version]
- Senobari, Z.; Karimi, G.; Jamialahmadi, K. Ellagitannins, promising pharmacological agents for the treatment of cancer stem cells. Phytother. Res. 2022, 36, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Bienkowska, K.J.; Hanley, C.J.; Thomas, G.J. Cancer-Associated Fibroblasts in Oral Cancer: A Current Perspective on Function and Potential for Therapeutic Targeting. Front. Oral Health 2021, 2, 686337. [Google Scholar] [CrossRef] [PubMed]
- Arneth, B. Tumor Microenvironment. Medicina 2019, 56, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, N.M.; Simon, M.C. The tumor microenvironment. Curr. Biol. 2020, 30, R921–R925. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Dai, Y. Tumor microenvironment and therapeutic response. Cancer Lett. 2017, 387, 61–68. [Google Scholar] [CrossRef]
- Togashi, Y.; Shitara, K.; Nishikawa, H. Regulatory T cells in cancer immunosuppression - implications for anticancer therapy. Nat. Rev. Clin. Oncol. 2019, 16, 356–371. [Google Scholar] [CrossRef]
- Sato, Y.; Passerini, L.; Piening, B.D.; Uyeda, M.J.; Goodwin, M.; Gregori, S.; Snyder, M.P.; Bertaina, A.; Roncarolo, M.G.; Bacchetta, R. Human-engineered Treg-like cells suppress FOXP3-deficient T cells but preserve adaptive immune responses. Clin. Transl. Immunol. 2020, 9, e1214. [Google Scholar] [CrossRef]
- Ding, X.; Peng, C.; Li, Y.; Liu, J.; Song, Y.; Cai, B.; Xiang, M.; Zhang, J.; Wang, Z.; Wang, L. Targeting Inhibition of Foxp3 by MMP2/9 Sensitive Short Peptide Linked P60 Fusion Protein 6(P60-MMPs) to Enhance Antitumor Immunity. Macromol. Biosci. 2020, 20, e2000098. [Google Scholar] [CrossRef] [PubMed]
- Sivori, S.; Della Chiesa, M.; Carlomagno, S.; Quatrini, L.; Munari, E.; Vacca, P.; Tumino, N.; Mariotti, F.R.; Mingari, M.C.; Pende, D.; et al. Inhibitory Receptors and Checkpoints in Human NK Cells, Implications for the Immunotherapy of Cancer. Front. Immunol. 2020, 11, 2156. [Google Scholar] [CrossRef] [PubMed]
- Sivori, S.; Vacca, P.; Del Zotto, G.; Munari, E.; Mingari, M.C.; Moretta, L. Human NK cells: Surface receptors, inhibitory checkpoints, and translational applications. Cell Mol. Immunol. 2019, 16, 430–441. [Google Scholar] [CrossRef] [PubMed]
- Song, K.; Minami, J.K.; Huang, A.; Dehkordi, S.R.; Lomeli, S.H.; Luebeck, J.; Goodman, M.H.; Moriceau, G.; Krijgsman, O.; Dharanipragada, P.; et al. Plasticity of Extrachromosomal and Intrachromosomal BRAF Amplifications in Overcoming Targeted Therapy Dosage Challenges. Cancer Discov. 2022, 12, 1046–1069. [Google Scholar] [CrossRef]
- Chauvin, J.M.; Zarour, H.M. TIGIT in cancer immunotherapy. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef]
- Darragh, L.B.; Karam, S.D. Amateur antigen-presenting cells in the tumor microenvironment. Mol. Carcinog. 2022, 61, 153–164. [Google Scholar] [CrossRef]
- Verneau, J.; Sautés-Fridman, C.; Sun, C.M. Dendritic cells in the tumor microenvironment: Prognostic and theranostic impact. Semin. Immunol. 2020, 48, 101410. [Google Scholar] [CrossRef]
- Ferraro, D.A.; Gaborit, N.; Maron, R.; Cohen-Dvashi, H.; Porat, Z.; Pareja, F.; Lavi, S.; Lindzen, M.; Ben-Chetrit, N.; Sela, M.; et al. Inhibition of triple-negative breast cancer models by combinations of antibodies to EGFR. Proc. Natl. Acad. Sci. USA 2013, 110, 1815–1820. [Google Scholar] [CrossRef] [Green Version]
- John, P.; Pulanco, M.C.; Galbo, P.M.; Wei, Y.; Ohaegbulam, K.C.; Zheng, D.; Zang, X. The immune checkpoint B7x expands tumor-infiltrating Tregs and promotes resistance to anti-CTLA-4 therapy. Nat. Commun. 2022, 13, 2506. [Google Scholar] [CrossRef]
- Danikowski, K.M.; Jayaraman, S.; Prabhakar, B.S. Regulatory T cells in multiple sclerosis and myasthenia gravis. J. Neuroinflamm. 2017, 14, 117. [Google Scholar] [CrossRef] [Green Version]
- Duraiswamy, J.; Kaluza, K.M.; Freeman, G.J.; Coukos, G. Dual blockade of PD-1 and CTLA-4 combined with tumor vaccine effectively restores T-cell rejection function in tumors. Cancer Res. 2013, 73, 3591–3603. [Google Scholar] [CrossRef] [Green Version]
- Koch, E.A.T.; Schaft, N.; Kummer, M.; Berking, C.; Schuler, G.; Hasumi, K.; Dörrie, J.; Schuler-Thurner, B. A One-Armed Phase I Dose Escalation Trial Design: Personalized Vaccination with IKKβ-Matured, RNA-Loaded Dendritic Cells for Metastatic Uveal Melanoma. Front. Immunol. 2022, 13, 785231. [Google Scholar] [CrossRef] [PubMed]
- Christodoulou, M.I.; Zaravinos, A. New Clinical Approaches and Emerging Evidence on Immune-Checkpoint Inhibitors as Anti-Cancer Therapeutics: CTLA-4 and PD-1 Pathways and Beyond. Crit. Rev. Immunol. 2019, 39, 379–408. [Google Scholar] [CrossRef]
- Ai, L.; Xu, A.; Xu, J. Roles of PD-1/PD-L1 Pathway: Signaling, Cancer, and Beyond. Adv. Exp. Med. Biol. 2020, 1248, 33–59. [Google Scholar] [CrossRef]
- Naidoo, J.; Page, D.B.; Li, B.T.; Connell, L.C.; Schindler, K.; Lacouture, M.E.; Postow, M.A.; Wolchok, J.D. Toxicities of the anti-PD-1 and anti-PD-L1 immune checkpoint antibodies. Ann. Oncol. 2015, 26, 2375–2391. [Google Scholar] [CrossRef]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef] [Green Version]
- Bader, J.E.; Voss, K.; Rathmell, J.C. Targeting Metabolism to Improve the Tumor Microenvironment for Cancer Immunotherapy. Mol. Cell. 2020, 78, 1019–1033. [Google Scholar] [CrossRef] [PubMed]
- Osipov, A.; Saung, M.T.; Zheng, L.; Murphy, A.G. Small molecule immunomodulation: The tumor microenvironment and overcoming immune escape. J. Immunother. Cancer 2019, 7, 224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahdavi Gorabi, A.; Sadat Ravari, M.; Sanaei, M.J.; Davaran, S.; Kesharwani, P.; Sahebkar, A. Immune checkpoint blockade in melanoma: Advantages, shortcomings and emerging roles of the nanoparticles. Int. Immunopharmacol. 2022, 113, 109300. [Google Scholar] [CrossRef] [PubMed]
- Morante, M.; Pandiella, A.; Crespo, P.; Herrero, A. Immune Checkpoint Inhibitors and RAS-ERK Pathway-Targeted Drugs as Combined Therapy for the Treatment of Melanoma. Biomolecules 2022, 12, 1562. [Google Scholar] [CrossRef]
- Goldinger, S.M.; Zimmer, L.; Schulz, C.; Ugurel, S.; Hoeller, C.; Kaehler, K.C.; Schadendorf, D.; Hassel, J.C.; Becker, J.; Hauschild, A.; et al. Upstream mitogen-activated protein kinase (MAPK) pathway inhibition: MEK inhibitor followed by a BRAF inhibitor in advanced melanoma patients. Eur. J. Cancer 2014, 50, 406–410. [Google Scholar] [CrossRef]
- Kreft, S.; Glutsch, V.; Zaremba, A.; Schummer, P.; Mohr, P.; Grimmelmann, I.; Gutzmer, R.; Meier, F.; Pföhler, C.; Sachse, M.M.; et al. MAPKinase inhibition after failure of immune checkpoint blockade in patients with advanced melanoma - An evaluation of the multicenter prospective skin cancer registry ADOREG. Eur. J. Cancer 2022, 167, 32–41. [Google Scholar] [CrossRef]
- Schirmer, M.; Garner, A.; Vlamakis, H.; Xavier, R.J. Microbial genes and pathways in inflammatory bowel disease. Nat. Rev. Microbiol. 2019, 17, 497–511. [Google Scholar] [CrossRef]
- Lam, K.C.; Araya, R.E.; Huang, A.; Chen, Q.; Di Modica, M.; Rodrigues, R.R.; Lopès, A.; Johnson, S.B.; Schwarz, B.; Bohrnsen, E.; et al. Microbiota triggers STING-type I IFN-dependent monocyte reprogramming of the tumor microenvironment. Cell 2021, 184, 5338–5356.e5321. [Google Scholar] [CrossRef]
- Thomas, S.C.; Madaan, T.; Kamble, N.S.; Siddiqui, N.A.; Pauletti, G.M.; Kotagiri, N. Engineered Bacteria Enhance Immunotherapy and Targeted Therapy through Stromal Remodeling of Tumors. Adv. Healthc. Mater 2022, 11, e2101487. [Google Scholar] [CrossRef]
- Fukuhara, H.; Ino, Y.; Todo, T. Oncolytic virus therapy: A new era of cancer treatment at dawn. Cancer Sci. 2016, 107, 1373–1379. [Google Scholar] [CrossRef]
- Goradel, N.H.; Baker, A.T.; Arashkia, A.; Ebrahimi, N.; Ghorghanlu, S.; Negahdari, B. Oncolytic virotherapy: Challenges and solutions. Curr. Probl. Cancer 2021, 45, 100639. [Google Scholar] [CrossRef] [PubMed]
- Russell, S.J.; Peng, K.W.; Bell, J.C. Oncolytic virotherapy. Nat. Biotechnol. 2012, 30, 658–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gentile, C.; Finizio, A.; Froechlich, G.; D’Alise, A.M.; Cotugno, G.; Amiranda, S.; Nicosia, A.; Scarselli, E.; Zambrano, N.; Sasso, E. Generation of a Retargeted Oncolytic. Int. J. Mol. Sci. 2021, 22, 13521. [Google Scholar] [CrossRef]
- Yu, L.; Sun, M.; Zhang, Q.; Zhou, Q.; Wang, Y. Harnessing the immune system by targeting immune checkpoints: Providing new hope for Oncotherapy. Front. Immunol. 2022, 13, 982026. [Google Scholar] [CrossRef]
- Gujar, S.A.; Marcato, P.; Pan, D.; Lee, P.W. Reovirus virotherapy overrides tumor antigen presentation evasion and promotes protective antitumor immunity. Mol. Cancer Ther. 2010, 9, 2924–2933. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kharouf, N.; Flanagan, T.W.; Hassan, S.-Y.; Shalaby, H.; Khabaz, M.; Hassan, S.-L.; Megahed, M.; Haikel, Y.; Santourlidis, S.; Hassan, M. Tumor Microenvironment as a Therapeutic Target in Melanoma Treatment. Cancers 2023, 15, 3147. https://doi.org/10.3390/cancers15123147
Kharouf N, Flanagan TW, Hassan S-Y, Shalaby H, Khabaz M, Hassan S-L, Megahed M, Haikel Y, Santourlidis S, Hassan M. Tumor Microenvironment as a Therapeutic Target in Melanoma Treatment. Cancers. 2023; 15(12):3147. https://doi.org/10.3390/cancers15123147
Chicago/Turabian StyleKharouf, Naji, Thomas W. Flanagan, Sofie-Yasmin Hassan, Hosam Shalaby, Marla Khabaz, Sarah-Lilly Hassan, Mosaad Megahed, Youssef Haikel, Simeon Santourlidis, and Mohamed Hassan. 2023. "Tumor Microenvironment as a Therapeutic Target in Melanoma Treatment" Cancers 15, no. 12: 3147. https://doi.org/10.3390/cancers15123147