
Regulations of Tumor Microenvironment by Prostaglandins

Abstract
:Simple Summary
Abstract
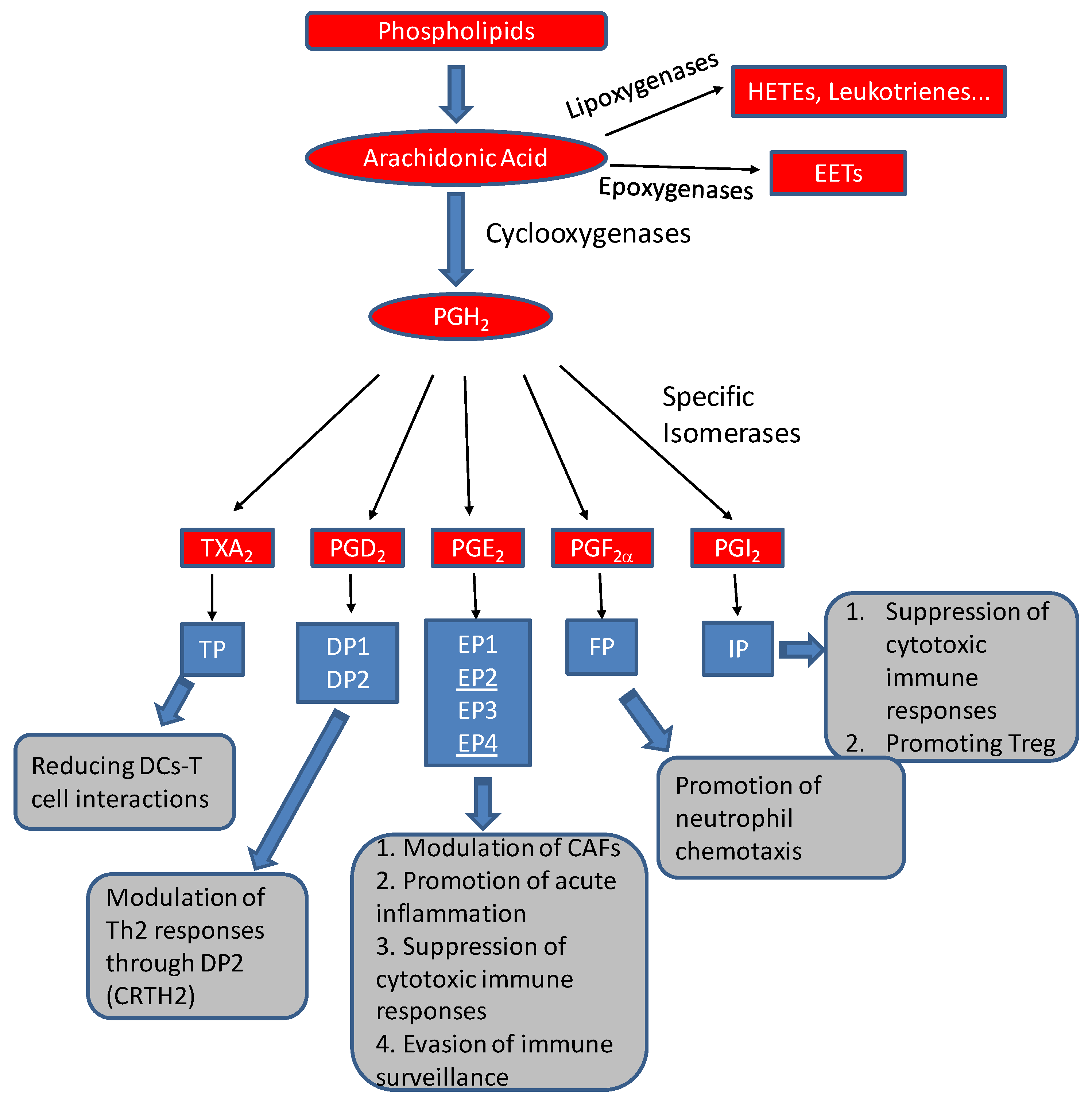
1. Prostaglandins: An Overview
2. Tumor Microenvironment in Carcinogenesis
3. Prostaglandin Regulation of Cancer-Associated Fibroblasts
4. Prostaglandin Regulation of Immune Constituents within the TME
4.1. Overview of Immune Phenotypes of the TME
4.2. Prostaglandin Regulation of Immune Components in TMEs
4.2.1. Prostaglandin E2
4.2.2. Prostaglandin I2
4.2.3. Prostaglandin D2
4.2.4. Thromboxane A2
4.2.5. Prostaglandin F2α
5. Role of Cyclooxygenases and Prostaglandins in Tumor Evasion of Immune Surveillance
6. Translational Potential and Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Funk, C.D. Prostaglandins and Leukotrienes: Advances in Eicosanoid Biology. Science 2001, 294, 1871–1875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinohara, H.; Balboa, M.A.; Johnson, C.A.; Balsinde, J.; Dennis, E. Regulation of Delayed Prostaglandin Production in Activated P388D1 Macrophages by Group IV Cytosolic and Group V Secretory Phospholipase A2s. J. Biol. Chem. 1999, 274, 12263–12268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hara, S.; Miyata, A.; Yokoyama, C.; Inoue, H.; Brugger, R.; Lottspeich, F.; Ullrich, V.; Tanabe, T. Isolation and molecular cloning of prostacyclin synthase from bovine endothelial cells. J. Biol. Chem. 1994, 269, 19897–19903. [Google Scholar] [CrossRef] [PubMed]
- Kuwamoto, S.; Inoue, H.; Tone, Y.; Izumi, Y.; Tanabe, T. Inverse gene expression of prostacyclin and thromboxane synthases in resident and activated peritoneal macrophages1. FEBS Lett. 1997, 409, 242–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, T.; Watanabe, K.; Kanaoka, Y.; Sato, T.; Hayaishi, O. Induction of Hematopoietic Prostaglandin D Synthase in Human Megakaryocytic Cells by Phorbol Ester. Biochem. Biophys. Res. Commun. 1997, 241, 288–293. [Google Scholar] [CrossRef]
- Jakobsson, P.-J.; Thorén, S.; Morgenstern, R.; Samuelsson, B. Identification of human prostaglandin E synthase: A microsomal, glutathione-dependent, inducible enzyme, constituting a potential novel drug target. Proc. Natl. Acad. Sci. USA 1999, 96, 7220–7225. [Google Scholar] [CrossRef] [Green Version]
- Bonvalet, J.P.; Pradelles, P.; Farman, N. Segmental synthesis and actions of prostaglandins along the nephron. Am. J. Physiol. Content 1987, 253, F377–F387. [Google Scholar] [CrossRef]
- Smith, W.L. Prostanoid biosynthesis and mechanisms of action. Am. J. Physiol. Physiol. 1992, 263, F181–F191. [Google Scholar] [CrossRef] [Green Version]
- Narumiya, S.; Sugimoto, Y.; Ushikubi, F.; Alexanian, A.; Sorokin, A.; Fujii, N.; Singh, M.S.; Halili, L.; Boulay, P.; Sigal, R.J.; et al. Prostanoid Receptors: Structures, Properties, and Functions. Physiol. Rev. 1999, 79, 1193–1226. [Google Scholar] [CrossRef] [Green Version]
- Nie, D.; Che, M.; Zacharek, A.; Qiao, Y.; Li, L.; Li, X.; Lamberti, M.; Tang, K.; Cai, Y.; Guo, Y.; et al. Differential Expression of Thromboxane Synthase in Prostate Carcinoma: Role in Tumor Cell Motility. Am. J. Pathol. 2004, 164, 429–439. [Google Scholar] [CrossRef] [Green Version]
- Hu, M.; Peluffo, G.; Chen, H.; Gelman, R.; Schnitt, S.; Polyak, K. Role of COX-2 in epithelial–stromal cell interactions and progression of ductal carcinoma in situ of the breast. Proc. Natl. Acad. Sci. USA 2009, 106, 3372–3377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finetti, F.; Travelli, C.; Ercoli, J.; Colombo, G.; Buoso, E.; Trabalzini, L. Prostaglandin E2 and Cancer: Insight into Tumor Progression and Immunity. Biology 2020, 9, 434. [Google Scholar] [CrossRef]
- Kobayashi, K.; Omori, K.; Murata, T. Role of prostaglandins in tumor microenvironment. Cancer Metastasis Rev. 2018, 37, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Zelenay, S.; Van Der Veen, A.G.; Böttcher, J.P.; Snelgrove, K.J.; Rogers, N.; Acton, S.E.; Chakravarty, P.; Girotti, M.R.; Marais, R.; Quezada, S.A.; et al. Faculty Opinions recommendation of Cyclooxygenase-Dependent Tumor Growth through Evasion of Immunity. Cell 2015, 162, 1257–1270. [Google Scholar] [CrossRef] [Green Version]
- Brash, A.R.; Boeglin, W.E.; Chang, M.S. Discovery of a second 15 S -lipoxygenase in humans. Proc. Natl. Acad. Sci. USA 1997, 94, 6148–6152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Y.; Wang, M.-T.; Chen, Y.; Yang, D.; Che, M.; Honn, K.V.; Akers, G.D.; Johnson, S.R.; Nie, D. Downregulation of vascular endothelial growth factor and induction of tumor dormancy by 15-lipoxygenase-2 in prostate cancer. Int. J. Cancer 2009, 124, 1545–1551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [Green Version]
- Joyce, J.A.; Pollard, J.W. Microenvironmental regulation of metastasis. Nat. Rev. Cancer 2008, 9, 239–252. [Google Scholar] [CrossRef]
- Shiga, K.; Hara, M.; Nagasaki, T.; Sato, T.; Takahashi, H.; Takeyama, H. Cancer-Associated Fibroblasts: Their Characteristics and Their Roles in Tumor Growth. Cancers 2015, 7, 2443–2458. [Google Scholar] [CrossRef]
- Wang, M.; Zhao, J.; Zhang, L.; Wei, F.; Lian, Y.; Wu, Y.; Gong, Z.; Zhang, S.; Zhou, J.; Cao, K.; et al. Role of tumor microenvironment in tumorigenesis. J. Cancer 2017, 8, 761–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witz, I.P. The Tumor Microenvironment: The Making of a Paradigm. Cancer Microenviron. 2009, 2, 9–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jara-Gutiérrez, Á.; Baladrón, V. The Role of Prostaglandins in Different Types of Cancer. Cells 2021, 10, 1487. [Google Scholar] [CrossRef] [PubMed]
- Parsonage, G.; Filer, A.D.; Haworth, O.; Nash, G.B.; Rainger, G.E.; Salmon, M.; Buckley, C.D. A stromal address code defined by fibroblasts. Trends Immunol. 2005, 26, 150–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomasek, J.J.; Gabbiani, G.; Hinz, B.; Chaponnier, C.; Brown, R.A. Faculty Opinions recommendation of Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat. Rev. Mol. Cell Biol. 2019, 3, 349–363. [Google Scholar] [CrossRef]
- Plikus, M.V.; Wang, X.; Sinha, S.; Forte, E.; Thompson, S.M.; Herzog, E.L.; Driskell, R.R.; Rosenthal, N.; Biernaskie, J.; Horsely, V. Fibroblasts: Origins, definitions, and functions in health and disease. Cell 2021, 184, 3852–3872. [Google Scholar] [CrossRef]
- Kalluri, R.; Zeisberg, M. Fibroblasts in cancer. Nat. Rev. Cancer 2006, 6, 392–401. [Google Scholar] [CrossRef]
- Li, B.; Wang, J.H.-C. Fibroblasts and myofibroblasts in wound healing: Force generation and measurement. J. Tissue Viability 2011, 20, 108–120. [Google Scholar] [CrossRef] [Green Version]
- Bhowmick, N.A.; Neilson, E.G.; Moses, H.L. Stromal fibroblasts in cancer initiation and progression. Nature 2004, 432, 332–337. [Google Scholar] [CrossRef] [Green Version]
- Feghali, C.A.; Wright, T.M. Cytokines in acute and chronic inflammation. Front. Biosci. 1997, 2, d12–d26. [Google Scholar]
- Fukumura, D.; Xavier, R.; Sugiura, T.; Chen, Y.; Park, E.-C.; Lu, N.; Selig, M.; Nielsen, G.; Taksir, T.; Jain, R.K.; et al. Tumor Induction of VEGF Promoter Activity in Stromal Cells. Cell 1998, 94, 715–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olumi, A.F.; Grossfeld, G.D.; Hayward, S.W.; Carroll, P.R.; Cunha, G.R.; Hein, P.; Tlsty, T.D. Carcinoma-associated fibroblasts direct tumor progression of initiated human prostatic epithelium. Cancer Res. 1999, 59, 5002–5011. [Google Scholar] [PubMed]
- Orimo, A.; Gupta, P.B.; Sgroi, D.C.; Arenzana-Seisdedos, F.; Delaunay, T.; Naeem, R.; Carey, V.J.; Richardson, A.L.; Weinberg, R.A. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 2005, 121, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Busch, S.; Acar, A.; Magnusson, Y.; Gregersson, P.; Rydén, L.; Landberg, G. TGF-beta receptor type-2 expression in cancer-associated fibroblasts regulates breast cancer cell growth and survival and is a prognostic marker in pre-menopausal breast cancer. Oncogene 2013, 34, 27–38. [Google Scholar] [CrossRef]
- Ma, X.; Aoki, T.; Tsuruyama, T.; Narumiya, S. Definition of Prostaglandin E2–EP2 Signals in the Colon Tumor Microenvironment That Amplify Inflammation and Tumor Growth. Cancer Res. 2015, 75, 2822–2832. [Google Scholar] [CrossRef] [Green Version]
- Chell, S.D.; Witherden, I.R.; Dobson, R.R.; Moorghen, M.; Herman, A.A.; Qualtrough, D.; Williams, A.C.; Paraskeva, C. Faculty Opinions recommendation of Increased EP4 receptor expression in colorectal cancer progression promotes cell growth and anchorage independence. Cancer Res. 2006, 66, 3106–3113. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Aladelokun, O.; Ideta, T.; Giardina, C.; Ellis, L.M.; Rosenberg, D.W. Inhibition of PGE(2)/EP4 receptor signaling enhances oxaliplatin efficacy in resistant colon cancer cells through modulation of oxidative stress. Sci. Rep. 2019, 9, 4954. [Google Scholar] [CrossRef] [Green Version]
- Gong, Z.; Li, Q.; Shi, J.; Wei, J.; Li, P.; Chang, C.-H.; Shultz, L.D.; Ren, G. Lung fibroblasts facilitate pre-metastatic niche formation by remodeling the local immune microenvironment. Immunity 2022, 55, 1483–1500.e9. [Google Scholar] [CrossRef]
- Engblom, C.; Pfirschke, C.; Mikael, J.P. FThe role of myeloid cells in cancer therapies. Nat. Rev. Cancer 2016, 16, 447–462. [Google Scholar] [CrossRef]
- Gajewski, T.F.; Schreiber, H.; Fu, Y.-X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013, 14, 1014–1022. [Google Scholar] [CrossRef] [Green Version]
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef] [Green Version]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef]
- Page, D.B.; Postow, M.A.; Callahan, M.K.; Allison, J.P.; Wolchok, J.D. Immune Modulation in Cancer with Antibodies. Annu. Rev. Med. 2014, 65, 185–202. [Google Scholar] [CrossRef]
- Hinshaw, D.C.; Shevde, L.A. The tumor microenvironment innately modulates cancer progression. Cancer Res. 2019, 79, 4557–4566. [Google Scholar] [CrossRef] [Green Version]
- Zou, W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat. Rev. Cancer 2005, 5, 263–274. [Google Scholar] [CrossRef]
- Jian, H.; Fangrong, S.; Haitao, H.; Chunhua, L.; Guangbo, Z. Th1high in tumor microenvironment is an indicator of poor prognosis for patients with NSCLC. Oncotarget 2017, 8, 13116–13125. [Google Scholar] [CrossRef] [Green Version]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Yang, T.H.O.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.e14. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Cabalag, C.S.; Clemons, N.J.; DuBois, R.N. Cyclooxygenases and Prostaglandins in Tumor Immunology and Microenvironment of Gastrointestinal Cancer. Gastroenterology 2021, 161, 1813–1829. [Google Scholar] [CrossRef]
- Mizuno, R.; Kawada, K.; Sakai, Y. Prostaglandin E2/EP Signaling in the Tumor Microenvironment of Colorectal Cancer. Int. J. Mol. Sci. 2019, 20, 6254. [Google Scholar] [CrossRef] [Green Version]
- Duffin, R.; O’Connor, R.A.; Crittenden, S.; Forster, T.; Yu, C.; Zheng, X.; Smyth, D.; Robb, C.T.; Rossi, F.; Skouras, C.; et al. Prostaglandin E(2) constrains systemic inflammation through an innate lymphoid cell-IL-22 axis. Science 2016, 351, 1333–1338. [Google Scholar] [CrossRef] [Green Version]
- Walker, W.; Rotondo, D. Prostaglandin E2 is a potent regulator of interleukin-12- and interleukin-18-induced natural killer cell interferon-gamma synthesis. Immunology 2004, 111, 298–305. [Google Scholar] [CrossRef]
- Joshi, P.C.; Zhou, X.; Cuchens, M.; Jones, Q. Prostaglandin E2 suppressed IL-15-mediated human NK cell function through down-regulation of common gamma-chain. J. Immunol. 2001, 166, 885–891. [Google Scholar] [CrossRef] [Green Version]
- Yakar, I.; Melamed, R.; Shakhar, G.; Shakhar, K.; Rosenne, E.; Abudarham, N.; Page, G.G.; Ben-Eliyahu, S. Prostaglandin E2 Suppresses NK Activity In Vivo and Promotes Postoperative Tumor Metastasis in Rats. Ann. Surg. Oncol. 2003, 10, 469–479. [Google Scholar] [CrossRef]
- Sharma, S.; Stolina, M.; Yang, S.-C.; Baratelli, F.; Lin, J.F.; Atianzar, K.; Luo, J.; Zhu, L.; Lin, Y.; Huang, M.; et al. Tumor cyclooxygenase 2-dependent suppression of dendritic cell function. Clin. Cancer Res. 2003, 9, 961–968. [Google Scholar]
- Heusinkveld, M.; de Vos van Steenwijk, P.J.; Goedemans, R.; Ramwadhdoebe, T.H.; Gorter, A.; Welters, M.J.P.; van Hall, T.; Van Der Burg, S.H. M2 Macrophages Induced by Prostaglandin E2 and IL-6 from Cervical Carcinoma Are Switched to Activated M1 Macrophages by CD4+ Th1 Cells. J. Immunol. 2011, 187, 1157–1165. [Google Scholar] [CrossRef] [Green Version]
- Obermajer, N.; Muthuswamy, R.; Lesnock, J.; Edwards, R.P.; Kalinski, P. Positive feedback between PGE2 and COX2 redirects the differentiation of human dendritic cells toward stable myeloid-derived suppressor cells. Blood 2011, 118, 5498–5505. [Google Scholar] [CrossRef] [Green Version]
- Gustafsson, K.; Ingelsten, M.; Bergqvist, L.; Nyström, J.; Andersson, B.; Karlsson-Parra, A. Recruitment and activation of natural killer cells in vitro by a human dendritic cell vaccine. Cancer Res. 2008, 68, 5965–5971. [Google Scholar] [CrossRef] [Green Version]
- Betz, M.; Fox, B.S. Prostaglandin E2 inhibits production of Th1 lymphokines but not of Th2 lymphokines. J. Immunol. 1991, 146, 108–113. [Google Scholar] [CrossRef]
- Snijdewint, F.G.; Kaliński, P.; Wierenga, E.; Bos, J.D.; Kapsenberg, M.L. Prostaglandin E2 differentially modulates cytokine secretion profiles of human T helper lymphocytes. J. Immunol. 1993, 150, 5321–5329. [Google Scholar] [CrossRef]
- Kaliński, P.; Vieira, P.L.; Schuitemaker, J.H.N.; De Jong, E.C.; Kapsenberg, M.L. Prostaglandin E2 is a selective inducer of interleukin-12 p40 (IL-12p40) production and an inhibitor of bioactive IL-12p70 heterodimer. Blood 2001, 97, 3466–3469. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.Y.; Wang, K.; McDyer, J.F.; Seder, R. Prostaglandin E2 and dexamethasone inhibit IL-12 receptor expression and IL-12 responsiveness. J. Immunol. 1998, 161, 2723–2730. [Google Scholar] [CrossRef] [PubMed]
- Smits, H.H.; van Rietschoten, J.G.I.; Hilkens, C.M.U.; Sayilir, R.; Stiekema, F.; Kapsenberg, M.L.; Wierenga, E.A. IL-12-induced reversal of human Th2 cells is accompanied by full restoration of IL-12 responsiveness and loss of GATA-3 expression. Eur. J. Immunol. 2001, 31, 1055–1065. [Google Scholar] [CrossRef] [PubMed]
- Thumkeo, D.; Punyawatthananukool, S.; Prasongtanakij, S.; Matsuura, R.; Arima, K.; Nie, H.; Yamamoto, R.; Aoyama, N.; Hamaguchi, H.; Sugahara, S.; et al. PGE(2)-EP2/EP4 signaling elicits immunosuppression by driving the mregDC-Treg axis in inflammatory tumor microenvironment. Cell Rep. 2022, 39, 110914. [Google Scholar] [CrossRef]
- Hashimoto, K.; Graham, B.S.; Geraci, M.W.; FitzGerald, G.A.; Egan, K.; Zhou, W.; Goleniewska, K.; O’Neal, J.F.; Morrow, J.D.; Durbin, R.K.; et al. Signaling through the Prostaglandin I 2 Receptor IP Protects against Respiratory Syncytial Virus-Induced Illness. J. Virol. 2004, 78, 10303–10309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, T.-H.; Gau, R.-J.; Chen, Y.-F.; Shen, H.-H.; Lin, C.T.-Y.; Chen, S.-L.; Suen, J.-L. Dendritic cells treated with a prostaglandin I2 analog, iloprost, promote antigen-specific regulatory T cell differentiation in mice. Int. Immunopharmacol. 2019, 79, 106106. [Google Scholar] [CrossRef]
- Dai, D.; Chen, B.; Feng, Y.; Wang, W.; Jiang, Y.; Huang, H.; Liu, J. Prognostic value of prostaglandin I2 synthase and its correlation with tumor-infiltrating immune cells in lung cancer, ovarian cancer, and gastric cancer. Aging 2020, 12, 9658–9685. [Google Scholar] [CrossRef]
- Hirai, H.; Tanaka, K.; Yoshie, O.; Ogawa, K.; Kenmotsu, K.; Takamori, Y.; Ichimasa, M.; Sugamura, K.; Nakamura, M.; Takano, S.; et al. Prostaglandin D2 Selectively Induces Chemotaxis in T Helper Type 2 Cells, Eosinophils, and Basophils via Seven-Transmembrane Receptor Crth2. J. Exp. Med. 2001, 193, 255–262. [Google Scholar] [CrossRef]
- Xue, L.; Salimi, M.; Panse, I.; Mjösberg, J.M.; McKenzie, A.N.; Spits, H.; Klenerman, P.; Ogg, G. Prostaglandin D2 activates group 2 innate lymphoid cells through chemoattractant receptor-homologous molecule expressed on TH2 cells. J. Allergy Clin. Immunol. 2014, 133, 1184–1194.e7. [Google Scholar] [CrossRef] [Green Version]
- Kabashima, K.; Murata, T.; Tanaka, H.; Matsuoka, T.; Sakata, D.; Yoshida, N.; Katagiri, K.; Kinashi, T.; Tanaka, T.; Miyasaka, M.; et al. Thromboxane A2 modulates interaction of dendritic cells and T cells and regulates acquired immunity. Nat. Immunol. 2003, 4, 694–701. [Google Scholar] [CrossRef]
- Abramovitz, M.; Adam, M.; Boie, Y.; Carrière, M.-C.; Denis, D.; Godbout, C.; Lamontagne, S.; Rochette, C.; Sawyer, N.; Tremblay, N.M.; et al. The utilization of recombinant prostanoid receptors to determine the affinities and selectivities of prostaglandins and related analogs. Biochim. Biophys. Acta 2000, 1483, 285–293. [Google Scholar] [CrossRef]
- Sugimoto, Y.; Yamasaki, A.; Segi, E.; Tsuboi, K.; Aze, Y.; Nishimura, T.; Oida, H.; Yoshida, N.; Tanaka, T.; Katsuyama, M.; et al. Failure of Parturition in Mice Lacking the Prostaglandin F Receptor. Science 1997, 277, 681–683. [Google Scholar] [CrossRef] [PubMed]
- Ricciotti, E.; FitzGerald, G.A. Prostaglandins and inflammation. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 986–1000. [Google Scholar] [CrossRef] [PubMed]
- Alexander, C.L.; Miller, S.J.; Abel, S.R. Prostaglandin analog treatment of glaucoma and ocular hypertension. Ann. Pharmacother. 2002, 36, 504–511. [Google Scholar] [CrossRef] [PubMed]
- Basu, S. Oxidative Injury Induced Cyclooxygenase Activation in Experimental Hepatotoxicity. Biochem. Biophys. Res. Commun. 1999, 254, 764–767. [Google Scholar] [CrossRef]
- Wallace, A.E.; Sales, K.J.; Catalano, R.D.; Anderson, R.A.; Williams, A.R.; Wilson, M.R.; Schwarze, J.; Wang, H.; Rossi, A.G.; Jabbour, H.N. Prostaglandin F2α-F-Prostanoid Receptor Signaling Promotes Neutrophil Chemotaxis via Chemokine (C-X-C Motif) Ligand 1 in Endometrial Adenocarcinoma. Cancer Res. 2009, 69, 5726–5733. [Google Scholar] [CrossRef] [Green Version]
- Maehara, T.; Fujimori, K. Inhibition of Prostaglandin F(2)(alpha) Receptors Exaggerates HCl-Induced Lung Inflammation in Mice. Int. J. Mol. Sci. 2021, 22, 12843. [Google Scholar] [CrossRef]
- Vinay, D.S.; Ryan, E.P.; Pawelec, G.; Talib, W.H.; Stagg, J.; Elkord, E.; Lichtor, T.; Decker, W.K.; Whelan, R.L.; Kumara, H.M.C.S.; et al. Immune evasion in cancer: Mechanistic basis and therapeutic strategies. Semin. Cancer Biol. 2015, 35, S185–S198. [Google Scholar] [CrossRef]
- Shklovskaya, E.; Rizos, H. Spatial and Temporal Changes in PD-L1 Expression in Cancer: The Role of Genetic Drivers, Tumor Microenvironment and Resistance to Therapy. Int. J. Mol. Sci. 2020, 21, 7139. [Google Scholar] [CrossRef]
- Pu, Y.; Ji, Q. Tumor-Associated Macrophages Regulate PD-1/PD-L1 Immunosuppression. Front. Immunol. 2022, 13, 874589. [Google Scholar] [CrossRef]
- Chen, S.; Crabill, G.A.; Pritchard, T.S.; McMiller, T.L.; Wei, P.; Pardoll, D.M.; Pan, F.; Topalian, S.L. Mechanisms regulating PD-L1 expression on tumor and immune cells. J. Immunother. Cancer 2019, 7, 305. [Google Scholar] [CrossRef]
- Prima, V.; Kaliberova, L.N.; Kaliberov, S.; Curiel, D.T.; Kusmartsev, S. COX2/mPGES1/PGE2 pathway regulates PD-L1 expression in tumor-associated macrophages and myeloid-derived suppressor cells. Proc. Natl. Acad. Sci. USA 2017, 114, 1117–1122. [Google Scholar] [CrossRef] [Green Version]
- Wei, J.; Zhang, J.; Wang, D.; Cen, B.; Lang, J.D.; DuBois, R.N. The COX-2–PGE2 Pathway Promotes Tumor Evasion in Colorectal Adenomas. Cancer Prev. Res. 2022, 15, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, L.; Kang, D.; Yang, D.; Tang, Y. Activation of PGE2/EP2 and PGE2/EP4 signaling pathways positively regulate the level of PD-1 in infiltrating CD8(+) T cells in patients with lung cancer. Oncol. Lett. 2018, 15, 552–558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kundu, N.; Ma, X.; Holt, D.; Goloubeva, O.; Ostrand-Rosenberg, S.; Fulton, A.M. Antagonism of the prostaglandin E receptor EP4 inhibits metastasis and enhances NK function. Breast Cancer Res. Treat. 2009, 117, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, E.P.; Markosyan, N.; Connolly, E.; Lawson, J.A.; Li, X.; Grant, G.; Grosser, T.; Fitzgerald, G.A.; Smyth, E.M. Myeloid Cell COX-2 deletion reduces mammary tumor growth through enhanced cytotoxic T-lymphocyte function. Carcinogenesis 2014, 35, 1788–1797. [Google Scholar] [CrossRef]
- Na, Y.-R.; Yoon, Y.-N.; Son, D.-I.; Seok, S.-H. Cyclooxygenase-2 Inhibition Blocks M2 Macrophage Differentiation and Suppresses Metastasis in Murine Breast Cancer Model. PLoS ONE 2013, 8, e63451. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Gene Name | R2 with CD274 (Log Scale) | Spearman Coefficient | Pearson Coefficient | |
|---|---|---|---|---|
| COX1 | PTGS1 | 0.17 | 0.43, p = 8.41 × 10−24 | 0.41, p = 1.75 × 10−22 |
| COX2 | PTGS2 | 0 | −0.04, p = 0.313 | −0.02, p = 0.679 |
| PGE2 pathway | ||||
| mPGES1 | PTGES | 0 | 0, p = 0.966 | −0.01, p = 0.741 |
| PTGES2 | PTGES2 | 0.01 | −0.13, p = 3.79 × 10−3 | −0.11, p = 0.0112 |
| PTGES3 | PTGES3 | 0 | 0.02, p = 0.578 | 0.04, p = 0.423 |
| EP1 | PTGER1 | 0 | 0.01, p = 0.842 | 0.01, p = 0.755 |
| EP2 | PTGER2 | 0.1 | 0.34, p = 2.80 × 10−15 | 0.31, p = 7.61 × 10−13 |
| EP3 | PTGER3 | 0 | −0.00, p = 0.977 | −0.03, p = 0.467 |
| EP4 | PTGER4 | 0.22 | 0.49, p = 1.83 × 10−32 | 0.47, p = 9.52 × 10−29 |
| PGD2 pathway | ||||
| PTGDS | PTGDS | 0.03 | 0.21, p = 1.053 × 10−6 | 0.19, p = 2.551 × 10−5 |
| DP | PTGDR | 0.11 | 0.37, p = 9.28 × 10−18 | 0.33, p = 7.73 × 10−15 |
| DP2 | PTGDR2 | 0.01 | −0.10, p = 0.0261 | −0.12, p = 6.516 × 10−3 |
| TXA2 pathway | ||||
| TBXAS1 | TBXAS1 | 0.05 | 0.23, p = 2.20 × 10−7 | 0.23, p = 2.44 × 10−7 |
| TP | TBXA2R | 0.04 | 0.17, p = 1.070 × 10−4 | 0.19, p = 1.580 × 10−5 |
| PGI2 pathway | ||||
| PGI2 synthase | PTGIS | 0.02 | 0.19, p = 1.220 × 10−5 | 0.15, p = 9.574 × 10−4 |
| IP | PTGIR | 0.03 | 0.19, p = 2.412 × 10−5 | 0.16, p = 3.604 × 10−4 |
| PGF2a pathway | ||||
| PGF synthase (predicted) | PRXL2B | 0 | −0.05, p = 0.216 | −0.04, p = 0.419 |
| FP | PTGFR | 0.03 | 0.20, p = 4.658 × 10−6 | 0.17, p = 9.216 × 10−5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nie, J.Z.; Wang, M.-T.; Nie, D. Regulations of Tumor Microenvironment by Prostaglandins. Cancers 2023, 15, 3090. https://doi.org/10.3390/cancers15123090
Nie JZ, Wang M-T, Nie D. Regulations of Tumor Microenvironment by Prostaglandins. Cancers. 2023; 15(12):3090. https://doi.org/10.3390/cancers15123090
Chicago/Turabian StyleNie, Jeffrey Z., Man-Tzu Wang, and Daotai Nie. 2023. "Regulations of Tumor Microenvironment by Prostaglandins" Cancers 15, no. 12: 3090. https://doi.org/10.3390/cancers15123090