

Overview of Molecular Detection Technologies for MET in Lung Cancer
Abstract
:Simple Summary
Abstract
1. Introduction
2. MET Receptor
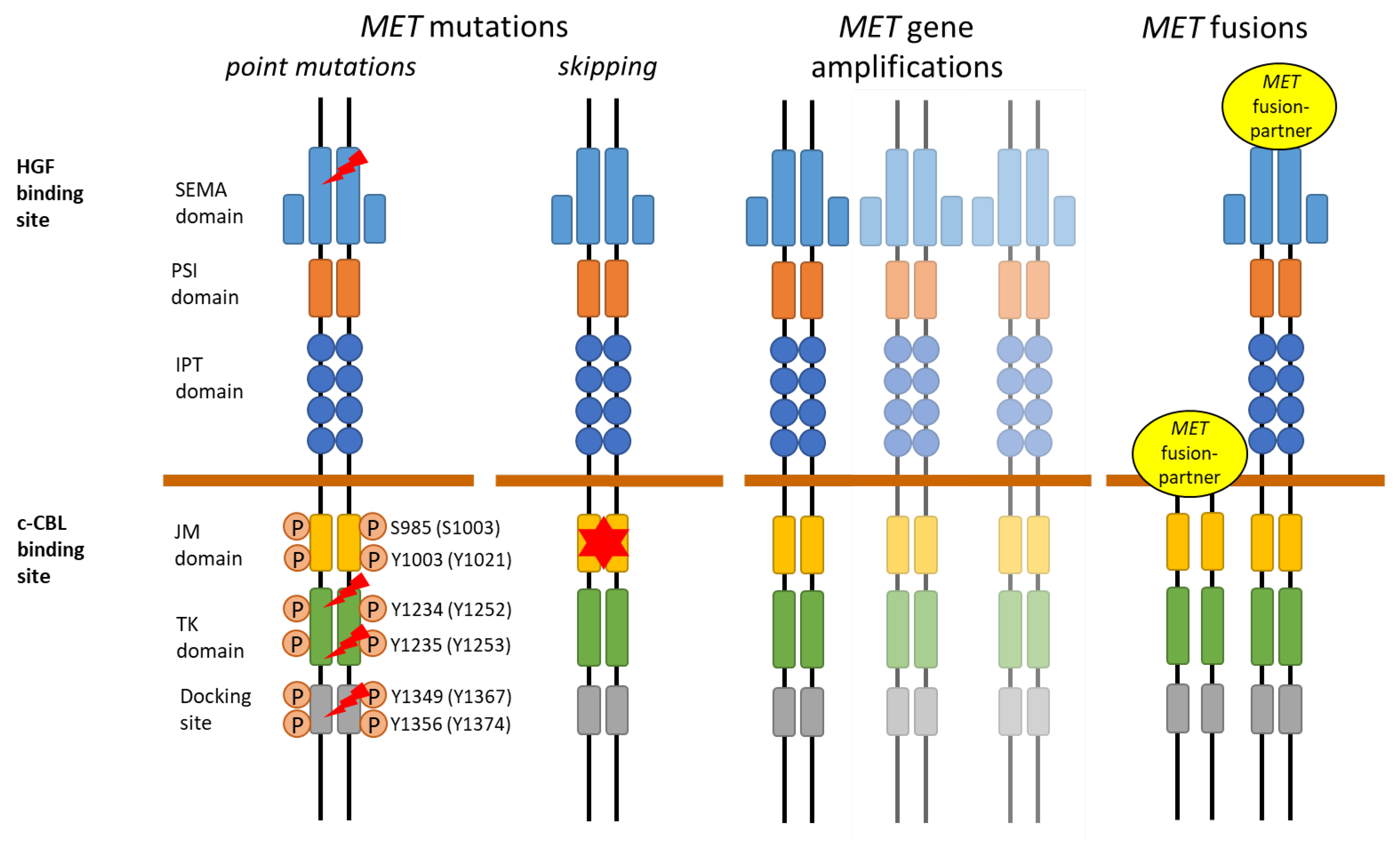
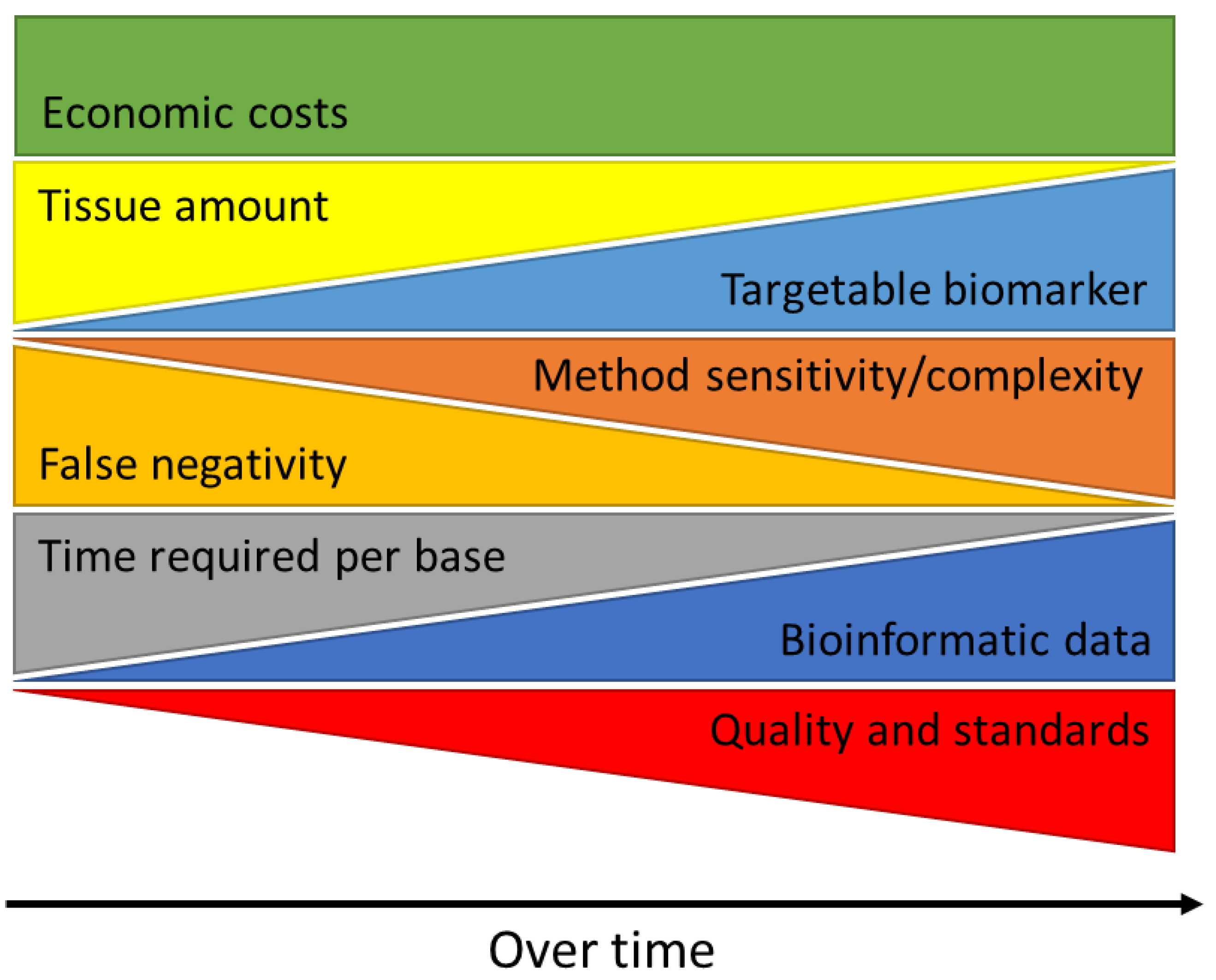
3. MET Aberrations in Lung Cancer
3.1. MET Overexpression

{kind=link}
{kind=link}
| MET Aberration | Detection Technique | Tested Material | Evaluation Criteria | Advantages | Disadvantages |
|---|---|---|---|---|---|
| MET overexpression [14,15,16,17,18,19,20,21,22,23] | IHC antibodies | FFPE slide | Semi-quantitative score 0–3+ | Technique widely used and available, fast and cheap | Observer-dependent, tissue sectioning artefacts, new FFPE slide for every analysis, no consensus on scoring system and cutoff |
| MET exon 14 skipping [25,26,27,28,29,30,31,32,33,34,35,36,37,38,39] | RNA NGS (amplicon-, AMP-, or hybridization-based) | RNA from FFPE or fresh frozen material | Mutation, coverage, MAF, fusion product of exon 13 and 15 | Sensitive, reliable, direct detection of alternative splicing, multiplexing | RNA degradation, underlying mutation cannot be determined |
| RT-PCR | RNA from FFPE or fresh frozen material | Fusion product of exon 13 and 15 | Sensitive, reliable, direct detection of alternative splicing, fast turnaround time, widely used and available | RNA degradation, underlying mutation cannot be determined, targeted mutations only | |
| MET exon 14 skipping mutations and point mutations [33,35,37,40,41,42,43,44] | DNA NGS (amplicon- or hybridization-based) | DNA from FFPE, fresh frozen material, or liquid biopsy | Mutation, coverage, VAF | Sensitive, reliable, detection of exact mutation, multiplexing | No assessment of splicing effect |
| Sanger sequencing | DNA from FFPE, fresh frozen material, or liquid biopsy | Mutation, VAF | Detection of exact mutation, fast turnaround time, widely used and available | Sensitivity, single assay for each target, no assessment of splicing effect | |
| MET amplifications [5,41,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61] | FISH | FFPE slides | MET GCN, MET/CEN7 ratio | Technique widely used and available, detection of focal amplification, polysomy, and chromosome duplications | Observer-dependent, tissue sectioning artefacts, new FFPE slide for every analysis, no consensus on scoring system and cutoff |
| DNA NGS (amplicon- or hybridization-based) | DNA from FFPE, fresh frozen material, or liquid biopsy | Mutation, coverage, VAF | Sensitive, DNA from FFPE easily accessible, multiplexing | High number of false negatives, no standardized cutoff or bioinformatics, no morphological correlation | |
| Other DNA-based technologies (ddPCR, NanoString nCounter technology) | DNA from FFPE, fresh frozen material, or liquid biopsy | Expression, GCN | DNA from FFPE easily accessible | High number of false negatives, no morphological correlation, no standardized cutoff, large amounts of DNA needed | |
| MET fusions [35,39,62,63,64,65,66,67,68,69,70,71] | RNA NGS (AMP- or hybridization-based) | RNA from FFPE or fresh frozen material | Fusionreads, 3′-5′ imbalance | Sensitive, reliable detection of known and novel fusion partners, multiplexing | RNA degradation |
| DNA NGS (Hybridization-based) | DNA from FFPE | Fusionreads, 3′-5′ imbalance, coverage, | DNA from FFPE easily accessible, detection of known and novel fusion partners if region is covered, multiplexing | False negative results, novel fusions are problematic due to location of fusion break point | |
| FISH | FFPE slides | n.a. break-apart events | Technique widely used and available | No standardized assay available, observer-dependent, tissue sectioning artefacts, new FFPE slide for every analysis | |
| RT-PCR | RNA from FFPE | Fusion product | Technique widely used and available | No standardized assay available, only for known fusion partners, RNA degradation |
3.2. MET Mutations
3.2.1. MET Exon 14 Skipping
Parallel Sequencing (NGS) Multigene Assays
Single Gene Analyzes
3.2.2. Other MET Mutations
3.3. MET Gene Amplification and Gene Copy Number Alterations
3.3.1. Fluorescence In Situ Hybridization (FISH)
3.3.2. DNA-Based Methods
3.4. MET Fusions
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Fujino, T.; Suda, K.; Mitsudomi, T. Lung Cancer with MET exon 14 Skipping Mutation: Genetic Feature, Current Treatments, and Future Challenges. Lung Cancer 2021, 12, 35–50. [Google Scholar] [CrossRef] [PubMed]
- Gelsomino, F.; Rossi, G.; Tiseo, M. MET and Small-Cell Lung Cancer. Cancers 2014, 6, 2100–2115. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Luo, J.; Chang, J.; Rekhtman, N.; Arcila, M.; Drilon, A. MET-dependent solid tumours-molecular diagnosis and targeted therapy. Nat. Rev. Clin. Oncol. 2020, 17, 569–587. [Google Scholar] [CrossRef]
- Lee, M.; Jain, P.; Wang, F.; Ma, P.C.; Borczuk, A.; Halmos, B. MET alterations and their impact on the future of non-small cell lung cancer (NSCLC) targeted therapies. Expert Opin. Targets 2021, 25, 249–268. [Google Scholar] [CrossRef] [PubMed]
- Jørgensen, J.T.; Mollerup, J. Companion Diagnostics and Predictive Biomarkers for MET-Targeted Therapy in NSCLC. Cancers 2022, 14, 2150. [Google Scholar] [CrossRef]
- Organ, S.L.; Tsao, M.S. An overview of the c-MET signaling pathway. Ther. Adv. Med. Oncol. 2011, 3, S7–S19. [Google Scholar] [CrossRef]
- Cecchi, F.; Rabe, D.C.; Bottaro, D.P. Targeting the HGF/Met signaling pathway in cancer therapy. Expert Opin. Targets 2012, 16, 553–572. [Google Scholar] [CrossRef]
- Kawaguchi, M.; Kataoka, H. Mechanisms of Hepatocyte Growth Factor Activation in Cancer Tissues. Cancers 2014, 6, 1890–1904. [Google Scholar] [CrossRef]
- Eder, J.P.; Vande Woude, G.F.; Boerner, S.A.; LoRusso, P.M. Novel therapeutic inhibitors of the c-Met signaling pathway in cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 2207–2214. [Google Scholar] [CrossRef]
- Catenacci, D.V.; Ang, A.; Liao, W.L.; Shen, J.; O’Day, E.; Loberg, R.D.; Cecchi, F.; Hembrough, T.; Ruzzo, A.; Graziano, F. MET tyrosine kinase receptor expression and amplification as prognostic biomarkers of survival in gastroesophageal adenocarcinoma. Cancer 2017, 123, 1061–1070. [Google Scholar] [CrossRef]
- Gayyed, M.F.; Abd El-Maqsoud, N.M.; El-Hameed El-Heeny, A.A.; Mohammed, M.F. c-MET expression in colorectal adenomas and primary carcinomas with its corresponding metastases. J. Gastrointest. Oncol. 2015, 6, 618–627. [Google Scholar] [CrossRef] [PubMed]
- Graveel, C.R.; Tolbert, D.; Vande Woude, G.F. MET: A critical player in tumorigenesis and therapeutic target. Cold Spring Harb. Perspect. Biol. 2013, 5, a009209. [Google Scholar] [CrossRef] [PubMed]
- Recondo, G.; Che, J.; Janne, P.A.; Awad, M.M. Targeting MET Dysregulation in Cancer. Cancer Discov. 2020, 10, 922–934. [Google Scholar] [CrossRef] [PubMed]
- Watermann, I.; Schmitt, B.; Stellmacher, F.; Muller, J.; Gaber, R.; Kugler, C.; Reinmuth, N.; Huber, R.M.; Thomas, M.; Zabel, P.; et al. Improved diagnostics targeting c-MET in non-small cell lung cancer: Expression, amplification and activation? Diagn. Pathol. 2015, 10, 130. [Google Scholar] [CrossRef]
- Guo, R.; Berry, L.D.; Aisner, D.L.; Sheren, J.; Boyle, T.; Bunn, P.A., Jr.; Johnson, B.E.; Kwiatkowski, D.J.; Drilon, A.; Sholl, L.M.; et al. MET IHC Is a Poor Screen for MET Amplification or MET Exon 14 Mutations in Lung Adenocarcinomas: Data from a Tri-Institutional Cohort of the Lung Cancer Mutation Consortium. J. Thorac. Oncol. 2019, 14, 1666–1671. [Google Scholar] [CrossRef] [PubMed]
- Salgia, R. MET in Lung Cancer: Biomarker Selection Based on Scientific Rationale. Mol. Cancer Ther. 2017, 16, 555–565. [Google Scholar] [CrossRef]
- Spigel, D.R.; Edelman, M.J.; O’Byrne, K.; Paz-Ares, L.; Mocci, S.; Phan, S.; Shames, D.S.; Smith, D.; Yu, W.; Paton, V.E.; et al. Results From the Phase III Randomized Trial of Onartuzumab Plus Erlotinib Versus Erlotinib in Previously Treated Stage IIIB or IV Non-Small-Cell Lung Cancer: METLung. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2017, 35, 412–420. [Google Scholar] [CrossRef]
- Ryan, C.J.; Rosenthal, M.; Ng, S.; Alumkal, J.; Picus, J.; Gravis, G.; Fizazi, K.; Forget, F.; Machiels, J.P.; Srinivas, S.; et al. Targeted MET inhibition in castration-resistant prostate cancer: A randomized phase II study and biomarker analysis with rilotumumab plus mitoxantrone and prednisone. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 215–224. [Google Scholar] [CrossRef]
- Iveson, T.; Donehower, R.C.; Davidenko, I.; Tjulandin, S.; Deptala, A.; Harrison, M.; Nirni, S.; Lakshmaiah, K.; Thomas, A.; Jiang, Y.; et al. Rilotumumab in combination with epirubicin, cisplatin, and capecitabine as first-line treatment for gastric or oesophagogastric junction adenocarcinoma: An open-label, dose de-escalation phase 1b study and a double-blind, randomised phase 2 study. Lancet. Oncol. 2014, 15, 1007–1018. [Google Scholar] [CrossRef]
- Srivastava, A.K.; Hollingshead, M.G.; Weiner, J.; Navas, T.; Evrard, Y.A.; Khin, S.A.; Ji, J.J.; Zhang, Y.; Borgel, S.; Pfister, T.D.; et al. Pharmacodynamic Response of the MET/HGF Receptor to Small-Molecule Tyrosine Kinase Inhibitors Examined with Validated, Fit-for-Clinic Immunoassays. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 3683–3694. [Google Scholar] [CrossRef]
- Neal, J.W.; Dahlberg, S.E.; Wakelee, H.A.; Aisner, S.C.; Bowden, M.; Huang, Y.; Carbone, D.P.; Gerstner, G.J.; Lerner, R.E.; Rubin, J.L.; et al. Erlotinib, cabozantinib, or erlotinib plus cabozantinib as second-line or third-line treatment of patients with EGFR wild-type advanced non-small-cell lung cancer (ECOG-ACRIN 1512): A randomised, controlled, open-label, multicentre, phase 2 trial. Lancet. Oncol. 2016, 17, 1661–1671. [Google Scholar] [CrossRef] [PubMed]
- Schuler, M.H.; Berardi, R.; Lim, W.-T.; Geel, R.V.; Jonge, M.J.D.; Bauer, T.M.; Azaro, A.; Gottfried, M.; Han, J.-Y.; Lee, D.H.; et al. Phase (Ph) I study of the safety and efficacy of the cMET inhibitor capmatinib (INC280) in patients (pts) with advanced cMET+ non-small cell lung cancer (NSCLC). J. Clin. Oncol. 2016, 34, 9067. [Google Scholar] [CrossRef]
- Finocchiaro, G.; Toschi, L.; Gianoncelli, L.; Baretti, M.; Santoro, A. Prognostic and predictive value of MET deregulation in non-small cell lung cancer. Ann. Transl. Med. 2015, 3, 83. [Google Scholar] [CrossRef] [PubMed]
- Tong, J.H.; Yeung, S.F.; Chan, A.W.; Chung, L.Y.; Chau, S.L.; Lung, R.W.; Tong, C.Y.; Chow, C.; Tin, E.K.; Yu, Y.H.; et al. MET Amplification and Exon 14 Splice Site Mutation Define Unique Molecular Subgroups of Non-Small Cell Lung Carcinoma with Poor Prognosis. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 3048–3056. [Google Scholar] [CrossRef]
- Frampton, G.M.; Ali, S.M.; Rosenzweig, M.; Chmielecki, J.; Lu, X.; Bauer, T.M.; Akimov, M.; Bufill, J.A.; Lee, C.; Jentz, D.; et al. Activation of MET via diverse exon 14 splicing alterations occurs in multiple tumor types and confers clinical sensitivity to MET inhibitors. Cancer Discov. 2015, 5, 850–859. [Google Scholar] [CrossRef]
- Kim, S.Y.; Yin, J.; Bohlman, S.; Walker, P.; Dacic, S.; Kim, C.; Khan, H.; Liu, S.V.; Ma, P.C.; Nagasaka, M.; et al. Characterization of MET Exon 14 Skipping Alterations (in NSCLC) and Identification of Potential Therapeutic Targets Using Whole Transcriptome Sequencing. JTO Clin. Res. Rep. 2022, 3, 100381. [Google Scholar] [CrossRef]
- Paik, P.K.; Drilon, A.; Fan, P.D.; Yu, H.; Rekhtman, N.; Ginsberg, M.S.; Borsu, L.; Schultz, N.; Berger, M.F.; Rudin, C.M.; et al. Response to MET inhibitors in patients with stage IV lung adenocarcinomas harboring MET mutations causing exon 14 skipping. Cancer Discov. 2015, 5, 842–849. [Google Scholar] [CrossRef]
- Onozato, R.; Kosaka, T.; Kuwano, H.; Sekido, Y.; Yatabe, Y.; Mitsudomi, T. Activation of MET by gene amplification or by splice mutations deleting the juxtamembrane domain in primary resected lung cancers. J. Thorac. Oncol. 2009, 4, 5–11. [Google Scholar] [CrossRef]
- Cheng, D.T.; Mitchell, T.N.; Zehir, A.; Shah, R.H.; Benayed, R.; Syed, A.; Chandramohan, R.; Liu, Z.Y.; Won, H.H.; Scott, S.N.; et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A Hybridization Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology. J. Mol. Diagn. JMD 2015, 17, 251–264. [Google Scholar] [CrossRef]
- Das, R.; Jakubowski, M.A.; Spildener, J.; Cheng, Y.W. Identification of Novel MET Exon 14 Skipping Variants in Non-Small Cell Lung Cancer Patients: A Prototype Workflow Involving in Silico Prediction and RT-PCR. Cancers 2022, 14, 4814. [Google Scholar] [CrossRef]
- Davies, K.D.; Lomboy, A.; Lawrence, C.A.; Yourshaw, M.; Bocsi, G.T.; Camidge, D.R.; Aisner, D.L. DNA-Based versus RNA-Based Detection of MET Exon 14 Skipping Events in Lung Cancer. J. Thorac. Oncol. 2019, 14, 737–741. [Google Scholar] [CrossRef] [PubMed]
- Depoilly, T.; Garinet, S.; van Kempen, L.C.; Schuuring, E.; Clave, S.; Bellosillo, B.; Ercolani, C.; Buglioni, S.; Siemanowski, J.; Merkelbach-Bruse, S.; et al. Multicenter Evaluation of the Idylla GeneFusion in Non-Small-Cell Lung Cancer. J. Mol. Diagn. JMD 2022, 24, 1021–1030. [Google Scholar] [CrossRef] [PubMed]
- Drilon, A.; Wang, L.; Arcila, M.E.; Balasubramanian, S.; Greenbowe, J.R.; Ross, J.S.; Stephens, P.; Lipson, D.; Miller, V.A.; Kris, M.G.; et al. Broad, Hybrid Capture-Based Next-Generation Sequencing Identifies Actionable Genomic Alterations in Lung Adenocarcinomas Otherwise Negative for Such Alterations by Other Genomic Testing Approaches. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 3631–3639. [Google Scholar] [CrossRef] [PubMed]
- Heydt, C.; Fassunke, J.; Kunstlinger, H.; Ihle, M.A.; Konig, K.; Heukamp, L.C.; Schildhaus, H.U.; Odenthal, M.; Buttner, R.; Merkelbach-Bruse, S. Comparison of pre-analytical FFPE sample preparation methods and their impact on massively parallel sequencing in routine diagnostics. PLoS ONE 2014, 9, e104566. [Google Scholar] [CrossRef]
- Heydt, C.; Wölwer, C.B.; Velazquez Camacho, O.; Wagener-Ryczek, S.; Pappesch, R.; Siemanowski, J.; Rehker, J.; Haller, F.; Agaimy, A.; Worm, K.; et al. Detection of gene fusions using targeted next-generation sequencing: A comparative evaluation. BMC Med. Genom. 2021, 14, 62. [Google Scholar] [CrossRef]
- Poirot, B.; Doucet, L.; Benhenda, S.; Champ, J.; Meignin, V.; Lehmann-Che, J. MET Exon 14 Alterations and New Resistance Mutations to Tyrosine Kinase Inhibitors: Risk of Inadequate Detection with Current Amplicon-Based NGS Panels. J. Thorac. Oncol. 2017, 12, 1582–1587. [Google Scholar] [CrossRef]
- Suh, J.H.; Johnson, A.; Albacker, L.; Wang, K.; Chmielecki, J.; Frampton, G.; Gay, L.; Elvin, J.A.; Vergilio, J.A.; Ali, S.; et al. Comprehensive Genomic Profiling Facilitates Implementation of the National Comprehensive Cancer Network Guidelines for Lung Cancer Biomarker Testing and Identifies Patients Who May Benefit From Enrollment in Mechanism-Driven Clinical Trials. Oncologist 2016, 21, 684–691. [Google Scholar] [CrossRef]
- Sui, J.S.Y.; Finn, S.P.; Gray, S.G. Detection of MET Exon 14 Skipping Alterations in Lung Cancer Clinical Samples Using a PCR-Based Approach. Methods Mol. Biol. 2021, 2279, 145–155. [Google Scholar] [CrossRef]
- Zheng, Z.; Liebers, M.; Zhelyazkova, B.; Cao, Y.; Panditi, D.; Lynch, K.D.; Chen, J.; Robinson, H.E.; Shim, H.S.; Chmielecki, J.; et al. Anchored multiplex PCR for targeted next-generation sequencing. Nat. Med. 2014, 20, 1479–1484. [Google Scholar] [CrossRef]
- Albiges, L.; Guegan, J.; Le Formal, A.; Verkarre, V.; Rioux-Leclercq, N.; Sibony, M.; Bernhard, J.C.; Camparo, P.; Merabet, Z.; Molinie, V.; et al. MET is a potential target across all papillary renal cell carcinomas: Result from a large molecular study of pRCC with CGH array and matching gene expression array. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 3411–3421. [Google Scholar] [CrossRef]
- Chen, Y.-C.; Seifuddin, F.; Nguyen, C.; Yang, Z.; Chen, W.; Yan, C.; Chen, Q.; Wang, C.; Xiao, W.; Pirooznia, M.; et al. Comprehensive Assessment of Somatic Copy Number Variation Calling Using Next-Generation Sequencing Data. bioRxiv 2021. [Google Scholar] [CrossRef]
- Pal, S.K.; Ali, S.M.; Yakirevich, E.; Geynisman, D.M.; Karam, J.A.; Elvin, J.A.; Frampton, G.M.; Huang, X.; Lin, D.I.; Rosenzweig, M.; et al. Characterization of Clinical Cases of Advanced Papillary Renal Cell Carcinoma via Comprehensive Genomic Profiling. Eur. Urol. 2018, 73, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Pfarr, N.; Stenzinger, A.; Penzel, R.; Warth, A.; Dienemann, H.; Schirmacher, P.; Weichert, W.; Endris, V. High-throughput diagnostic profiling of clinically actionable gene fusions in lung cancer. Genes Chromosomes Cancer 2016, 55, 30–44. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, L.; Duh, F.M.; Chen, F.; Kishida, T.; Glenn, G.; Choyke, P.; Scherer, S.W.; Zhuang, Z.; Lubensky, I.; Dean, M.; et al. Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas. Nat. Genet. 1997, 16, 68–73. [Google Scholar] [CrossRef]
- Budczies, J.; Pfarr, N.; Stenzinger, A.; Treue, D.; Endris, V.; Ismaeel, F.; Bangemann, N.; Blohmer, J.U.; Dietel, M.; Loibl, S.; et al. Ioncopy: A novel method for calling copy number alterations in amplicon sequencing data including significance assessment. Oncotarget 2016, 7, 13236–13247. [Google Scholar] [CrossRef]
- Camidge, D.R.; Otterson, G.A.; Clark, J.W.; Ignatius Ou, S.H.; Weiss, J.; Ades, S.; Shapiro, G.I.; Socinski, M.A.; Murphy, D.A.; Conte, U.; et al. Crizotinib in Patients With MET-Amplified NSCLC. J. Thorac. Oncol. 2021, 16, 1017–1029. [Google Scholar] [CrossRef]
- Caparica, R.; Yen, C.T.; Coudry, R.; Ou, S.I.; Varella-Garcia, M.; Camidge, D.R.; de Castro, G., Jr. Responses to Crizotinib Can Occur in High-Level MET-Amplified Non-Small Cell Lung Cancer Independent of MET Exon 14 Alterations. J. Thorac. Oncol. 2017, 12, 141–144. [Google Scholar] [CrossRef]
- Cappuzzo, F.; Marchetti, A.; Skokan, M.; Rossi, E.; Gajapathy, S.; Felicioni, L.; Del Grammastro, M.; Sciarrotta, M.G.; Buttitta, F.; Incarbone, M.; et al. Increased MET gene copy number negatively affects survival of surgically resected non-small-cell lung cancer patients. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2009, 27, 1667–1674. [Google Scholar] [CrossRef]
- Dagogo-Jack, I.; Moonsamy, P.; Gainor, J.F.; Lennerz, J.K.; Piotrowska, Z.; Lin, J.J.; Lennes, I.T.; Sequist, L.V.; Shaw, A.T.; Goodwin, K.; et al. A Phase 2 Study of Capmatinib in Patients With MET-Altered Lung Cancer Previously Treated With a MET Inhibitor. J. Thorac. Oncol. 2021, 16, 850–859. [Google Scholar] [CrossRef]
- Go, H.; Jeon, Y.K.; Park, H.J.; Sung, S.W.; Seo, J.W.; Chung, D.H. High MET gene copy number leads to shorter survival in patients with non-small cell lung cancer. J. Thorac. Oncol. 2010, 5, 305–313. [Google Scholar] [CrossRef]
- Gusnanto, A.; Wood, H.M.; Pawitan, Y.; Rabbitts, P.; Berri, S. Correcting for cancer genome size and tumour cell content enables better estimation of copy number alterations from next-generation sequence data. Bioinformatics 2011, 28, 40–47. [Google Scholar] [CrossRef]
- Heydt, C.; Becher, A.K.; Wagener-Ryczek, S.; Ball, M.; Schultheis, A.M.; Schallenberg, S.; Rüsseler, V.; Büttner, R.; Merkelbach-Bruse, S. Comparison of in situ and extraction-based methods for the detection of MET amplifications in solid tumors. Comput. Struct. Biotechnol. J. 2019, 17, 1339–1347. [Google Scholar] [CrossRef] [PubMed]
- Jardim, D.L.; Tang, C.; Gagliato Dde, M.; Falchook, G.S.; Hess, K.; Janku, F.; Fu, S.; Wheler, J.J.; Zinner, R.G.; Naing, A.; et al. Analysis of 1,115 patients tested for MET amplification and therapy response in the MD Anderson Phase I Clinic. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 6336–6345. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.E.; Kim, M.A.; Lee, H.S.; Jung, E.J.; Yang, H.K.; Lee, B.L.; Bang, Y.J.; Kim, W.H. MET in gastric carcinomas: Comparison between protein expression and gene copy number and impact on clinical outcome. Br. J. Cancer 2012, 107, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Overbeck, T.R.; Cron, D.A.; Schmitz, K.; Rittmeyer, A.; Korber, W.; Hugo, S.; Schnalke, J.; Lukat, L.; Hugo, T.; Hinterthaner, M.; et al. Top-level MET gene copy number gain defines a subtype of poorly differentiated pulmonary adenocarcinomas with poor prognosis. Transl. Lung Cancer Res. 2020, 9, 603–616. [Google Scholar] [CrossRef]
- Paik, P.K.; Felip, E.; Veillon, R.; Sakai, H.; Cortot, A.B.; Garassino, M.C.; Mazieres, J.; Viteri, S.; Senellart, H.; Van Meerbeeck, J.; et al. Tepotinib in Non-Small-Cell Lung Cancer with MET Exon 14 Skipping Mutations. N. Engl. J. Med. 2020, 383, 931–943. [Google Scholar] [CrossRef]
- Peng, L.X.; Jie, G.L.; Li, A.N.; Liu, S.Y.; Sun, H.; Zheng, M.M.; Zhou, J.Y.; Zhang, J.T.; Zhang, X.C.; Zhou, Q.; et al. MET amplification identified by next-generation sequencing and its clinical relevance for MET inhibitors. Exp. Hematol. Oncol. 2021, 10, 52. [Google Scholar] [CrossRef]
- Schildhaus, H.U.; Schultheis, A.M.; Ruschoff, J.; Binot, E.; Merkelbach-Bruse, S.; Fassunke, J.; Schulte, W.; Ko, Y.D.; Schlesinger, A.; Bos, M.; et al. MET amplification status in therapy-naive adeno- and squamous cell carcinomas of the lung. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 907–915. [Google Scholar] [CrossRef]
- Schmitt, C.; Schulz, A.A.; Winkelmann, R.; Smith, K.; Wild, P.J.; Demes, M. Comparison of MET gene amplification analysis by next-generation sequencing and fluorescence in situ hybridization. Oncotarget 2021, 12, 2273–2282. [Google Scholar] [CrossRef]
- Schubart, C.; Stohr, R.; Togel, L.; Fuchs, F.; Sirbu, H.; Seitz, G.; Seggewiss-Bernhardt, R.; Leistner, R.; Sterlacci, W.; Vieth, M.; et al. MET Amplification in Non-Small Cell Lung Cancer (NSCLC)-A Consecutive Evaluation Using Next-Generation Sequencing (NGS) in a Real-World Setting. Cancers 2021, 13, 5023. [Google Scholar] [CrossRef]
- Wolf, J.; Seto, T.; Han, J.Y.; Reguart, N.; Garon, E.B.; Groen, H.J.M.; Tan, D.S.W.; Hida, T.; de Jonge, M.; Orlov, S.V.; et al. Capmatinib in MET Exon 14-Mutated or MET-Amplified Non-Small-Cell Lung Cancer. New Engl. J. Med. 2020, 383, 944–957. [Google Scholar] [CrossRef] [PubMed]
- Benayed, R.; Offin, M.; Mullaney, K.; Sukhadia, P.; Rios, K.; Desmeules, P.; Ptashkin, R.; Won, H.; Chang, J.; Halpenny, D.; et al. High Yield of RNA Sequencing for Targetable Kinase Fusions in Lung Adenocarcinomas with No Mitogenic Driver Alteration Detected by DNA Sequencing and Low Tumor Mutation Burden. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 4712–4722. [Google Scholar] [CrossRef] [PubMed]
- Duplaquet, L.; Kherrouche, Z.; Baldacci, S.; Jamme, P.; Cortot, A.B.; Copin, M.C.; Tulasne, D. The multiple paths towards MET receptor addiction in cancer. Oncogene 2018, 37, 3200–3215. [Google Scholar] [CrossRef]
- Gow, C.H.; Liu, Y.N.; Li, H.Y.; Hsieh, M.S.; Chang, S.H.; Luo, S.C.; Tsai, T.H.; Chen, P.L.; Tsai, M.F.; Shih, J.Y. Oncogenic Function of a KIF5B-MET Fusion Variant in Non-Small Cell Lung Cancer. Neoplasia 2018, 20, 838–847. [Google Scholar] [CrossRef] [PubMed]
- International Cancer Genome Consortium PedBrain Tumor Project. Recurrent MET fusion genes represent a drug target in pediatric glioblastoma. Nat. Med. 2016, 22, 1314–1320. [Google Scholar] [CrossRef] [PubMed]
- Kim, P.; Jia, P.; Zhao, Z. Kinase impact assessment in the landscape of fusion genes that retain kinase domains: A pan-cancer study. Brief. Bioinform. 2018, 19, 450–460. [Google Scholar] [CrossRef]
- Pan, Y.; Zhang, Y.; Ye, T.; Zhao, Y.; Gao, Z.; Yuan, H.; Zheng, D.; Zheng, S.; Li, H.; Li, Y.; et al. Detection of Novel NRG1, EGFR, and MET Fusions in Lung Adenocarcinomas in the Chinese Population. J. Thorac. Oncol. 2019, 14, 2003–2008. [Google Scholar] [CrossRef]
- Plenker, D.; Bertrand, M.; de Langen, A.J.; Riedel, R.; Lorenz, C.; Scheel, A.H.; Muller, J.; Bragelmann, J.; Dassler-Plenker, J.; Kobe, C.; et al. Structural Alterations of MET Trigger Response to MET Kinase Inhibition in Lung Adenocarcinoma Patients. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 1337–1343. [Google Scholar] [CrossRef]
- Siemanowski, J.; Heydt, C.; Merkelbach-Bruse, S. Predictive molecular pathology of lung cancer in Germany with focus on gene fusion testing: Methods and quality assurance. Cancer Cytopathol. 2020, 128, 611–621. [Google Scholar] [CrossRef]
- Vigna, E.; Gramaglia, D.; Longati, P.; Bardelli, A.; Comoglio, P.M. Loss of the exon encoding the juxtamembrane domain is essential for the oncogenic activation of TPR-MET. Oncogene 1999, 18, 4275–4281. [Google Scholar] [CrossRef]
- Zhu, Y.C.; Wang, W.X.; Xu, C.W.; Zhang, Q.X.; Du, K.Q.; Chen, G.; Lv, T.F.; Song, Y. Identification of a novel crizotinib-sensitive MET-ATXN7L1 gene fusion variant in lung adenocarcinoma by next generation sequencing. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2018, 29, 2392–2393. [Google Scholar] [CrossRef] [PubMed]
- Peschard, P.; Fournier, T.M.; Lamorte, L.; Naujokas, M.A.; Band, H.; Langdon, W.Y.; Park, M. Mutation of the c-Cbl TKB domain binding site on the Met receptor tyrosine kinase converts it into a transforming protein. Mol. Cell. 2001, 8, 995–1004. [Google Scholar] [CrossRef] [PubMed]
- Awad, M.M.; Oxnard, G.R.; Jackman, D.M.; Savukoski, D.O.; Hall, D.; Shivdasani, P.; Heng, J.C.; Dahlberg, S.E.; Janne, P.A.; Verma, S.; et al. MET Exon 14 Mutations in Non-Small-Cell Lung Cancer Are Associated With Advanced Age and Stage-Dependent MET Genomic Amplification and c-Met Overexpression. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2016, 34, 721–730. [Google Scholar] [CrossRef] [PubMed]
- Krishnaswamy, S.; Kanteti, R.; Duke-Cohan, J.S.; Loganathan, S.; Liu, W.; Ma, P.C.; Sattler, M.; Singleton, P.A.; Ramnath, N.; Innocenti, F.; et al. Ethnic differences and functional analysis of MET mutations in lung cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 5714–5723. [Google Scholar] [CrossRef]
- Tsai, J.M.; Hata, A.N.; Lennerz, J.K. MET D1228N and D1246N are the Same Resistance Mutation in MET Exon 14 Skipping. Oncol. 2021, 26, e2297–e2301. [Google Scholar] [CrossRef]
- Li, M.M.; Datto, M.; Duncavage, E.J.; Kulkarni, S.; Lindeman, N.I.; Roy, S.; Tsimberidou, A.M.; Vnencak-Jones, C.L.; Wolff, D.J.; Younes, A.; et al. Standards and Guidelines for the Interpretation and Reporting of Sequence Variants in Cancer: A Joint Consensus Recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J. Mol. Diagn. 2017, 19, 4–23. [Google Scholar] [CrossRef]
- Davies, K.D.; Ritterhouse, L.L.; Snow, A.N.; Sidiropoulos, N. MET Exon 14 Skipping Mutations: Essential Considerations for Current Management of Non-Small-Cell Lung Cancer. J. Mol. Diagn. JMD 2022, 24, 841–843. [Google Scholar] [CrossRef]
- Feng, Y.; Thiagarajan, P.S.; Ma, P.C. MET signaling: Novel targeted inhibition and its clinical development in lung cancer. J. Thorac. Oncol. 2012, 7, 459–467. [Google Scholar] [CrossRef]
- Ma, P.C.; Tretiakova, M.S.; MacKinnon, A.C.; Ramnath, N.; Johnson, C.; Dietrich, S.; Seiwert, T.; Christensen, J.G.; Jagadeeswaran, R.; Krausz, T.; et al. Expression and mutational analysis of MET in human solid cancers. Genes Chromosomes Cancer 2008, 47, 1025–1037. [Google Scholar] [CrossRef]
- Jeffers, M.; Schmidt, L.; Nakaigawa, N.; Webb, C.P.; Weirich, G.; Kishida, T.; Zbar, B.; Vande Woude, G.F. Activating mutations for the met tyrosine kinase receptor in human cancer. Proc. Natl. Acad. Sci. USA. 1997, 94, 11445–11450. [Google Scholar] [CrossRef]
- Heist, R.S.; Sequist, L.V.; Borger, D.; Gainor, J.F.; Arellano, R.S.; Le, L.P.; Dias-Santagata, D.; Clark, J.W.; Engelman, J.A.; Shaw, A.T.; et al. Acquired Resistance to Crizotinib in NSCLC with MET Exon 14 Skipping. J. Thorac. Oncol. 2016, 11, 1242–1245. [Google Scholar] [CrossRef] [PubMed]
- Park, M.; Dean, M.; Cooper, C.S.; Schmidt, M.; O’Brien, S.J.; Blair, D.G.; Vande Woude, G.F. Mechanism of met oncogene activation. Cell 1986, 45, 895–904. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heydt, C.; Ihle, M.A.; Merkelbach-Bruse, S. Overview of Molecular Detection Technologies for MET in Lung Cancer. Cancers 2023, 15, 2932. https://doi.org/10.3390/cancers15112932
Heydt C, Ihle MA, Merkelbach-Bruse S. Overview of Molecular Detection Technologies for MET in Lung Cancer. Cancers. 2023; 15(11):2932. https://doi.org/10.3390/cancers15112932
Chicago/Turabian StyleHeydt, Carina, Michaela Angelika Ihle, and Sabine Merkelbach-Bruse. 2023. "Overview of Molecular Detection Technologies for MET in Lung Cancer" Cancers 15, no. 11: 2932. https://doi.org/10.3390/cancers15112932