
A Comprehensive Transcriptional Signature in Pancreatic Ductal Adenocarcinoma Reveals New Insights into the Immune and Desmoplastic Microenvironments
 , , , , , and
, , , , , and
Abstract
:Simple Summary
Abstract
1. Introduction
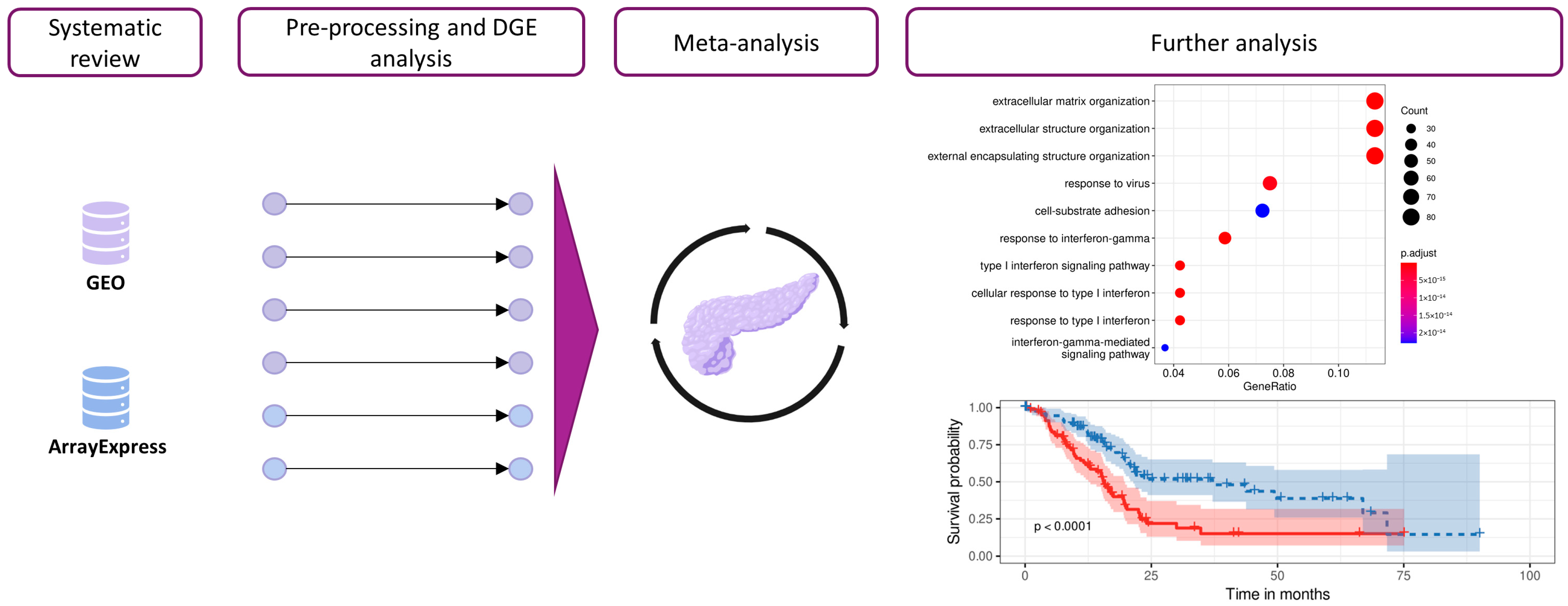
2. Materials and Methods
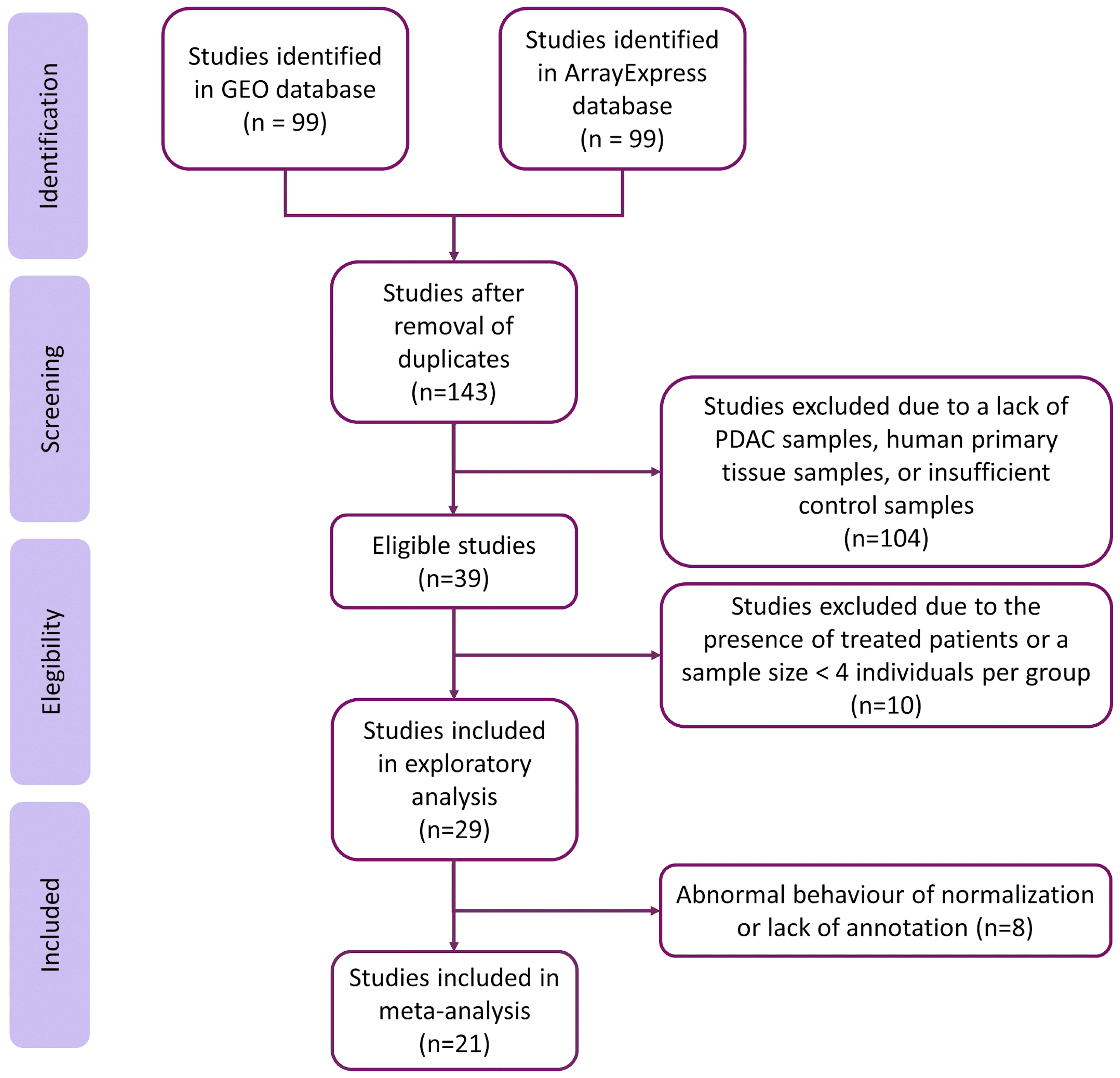
2.1. Study Search and Selection
- Normal and PDAC samples available.
- RNA extracted directly from human pancreas biopsies.
- Patients had not undergone treatment before biopsy.
- Sample size > 4 for PDAC and control groups.
2.2. Individual Preprocessing and Analysis
2.3. Gene Expression Meta-Analysis
2.4. Web Tool
2.5. Survival Analysis
3. Results
3.1. Systematic Review
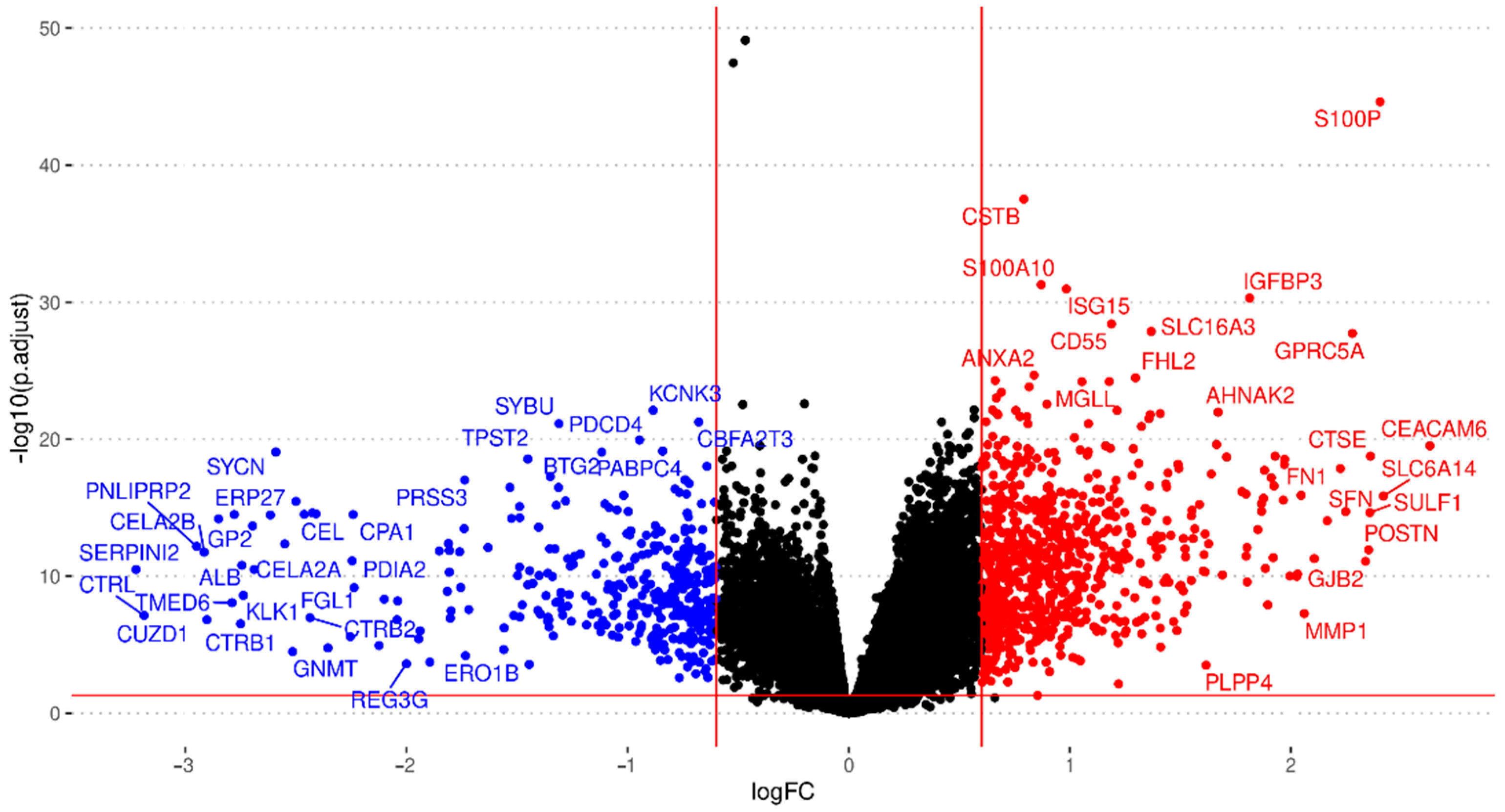
3.2. Integration of Differential Expression Profiles
3.3. Interactive Tool for Results Visualization
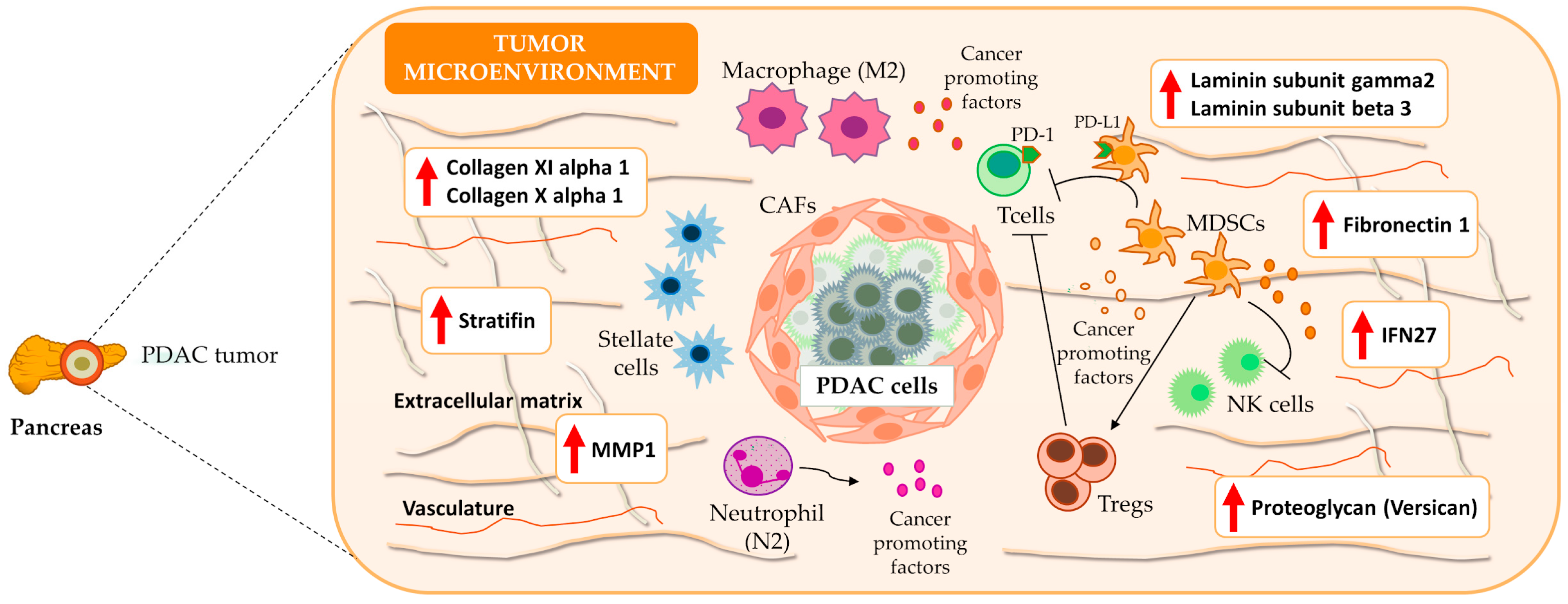
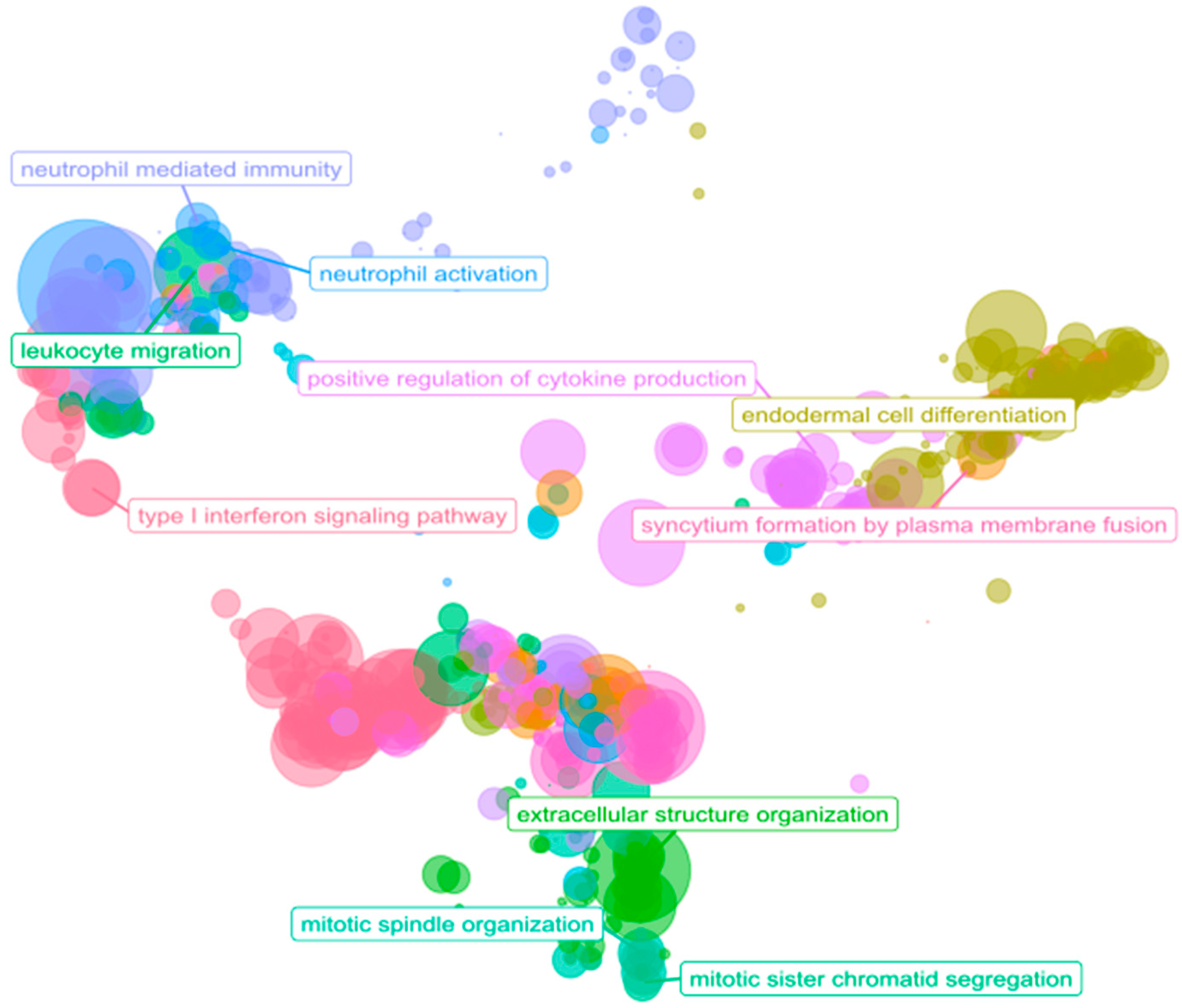
3.4. Immune System: A Functional Overview in PDAC
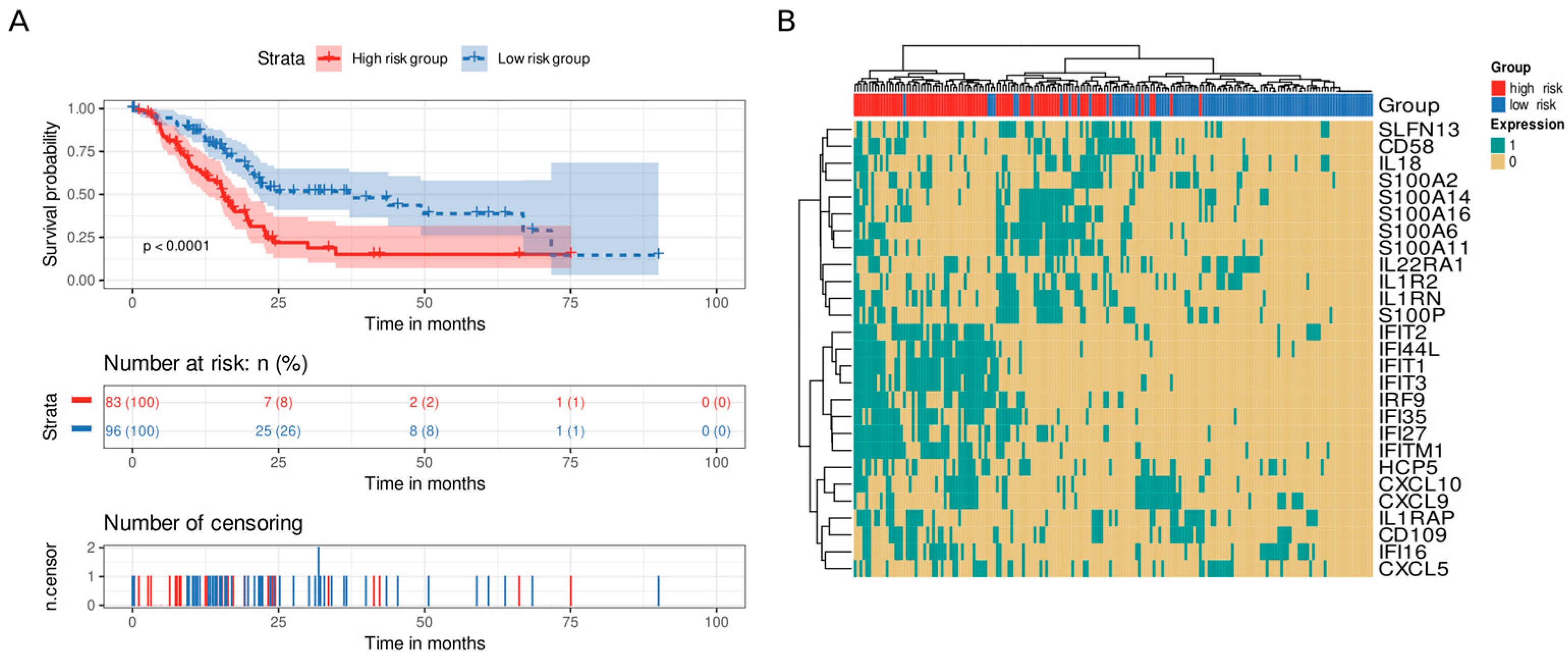
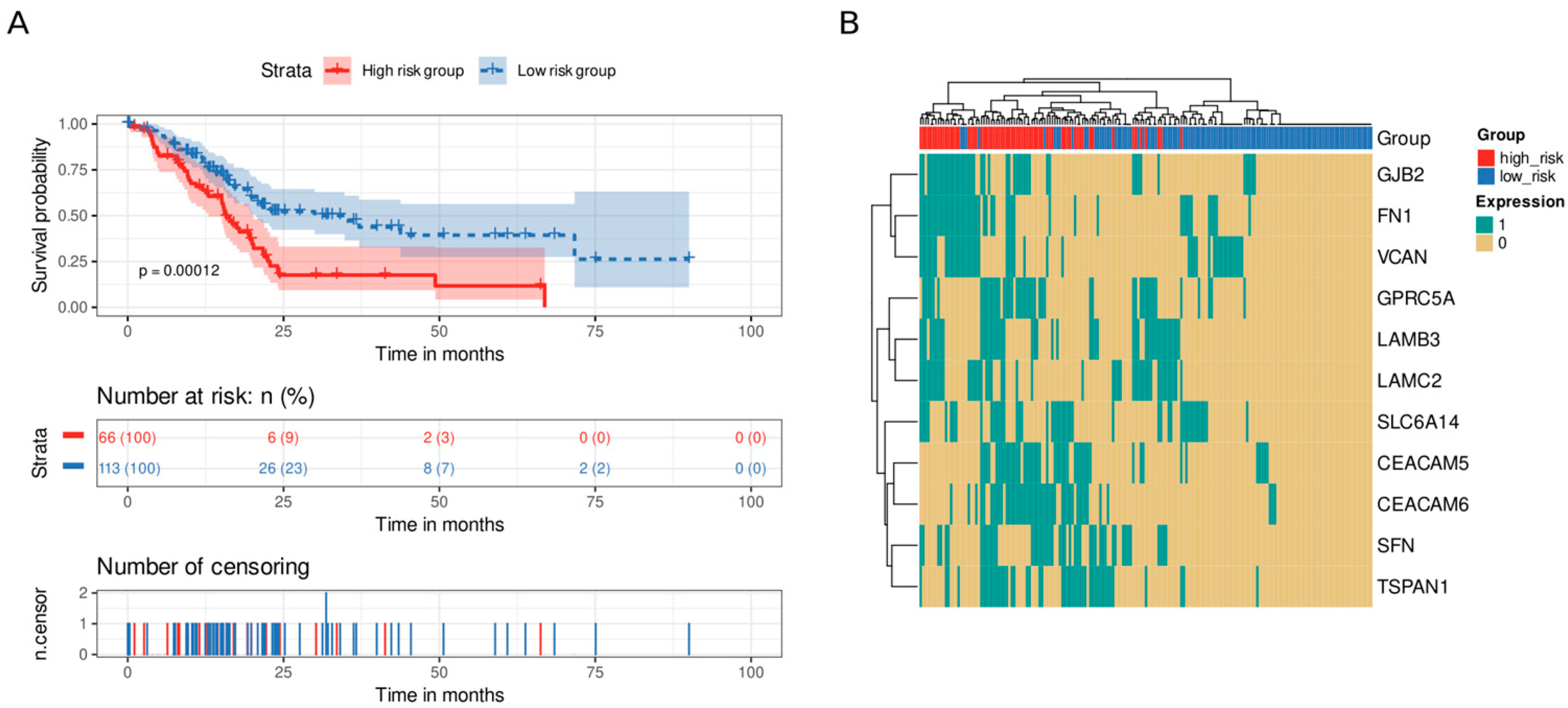
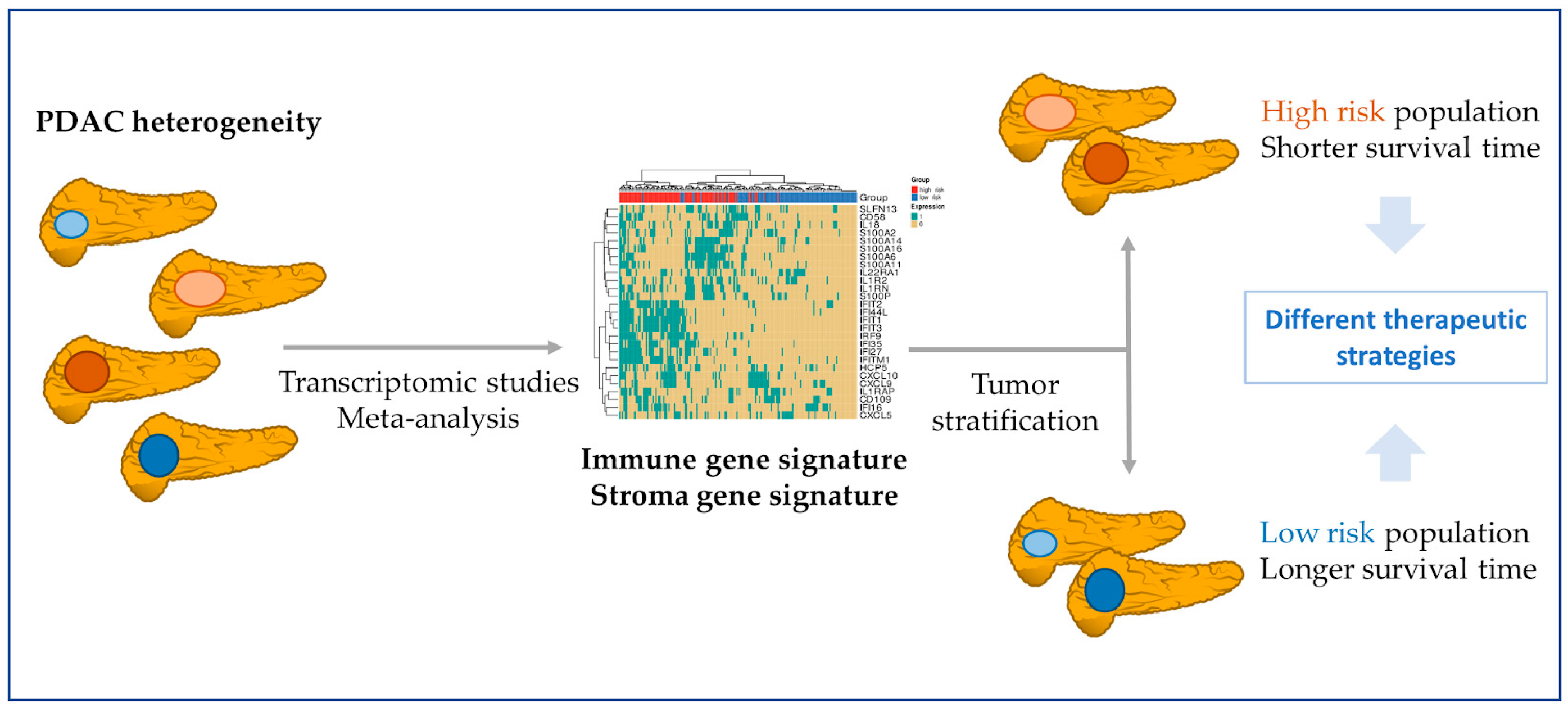
3.5. Immune and Stromal Survival Signatures Impact PDAC Prognosis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef]
- Park, W.; Chawla, A.; O’Reilly, E.M. Pancreatic Cancer: A Review. JAMA 2021, 326, 851–862. [Google Scholar] [CrossRef] [PubMed]
- Henriksen, A.; Dyhl-Polk, A.; Chen, I.; Nielsen, D. Checkpoint Inhibitors in Pancreatic Cancer. Cancer Treat. Rev. 2019, 78, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Nezhad Shamohammadi, F.; Yazdanifar, M.; Oraei, M.; Kazemi, M.H.; Roohi, A.; Mahya Shariat Razavi, S.; Rezaei, F.; Parvizpour, F.; Karamlou, Y.; Namdari, H. Controversial Role of Γδ T Cells in Pancreatic Cancer. Int. Immunopharmacol. 2022, 108, 108895. [Google Scholar] [CrossRef] [PubMed]
- Ullman, N.A.; Burchard, P.R.; Dunne, R.F.; Linehan, D.C. Immunologic Strategies in Pancreatic Cancer: Making Cold Tumors Hot. J. Clin. Oncol. 2022, 40, 2789–2805. [Google Scholar] [CrossRef]
- Dong, C.; Dang, D.; Zhao, X.; Wang, Y.; Wang, Z.; Zhang, C. Integrative Characterization of the Role of IL27 In Melanoma Using Bioinformatics Analysis. Front. Immunol. 2021, 12, 713001. [Google Scholar] [CrossRef]
- Ostios-Garcia, L.; Villamayor, J.; Garcia-Lorenzo, E.; Vinal, D.; Feliu, J. Understanding the Immune Response and the Current Landscape of Immunotherapy in Pancreatic Cancer. World J. Gastroenterol. 2021, 27, 6775–6793. [Google Scholar] [CrossRef]
- Di Federico, A.; Mosca, M.; Pagani, R.; Carloni, R.; Frega, G.; De Giglio, A.; Rizzo, A.; Ricci, D.; Tavolari, S.; Di Marco, M.; et al. Immunotherapy in Pancreatic Cancer: Why Do We Keep Failing? A Focus on Tumor Immune Microenvironment, Predictive Biomarkers and Treatment Outcomes. Cancers 2022, 14, 2429. [Google Scholar] [CrossRef]
- Moffitt, R.; Marayati, R.; Flate, E.; Volmar, K.E.; Herrera Loeza, S.G.; Hoadley, K.A.; Rashid, N.U.; Williams, L.A.; Eaton, S.C.; Chung, A.H.; et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat Genet. 2015, 47, 1168–1178. [Google Scholar] [CrossRef]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.; Gingras, M.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.C.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2021. [Google Scholar]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI Gene Expression and Hybridization Array Data Repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Athar, A.; Füllgrabe, A.; George, N.; Iqbal, H.; Huerta, L.; Ali, A.; Snow, C.; Fonseca, N.A.; Petryszak, R.; Papatheodorou, I.; et al. ArrayExpress Update—From Bulk to Single-Cell Expression Data. Nucleic Acids Res. 2019, 47, D711–D715. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network; Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.M.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer Analysis Project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef] [PubMed]
- Moher, D.; Liberati, A.; Tetzlaff, J.; Altman, D.G. PRISMA Group Preferred Reporting Items for Systematic Reviews and Meta-Analyses: The PRISMA Statement. PLoS Med. 2009, 6, e1000097. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor Package for Differential Expression Analysis of Digital Gene Expression Data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Robinson, M.D.; Oshlack, A. A Scaling Normalization Method for Differential Expression Analysis of RNA-Seq Data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma Powers Differential Expression Analyses for RNA-Sequencing and Microarray Studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- DerSimonian, R.; Laird, N. Meta-Analysis in Clinical Trials. Control. Clin. Trials 1986, 7, 177–188. [Google Scholar] [CrossRef]
- Sterne, J.A.; Egger, M. Funnel Plots for Detecting Bias in Meta-Analysis: Guidelines on Choice of Axis. J. Clin. Epidemiol. 2001, 54, 1046–1055. [Google Scholar] [CrossRef]
- Viechtbauer, W. Conducting Meta-Analyses in R with the Metafor Package. J. Stat. Softw. 2010, 36, 1–48. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. ClusterProfiler: An R Package for Comparing Biological Themes among Gene Clusters. Omics J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. ClusterProfiler 4.0: A Universal Enrichment Tool for Interpreting Omics Data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; He, Q.-Y. ReactomePA: An R/Bioconductor Package for Reactome Pathway Analysis and Visualization. Mol. Biosyst. 2016, 12, 477–479. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the Unification of Biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Gene Ontology Consortium. The Gene Ontology Resource: Enriching a GOld Mine. Nucleic Acids Res. 2021, 49, D325–D334. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, M.; Jassal, B.; Stephan, R.; Milacic, M.; Rothfels, K.; Senff-Ribeiro, A.; Griss, J.; Sevilla, C.; Matthews, L.; Gong, C.; et al. The Reactome Pathway Knowledgebase 2022. Nucleic Acids Res. 2022, 50, D687–D692. [Google Scholar] [CrossRef]
- Sayols, S. rrvgo: A Bioconductor Package to Reduce and Visualize Gene Ontology Terms. microPubl. Biol. 2023. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The CBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Hessmann, E.; Buchholz, S.M.; Demir, I.E.; Singh, S.K.; Gress, T.M.; Ellenrieder, V.; Neesse, A. Microenvironmental Determinants of Pancreatic Cancer. Physiol. Rev. 2020, 100, 1707–1751. [Google Scholar] [CrossRef]
- Whatcott, C.J.; Diep, C.H.; Jiang, P.; Watanabe, A.; LoBello, J.; Sima, C.; Hostetter, G.; Shepard, H.M.; Von Hoff, D.D.; Han, H. Desmoplasia in Primary Tumors and Metastatic Lesions of Pancreatic Cancer. Clin. Cancer Res. 2015, 21, 3561–3568. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Liu, Y.; Hu, M.; Wang, M.; Liu, X.; Huang, L. Relaxin Gene Delivery Modulates Macrophages to Resolve Cancer Fibrosis and Synergizes with Immune Checkpoint Blockade Therapy. Sci. Adv. 2021, 7, eabb6596. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Zhao, H.; Guo, Y.; Zhang, K.; Shang, F.; Liu, T. Bioinformatics-Based Analysis: Noncoding RNA-Mediated COL10A1 Is Associated with Poor Prognosis and Immune Cell Infiltration in Pancreatic Cancer. J. Healthc. Eng. 2022, 2022, 7904982. [Google Scholar] [CrossRef]
- Wang, H.; Ren, R.; Yang, Z.; Cai, J.; Du, S.; Shen, X. The COL11A1/Akt/CREB Signaling Axis Enables Mitochondrial-Mediated Apoptotic Evasion to Promote Chemoresistance in Pancreatic Cancer Cells through Modulating BAX/BCL-2 Function. J. Cancer 2021, 12, 1406–1420. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Liu, X.; Zheng, H.; Wang, H.; Hong, D. Integrated Bioinformatics Analysis Identified COL11A1 as an Immune Infiltrates Correlated Prognosticator in Pancreatic Adenocarcinoma. Int. Immunopharmacol. 2021, 90, 106982. [Google Scholar] [CrossRef] [PubMed]
- Neuzillet, C.; Nicolle, R.; Raffenne, J.; Tijeras-Raballand, A.; Brunel, A.; Astorgues-Xerri, L.; Vacher, S.; Arbateraz, F.; Fanjul, M.; Hilmi, M.; et al. Periostin- and Podoplanin-Positive Cancer-Associated Fibroblast Subtypes Cooperate to Shape the Inflamed Tumor Microenvironment in Aggressive Pancreatic Adenocarcinoma. J. Pathol. 2022, 258, 408–425. [Google Scholar] [CrossRef]
- Jiang, J.; Liu, H.-L.; Liu, Z.-H.; Tan, S.-W.; Wu, B. Identification of Cystatin SN as a Novel Biomarker for Pancreatic Cancer. Tumour Biol. 2015, 36, 3903–3910. [Google Scholar] [CrossRef]
- Chavez-Muñoz, C.; Morse, J.; Kilani, R.; Ghahary, A. Primary Human Keratinocytes Externalize Stratifin Protein via Exosomes. J. Cell. Biochem. 2008, 104, 2165–2173. [Google Scholar] [CrossRef]
- Mohamed, M.M.; Sloane, B.F. Cysteine Cathepsins: Multifunctional Enzymes in Cancer. Nat. Rev. Cancer 2006, 6, 764–775. [Google Scholar] [CrossRef]
- Berquin, I.M.; Sloane, B.F. Cathepsin B Expression in Human Tumors. Adv. Exp. Med. Biol. 1996, 389, 281–294. [Google Scholar] [CrossRef]
- Atkinson, S.P.; Andreu, Z.; Vicent, M.J. Polymer Therapeutics: Biomarkers and New Approaches for Personalized Cancer Treatment. J. Pers. Med. 2018, 8, 6. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Sha, W.; Liu, Z.; Chi, C.-W. Effect of Chymotrypsin C and Related Proteins on Pancreatic Cancer Cell Migration. Acta Biochim. Biophys. Sin. 2011, 43, 362–371. [Google Scholar] [CrossRef] [PubMed]
- González-Titos, A.; Hernández-Camarero, P.; Barungi, S.; Marchal, J.A.; Kenyon, J.; Perán, M. Trypsinogen and Chymotrypsinogen: Potent Anti-Tumor Agents. Expert Opin. Biol. Ther. 2021, 21, 1609–1621. [Google Scholar] [CrossRef] [PubMed]
- Makkouk, A.; Weiner, G.J. Cancer Immunotherapy and Breaking Immune Tolerance: New Approaches to an Old Challenge. Cancer Res. 2015, 75, 5–10. [Google Scholar] [CrossRef]
- Liu, Y.-T.; Sun, Z.-J. Turning Cold Tumors into Hot Tumors by Improving T-Cell Infiltration. Theranostics 2021, 11, 5365–5386. [Google Scholar] [CrossRef]
- Rubin, S.J.S.; Sojwal, R.S.; Gubatan, J.; Rogalla, S. The Tumor Immune Microenvironment in Pancreatic Ductal Adenocarcinoma: Neither Hot nor Cold. Cancers 2022, 14, 4236. [Google Scholar] [CrossRef]
- Zhuang, H.; Chen, X.; Dong, F.; Zhang, Z.; Zhou, Z.; Ma, Z.; Huang, S.; Chen, B.; Zhang, C.; Hou, B. Prognostic Values and Immune Suppression of the S100A Family in Pancreatic Cancer. J. Cell. Mol. Med. 2021, 25, 3006–3018. [Google Scholar] [CrossRef]
- Fujisawa, M.; Kanda, T.; Shibata, T.; Sasaki, R.; Masuzaki, R.; Matsumoto, N.; Nirei, K.; Imazu, H.; Kuroda, K.; Sugitani, M.; et al. Involvement of the Interferon Signaling Pathways in Pancreatic Cancer Cells. Anticancer Res. 2020, 40, 4445–4455. [Google Scholar] [CrossRef]
- Herremans, K.M.; Szymkiewicz, D.D.; Riner, A.N.; Bohan, R.P.; Tushoski, G.W.; Davidson, A.M.; Lou, X.; Leong, M.C.; Dean, B.D.; Gerber, M.; et al. The Interleukin-1 Axis and the Tumor Immune Microenvironment in Pancreatic Ductal Adenocarcinoma. Neoplasia 2022, 28, 100789. [Google Scholar] [CrossRef]
- Yuan, B.; Guan, Q.; Yan, T.; Zhang, X.; Xu, W.; Li, J. LncRNA HCP5 Regulates Pancreatic Cancer Progression by MiR-140-5p/CDK8 Axis. Cancer Biother. Radiopharm. 2020, 35, 711–719. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, J.; Dong, L.; Xia, L.; Zhu, H.; Li, Z.; Yu, X. Long Noncoding RNA HCP5 Regulates Pancreatic Cancer Gemcitabine (GEM) Resistance By Sponging Hsa-MiR-214-3p To Target HDGF. OncoTargets Ther. 2019, 12, 8207–8216. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Chen, S.; Liang, J.; Hao, T.; Wang, H.; Liu, G.; Jin, X.; Li, H.; Zhang, J.; Zhang, C.; et al. Schlafen Family Is a Prognostic Biomarker and Corresponds with Immune Infiltration in Gastric Cancer. Front. Immunol. 2022, 13, 922138. [Google Scholar] [CrossRef]
- Rodolosse, A.; Chalaux, E.; Adell, T.; Hagège, H.; Skoudy, A.; Real, F.X. PTF1alpha/P48 Transcription Factor Couples Proliferation and Differentiation in the Exocrine Pancreas [Corrected]. Gastroenterology 2004, 127, 937–949. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Wang, B.; Yi, K.; Lei, Q.; Wang, G.; Xu, X. IFI35 Is Involved in the Regulation of the Radiosensitivity of Colorectal Cancer Cells. Cancer Cell Int. 2021, 21, 290. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Zhan, M.; Zhang, Y.; Huang, S.; Xu, S.; Huang, X.; He, M.; Yao, Y.; Man, M.; Wang, J. PLZF Inhibits Proliferation and Metastasis of Gallbladder Cancer by Regulating IFIT2. Cell Death Dis. 2018, 9, 71. [Google Scholar] [CrossRef]
- Ho, W.J.; Jaffee, E.M.; Zheng, L. The Tumour Microenvironment in Pancreatic Cancer—Clinical Challenges and Opportunities. Nat. Rev. Clin. Oncol. 2020, 17, 527–540. [Google Scholar] [CrossRef]
- Hosein, A.N.; Brekken, R.A.; Maitra, A. Pancreatic Cancer Stroma: An Update on Therapeutic Targeting Strategies. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 487–505. [Google Scholar] [CrossRef]
- Zińczuk, J.; Zaręba, K.; Romaniuk, W.; Kamińska, D.; Nizioł, M.; Baszun, M.; Kędra, B.; Guzińska-Ustymowicz, K.; Pryczynicz, A. Expression of Chosen Carcinoembryonic-Related Cell Adhesion Molecules in Pancreatic Intraepithelial Neoplasia (PanIN) Associated with Chronic Pancreatitis and Pancreatic Ductal Adenocarcinoma (PDAC). Int. J. Med. Sci. 2019, 16, 583–592. [Google Scholar] [CrossRef]
- Rizeq, B.; Zakaria, Z.; Ouhtit, A. Towards Understanding the Mechanisms of Actions of Carcinoembryonic Antigen-Related Cell Adhesion Molecule 6 in Cancer Progression. Cancer Sci. 2018, 109, 33–42. [Google Scholar] [CrossRef]
- Han, Z.-W.; Lyv, Z.-W.; Cui, B.; Wang, Y.-Y.; Cheng, J.-T.; Zhang, Y.; Cai, W.-Q.; Zhou, Y.; Ma, Z.-W.; Wang, X.-W.; et al. The Old CEACAMs Find Their New Role in Tumor Immunotherapy. Investig. New Drugs 2020, 38, 1888–1898. [Google Scholar] [CrossRef]
- Okada, Y.; Takahashi, N.; Takayama, T.; Goel, A. LAMC2 Promotes Cancer Progression and Gemcitabine Resistance through Modulation of EMT and ATP-Binding Cassette Transporters in Pancreatic Ductal Adenocarcinoma. Carcinogenesis 2021, 42, 546–556. [Google Scholar] [CrossRef]
- Zhang, H.; Pan, Y.-Z.; Cheung, M.; Cao, M.; Yu, C.; Chen, L.; Zhan, L.; He, Z.-W.; Sun, C.-Y. LAMB3 Mediates Apoptotic, Proliferative, Invasive, and Metastatic Behaviors in Pancreatic Cancer by Regulating the PI3K/Akt Signaling Pathway. Cell Death Dis. 2019, 10, 230. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Li, D.; Liu, G.; Xiao, E.; Mu, S.; Pan, Y.; Qin, F.; Zhai, Y.; Duan, S.; Li, D.; et al. Identification of Critical Pathways and Potential Key Genes in Poorly Differentiated Pancreatic Adenocarcinoma. OncoTargets Ther. 2021, 14, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.; Mahadevan, D. Emerging Role and Targeting of Carcinoembryonic Antigen-Related Cell Adhesion Molecule 6 (CEACAM6) in Human Malignancies. Clin. Cancer Drugs 2015, 2, 100–111. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Chen, G.; Li, J.; Wen, W.; Gong, J.; Fu, J. Comprehensive Analysis of Abnormal Expression, Prognostic Value and Oncogenic Role of the Hub Gene FN1 in Pancreatic Ductal Adenocarcinoma via Bioinformatic Analysis and in Vitro Experiments. PeerJ 2021, 9, e12141. [Google Scholar] [CrossRef] [PubMed]
- Fukuhisa, H.; Seki, N.; Idichi, T.; Kurahara, H.; Yamada, Y.; Toda, H.; Kita, Y.; Kawasaki, Y.; Tanoue, K.; Mataki, Y.; et al. Gene Regulation by Antitumor MiR-130b-5p in Pancreatic Ductal Adenocarcinoma: The Clinical Significance of Oncogenic EPS8. J. Hum. Genet. 2019, 64, 521–534. [Google Scholar] [CrossRef]
- Jahny, E.; Yang, H.; Liu, B.; Jahnke, B.; Lademann, F.; Knösel, T.; Rümmele, P.; Grützmann, R.; Aust, D.E.; Pilarsky, C.; et al. The G Protein-Coupled Receptor RAI3 Is an Independent Prognostic Factor for Pancreatic Cancer Survival and Regulates Proliferation via STAT3 Phosphorylation. PLoS ONE 2017, 12, e0170390. [Google Scholar] [CrossRef]
- Islam, S.; Kitagawa, T.; Baron, B.; Abiko, Y.; Chiba, I.; Kuramitsu, Y. ITGA2, LAMB3, and LAMC2 May Be the Potential Therapeutic Targets in Pancreatic Ductal Adenocarcinoma: An Integrated Bioinformatics Analysis. Sci. Rep. 2021, 11, 10563. [Google Scholar] [CrossRef]
- Lin, H.; Yang, P.; Li, B.; Chang, Y.; Chen, Y.; Li, Y.; Liu, K.; Liang, X.; Chen, T.; Dai, Y.; et al. S100A10 Promotes Pancreatic Ductal Adenocarcinoma Cells Proliferation, Migration and Adhesion through JNK/LAMB3-LAMC2 Axis. Cancers 2022, 15, 202. [Google Scholar] [CrossRef]
- Robin, F.; Angenard, G.; Cano, L.; Courtin-Tanguy, L.; Gaignard, E.; Khene, Z.-E.; Bergeat, D.; Clément, B.; Boudjema, K.; Coulouarn, C.; et al. Molecular Profiling of Stroma Highlights Stratifin as a Novel Biomarker of Poor Prognosis in Pancreatic Ductal Adenocarcinoma. Br. J. Cancer 2020, 123, 72–80. [Google Scholar] [CrossRef]
- Schniers, B.K.; Wachtel, M.S.; Sharma, M.; Korac, K.; Rajasekaran, D.; Yang, S.; Sniegowski, T.; Ganapathy, V.; Bhutia, Y.D. Deletion of Slc6a14 Reduces Cancer Growth and Metastatic Spread and Improves Survival in KPC Mouse Model of Spontaneous Pancreatic Cancer. Biochem. J. 2022, 479, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Liang, Y.; Zhou, L.; Yan, Y.; Liu, N.; Zhang, R.; Huang, Y.; Wang, M.; Tang, Y.; Ali, D.W.; et al. TSPAN1 Promotes Autophagy Flux and Mediates Cooperation between WNT-CTNNB1 Signaling and Autophagy via the MIR454-FAM83A-TSPAN1 Axis in Pancreatic Cancer. Autophagy 2021, 17, 3175–3195. [Google Scholar] [CrossRef] [PubMed]
- Goonesekere, N.C.W.; Wang, X.; Ludwig, L.; Guda, C. A Meta Analysis of Pancreatic Microarray Datasets Yields New Targets as Cancer Genes and Biomarkers. PLoS ONE 2014, 9, e93046. [Google Scholar] [CrossRef] [PubMed]
- Irigoyen, A.; Jimenez-Luna, C.; Benavides, M.; Caba, O.; Gallego, J.; Ortuño, F.M.; Guillen-Ponce, C.; Rojas, I.; Aranda, E.; Torres, C.; et al. Integrative Multi-Platform Meta-Analysis of Gene Expression Profiles in Pancreatic Ductal Adenocarcinoma Patients for Identifying Novel Diagnostic Biomarkers. PLoS ONE 2018, 13, e0194844. [Google Scholar] [CrossRef]
- Normand, S.L. Meta-Analysis: Formulating, Evaluating, Combining, and Reporting. Stat. Med. 1999, 18, 321–359. [Google Scholar] [CrossRef]
- Higgins, J.P.T.; Thomas, J.; Chandler, J.; Cumpston, M.; Li, T.; Page, M.J.; Welch, V.A. Cochrane Handbook for Systematic Reviews of Interventions; John Wiley & Sons: Hoboken, NJ, USA, 2019; ISBN 978-1-119-53661-1. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Gene Name | Expression Level | Function |
|---|---|---|---|
| CEACAM6 | CEA cell adhesion molecule 6 | UP | EMR |
| SLC6A14 | Solute carrier family 6 member 14 | UP | EMR |
| S100P | S100 calcium-binding protein P | UP | EMR |
| CTSE | Cathepsin E | UP | EMR |
| SULF1 | Sulfatase 1 | UP | EMR |
| POSTN | Periostin | UP | EMR |
| GJB2 | Gap junction protein beta 2 | UP | EMR |
| GPRC5A | G protein-coupled receptor class C group 5 member A | UP | EMR |
| SFN | Stratifin | UP | EMR |
| FN1 | Fibronectin 1 | UP | EMR |
| LAMC2 | Laminin subunit gamma 2 | UP | EMR |
| CEACAM5 | CEA cell adhesion molecule 5 | UP | EMR |
| MMP1 | Matrix metallopeptidase 1 | UP | EMR |
| COL11A1 | Collagen type XI alpha 1 chain | UP | EMR |
| TSPAN1 | Tetraspanin 1 | UP | EMR |
| IFI27 | Interferon alpha inducible Protein 27 | UP | IS |
| CST1 | Cystatin SN | UP | EMT |
| LAMB3 | Laminin subunit beta 3 | UP | EMR |
| COL10A1 | Collagen type X alpha 1 chain | UP | EMR |
| VCAN | Versican | UP | EMR |
| CTRB2 | Chymotrypsinogen B2 | DOWN | EMR |
| PLA2G1B | Phospholipase A2 group IB | DOWN | Metabolism |
| CTRC | Chymotrypsin C | DOWN | EMR |
| GNMT | Glycine N-methyltransferase | DOWN | Metabolism |
| AQP8 | Aquaporin 8 | DOWN | H2O2 transport |
| SYCN | Syncolin | DOWN | Exocytosis |
| CPA2 | Carboxypeptidase A2 | DOWN | Metabolism |
| CELA2A | Chymotrypsin-like elastase 2A | DOWN | EMR |
| GP2 | Glycoprotein 2 | DOWN | Metabolism |
| KLK1 | Kallikrein 1 | DOWN | Serine protease |
| ALB | Albumin | DOWN | Oncotic pressure |
| CTRB1 | Chymotrypsinogen B1 | DOWN | EMR |
| ERP27 | Endoplasmic reticulum protein 27 | DOWN | Lipid and protein synthesis |
| TMED6 | Transmembrane p24 trafficking protein 6 | DOWN | Insulin secretion |
| PNLIPRP1 | Pancreatic lipase-related protein 1 | DOWN | Metabolism |
| CUZD1 | CUB and zona pellucida-like domain 1 | DOWN | EMR and IS |
| CELA2B | Chymotrypsin-like elastase 2B | DOWN | EMR |
| PNLIPRP2 | Pancreatic lipase-related protein 2 | DOWN | Metabolism |
| CTRL | Chymotrypsin-like | DOWN | EMR |
| SERPINI2 | Serpin family I member 2 | DOWN | Protease inhibitor |
| Functional Group | Genes |
|---|---|
| HLA | HLA-F, HLA-DRB5, HLA-B, HLA-A, HCP5, HLA-DRA, HLA-DPA1, HLA-DQB1, HLA-DQA1, HLA-DMB, HLA-DRB1, HLA-G, HLA-DPB1, SLFN12, SLFN13, and SLFN11 |
| Interleukin | IL1R2, IL1RN, IL1RAP, IL7R, IL2RG, IRAK3, IL18, LIF, and IL22RA1 |
| CD | CD58, CD109, CD52, CD53, CD74, CD14, CCDC80, CCDC141, CCDC69, DCDC2, and PDCD4 |
| Interferon | IFI27, IFI44L, IFI6, STING1, IFI16, IFITM1, ISG20, IFIT1, IFIT3, IFITM2, IRF9, IFIT2, IFNGR2, IFITM3, and IFI35 |
| Chemokine | CCL20, CCL18, CXCL10, CXCL5, CXCL8, CXCR4, CKLF, CXCL9, CXCL3, CXCL14, and CXCL12 |
| S100 | S100P, S100A6, S100A2, S100A16, S100A11, S100A4, S100A14, and S100A10 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pérez-Díez, I.; Andreu, Z.; Hidalgo, M.R.; Perpiñá-Clérigues, C.; Fantín, L.; Fernandez-Serra, A.; de la Iglesia-Vaya, M.; Lopez-Guerrero, J.A.; García-García, F. A Comprehensive Transcriptional Signature in Pancreatic Ductal Adenocarcinoma Reveals New Insights into the Immune and Desmoplastic Microenvironments. Cancers 2023, 15, 2887. https://doi.org/10.3390/cancers15112887
Pérez-Díez I, Andreu Z, Hidalgo MR, Perpiñá-Clérigues C, Fantín L, Fernandez-Serra A, de la Iglesia-Vaya M, Lopez-Guerrero JA, García-García F. A Comprehensive Transcriptional Signature in Pancreatic Ductal Adenocarcinoma Reveals New Insights into the Immune and Desmoplastic Microenvironments. Cancers. 2023; 15(11):2887. https://doi.org/10.3390/cancers15112887
Chicago/Turabian StylePérez-Díez, Irene, Zoraida Andreu, Marta R. Hidalgo, Carla Perpiñá-Clérigues, Lucía Fantín, Antonio Fernandez-Serra, María de la Iglesia-Vaya, José A. Lopez-Guerrero, and Francisco García-García. 2023. "A Comprehensive Transcriptional Signature in Pancreatic Ductal Adenocarcinoma Reveals New Insights into the Immune and Desmoplastic Microenvironments" Cancers 15, no. 11: 2887. https://doi.org/10.3390/cancers15112887