

Follicular Helper and Regulatory T Cells Drive the Development of Spontaneous Epstein–Barr Virus Lymphoproliferative Disorder
 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Participants
2.2. Mice
2.3. Donor Screening in the Hu-PBL-SCID
2.4. Quantitative Analysis of EBV DNA in Samples via qPCR
2.5. Elispot Assay
2.6. Mass Cytometry
2.7. Cell Sorting and RNA Isolation
2.8. Ampliseq Transcriptome Analysis
2.9. Depletion of the Th Cell Subsets
2.10. Statistical Analyses
3. Results
3.1. Identification of EBV-LPD High Incidence (HI) and No Incidence (NI) Donors
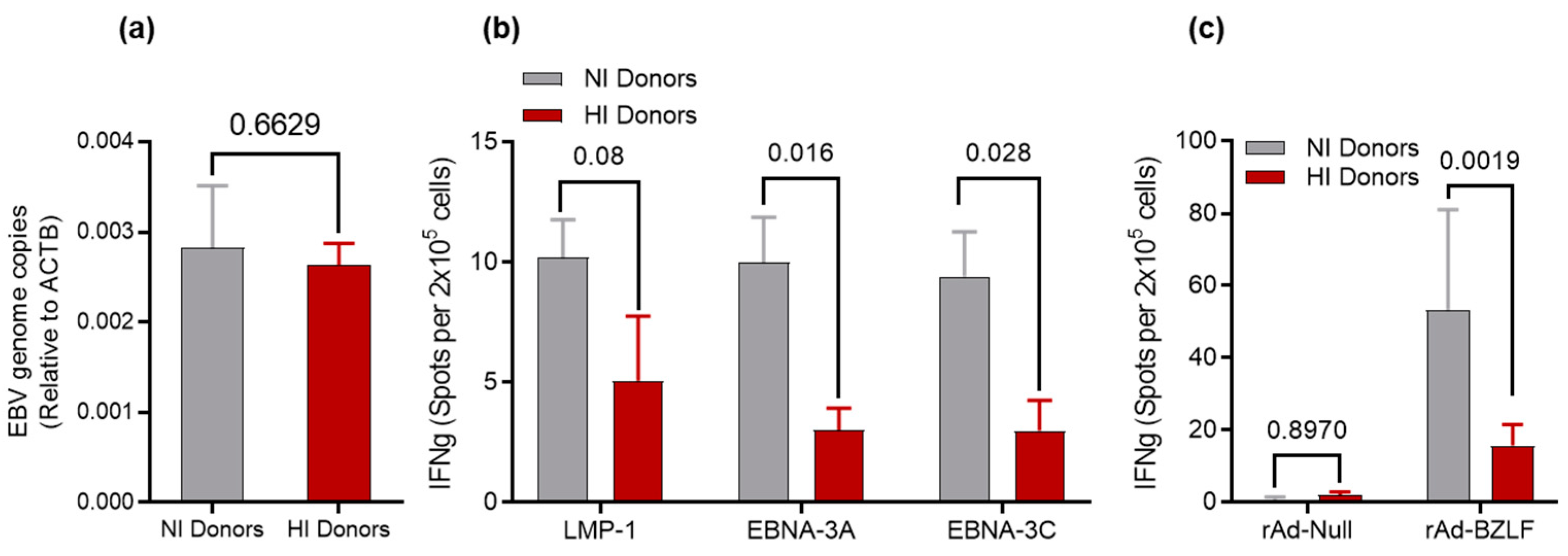
3.2. Determination of EBV DNA Content for HI and NI Donors
3.3. Recall Responses to EBV Antigens
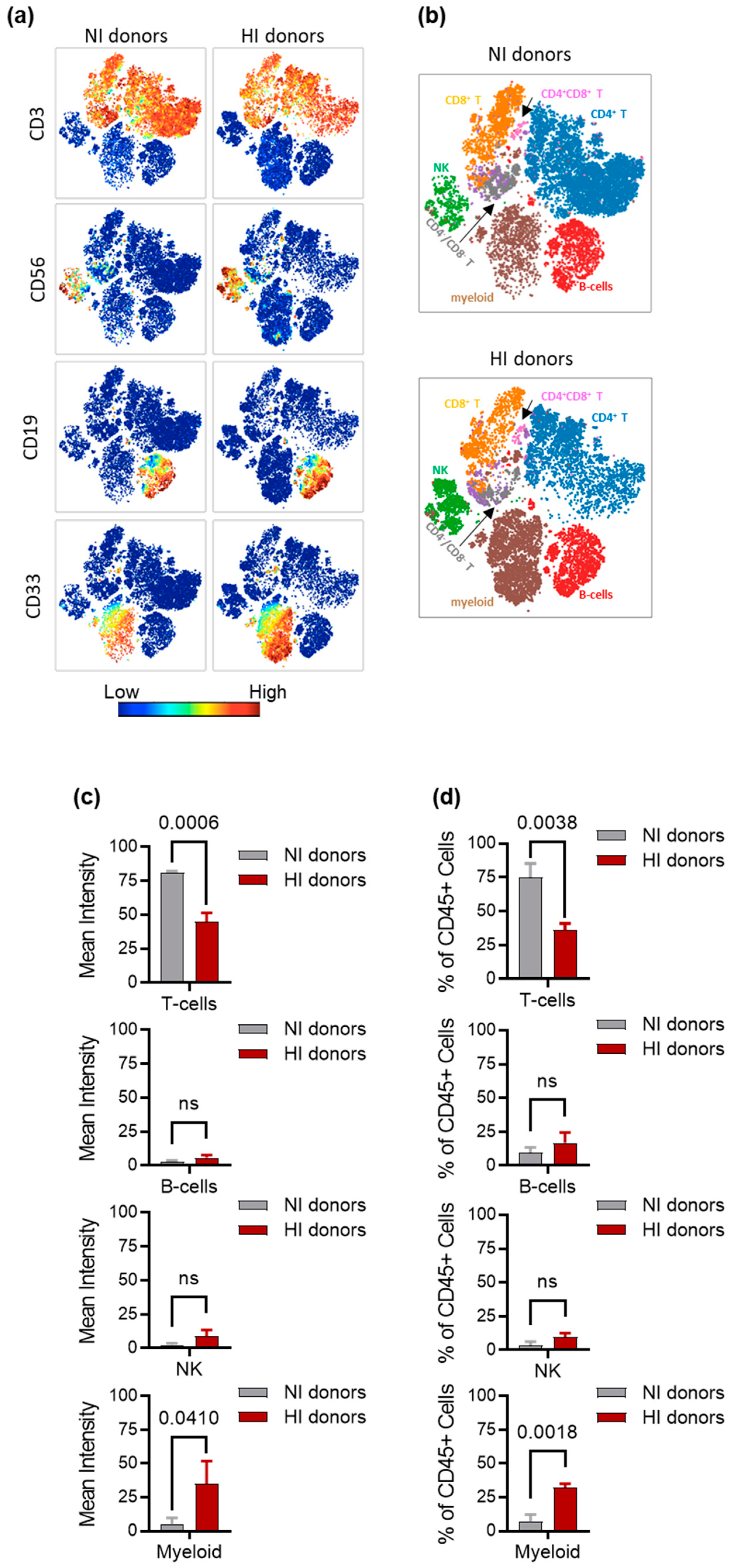
3.4. Characterization of Immune Cell Subsets in NI and HI Donors
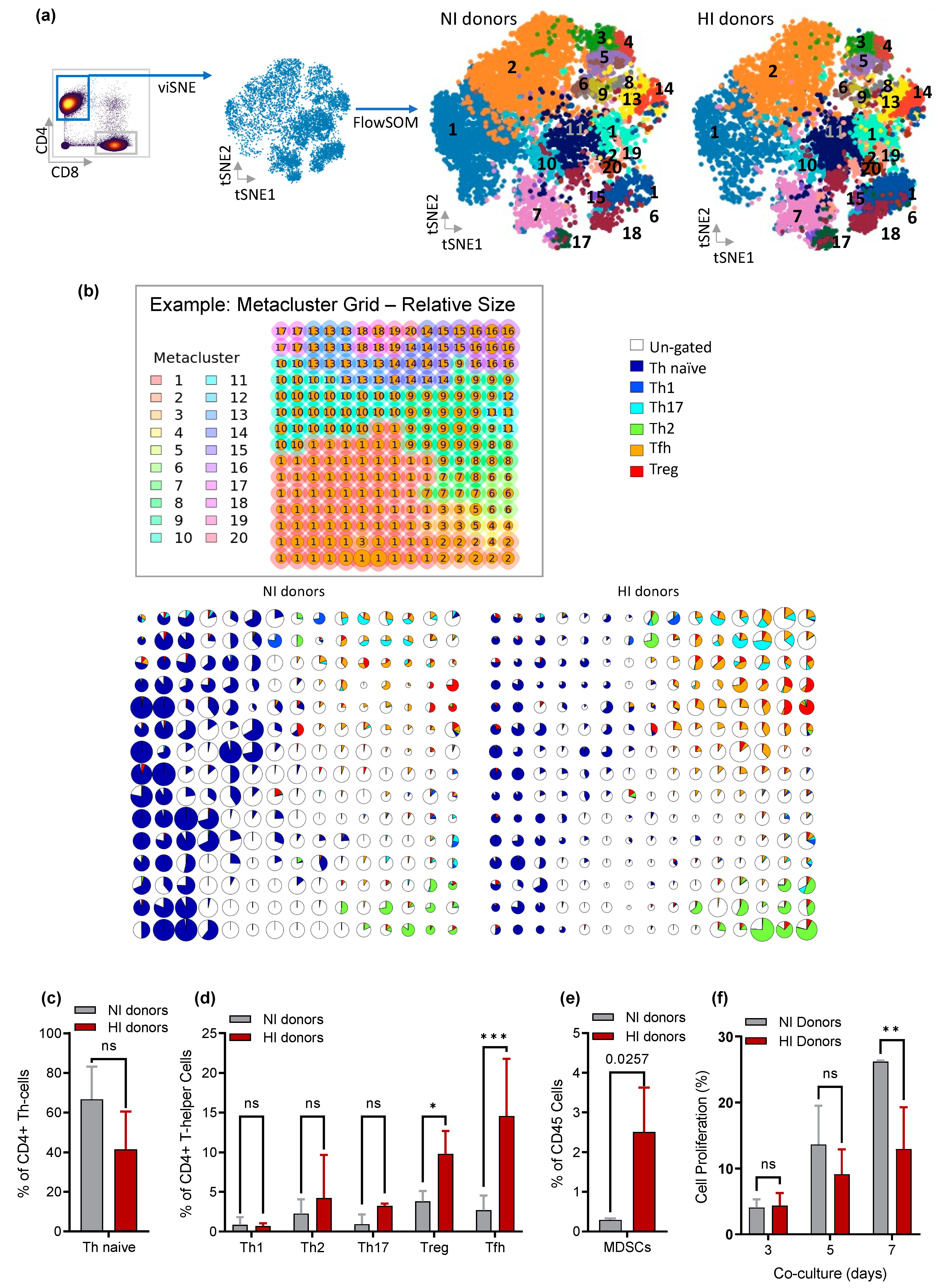
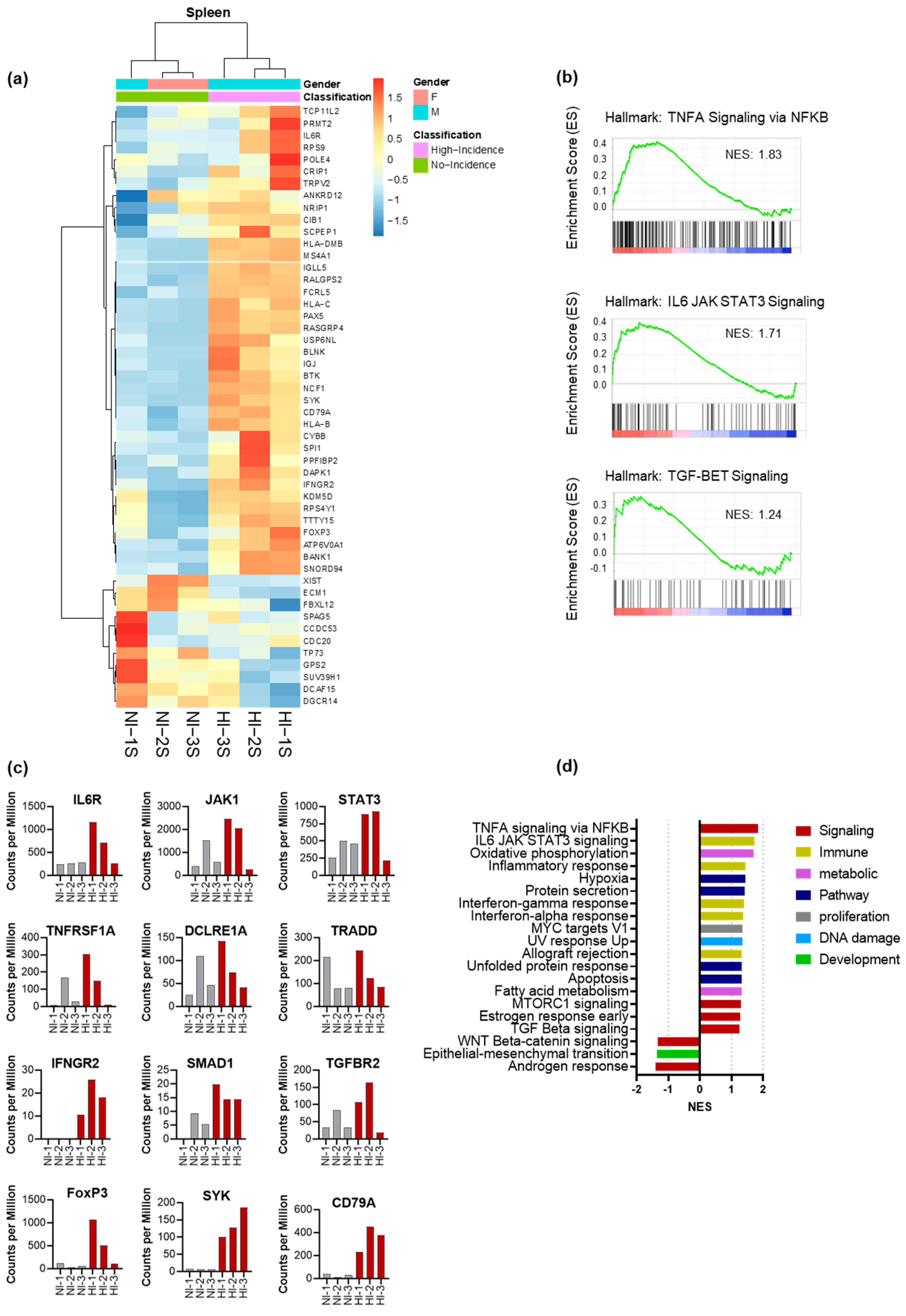
3.5. CD4+ Th Cell from HI Genetic Signature: Correlated with Treg and Tfh Expansion and the Risk for the Development of EBV-LPD
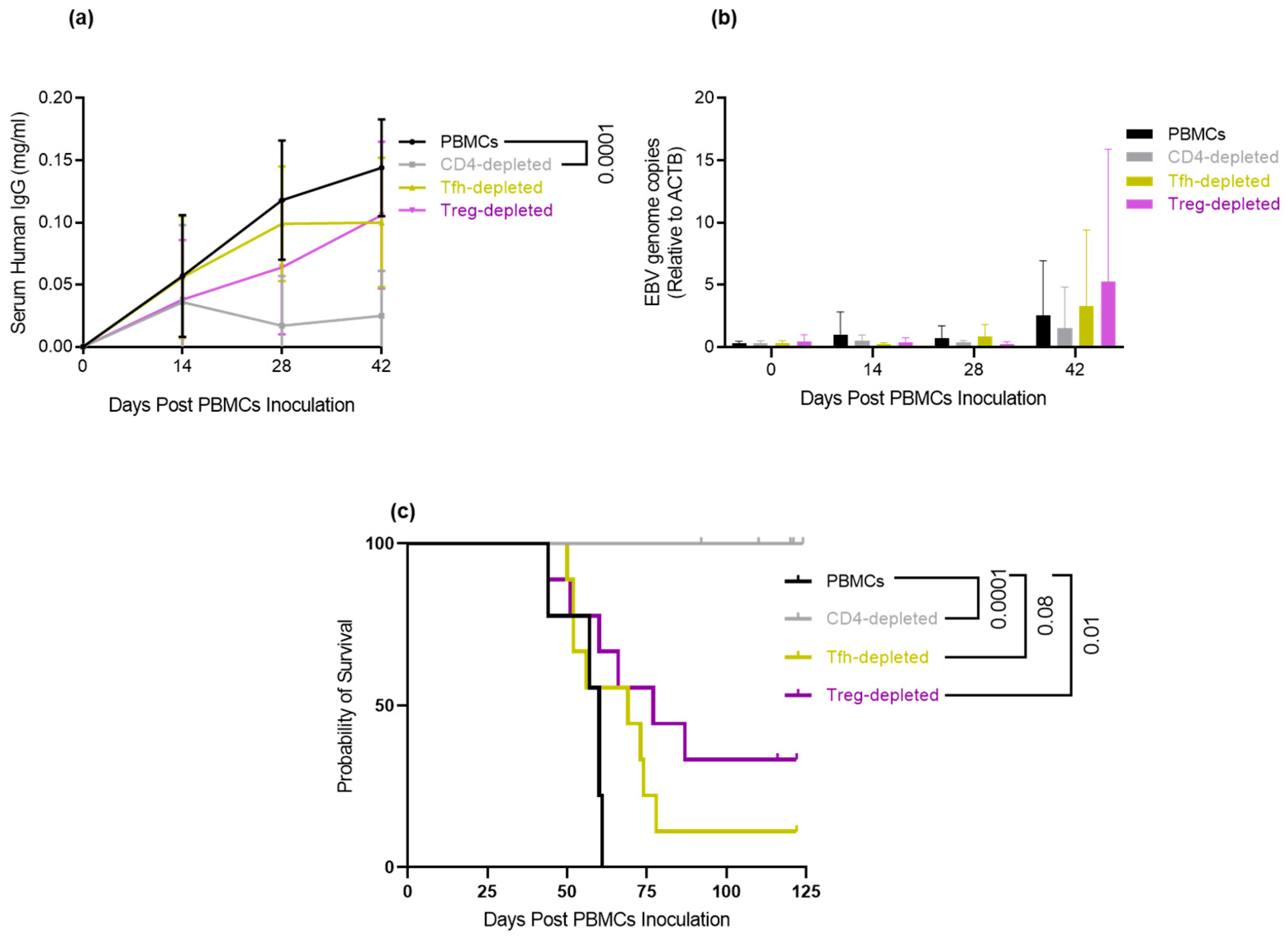
3.6. Depletion of Tfh and Treg: Delays/Prevents EBV-LPD and Improves Survival in Hu-PBL-SCID Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Epstein, M.A.; Achong, B.G.; Barr, Y.M. Virus Particles in Cultured Lymphoblasts from Burkitt’s Lymphoma. Lancet 1964, 283, 702–703. [Google Scholar] [CrossRef]
- Hewetson, J.F.; Rocchi, G.; Henle, W.; Henle, G. Neutralizing antibodies to Epstein-Barr virus in healthy populations and patients with infectious mononucleosis. J. Infect. Dis. 1973, 128, 283–289. [Google Scholar] [CrossRef]
- Thorley-Lawson, D.A. EBV Persistence—Introducing the Virus. Curr. Top. Microbiol. Immunol. 2015, 390, 151–209. [Google Scholar] [CrossRef] [Green Version]
- Shindiapina, P.; Ahmed, E.H.; Mozhenkova, A.; Abebe, T.; Baiocchi, R.A. Immunology of EBV-Related Lymphoproliferative Disease in HIV-Positive Individuals. Front. Oncol. 2020, 10, 1723. [Google Scholar] [CrossRef]
- Park, S.; Lee, J.; Ko, Y.H.; Han, A.; Jun, H.J.; Lee, S.C.; Hwang, I.G.; Park, Y.H.; Ahn, J.S.; Jung, C.W.; et al. The impact of Epstein-Barr virus status on clinical outcome in diffuse large B-cell lymphoma. Blood 2007, 110, 972–978. [Google Scholar] [CrossRef]
- Ramos, J.C.; Sparano, J.A.; Chadburn, A.; Reid, E.G.; Ambinder, R.F.; Siegel, E.R.; Moore, P.C.; Rubinstein, P.G.; Durand, C.M.; Cesarman, E.; et al. Impact of Myc in HIV-associated non-Hodgkin lymphomas treated with EPOCH and outcomes with vorinostat (AMC-075 trial). Blood 2020, 136, 1284–1297. [Google Scholar] [CrossRef]
- Taylor, A.L.; Marcus, R.; Bradley, J.A. Post-transplant lymphoproliferative disorders (PTLD) after solid organ transplantation. Crit. Rev. Oncol. Hematol. 2005, 56, 155–167. [Google Scholar] [CrossRef]
- Nourse, J.P.; Jones, K.; Gandhi, M.K. Epstein-Barr Virus-related post-transplant lymphoproliferative disorders: Pathogenetic insights for targeted therapy. Am. J. Transplant. Off J. Am. Soc. Transplant. Am. Soc. Transpl. Surg. 2011, 11, 888–895. [Google Scholar] [CrossRef]
- Trappe, R.U.; Dierickx, D.; Zimmermann, H.; Morschhauser, F.; Mollee, P.; Zaucha, J.M.; Dreyling, M.H.; Dührsen, U.; Reinke, P.; Verhoef, G.; et al. Response to Rituximab Induction Is a Predictive Marker in B-Cell Post-Transplant Lymphoproliferative Disorder and Allows Successful Stratification Into Rituximab or R-CHOP Consolidation in an International, Prospective, Multicenter Phase II Trial. J. Clin. Oncol. Off J. Am. Soc. Clin. Oncol. 2017, 35, 536–543. [Google Scholar] [CrossRef] [Green Version]
- Mosier, D.E.; Picchio, G.R.; Kirven, M.B.; Garnier, J.L.; Torbett, B.E.; Baird, S.M.; Kobayashi, R.; Kipps, T.J. EBV-induced human B cell lymphomas in hu-PBL-SCID mice. AIDS Res. Hum. Retrovir. 1992, 8, 735–740. [Google Scholar]
- Ma, S.D.; Xu, X.; Plowshay, J.; Ranheim, E.A.; Burlingham, W.J.; Jensen, J.L.; Asimakopoulos, F.; Tang, W.; Gulley, M.L.; Cesarman, E.; et al. LMP1-deficient Epstein-Barr virus mutant requires T cells for lymphomagenesis. J. Clin. Investig. 2015, 125, 304–315. [Google Scholar] [CrossRef] [Green Version]
- Veronese, M.L.; Veronesi, A.; D’Andrea, E.; Del Mistro, A.; Indraccolo, S.; Mazza, M.R.; Mion, M.; Zamarchi, R.; Menin, C.; Panozzo, M.; et al. Lymphoproliferative disease in human peripheral blood mononuclear cell-injected SCID mice. I. T lymphocyte requirement for B cell tumor generation. J. Exp. Med. 1992, 176, 1763–1767. [Google Scholar] [CrossRef] [Green Version]
- Baiocchi, R.A.; Caligiuri, M.A. Low-dose interleukin 2 prevents the development of Epstein-Barr virus (EBV)-associated lymphoproliferative disease in scid/scid mice reconstituted i.p. with EBV-seropositive human peripheral blood lymphocytes. Proc. Natl. Acad. Sci. USA 1994, 91, 5577–5581. [Google Scholar] [CrossRef] [Green Version]
- Picchio, G.R.; Kobayashi, R.; Kirven, M.; Baird, S.M.; Kipps, T.J.; Mosier, D.E. Heterogeneity among Epstein-Barr virus-seropositive donors in the generation of immunoblastic B-cell lymphomas in SCID mice receiving human peripheral blood leukocyte grafts. Cancer Res. 1992, 52, 2468–2477. [Google Scholar]
- Ahmed, E.H.; Baiocchi, R.A. Murine Models of Epstein-Barr Virus-Associated Lymphomagenesis. ILAR J. 2016, 57, 55–62. [Google Scholar] [CrossRef] [Green Version]
- Swain, S.L.; McKinstry, K.K.; Strutt, T.M. Expanding roles for CD4(+) T cells in immunity to viruses. Nat. Rev. Immunol. 2012, 12, 136–148. [Google Scholar] [CrossRef]
- Laidlaw, B.J.; Craft, J.E.; Kaech, S.M. The multifaceted role of CD4(+) T cells in CD8(+) T cell memory. Nat. Rev. Immunol. 2016, 16, 102–111. [Google Scholar] [CrossRef] [Green Version]
- Gershon, R.K.; Kondo, K. Cell interactions in the induction of tolerance: The role of thymic lymphocytes. Immunology 1970, 18, 723–737. [Google Scholar]
- Gershon, R.K.; Kondo, K. Infectious immunological tolerance. Immunology 1971, 21, 903–914. [Google Scholar]
- Li, J.; Qian, C.N.; Zeng, Y.X. Regulatory T cells and EBV associated malignancies. Int. Immunopharmacol. 2009, 9, 590–592. [Google Scholar] [CrossRef]
- Zhang, N.-N.; Chen, J.-N.; Xiao, L.; Tang, F.; Zhang, Z.-G.; Zhang, Y.-W.; Feng, Z.-Y.; Jiang, Y.; Shao, C.-K. Accumulation Mechanisms of CD4+CD25+FOXP3+ Regulatory T Cells in EBV-associated Gastric Carcinoma. Sci. Rep. 2015, 5, 18057. [Google Scholar] [CrossRef] [Green Version]
- Vinuesa, C.G.; Linterman, M.A.; Yu, D.; MacLennan, I.C. Follicular Helper T Cells. Annu. Rev. Immunol. 2016, 34, 335–368. [Google Scholar] [CrossRef]
- Crotty, S. T follicular helper cell differentiation, function, and roles in disease. Immunity 2014, 41, 529–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crotty, S. T Follicular Helper Cell Biology: A Decade of Discovery and Diseases. Immunity 2019, 50, 1132–1148. [Google Scholar] [CrossRef]
- Mintz, M.A.; Cyster, J.G. T follicular helper cells in germinal center B cell selection and lymphomagenesis. Immunol. Rev. 2020, 296, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, E.H.; Brooks, E.; Sloan, S.; Schlotter, S.; Jeney, F.; Hale, C.; Mao, C.; Zhang, X.; McLaughlin, E.; Shindiapina, P.; et al. Targeted Delivery of BZLF1 to DEC205 Drives EBV-Protective Immunity in a Spontaneous Model of EBV-Driven Lymphoproliferative Disease. Vaccines 2021, 9, 555. [Google Scholar] [CrossRef] [PubMed]
- Steven, N.M.; Annels, N.E.; Kumar, A.; Leese, A.M.; Kurilla, M.G.; Rickinson, A.B. Immediate early and early lytic cycle proteins are frequent targets of the Epstein-Barr virus-induced cytotoxic T cell response. J. Exp. Med. 1997, 185, 1605–1617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pudney, V.A.; Leese, A.M.; Rickinson, A.B.; Hislop, A.D. CD8+ immunodominance among Epstein-Barr virus lytic cycle antigens directly reflects the efficiency of antigen presentation in lytically infected cells. J. Exp. Med. 2005, 201, 349–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murata, T. Regulation of Epstein-Barr virus reactivation from latency. Microbiol. Immunol. 2014, 58, 307–317. [Google Scholar] [CrossRef]
- Ma, S.D.; Hegde, S.; Young, K.H.; Sullivan, R.; Rajesh, D.; Zhou, Y.; Jankowska-Gan, E.; Burlingham, W.J.; Sun, X.; Gulley, M.L.; et al. A new model of Epstein-Barr virus infection reveals an important role for early lytic viral protein expression in the development of lymphomas. J. Virol. 2011, 85, 165–177. [Google Scholar] [CrossRef] [Green Version]
- Germini, D.; Sall, F.B.; Shmakova, A.; Wiels, J.; Dokudovskaya, S.; Drouet, E.; Vassetzky, Y. Oncogenic Properties of the EBV ZEBRA Protein. Cancers 2020, 12, 1479. [Google Scholar] [CrossRef]
- Hartlage, A.S.; Liu, T.; Patton, J.T.; Garman, S.L.; Zhang, X.; Kurt, H.; Lozanski, G.; Lustberg, M.E.; Caligiuri, M.A.; Baiocchi, R.A. The Epstein-Barr Virus Lytic Protein BZLF1 as a Candidate Target Antigen for Vaccine Development. Cancer Immunol. Res. 2015, 3, 787–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leipold, M.D.; Newell, E.W.; Maecker, H.T. Multiparameter Phenotyping of Human PBMCs Using Mass Cytometry. Methods Mol. Biol. 2015, 1343, 81–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bandura, D.R.; Baranov, V.I.; Ornatsky, O.I.; Antonov, A.; Kinach, R.; Lou, X.; Pavlov, S.; Vorobiev, S.; Dick, J.E.; Tanner, S.D. Mass Cytometry: Technique for Real Time Single Cell Multitarget Immunoassay Based on Inductively Coupled Plasma Time-of-Flight Mass Spectrometry. Anal. Chem. 2009, 81, 6813–6822. [Google Scholar] [CrossRef] [PubMed]
- Bjornson, Z.B.; Nolan, G.P.; Fantl, W.J. Single-cell mass cytometry for analysis of immune system functional states. Curr. Opin. Immunol. 2013, 25, 484–494. [Google Scholar] [CrossRef] [Green Version]
- Bendall, S.C.; Simonds, E.F.; Qiu, P.; Amir, E.-A.D.; Krutzik, P.O.; Finck, R.; Bruggner, R.V.; Melamed, R.; Trejo, A.; Ornatsky, O.I.; et al. Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science 2011, 332, 687–696. [Google Scholar] [CrossRef] [Green Version]
- Spitzer, M.H.; Nolan, G.P. Mass Cytometry: Single Cells, Many Features. Cell 2016, 165, 780–791. [Google Scholar] [CrossRef] [Green Version]
- Finck, R.; Simonds, E.F.; Jager, A.; Krishnaswamy, S.; Sachs, K.; Fantl, W.; Pe’er, D.; Nolan, G.P.; Bendall, S.C. Normalization of mass cytometry data with bead standards. Cytom. Part A J. Int. Soc. Anal. Cytol. 2013, 83, 483–494. [Google Scholar] [CrossRef]
- Amir, E.A.D.; Davis, K.L.; Tadmor, M.D.; Simonds, E.F.; Levine, J.H.; Bendall, S.C.; Shenfeld, D.K.; Krishnaswamy, S.; Nolan, G.P.; Pe’er, D. viSNE enables visualization of high dimensional single-cell data and reveals phenotypic heterogeneity of leukemia. Nat. Biotechnol. 2013, 31, 545–552. [Google Scholar] [CrossRef] [Green Version]
- Kimball, A.K.; Oko, L.M.; Bullock, B.L.; Nemenoff, R.A.; van Dyk, L.F.; Clambey, E.T. A Beginner’s Guide to Analyzing and Visualizing Mass Cytometry Data. J. Immunol. 2018, 200, 3–22. [Google Scholar] [CrossRef] [Green Version]
- Maecker, H.T.; McCoy, J.P.; Nussenblatt, R. Standardizing immunophenotyping for the Human Immunology Project. Nat. Rev. Immunol. 2012, 12, 191–200. [Google Scholar] [CrossRef] [Green Version]
- Pietrzak, M.; Papp, A.; Curtis, A.; Handelman, S.K.; Kataki, M.; Scharre, D.W.; Rempala, G.; Sadee, W. Gene expression profiling of brain samples from patients with Lewy body dementia. Biochem. Biophys. Res. Commun. 2016, 479, 875–880. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thangavelu, M.; Snyder, L.; Anastasi, J.; Le Beau, M.M.; Kirven, M.; Picchio, G.; Mosier, D.E.; Rowley, J.D. Cytogenetic characterization of B-cell lymphomas from severe combined immunodeficiency disease mice given injections of lymphocytes from Epstein-Barr virus-positive donors. Cancer Res. 1992, 52, 4678–4681. [Google Scholar] [PubMed]
- Mosier, D.E.; Gulizia, R.J.; Baird, S.M.; Wilson, D.B. Transfer of a functional human immune system to mice with severe combined immunodeficiency. Nature 1988, 335, 256–259. [Google Scholar] [CrossRef] [PubMed]
- Taylor, G.S.; Long, H.M.; Brooks, J.M.; Rickinson, A.B.; Hislop, A.D. The immunology of Epstein-Barr virus-induced disease. Annu. Rev. Immunol. 2015, 33, 787–821. [Google Scholar] [CrossRef]
- Pearson, T.; Greiner, D.L.; Shultz, L.D. Creation of “humanized” mice to study human immunity. Curr. Protoc. Immunol. 2008, 15, 15–21. [Google Scholar] [CrossRef] [Green Version]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [Green Version]
- Sunil-Chandra, N.P.; Efstathiou, S.; Arno, J.; Nash, A.A. Virological and pathological features of mice infected with murine gamma-herpesvirus 68. J. Gen. Virol. 1992, 73 Pt 9, 2347–2356. [Google Scholar] [CrossRef]
- Okano, M.; Taguchi, Y.; Nakamine, H.; Pirruccello, S.J.; Davis, J.R.; Beisel, K.W.; Kleveland, K.L.; Sanger, W.G.; Fordyce, R.R.; Purtilo, D.T. Characterization of Epstein-Barr virus-induced lymphoproliferation derived from human peripheral blood mononuclear cells transferred to severe combined immunodeficient mice. Am. J. Pathol. 1990, 137, 517–522. [Google Scholar]
- Rowe, M.; Young, L.S.; Crocker, J.; Stokes, H.; Henderson, S.; Rickinson, A.B. Epstein-Barr virus (EBV)-associated lymphoproliferative disease in the SCID mouse model: Implications for the pathogenesis of EBV-positive lymphomas in man. J. Exp. Med. 1991, 173, 147–158. [Google Scholar] [CrossRef]
- Baiocchi, R.A.; Ward, J.S.; Carrodeguas, L.; Eisenbeis, C.F.; Peng, R.; Roychowdhury, S.; Vourganti, S.; Sekula, T.; O’Brien, M.; Moeschberger, M.; et al. GM-CSF and IL-2 induce specific cellular immunity and provide protection against Epstein-Barr virus lymphoproliferative disorder. J. Clin. Investig. 2001, 108, 887–894. [Google Scholar] [CrossRef]
- Coles, R.E.; Boyle, T.J.; DiMaio, J.M.; Berend, K.R.; Via, D.F.; Lyerly, H.K. T cells or active Epstein-Barr virus infection in the development of lymphoproliferative disease in human B cell-injected severe combined immunodeficient mice. Ann. Surg. Oncol. 1994, 1, 405–410. [Google Scholar] [CrossRef]
- Clinchy, B.; Vitetta, E.S. The use of an anti-CD3 immunotoxin to prevent the development of lymphoproliferative disease in SCID/PBL mice. J. Immunol. Methods 1998, 218, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Burack, W.R.; Spence, J.M.; Spence, J.P.; Spence, S.A.; Rock, P.J.; Shenoy, G.N.; Shultz, L.D.; Bankert, R.B.; Bernstein, S.H. Patient-derived xenografts of low-grade B-cell lymphomas demonstrate roles of the tumor microenvironment. Blood Adv. 2017, 1, 1263–1273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breitfeld, D.; Ohl, L.; Kremmer, E.; Ellwart, J.; Sallusto, F.; Lipp, M.; Forster, R. Follicular B helper T cells express CXC chemokine receptor 5, localize to B cell follicles, and support immunoglobulin production. J. Exp. Med. 2000, 192, 1545–1552. [Google Scholar] [CrossRef] [Green Version]
- Qian, J.; Yu, Q.; Chen, G.; Wang, M.; Zhao, Z.; Zhang, Y.; Qiu, L. Altered ratio of circulating follicular regulatory T cells and follicular helper T cells during primary EBV infection. Clin. Exp. Med. 2020, 20, 373–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Zhou, Y.; Yu, Q.; Zhao, Z.; Wang, H.; Luo, X.; Chen, Y.; Zhu, Z.; Chen, G.; Wu, M.; et al. Higher Frequency of CD4+CXCR5+ICOS+PD1+ T Follicular Helper Cells in Patients With Infectious Mononucleosis. Medicine 2015, 94, e2061. [Google Scholar] [CrossRef]
- Eddahri, F.; Denanglaire, S.; Bureau, F.; Spolski, R.; Leonard, W.J.; Leo, O.; Andris, F. Interleukin-6/STAT3 signaling regulates the ability of naive T cells to acquire B-cell help capacities. Blood 2009, 113, 2426–2433. [Google Scholar] [CrossRef] [Green Version]
- Heawchaiyaphum, C.; Pientong, C.; Yoshiyama, H.; Iizasa, H.; Panthong, W.; Ekalaksananan, T. General Features and Novel Gene Signatures That Identify Epstein-Barr Virus-Associated Epithelial Cancers. Cancers 2021, 14, 31. [Google Scholar] [CrossRef]
- Gires, O.; Zimber-Strobl, U.; Gonnella, R.; Ueffing, M.; Marschall, G.; Zeidler, R.; Pich, D.; Hammerschmidt, W. Latent membrane protein 1 of Epstein-Barr virus mimics a constitutively active receptor molecule. EMBO J. 1997, 16, 6131–6140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Busch, L.K.; Bishop, G.A. The EBV transforming protein, latent membrane protein 1, mimics and cooperates with CD40 signaling in B lymphocytes. J. Immunol. 1999, 162, 2555–2561. [Google Scholar] [CrossRef]
- Kirk, A.D.; Harlan, D.M.; Armstrong, N.N.; Davis, T.A.; Dong, Y.; Gray, G.S.; Hong, X.; Thomas, D.; Fechner, J.H., Jr.; Knechtle, S.J. CTLA4-Ig and anti-CD40 ligand prevent renal allograft rejection in primates. Proc. Natl. Acad. Sci. USA 1997, 94, 8789–8794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malvezzi, P.; Jouve, T.; Rostaing, L. Costimulation Blockade in Kidney Transplantation: An Update. Transplantation 2016, 100, 2315–2323. [Google Scholar] [CrossRef]
- Hussain, S.K.; Zhu, W.; Chang, S.C.; Breen, E.C.; Vendrame, E.; Magpantay, L.; Widney, D.; Conn, D.; Sehl, M.; Jacobson, L.P.; et al. Serum levels of the chemokine CXCL13, genetic variation in CXCL13 and its receptor CXCR5, and HIV-associated non-hodgkin B-cell lymphoma risk. Cancer Epidemiol. Biomark. Prev. A Publ. Am. Assoc. Cancer Res. Cosponsored By Am. Soc. Prev. Oncol. 2013, 22, 295–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiffer, L.; Henke-Gendo, C.; Wilsdorf, N.; Hussein, K.; Pape, L.; Schmitt, C.; Haller, H.; Schiffer, M.; Klein, C.; Kreipe, H.; et al. CXCL13 as a novel marker for diagnosis and disease monitoring in pediatric PTLD. Am. J. Transplant. Off J. Am. Soc. Transplant. Am. Soc. Transpl. Surg. 2012, 12, 1610–1617. [Google Scholar] [CrossRef]
- Bluestone, J.A.; Mackay, C.R.; O’Shea, J.J.; Stockinger, B. The functional plasticity of T cell subsets. Nat. Rev. Immunol. 2009, 9, 811–816. [Google Scholar] [CrossRef] [Green Version]
- Bennett, C.L.; Christie, J.; Ramsdell, F.; Brunkow, M.E.; Ferguson, P.J.; Whitesell, L.; Kelly, T.E.; Saulsbury, F.T.; Chance, P.F.; Ochs, H.D. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat. Genet. 2001, 27, 20–21. [Google Scholar] [CrossRef]
- Brunkow, M.E.; Jeffery, E.W.; Hjerrild, K.A.; Paeper, B.; Clark, L.B.; Yasayko, S.A.; Wilkinson, J.E.; Galas, D.; Ziegler, S.F.; Ramsdell, F. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat. Genet. 2001, 27, 68–73. [Google Scholar] [CrossRef]
- Takeuchi, Y.; Nishikawa, H. Roles of regulatory T cells in cancer immunity. Int. Immunol. 2016, 28, 401–409. [Google Scholar] [CrossRef] [Green Version]
- Tao, H.; Mimura, Y.; Aoe, K.; Kobayashi, S.; Yamamoto, H.; Matsuda, E.; Okabe, K.; Matsumoto, T.; Sugi, K.; Ueoka, H. Prognostic potential of FOXP3 expression in non-small cell lung cancer cells combined with tumor-infiltrating regulatory T cells. Lung Cancer 2012, 75, 95–101. [Google Scholar] [CrossRef]
- Shang, B.; Liu, Y.; Jiang, S.J.; Liu, Y. Prognostic value of tumor-infiltrating FoxP3+ regulatory T cells in cancers: A systematic review and meta-analysis. Sci. Rep. 2015, 5, 15179. [Google Scholar] [CrossRef] [Green Version]
- Rushbrook, S.M.; Ward, S.M.; Unitt, E.; Vowler, S.L.; Lucas, M.; Klenerman, P.; Alexander, G.J. Regulatory T cells suppress in vitro proliferation of virus-specific CD8+ T cells during persistent hepatitis C virus infection. J. Virol. 2005, 79, 7852–7859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardoll, D. Does the immune system see tumors as foreign or self? Annu. Rev. Immunol. 2003, 21, 807–839. [Google Scholar] [CrossRef] [PubMed]
- Wingate, P.J.; McAulay, K.A.; Anthony, I.C.; Crawford, D.H. Regulatory T cell activity in primary and persistent Epstein-Barr virus infection. J. Med. Virol. 2009, 81, 870–877. [Google Scholar] [CrossRef]
- Dierksheide, J.E.; Baiocchi, R.A.; Ferketich, A.K.; Roychowdhury, S.; Pelletier, R.P.; Eisenbeis, C.F.; Caligiuri, M.A.; VanBuskirk, A.M. IFN-gamma gene polymorphisms associate with development of EBV+ lymphoproliferative disease in hu PBL-SCID mice. Blood 2005, 105, 1558–1565. [Google Scholar] [CrossRef] [PubMed]
- Baiocchi, R.A.; Ross, M.E.; Tan, J.C.; Chou, C.C.; Sullivan, L.; Haldar, S.; Monne, M.; Seiden, M.V.; Narula, S.K.; Sklar, J.; et al. Lymphomagenesis in the SCID-hu mouse involves abundant production of human interleukin-10. Blood 1995, 85, 1063–1074. [Google Scholar] [CrossRef] [Green Version]
- Fu, S.; Zhang, N.; Yopp, A.C.; Chen, D.; Mao, M.; Chen, D.; Zhang, H.; Ding, Y.; Bromberg, J.S. TGF-beta induces Foxp3 + T-regulatory cells from CD4 + CD25—Precursors. Am. J. Transplant. Off J. Am. Soc. Transplant. Am. Soc. Transpl. Surg. 2004, 4, 1614–1627. [Google Scholar] [CrossRef]
- Collins, P.J.; Fox, C.P.; George, L.; Pearce, H.; Ryan, G.; De Santo, C.; Mussai, F.; Lewis, D.; Long, H.; Shannon-Lowe, C. Characterizing EBV-associated lymphoproliferative diseases and the role of myeloid-derived suppressor cells. Blood 2021, 137, 203–215. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmed, E.H.; Lustberg, M.; Hale, C.; Sloan, S.; Mao, C.; Zhang, X.; Ozer, H.G.; Schlotter, S.; Smith, P.L.; Jeney, F.; et al. Follicular Helper and Regulatory T Cells Drive the Development of Spontaneous Epstein–Barr Virus Lymphoproliferative Disorder. Cancers 2023, 15, 3046. https://doi.org/10.3390/cancers15113046
Ahmed EH, Lustberg M, Hale C, Sloan S, Mao C, Zhang X, Ozer HG, Schlotter S, Smith PL, Jeney F, et al. Follicular Helper and Regulatory T Cells Drive the Development of Spontaneous Epstein–Barr Virus Lymphoproliferative Disorder. Cancers. 2023; 15(11):3046. https://doi.org/10.3390/cancers15113046
Chicago/Turabian StyleAhmed, Elshafa Hassan, Mark Lustberg, Claire Hale, Shelby Sloan, Charlene Mao, Xiaoli Zhang, Hatice Gulcin Ozer, Sarah Schlotter, Porsha L. Smith, Frankie Jeney, and et al. 2023. "Follicular Helper and Regulatory T Cells Drive the Development of Spontaneous Epstein–Barr Virus Lymphoproliferative Disorder" Cancers 15, no. 11: 3046. https://doi.org/10.3390/cancers15113046