
Mutant IDH in Gliomas: Role in Cancer and Treatment Options


Abstract
:Simple Summary
Abstract
1. Introduction
2. The Oncogenic Role of Mutant IDH in Tumour Formation and Progression
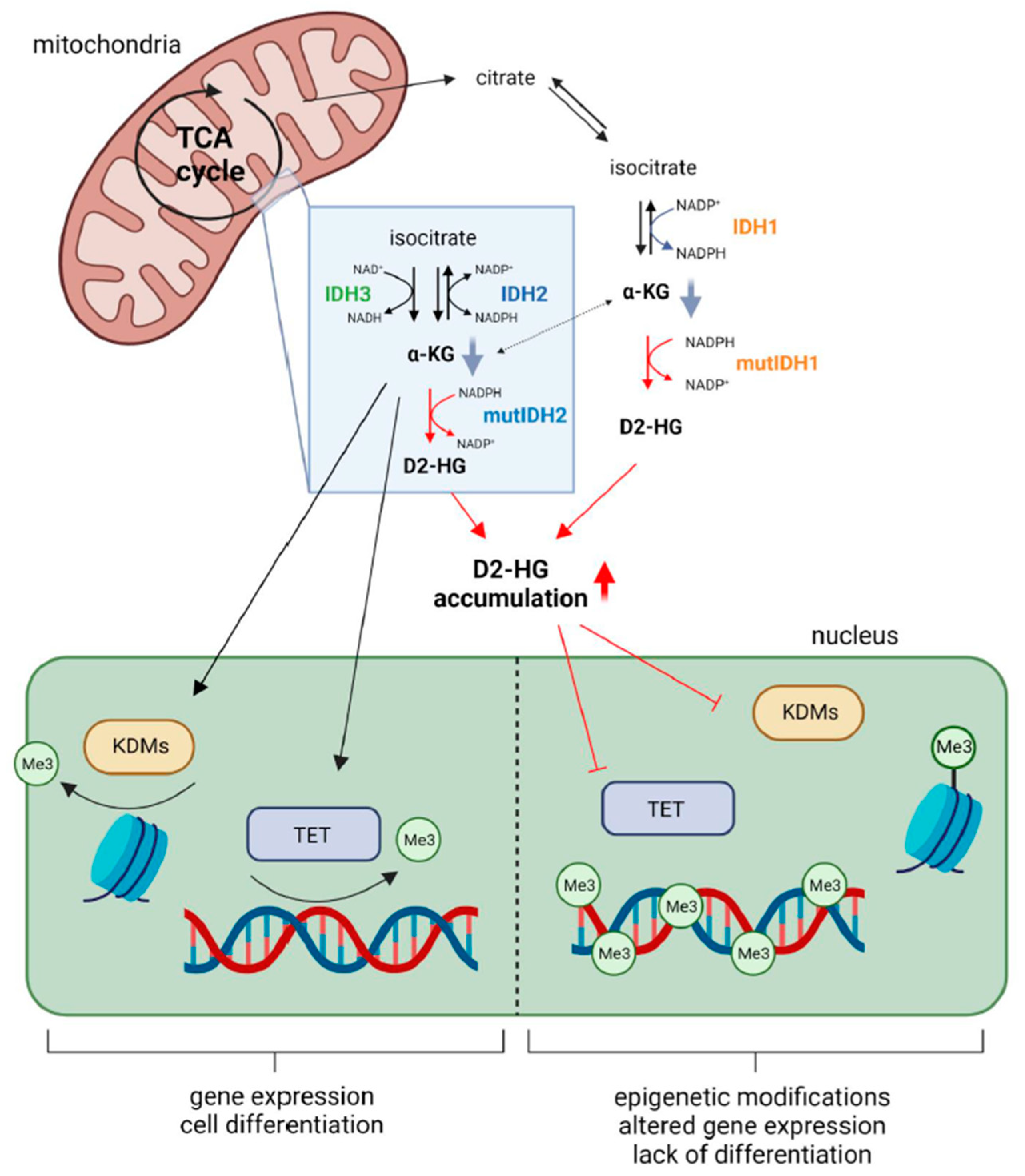
2.1. Metabolic Alterations
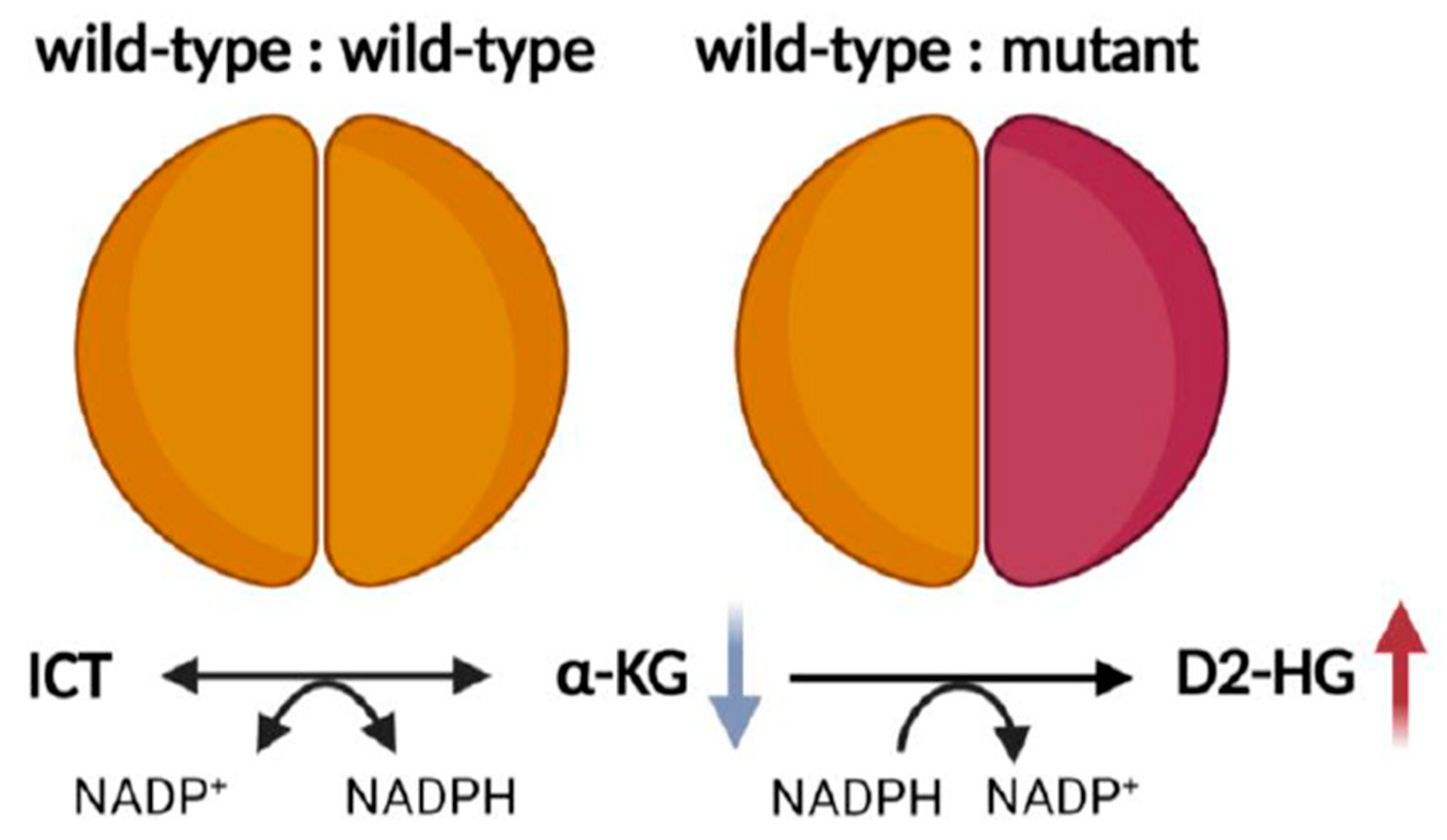
2.2. Redox Imbalance
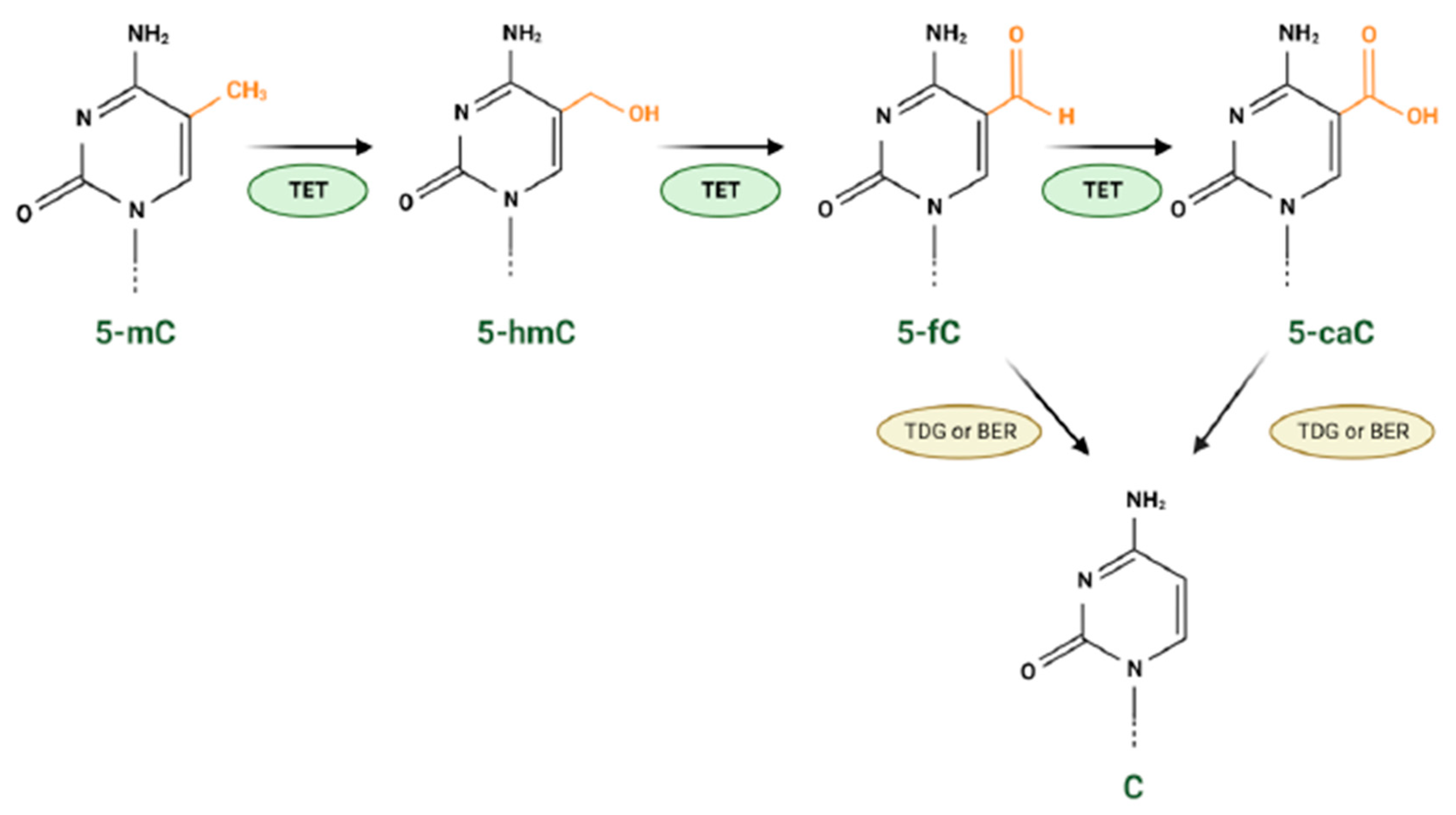
2.3. Epigenetic Modifications
2.4. Tumour Microenvironment
2.4.1. IDH Mutation and Tumour-Specific Immune Cells
2.4.2. IDH Mutation, Intra-Tumour Hypoxia and Angiogenesis
2.4.3. IDH Mutation and Tumour-Associated Epilepsy
2.5. The Role of IDH Mutation in Tumour Invasion
2.6. The Emerging Role of IDH3 in Cancer and Beyond
3. Novel Therapeutic Options for IDH Mutant Glioma
3.1. Direct Inhibition of Mutant IDH
3.2. IDH Vaccine
3.3. Modulating Epigenetic Alterations in IDH Mutant Gliomas
3.4. Inhibiting DNA Repair
3.5. Inhibiting Metabolic Pathways
3.6. Modulating Redox Homeostasis
3.7. Differentiation Therapy
4. Conclusions and Future Direction
Author Contributions
Funding
Conflicts of Interest
References
- Koh, H.J.; Lee, S.M.; Son, B.G.; Lee, S.H.; Ryoo, Z.Y.; Chang, K.T.; Park, J.W.; Park, D.C.; Song, B.J.; Veech, R.L.; et al. Cytosolic NADP+-dependent isocitrate dehydrogenase plays a key role in lipid metabolism. J. Biol. Chem. 2004, 279, 39968–39974. [Google Scholar] [CrossRef] [PubMed]
- Badur, M.G.; Muthusamy, T.; Parker, S.J.; Ma, S.; McBrayer, S.K.; Cordes, T.; Magana, J.H.; Guan, K.L.; Metallo, C.M. Oncogenic R132 IDH1 Mutations Limit NADPH for De Novo Lipogenesis through (D)2-Hydroxyglutarate Production in Fibrosarcoma Cells. Cell Rep. 2018, 25, 1680. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Jo, S.H.; Lee, S.M.; Koh, H.J.; Song, H.; Park, J.W.; Lee, W.H.; Huh, T.L. Role of NADP+-dependent isocitrate dehydrogenase (NADP+-ICDH) on cellular defence against oxidative injury by gamma-rays. Int. J. Radiat. Biol. 2004, 80, 635–642. [Google Scholar] [CrossRef] [PubMed]
- Joseph, J.W.; Jensen, M.V.; Ilkayeva, O.; Palmieri, F.; Alárcon, C.; Rhodes, C.J.; Newgard, C.B. The mitochondrial citrate/isocitrate carrier plays a regulatory role in glucose-stimulated insulin secretion. J. Biol. Chem. 2006, 281, 35624–35632. [Google Scholar] [CrossRef]
- Leighton, F.; Poole, B.; Lazarow, P.B.; De Duve, C. The synthesis and turnover of rat liver peroxisomes. I. Fractionation of peroxisome proteins. J. Cell Biol. 1969, 41, 521–535. [Google Scholar] [CrossRef]
- Yang, C.; Sudderth, J.; Dang, T.; Bachoo, R.M.; McDonald, J.G.; DeBerardinis, R.J. Glioblastoma cells require glutamate dehydrogenase to survive impairments of glucose metabolism or Akt signaling. Cancer Res. 2009, 69, 7986–7993. [Google Scholar] [CrossRef]
- Xu, X.; Zhao, J.; Xu, Z.; Peng, B.; Huang, Q.; Arnold, E.; Ding, J. Structures of human cytosolic NADP-dependent isocitrate dehydrogenase reveal a novel self-regulatory mechanism of activity. J. Biol. Chem. 2004, 279, 33946–33957. [Google Scholar] [CrossRef]
- Ma, T.; Peng, Y.; Huang, W.; Liu, Y.; Ding, J. The β and γ subunits play distinct functional roles in the α(2)βγ heterotetramer of human NAD-dependent isocitrate dehydrogenase. Sci. Rep. 2017, 7, 41882. [Google Scholar] [CrossRef]
- Hurley, J.H.; Dean, A.M.; Koshland, D.E., Jr.; Stroud, R.M. Catalytic mechanism of NADP(+)-dependent isocitrate dehydrogenase: Implications from the structures of magnesium-isocitrate and NADP+ complexes. Biochemistry 1991, 30, 8671–8678. [Google Scholar] [CrossRef]
- Gabriel, J.L.; Zervos, P.R.; Plaut, G.W. Activity of purified NAD-specific isocitrate dehydrogenase at modulator and substrate concentrations approximating conditions in mitochondria. Metab. Clin. Exp. 1986, 35, 661–667. [Google Scholar] [CrossRef]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef]
- Hartmann, C.; Meyer, J.; Balss, J.; Capper, D.; Mueller, W.; Christians, A.; Felsberg, J.; Wolter, M.; Mawrin, C.; Wick, W.; et al. Type and frequency of IDH1 and IDH2 mutations are related to astrocytic and oligodendroglial differentiation and age: A study of 1010 diffuse gliomas. Acta Neuropathol. 2009, 118, 469–474. [Google Scholar] [CrossRef]
- Balss, J.; Meyer, J.; Mueller, W.; Korshunov, A.; Hartmann, C.; von Deimling, A. Analysis of the IDH1 codon 132 mutation in brain tumors. Acta Neuropathol. 2008, 116, 597–602. [Google Scholar] [CrossRef]
- Ichimura, K.; Pearson, D.M.; Kocialkowski, S.; Bäcklund, L.M.; Chan, R.; Jones, D.T.W.; Collins, V.P. IDH1 mutations are present in the majority of common adult gliomas but rare in primary glioblastomas. Neuro-oncology 2009, 11, 341–347. [Google Scholar] [CrossRef]
- Watanabe, T.; Nobusawa, S.; Kleihues, P.; Ohgaki, H. IDH1 mutations are early events in the development of astrocytomas and oligodendrogliomas. Am. J. Pathol. 2009, 174, 1149–1153. [Google Scholar] [CrossRef]
- Wang, H.Y.; Tang, K.; Liang, T.Y.; Zhang, W.Z.; Li, J.Y.; Wang, W.; Hu, H.M.; Li, M.Y.; Wang, H.Q.; He, X.Z.; et al. The comparison of clinical and biological characteristics between IDH1 and IDH2 mutations in gliomas. J. Exp. Clin. Cancer Res. CR 2016, 35, 86. [Google Scholar] [CrossRef]
- Amary, M.F.; Bacsi, K.; Maggiani, F.; Damato, S.; Halai, D.; Berisha, F.; Pollock, R.; O’Donnell, P.; Grigoriadis, A.; Diss, T.; et al. IDH1 and IDH2 mutations are frequent events in central chondrosarcoma and central and periosteal chondromas but not in other mesenchymal tumours. J. Pathol. 2011, 224, 334–343. [Google Scholar] [CrossRef]
- Arai, M.; Nobusawa, S.; Ikota, H.; Takemura, S.; Nakazato, Y. Frequent IDH1/2 mutations in intracranial chondrosarcoma: A possible diagnostic clue for its differentiation from chordoma. Brain Tumor Pathol. 2012, 29, 201–206. [Google Scholar] [CrossRef]
- Lu, C.; Venneti, S.; Akalin, A.; Fang, F.; Ward, P.S.; Dematteo, R.G.; Intlekofer, A.M.; Chen, C.; Ye, J.; Hameed, M.; et al. Induction of sarcomas by mutant IDH2. Genes Dev. 2013, 27, 1986–1998. [Google Scholar] [CrossRef]
- Kato Kaneko, M.; Liu, X.; Oki, H.; Ogasawara, S.; Nakamura, T.; Saidoh, N.; Tsujimoto, Y.; Matsuyama, Y.; Uruno, A.; Sugawara, M.; et al. Isocitrate dehydrogenase mutation is frequently observed in giant cell tumor of bone. Cancer Sci. 2014, 105, 744–748. [Google Scholar] [CrossRef] [PubMed]
- Lugowska, I.; Teterycz, P.; Mikula, M.; Kulecka, M.; Kluska, A.; Balabas, A.; Piatkowska, M.; Wagrodzki, M.; Pienkowski, A.; Rutkowski, P.; et al. IDH1/2 Mutations Predict Shorter Survival in Chondrosarcoma. J. Cancer 2018, 9, 998–1005. [Google Scholar] [CrossRef] [PubMed]
- Cleven, A.H.G.; Suijker, J.; Agrogiannis, G.; Briaire-de Bruijn, I.H.; Frizzell, N.; Hoekstra, A.S.; Wijers-Koster, P.M.; Cleton-Jansen, A.-M.; Bovée, J.V.M.G. IDH1 or -2 mutations do not predict outcome and do not cause loss of 5-hydroxymethylcytosine or altered histone modifications in central chondrosarcomas. Clin. Sarcoma Res. 2017, 7, 8. [Google Scholar] [CrossRef] [PubMed]
- Tallegas, M.; Miquelestorena-Standley, É.; Labit-Bouvier, C.; Badoual, C.; Francois, A.; Gomez-Brouchet, A.; Aubert, S.; Collin, C.; Tallet, A.; de Pinieux, G. IDH mutation status in a series of 88 head and neck chondrosarcomas: Different profile between tumors of the skull base and tumors involving the facial skeleton and the laryngotracheal tract. Hum. Pathol. 2019, 84, 183–191. [Google Scholar] [CrossRef]
- Borger, D.R.; Tanabe, K.K.; Fan, K.C.; Lopez, H.U.; Fantin, V.R.; Straley, K.S.; Schenkein, D.P.; Hezel, A.F.; Ancukiewicz, M.; Liebman, H.M.; et al. Frequent mutation of isocitrate dehydrogenase (IDH)1 and IDH2 in cholangiocarcinoma identified through broad-based tumor genotyping. Oncologist 2012, 17, 72–79. [Google Scholar] [CrossRef]
- Kipp, B.R.; Voss, J.S.; Kerr, S.E.; Barr Fritcher, E.G.; Graham, R.P.; Zhang, L.; Highsmith, W.E.; Zhang, J.; Roberts, L.R.; Gores, G.J.; et al. Isocitrate dehydrogenase 1 and 2 mutations in cholangiocarcinoma. Hum. Pathol. 2012, 43, 1552–1558. [Google Scholar] [CrossRef]
- Wang, P.; Dong, Q.; Zhang, C.; Kuan, P.F.; Liu, Y.; Jeck, W.R.; Andersen, J.B.; Jiang, W.; Savich, G.L.; Tan, T.X.; et al. Mutations in isocitrate dehydrogenase 1 and 2 occur frequently in intrahepatic cholangiocarcinomas and share hypermethylation targets with glioblastomas. Oncogene 2013, 32, 3091–3100. [Google Scholar] [CrossRef]
- Jiao, Y.; Pawlik, T.M.; Anders, R.A.; Selaru, F.M.; Streppel, M.M.; Lucas, D.J.; Niknafs, N.; Guthrie, V.B.; Maitra, A.; Argani, P. Exome sequencing identifies frequent inactivating mutations in BAP1, ARID1A and PBRM1 in intrahepatic cholangiocarcinomas. Nat. Genet. 2013, 45, 1470–1473. [Google Scholar] [CrossRef]
- Ross, J.S.; Wang, K.; Gay, L.; Al-Rohil, R.; Rand, J.V.; Jones, D.M.; Lee, H.J.; Sheehan, C.E.; Otto, G.A.; Palmer, G.; et al. New Routes to Targeted Therapy of Intrahepatic Cholangiocarcinomas Revealed by Next-Generation Sequencing. Oncologist 2014, 19, 235–242. [Google Scholar] [CrossRef]
- Farshidfar, F.; Zheng, S.; Gingras, M.C.; Newton, Y.; Shih, J.; Robertson, A.G.; Hinoue, T.; Hoadley, K.A.; Gibb, E.A.; Roszik, J.; et al. Integrative Genomic Analysis of Cholangiocarcinoma Identifies Distinct IDH-Mutant Molecular Profiles. Cell Rep. 2017, 18, 2780–2794. [Google Scholar] [CrossRef]
- Lee, J.H.; Shin, D.H.; Park, W.Y.; Shin, N.; Kim, A.; Lee, H.J.; Kim, Y.K.; Choi, K.U.; Kim, J.Y.; Yang, Y.I.; et al. IDH1 R132C mutation is detected in clear cell hepatocellular carcinoma by pyrosequencing. World J. Surg. Oncol. 2017, 15, 82. [Google Scholar] [CrossRef]
- Nepal, C.; O’Rourke, C.J.; Oliveira, D.; Taranta, A.; Shema, S.; Gautam, P.; Calderaro, J.; Barbour, A.; Raggi, C.; Wennerberg, K.; et al. Genomic perturbations reveal distinct regulatory networks in intrahepatic cholangiocarcinoma. Hepatology (Baltim. Md.) 2018, 68, 949–963. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, Z.G.; Ding, Z.Y.; Dong, W.; Liang, H.F.; Chu, L.; Zhang, B.X.; Chen, X.P. IDH1 mutation correlates with a beneficial prognosis and suppresses tumor growth in IHCC. J. Surg. Res. 2018, 231, 116–125. [Google Scholar] [CrossRef]
- Mardis, E.R.; Ding, L.; Dooling, D.J.; Larson, D.E.; McLellan, M.D.; Chen, K.; Koboldt, D.C.; Fulton, R.S.; Delehaunty, K.D.; McGrath, S.D.; et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N. Engl. J. Med. 2009, 361, 1058–1066. [Google Scholar] [CrossRef]
- Ward, P.S.; Patel, J.; Wise, D.R.; Abdel-Wahab, O.; Bennett, B.D.; Coller, H.A.; Cross, J.R.; Fantin, V.R.; Hedvat, C.V.; Perl, A.E.; et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 2010, 17, 225–234. [Google Scholar] [CrossRef]
- Abbas, S.; Lugthart, S.; Kavelaars, F.G.; Schelen, A.; Koenders, J.E.; Zeilemaker, A.; van Putten, W.J.; Rijneveld, A.W.; Löwenberg, B.; Valk, P.J. Acquired mutations in the genes encoding IDH1 and IDH2 both are recurrent aberrations in acute myeloid leukemia: Prevalence and prognostic value. Blood 2010, 116, 2122–2126. [Google Scholar] [CrossRef]
- Marcucci, G.; Maharry, K.; Wu, Y.Z.; Radmacher, M.D.; Mrózek, K.; Margeson, D.; Holland, K.B.; Whitman, S.P.; Becker, H.; Schwind, S.; et al. IDH1 and IDH2 gene mutations identify novel molecular subsets within de novo cytogenetically normal acute myeloid leukemia: A Cancer and Leukemia Group B study. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2010, 28, 2348–2355. [Google Scholar] [CrossRef]
- Gross, S.; Cairns, R.A.; Minden, M.D.; Driggers, E.M.; Bittinger, M.A.; Jang, H.G.; Sasaki, M.; Jin, S.; Schenkein, D.P.; Su, S.M.; et al. Cancer-associated metabolite 2-hydroxyglutarate accumulates in acute myelogenous leukemia with isocitrate dehydrogenase 1 and 2 mutations. J. Exp. Med. 2010, 207, 339–344. [Google Scholar] [CrossRef]
- Schnittger, S.; Haferlach, C.; Ulke, M.; Alpermann, T.; Kern, W.; Haferlach, T. IDH1 mutations are detected in 6.6% of 1414 AML patients and are associated with intermediate risk karyotype and unfavorable prognosis in adults younger than 60 years and unmutated NPM1 status. Blood 2010, 116, 5486–5496. [Google Scholar] [CrossRef]
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; Vasanthakumar, A.; Fernandez, H.F.; et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef]
- Molenaar, R.J.; Thota, S.; Nagata, Y.; Patel, B.; Clemente, M.; Przychodzen, B.; Hirsh, C.; Viny, A.D.; Hosano, N.; Bleeker, F.E.; et al. Clinical and biological implications of ancestral and non-ancestral IDH1 and IDH2 mutations in myeloid neoplasms. Leukemia 2015, 29, 2134–2142. [Google Scholar] [CrossRef] [PubMed]
- Paschka, P.; Schlenk, R.F.; Gaidzik, V.I.; Habdank, M.; Krönke, J.; Bullinger, L.; Späth, D.; Kayser, S.; Zucknick, M.; Götze, K.; et al. IDH1 and IDH2 mutations are frequent genetic alterations in acute myeloid leukemia and confer adverse prognosis in cytogenetically normal acute myeloid leukemia with NPM1 mutation without FLT3 internal tandem duplication. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2010, 28, 3636–3643. [Google Scholar] [CrossRef] [PubMed]
- Cairns, R.A.; Iqbal, J.; Lemonnier, F.; Kucuk, C.; de Leval, L.; Jais, J.P.; Parrens, M.; Martin, A.; Xerri, L.; Brousset, P.; et al. IDH2 mutations are frequent in angioimmunoblastic T-cell lymphoma. Blood 2012, 119, 1901–1903. [Google Scholar] [CrossRef] [PubMed]
- Odejide, O.; Weigert, O.; Lane, A.A.; Toscano, D.; Lunning, M.A.; Kopp, N.; Kim, S.; van Bodegom, D.; Bolla, S.; Schatz, J.H.; et al. A targeted mutational landscape of angioimmunoblastic T-cell lymphoma. Blood 2014, 123, 1293–1296. [Google Scholar] [CrossRef] [PubMed]
- Sakata-Yanagimoto, M.; Enami, T.; Yoshida, K.; Shiraishi, Y.; Ishii, R.; Miyake, Y.; Muto, H.; Tsuyama, N.; Sato-Otsubo, A.; Okuno, Y.; et al. Somatic RHOA mutation in angioimmunoblastic T cell lymphoma. Nat. Genet. 2014, 46, 171–175. [Google Scholar] [CrossRef]
- Wang, C.; McKeithan, T.W.; Gong, Q.; Zhang, W.; Bouska, A.; Rosenwald, A.; Gascoyne, R.D.; Wu, X.; Wang, J.; Muhammad, Z.; et al. IDH2R172 mutations define a unique subgroup of patients with angioimmunoblastic T-cell lymphoma. Blood 2015, 126, 1741–1752. [Google Scholar] [CrossRef]
- Ward, P.S.; Cross, J.R.; Lu, C.; Weigert, O.; Abel-Wahab, O.; Levine, R.L.; Weinstock, D.M.; Sharp, K.A.; Thompson, C.B. Identification of additional IDH mutations associated with oncometabolite R(-)-2-hydroxyglutarate production. Oncogene 2012, 31, 2491–2498. [Google Scholar] [CrossRef]
- Fathi, A.T.; Sadrzadeh, H.; Comander, A.H.; Higgins, M.J.; Bardia, A.; Perry, A.; Burke, M.; Silver, R.; Matulis, C.R.; Straley, K.S.; et al. Isocitrate Dehydrogenase 1 (IDH1) Mutation in Breast Adenocarcinoma Is Associated With Elevated Levels of Serum and Urine 2-Hydroxyglutarate. Oncologist 2014, 19, 602–607. [Google Scholar] [CrossRef]
- Chiang, S.; Weigelt, B.; Wen, H.C.; Pareja, F.; Raghavendra, A.; Martelotto, L.G.; Burke, K.A.; Basili, T.; Li, A.; Geyer, F.C.; et al. IDH2 Mutations Define a Unique Subtype of Breast Cancer with Altered Nuclear Polarity. Cancer Res. 2016, 76, 7118–7129. [Google Scholar] [CrossRef]
- Lozada, J.R.; Basili, T.; Pareja, F.; Alemar, B.; Paula, A.D.C.; Gularte-Merida, R.; Giri, D.D.; Querzoli, P.; Cserni, G.; Rakha, E.A.; et al. Solid papillary breast carcinomas resembling the tall cell variant of papillary thyroid neoplasms (solid papillary carcinomas with reverse polarity) harbour recurrent mutations affecting IDH2 and PIK3CA: A validation cohort. Histopathology 2018, 73, 339–344. [Google Scholar] [CrossRef]
- Li-Chang, H.H.; Kasaian, K.; Ng, Y.; Lum, A.; Kong, E.; Lim, H.; Jones, S.J.; Huntsman, D.G.; Schaeffer, D.F.; Yip, S. Retrospective review using targeted deep sequencing reveals mutational differences between gastroesophageal junction and gastric carcinomas. BMC Cancer 2015, 15, 32. [Google Scholar] [CrossRef]
- Hartman, D.J.; Binion, D.; Regueiro, M.; Schraut, W.; Bahary, N.; Sun, W.; Nikiforova, M.; Pai, R.K. Isocitrate dehydrogenase-1 is mutated in inflammatory bowel disease-associated intestinal adenocarcinoma with low-grade tubuloglandular histology but not in sporadic intestinal adenocarcinoma. Am. J. Surg. Pathol. 2014, 38, 1147–1156. [Google Scholar] [CrossRef]
- Lopez, G.Y.; Reitman, Z.J.; Solomon, D.; Waldman, T.; Bigner, D.D.; McLendon, R.E.; Rosenberg, S.A.; Samuels, Y.; Yan, H. IDH1(R132) mutation identified in one human melanoma metastasis, but not correlated with metastases to the brain. Biochem. Biophys. Res. Commun. 2010, 398, 585–587. [Google Scholar] [CrossRef]
- Toth, L.N.; de Abreu, F.B.; Tafe, L.J. Non-small cell lung cancers with isocitrate dehydrogenase 1 or 2 (IDH1/2) mutations. Hum. Pathol. 2018, 78, 138–143. [Google Scholar] [CrossRef]
- Gaal, J.; Burnichon, N.; Korpershoek, E.; Roncelin, I.; Bertherat, J.r.m.; Plouin, P.-F.o.; de Krijger, R.R.; Gimenez-Roqueplo, A.-P.; Dinjens, W.N.M. Isocitrate Dehydrogenase Mutations Are Rare in Pheochromocytomas and Paragangliomas. J. Clin. Endocrinol. Metab. 2010, 95, 1274–1278. [Google Scholar] [CrossRef]
- Hinsch, A.; Brolund, M.; Hube-Magg, C.; Kluth, M.; Simon, R.; Möller-Koop, C.; Sauter, G.; Steurer, S.; Luebke, A.; Angerer, A. Immunohistochemically detected IDH1 R132H mutation is rare and mostly heterogeneous in prostate cancer. World J. Urol. 2018, 36, 877–882. [Google Scholar] [CrossRef]
- Kurek, K.C.; Pansuriya, T.C.; van Ruler, M.A.; van den Akker, B.; Luks, V.L.; Verbeke, S.L.; Kozakewich, H.P.; Sciot, R.; Lev, D.; Lazar, A.J.; et al. R132C IDH1 mutations are found in spindle cell hemangiomas and not in other vascular tumors or malformations. Am. J. Pathol. 2013, 182, 1494–1500. [Google Scholar] [CrossRef]
- Hemerly, J.P.; Bastos, A.U.; Cerutti, J.M. Identification of several novel non-p.R132 IDH1 variants in thyroid carcinomas. Eur. J. Endocrinol. 2010, 163, 747–755. [Google Scholar] [CrossRef]
- Murugan, A.K.; Bojdani, E.; Xing, M. Identification and functional characterization of isocitrate dehydrogenase 1 (IDH1) mutations in thyroid cancer. Biochem. Biophys. Res. Commun. 2010, 393, 555–559. [Google Scholar] [CrossRef]
- Al-Khallaf, H. Isocitrate dehydrogenases in physiology and cancer: Biochemical and molecular insight. Cell Biosci. 2017, 7, 37. [Google Scholar] [CrossRef]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009, 462, 739–744. [Google Scholar] [CrossRef] [PubMed]
- Pope, W.B.; Prins, R.M.; Albert Thomas, M.; Nagarajan, R.; Yen, K.E.; Bittinger, M.A.; Salamon, N.; Chou, A.P.; Yong, W.H.; Soto, H.; et al. Non-invasive detection of 2-hydroxyglutarate and other metabolites in IDH1 mutant glioma patients using magnetic resonance spectroscopy. J. Neuro-Oncol. 2012, 107, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.T.; et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, R.; Yeoh, K.K.; Tian, Y.M.; Hillringhaus, L.; Bagg, E.A.; Rose, N.R.; Leung, I.K.; Li, X.S.; Woon, E.C.; Yang, M.; et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 2011, 12, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Reitman, Z.J.; Jin, G.; Karoly, E.D.; Spasojevic, I.; Yang, J.; Kinzler, K.W.; He, Y.; Bigner, D.D.; Vogelstein, B.; Yan, H. Profiling the effects of isocitrate dehydrogenase 1 and 2 mutations on the cellular metabolome. Proc. Natl. Acad. Sci. USA 2011, 108, 3270–3275. [Google Scholar] [CrossRef]
- Zhou, L.; Wang, Z.; Hu, C.; Zhang, C.; Kovatcheva-Datchary, P.; Yu, D.; Liu, S.; Ren, F.; Wang, X.; Li, Y.; et al. Integrated Metabolomics and Lipidomics Analyses Reveal Metabolic Reprogramming in Human Glioma with IDH1 Mutation. J. Proteome Res. 2019, 18, 960–969. [Google Scholar] [CrossRef]
- Fack, F.; Tardito, S.; Hochart, G.; Oudin, A.; Zheng, L.; Fritah, S.; Golebiewska, A.; Nazarov, P.V.; Bernard, A.; Hau, A.C.; et al. Altered metabolic landscape in IDH-mutant gliomas affects phospholipid, energy, and oxidative stress pathways. EMBO Mol. Med. 2017, 9, 1681–1695. [Google Scholar] [CrossRef]
- Walsby-Tickle, J.; Gannon, J.; Hvinden, I.; Bardella, C.; Abboud, M.I.; Nazeer, A.; Hauton, D.; Pires, E.; Cadoux-Hudson, T.; Schofield, C.J.; et al. Anion-exchange chromatography mass spectrometry provides extensive coverage of primary metabolic pathways revealing altered metabolism in IDH1 mutant cells. Commun. Biol. 2020, 3, 247. [Google Scholar] [CrossRef]
- Wen, H.; Cho, H.R.; Yun, T.; Kim, H.; Park, C.K.; Lee, S.H.; Choi, S.H.; Park, S. Metabolomic comparison between cells over-expressing isocitrate dehydrogenase 1 and 2 mutants and the effects of an inhibitor on the metabolism. J. Neurochem. 2015, 132, 183–193. [Google Scholar] [CrossRef]
- Sasaki, M.; Knobbe, C.B.; Itsumi, M.; Elia, A.J.; Harris, I.S.; Chio, I.I.C.; Cairns, R.A.; McCracken, S.; Wakeham, A.; Haight, J.; et al. D-2-hydroxyglutarate produced by mutant IDH1 perturbs collagen maturation and basement membrane function. Genes Dev. 2012, 26, 2038–2049. [Google Scholar] [CrossRef]
- Bardella, C.; Al-Dalahmah, O.; Krell, D.; Brazauskas, P.; Al-Qahtani, K.; Tomkova, M.; Adam, J.; Serres, S.; Lockstone, H.; Freeman-Mills, L.; et al. Expression of Idh1(R132H) in the Murine Subventricular Zone Stem Cell Niche Recapitulates Features of Early Gliomagenesis. Cancer Cell 2016, 30, 578–594. [Google Scholar] [CrossRef]
- Tesileanu, C.M.S.; Vallentgoed, W.R.; Sanson, M.; Taal, W.; Clement, P.M.; Wick, W.; Brandes, A.A.; Baurain, J.F.; Chinot, O.L.; Wheeler, H.; et al. Non-IDH1-R132H IDH1/2 mutations are associated with increased DNA methylation and improved survival in astrocytomas, compared to IDH1-R132H mutations. Acta Neuropathol. 2021, 141, 945–957. [Google Scholar] [CrossRef]
- Pusch, S.; Schweizer, L.; Beck, A.C.; Lehmler, J.M.; Weissert, S.; Balss, J.; Miller, A.K.; von Deimling, A. D-2-Hydroxyglutarate producing neo-enzymatic activity inversely correlates with frequency of the type of isocitrate dehydrogenase 1 mutations found in glioma. Acta Neuropathol. Commun. 2014, 2, 19. [Google Scholar] [CrossRef]
- Avellaneda Matteo, D.; Grunseth, A.J.; Gonzalez, E.R.; Anselmo, S.L.; Kennedy, M.A.; Moman, P.; Scott, D.A.; Hoang, A.; Sohl, C.D. Molecular mechanisms of isocitrate dehydrogenase 1 (IDH1) mutations identified in tumors: The role of size and hydrophobicity at residue 132 on catalytic efficiency. J. Biol. Chem. 2017, 292, 7971–7983. [Google Scholar] [CrossRef]
- Borodovsky, A.; Seltzer, M.J.; Riggins, G.J. Altered cancer cell metabolism in gliomas with mutant IDH1 or IDH2. Curr. Opin. Oncol. 2012, 24, 83–89. [Google Scholar] [CrossRef]
- Ohka, F.; Ito, M.; Ranjit, M.; Senga, T.; Motomura, A.; Motomura, K.; Saito, K.; Kato, K.; Kato, Y.; Wakabayashi, T.; et al. Quantitative metabolome analysis profiles activation of glutaminolysis in glioma with IDH1 mutation. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2014, 35, 5911–5920. [Google Scholar] [CrossRef]
- Maus, A.; Peters, G.J. Glutamate and α-ketoglutarate: Key players in glioma metabolism. Amino Acids 2017, 49, 21–32. [Google Scholar] [CrossRef]
- Waitkus, M.S.; Pirozzi, C.J.; Moure, C.J.; Diplas, B.H.; Hansen, L.J.; Carpenter, A.B.; Yang, R.; Wang, Z.; Ingram, B.O.; Karoly, E.D.; et al. Adaptive Evolution of the GDH2 Allosteric Domain Promotes Gliomagenesis by Resolving IDH1(R132H)-Induced Metabolic Liabilities. Cancer Res. 2018, 78, 36–50. [Google Scholar] [CrossRef]
- Seltzer, M.J.; Bennett, B.D.; Joshi, A.D.; Gao, P.; Thomas, A.G.; Ferraris, D.V.; Tsukamoto, T.; Rojas, C.J.; Slusher, B.S.; Rabinowitz, J.D.; et al. Inhibition of glutaminase preferentially slows growth of glioma cells with mutant IDH1. Cancer Res. 2010, 70, 8981–8987. [Google Scholar] [CrossRef]
- Doherty, J.R.; Cleveland, J.L. Targeting lactate metabolism for cancer therapeutics. J. Clin. Investig. 2013, 123, 3685–3692. [Google Scholar] [CrossRef]
- Le, A.; Cooper, C.R.; Gouw, A.M.; Dinavahi, R.; Maitra, A.; Deck, L.M.; Royer, R.E.; Vander Jagt, D.L.; Semenza, G.L.; Dang, C.V. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc. Natl. Acad. Sci. USA 2010, 107, 2037–2042. [Google Scholar] [CrossRef] [PubMed]
- Khurshed, M.; Molenaar, R.J.; Lenting, K.; Leenders, W.P.; van Noorden, C.J.F. In silico gene expression analysis reveals glycolysis and acetate anaplerosis in IDH1 wild-type glioma and lactate and glutamate anaplerosis in IDH1-mutated glioma. Oncotarget 2017, 8, 49165–49177. [Google Scholar] [CrossRef]
- Chesnelong, C.; Chaumeil, M.M.; Blough, M.D.; Al-Najjar, M.; Stechishin, O.D.; Chan, J.A.; Pieper, R.O.; Ronen, S.M.; Weiss, S.; Luchman, H.A.; et al. Lactate dehydrogenase A silencing in IDH mutant gliomas. Neuro-oncology 2014, 16, 686–695. [Google Scholar] [CrossRef] [PubMed]
- Chaumeil, M.M.; Radoul, M.; Najac, C.; Eriksson, P.; Viswanath, P.; Blough, M.D.; Chesnelong, C.; Luchman, H.A.; Cairncross, J.G.; Ronen, S.M. Hyperpolarized (13)C MR imaging detects no lactate production in mutant IDH1 gliomas: Implications for diagnosis and response monitoring. NeuroImage. Clin. 2016, 12, 180–189. [Google Scholar] [CrossRef] [PubMed]
- Hvinden, I.C.; Cadoux-Hudson, T.; Schofield, C.J.; McCullagh, J.S.O. Metabolic adaptations in cancers expressing isocitrate dehydrogenase mutations. Cell Rep. Med. 2021, 2, 100469. [Google Scholar] [CrossRef]
- Jo, S.H.; Son, M.K.; Koh, H.J.; Lee, S.M.; Song, I.H.; Kim, Y.O.; Lee, Y.S.; Jeong, K.S.; Kim, W.B.; Park, J.W.; et al. Control of mitochondrial redox balance and cellular defense against oxidative damage by mitochondrial NADP+-dependent isocitrate dehydrogenase. J. Biol. Chem. 2001, 276, 16168–16176. [Google Scholar] [CrossRef]
- Itsumi, M.; Inoue, S.; Elia, A.J.; Murakami, K.; Sasaki, M.; Lind, E.F.; Brenner, D.; Harris, I.S.; Chio, I.; Afzal, S.; et al. Idh1 protects murine hepatocytes from endotoxin-induced oxidative stress by regulating the intracellular NADP(+)/NADPH ratio. Cell Death Differ. 2015, 22, 1837–1845. [Google Scholar] [CrossRef]
- Liu, Y.; Lu, Y.; Celiku, O.; Li, A.; Wu, Q.; Zhou, Y.; Yang, C. Targeting IDH1-Mutated Malignancies with NRF2 Blockade. J. Natl. Cancer Inst. 2019, 111, 1033–1041. [Google Scholar] [CrossRef]
- Behrend, L.; Henderson, G.; Zwacka, R.M. Reactive oxygen species in oncogenic transformation. Biochem. Soc. Trans. 2003, 31, 1441–1444. [Google Scholar] [CrossRef]
- Gilbert, M.R.; Liu, Y.; Neltner, J.; Pu, H.; Morris, A.; Sunkara, M.; Pittman, T.; Kyprianou, N.; Horbinski, C. Autophagy and oxidative stress in gliomas with IDH1 mutations. Acta Neuropathol. 2014, 127, 221–233. [Google Scholar] [CrossRef]
- Shi, J.; Sun, B.; Shi, W.; Zuo, H.; Cui, D.; Ni, L.; Chen, J. Decreasing GSH and increasing ROS in chemosensitivity gliomas with IDH1 mutation. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2015, 36, 655–662. [Google Scholar] [CrossRef]
- Mohrenz, I.V.; Antonietti, P.; Pusch, S.; Capper, D.; Balss, J.; Voigt, S.; Weissert, S.; Mukrowsky, A.; Frank, J.; Senft, C.; et al. Isocitrate dehydrogenase 1 mutant R132H sensitizes glioma cells to BCNU-induced oxidative stress and cell death. Apoptosis Int. J. Program. Cell Death 2013, 18, 1416–1425. [Google Scholar] [CrossRef]
- Cai, S.J.; Liu, Y.; Han, S.; Yang, C. Brusatol, an NRF2 inhibitor for future cancer therapeutic. Cell Biosci. 2019, 9, 45. [Google Scholar] [CrossRef]
- Grønbaek, K.; Hother, C.; Jones, P.A. Epigenetic changes in cancer. Acta Pathol. Microbiol. Immunol. Scand. 2007, 115, 1039–1059. [Google Scholar] [CrossRef]
- Kimura, H. Histone modifications for human epigenome analysis. J. Hum. Genet. 2013, 58, 439–445. [Google Scholar] [CrossRef]
- Unruh, D.; Zewde, M.; Buss, A.; Drumm, M.R.; Tran, A.N.; Scholtens, D.M.; Horbinski, C. Methylation and transcription patterns are distinct in IDH mutant gliomas compared to other IDH mutant cancers. Sci. Rep. 2019, 9, 8946. [Google Scholar] [CrossRef]
- Noushmehr, H.; Weisenberger, D.J.; Diefes, K.; Phillips, H.S.; Pujara, K.; Berman, B.P.; Pan, F.; Pelloski, C.E.; Sulman, E.P.; Bhat, K.P.; et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 2010, 17, 510–522. [Google Scholar] [CrossRef]
- Christensen, B.C.; Smith, A.A.; Zheng, S.; Koestler, D.C.; Houseman, E.A.; Marsit, C.J.; Wiemels, J.L.; Nelson, H.H.; Karagas, M.R.; Wrensch, M.R.; et al. DNA methylation, isocitrate dehydrogenase mutation, and survival in glioma. J. Natl. Cancer Inst. 2011, 103, 143–153. [Google Scholar] [CrossRef]
- Malta, T.M.; de Souza, C.F.; Sabedot, T.S.; Silva, T.C.; Mosella, M.S.; Kalkanis, S.N.; Snyder, J.; Castro, A.V.B.; Noushmehr, H. Glioma CpG island methylator phenotype (G-CIMP): Biological and clinical implications. Neuro-oncology 2018, 20, 608–620. [Google Scholar] [CrossRef]
- Tsukada, Y.; Fang, J.; Erdjument-Bromage, H.; Warren, M.E.; Borchers, C.H.; Tempst, P.; Zhang, Y. Histone demethylation by a family of JmjC domain-containing proteins. Nature 2006, 439, 811–816. [Google Scholar] [CrossRef]
- Kohli, R.M.; Zhang, Y. TET enzymes, TDG and the dynamics of DNA demethylation. Nature 2013, 502, 472–479. [Google Scholar] [CrossRef] [PubMed]
- Carrillo, J.A.; Lai, A.; Nghiemphu, P.L.; Kim, H.J.; Phillips, H.S.; Kharbanda, S.; Moftakhar, P.; Lalaezari, S.; Yong, W.; Ellingson, B.M.; et al. Relationship between tumor enhancement, edema, IDH1 mutational status, MGMT promoter methylation, and survival in glioblastoma. Am. J. Neuroradiol. 2012, 33, 1349–1355. [Google Scholar] [CrossRef] [PubMed]
- Duncan, C.G.; Barwick, B.G.; Jin, G.; Rago, C.; Kapoor-Vazirani, P.; Powell, D.R.; Chi, J.T.; Bigner, D.D.; Vertino, P.M.; Yan, H. A heterozygous IDH1R132H/WT mutation induces genome-wide alterations in DNA methylation. Genome Res. 2012, 22, 2339–2355. [Google Scholar] [CrossRef] [PubMed]
- Turcan, S.; Makarov, V.; Taranda, J.; Wang, Y.; Fabius, A.W.M.; Wu, W.; Zheng, Y.; El-Amine, N.; Haddock, S.; Nanjangud, G.; et al. Mutant-IDH1-dependent chromatin state reprogramming, reversibility, and persistence. Nat. Genet. 2018, 50, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Linninger, A.; Hartung, G.A.; Liu, B.P.; Mirkov, S.; Tangen, K.; Lukas, R.V.; Unruh, D.; James, C.D.; Sarkaria, J.N.; Horbinski, C. Modeling the diffusion of D-2-hydroxyglutarate from IDH1 mutant gliomas in the central nervous system. Neuro-oncology 2018, 20, 1197–1206. [Google Scholar] [CrossRef]
- Kalluri, A.L.; Shah, P.P.; Lim, M. The Tumor Immune Microenvironment in Primary CNS Neoplasms: A Review of Current Knowledge and Therapeutic Approaches. Int. J. Mol. Sci. 2023, 24, 2020. [Google Scholar] [CrossRef]
- Amankulor, N.M.; Kim, Y.; Arora, S.; Kargl, J.; Szulzewsky, F.; Hanke, M.; Margineantu, D.H.; Rao, A.; Bolouri, H.; Delrow, J.; et al. Mutant IDH1 regulates the tumor-associated immune system in gliomas. Genes Dev. 2017, 31, 774–786. [Google Scholar] [CrossRef]
- Kohanbash, G.; Carrera, D.A.; Shrivastav, S.; Ahn, B.J.; Jahan, N.; Mazor, T.; Chheda, Z.S.; Downey, K.M.; Watchmaker, P.B.; Beppler, C.; et al. Isocitrate dehydrogenase mutations suppress STAT1 and CD8+ T cell accumulation in gliomas. J. Clin. Investig. 2017, 127, 1425–1437. [Google Scholar] [CrossRef]
- Zhang, X.; Rao, A.; Sette, P.; Deibert, C.; Pomerantz, A.; Kim, W.J.; Kohanbash, G.; Chang, Y.; Park, Y.; Engh, J.; et al. IDH mutant gliomas escape natural killer cell immune surveillance by downregulation of NKG2D ligand expression. Neuro-oncology 2016, 18, 1402–1412. [Google Scholar] [CrossRef]
- Tang, F.; Pan, Z.; Wang, Y.; Lan, T.; Wang, M.; Li, F.; Quan, W.; Liu, Z.; Wang, Z.; Li, Z. Advances in the Immunotherapeutic Potential of Isocitrate Dehydrogenase Mutations in Glioma. Neurosci. Bull. 2022, 38, 1069–1084. [Google Scholar] [CrossRef]
- Poon, C.C.; Gordon, P.M.K.; Liu, K.; Yang, R.; Sarkar, S.; Mirzaei, R.; Ahmad, S.T.; Hughes, M.L.; Yong, V.W.; Kelly, J.J.P. Differential microglia and macrophage profiles in human IDH-mutant and -wild type glioblastoma. Oncotarget 2019, 10, 3129–3143. [Google Scholar] [CrossRef]
- Friedrich, M.; Sankowski, R.; Bunse, L.; Kilian, M.; Green, E.; Ramallo Guevara, C.; Pusch, S.; Poschet, G.; Sanghvi, K.; Hahn, M.; et al. Tryptophan metabolism drives dynamic immunosuppressive myeloid states in IDH-mutant gliomas. Nat. Cancer 2021, 2, 723–740. [Google Scholar] [CrossRef]
- Ma, D.; Zhan, D.; Fu, Y.; Wei, S.; Lal, B.; Wang, J.; Li, Y.; Lopez-Bertoni, H.; Yalcin, F.; Dzaye, O.; et al. Mutant IDH1 promotes phagocytic function of microglia/macrophages in gliomas by downregulating ICAM1. Cancer Lett. 2021, 517, 35–45. [Google Scholar] [CrossRef]
- Zhang, L.; Sorensen, M.D.; Kristensen, B.W.; Reifenberger, G.; McIntyre, T.M.; Lin, F. D-2-Hydroxyglutarate Is an Intercellular Mediator in IDH-Mutant Gliomas Inhibiting Complement and T Cells. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 5381–5391. [Google Scholar] [CrossRef]
- Yang, Q.; Hao, J.; Chi, M.; Wang, Y.; Li, J.; Huang, J.; Zhang, J.; Zhang, M.; Lu, J.; Zhou, S.; et al. D2HGDH-mediated D2HG catabolism enhances the anti-tumor activities of CAR-T cells in an immunosuppressive microenvironment. Mol. Ther. J. Am. Soc. Gene Ther. 2022, 30, 1188–1200. [Google Scholar] [CrossRef]
- Mortazavi, A.; Fayed, I.; Bachani, M.; Dowdy, T.; Jahanipour, J.; Khan, A.; Owotade, J.; Walbridge, S.; Inati, S.K.; Steiner, J.; et al. IDH-mutated gliomas promote epileptogenesis through d-2-hydroxyglutarate-dependent mTOR hyperactivation. Neuro-oncology 2022, 24, 1423–1435. [Google Scholar] [CrossRef]
- Notarangelo, G.; Spinelli, J.B.; Perez, E.M.; Baker, G.J.; Kurmi, K.; Elia, I.; Stopka, S.A.; Baquer, G.; Lin, J.R.; Golby, A.J.; et al. Oncometabolite d-2HG alters T cell metabolism to impair CD8(+) T cell function. Science 2022, 377, 1519–1529. [Google Scholar] [CrossRef]
- Zhao, S.; Lin, Y.; Xu, W.; Jiang, W.; Zha, Z.; Wang, P.; Yu, W.; Li, Z.; Gong, L.; Peng, Y.; et al. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha. Science 2009, 324, 261–265. [Google Scholar] [CrossRef]
- Metellus, P.; Colin, C.; Taieb, D.; Guedj, E.; Nanni-Metellus, I.; de Paula, A.M.; Colavolpe, C.; Fuentes, S.; Dufour, H.; Barrie, M.; et al. IDH mutation status impact on in vivo hypoxia biomarkers expression: New insights from a clinical, nuclear imaging and immunohistochemical study in 33 glioma patients. J. Neuro-Oncol. 2011, 105, 591–600. [Google Scholar] [CrossRef]
- Williams, S.C.; Karajannis, M.A.; Chiriboga, L.; Golfinos, J.G.; von Deimling, A.; Zagzag, D. R132H-mutation of isocitrate dehydrogenase-1 is not sufficient for HIF-1α upregulation in adult glioma. Acta Neuropathol. 2011, 121, 279–281. [Google Scholar] [CrossRef]
- Losman, J.A.; Kaelin, W.G.; Jr. What a difference a hydroxyl makes: Mutant IDH, (R)-2-hydroxyglutarate, and cancer. Genes Dev. 2013, 27, 836–852. [Google Scholar] [CrossRef] [PubMed]
- Koivunen, P.; Lee, S.; Duncan, C.G.; Lopez, G.; Lu, G.; Ramkissoon, S.; Losman, J.A.; Joensuu, P.; Bergmann, U.; Gross, S.; et al. Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature 2012, 483, 484–488. [Google Scholar] [CrossRef] [PubMed]
- Polívka, J., Jr.; Pešta, M.; Pitule, P.; Hes, O.; Holubec, L.; Polívka, J.; Kubíková, T.; Tonar, Z. IDH1 mutation is associated with lower expression of VEGF but not microvessel formation in glioblastoma multiforme. Oncotarget 2018, 9, 16462–16476. [Google Scholar] [CrossRef] [PubMed]
- Kickingereder, P.; Sahm, F.; Radbruch, A.; Wick, W.; Heiland, S.; Deimling, A.; Bendszus, M.; Wiestler, B. IDH mutation status is associated with a distinct hypoxia/angiogenesis transcriptome signature which is non-invasively predictable with rCBV imaging in human glioma. Sci. Rep. 2015, 5, 16238. [Google Scholar] [CrossRef] [PubMed]
- Yalaza, C.; Ak, H.; Cagli, M.S.; Ozgiray, E.; Atay, S.; Aydin, H.H. R132H Mutation in IDH1 Gene is Associated with Increased Tumor HIF1-Alpha and Serum VEGF Levels in Primary Glioblastoma Multiforme. Ann. Clin. Lab. Sci. 2017, 47, 362–364. [Google Scholar]
- Liubinas, S.V.; D’Abaco, G.M.; Moffat, B.M.; Gonzales, M.; Feleppa, F.; Nowell, C.J.; Gorelik, A.; Drummond, K.J.; O’Brien, T.J.; Kaye, A.H.; et al. IDH1 mutation is associated with seizures and protoplasmic subtype in patients with low-grade gliomas. Epilepsia 2014, 55, 1438–1443. [Google Scholar] [CrossRef]
- Chen, H.; Judkins, J.; Thomas, C.; Wu, M.; Khoury, L.; Benjamin, C.G.; Pacione, D.; Golfinos, J.G.; Kumthekar, P.; Ghamsari, F.; et al. Mutant IDH1 and seizures in patients with glioma. Neurology 2017, 88, 1805–1813. [Google Scholar] [CrossRef]
- Correia, C.E.; Umemura, Y.; Flynn, J.R.; Reiner, A.S.; Avila, E.K. Pharmacoresistant seizures and IDH mutation in low-grade gliomas. Neuro-Oncol. Adv. 2021, 3, vdab146. [Google Scholar] [CrossRef]
- Chen, J.; Yang, J.; Cao, P. The Evolving Landscape in the Development of Isocitrate Dehydrogenase Mutant Inhibitors. Mini Rev. Med. Chem. 2016, 16, 1344–1358. [Google Scholar] [CrossRef]
- Kölker, S.; Pawlak, V.; Ahlemeyer, B.; Okun, J.G.; Hörster, F.; Mayatepek, E.; Krieglstein, J.; Hoffmann, G.F.; Köhr, G. NMDA receptor activation and respiratory chain complex V inhibition contribute to neurodegeneration in d-2-hydroxyglutaric aciduria. Eur. J. Neurosci. 2002, 16, 21–28. [Google Scholar] [CrossRef]
- Armstrong, T.S.; Grant, R.; Gilbert, M.R.; Lee, J.W.; Norden, A.D. Epilepsy in glioma patients: Mechanisms, management, and impact of anticonvulsant therapy. Neuro-oncology 2016, 18, 779–789. [Google Scholar] [CrossRef]
- Lange, F.; Hörnschemeyer, J.; Kirschstein, T. Glutamatergic Mechanisms in Glioblastoma and Tumor-Associated Epilepsy. Cells 2021, 10, 1226. [Google Scholar] [CrossRef]
- Andronesi, O.C.; Kim, G.S.; Gerstner, E.; Batchelor, T.; Tzika, A.A.; Fantin, V.R.; Vander Heiden, M.G.; Sorensen, A.G. Detection of 2-hydroxyglutarate in IDH-mutated glioma patients by in vivo spectral-editing and 2D correlation magnetic resonance spectroscopy. Sci. Transl. Med. 2012, 4, 116ra114. [Google Scholar] [CrossRef]
- Herman, M.A.; Nahir, B.; Jahr, C.E. Distribution of extracellular glutamate in the neuropil of hippocampus. PLoS ONE 2011, 6, e26501. [Google Scholar] [CrossRef]
- Baldock, A.L.; Yagle, K.; Born, D.E.; Ahn, S.; Trister, A.D.; Neal, M.; Johnston, S.K.; Bridge, C.A.; Basanta, D.; Scott, J.; et al. Invasion and proliferation kinetics in enhancing gliomas predict IDH1 mutation status. Neuro-oncology 2014, 16, 779–786. [Google Scholar] [CrossRef]
- Molenaar, R.J.; Radivoyevitch, T.; Maciejewski, J.P.; van Noorden, C.J.; Bleeker, F.E. The driver and passenger effects of isocitrate dehydrogenase 1 and 2 mutations in oncogenesis and survival prolongation. Biochim. Biophys. Acta 2014, 1846, 326–341. [Google Scholar] [CrossRef]
- van Lith, S.A.; Molenaar, R.; van Noorden, C.J.; Leenders, W.P. Tumor cells in search for glutamate: An alternative explanation for increased invasiveness of IDH1 mutant gliomas. Neuro-oncology 2014, 16, 1669–1670. [Google Scholar] [CrossRef]
- Colvin, H.; Nishida, N.; Konno, M.; Haraguchi, N.; Takahashi, H.; Nishimura, J.; Hata, T.; Kawamoto, K.; Asai, A.; Tsunekuni, K.; et al. Oncometabolite D-2-Hydroxyglurate Directly Induces Epithelial-Mesenchymal Transition and is Associated with Distant Metastasis in Colorectal Cancer. Sci. Rep. 2016, 6, 36289. [Google Scholar] [CrossRef]
- Lu, J.; Li, D.; Zeng, Y.; Wang, H.; Feng, W.; Qi, S.; Yu, L. IDH1 mutation promotes proliferation and migration of glioma cells via EMT induction. Off. J. Balk. Union Oncol. 2019, 24, 2458–2464. [Google Scholar]
- Cui, D.; Ren, J.; Shi, J.; Feng, L.; Wang, K.; Zeng, T.; Jin, Y.; Gao, L. R132H mutation in IDH1 gene reduces proliferation, cell survival and invasion of human glioma by downregulating Wnt/β-catenin signaling. Int. J. Biochem. Cell Biol. 2016, 73, 72–81. [Google Scholar] [CrossRef]
- Ramachandran, N.; Colman, R.F. Chemical characterization of distinct subunits of pig heart DPN-specific isocitrate dehydrogenase. J. Biol. Chem. 1980, 255, 8859–8864. [Google Scholar] [CrossRef] [PubMed]
- Barnes, L.D.; Kuehn, G.D.; Atkinson, D.E. Yeast diphosphopyridine nucleotide specific isocitrate dehydrogenase. Purification and some properties. Biochemistry 1971, 10, 3939–3944. [Google Scholar] [CrossRef] [PubMed]
- McDonough, M.A.; Loenarz, C.; Chowdhury, R.; Clifton, I.J.; Schofield, C.J. Structural studies on human 2-oxoglutarate dependent oxygenases. Curr. Opin. Struct. Biol. 2010, 20, 659–672. [Google Scholar] [CrossRef] [PubMed]
- Nagaraj, R.; Sharpley, M.S.; Chi, F.; Braas, D.; Zhou, Y.; Kim, R.; Clark, A.T.; Banerjee, U. Nuclear Localization of Mitochondrial TCA Cycle Enzymes as a Critical Step in Mammalian Zygotic Genome Activation. Cell 2017, 168, 210–223.e211. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Long, Q.; Wu, H.; Zhou, Y.; Duan, L.; Yuan, H.; Ding, Y.; Huang, Y.; Wu, Y.; Huang, J.; et al. Nuclear localization of mitochondrial TCA cycle enzymes modulates pluripotency via histone acetylation. Nat. Commun. 2022, 13, 7414. [Google Scholar] [CrossRef]
- Liu, X.; Si, W.; He, L.; Yang, J.; Peng, Y.; Ren, J.; Liu, X.; Jin, T.; Yu, H.; Zhang, Z.; et al. The existence of a nonclassical TCA cycle in the nucleus that wires the metabolic-epigenetic circuitry. Signal Transduct. Target. Ther. 2021, 6, 375. [Google Scholar] [CrossRef]
- Hartong, D.T.; Dange, M.; McGee, T.L.; Berson, E.L.; Dryja, T.P.; Colman, R.F. Insights from retinitis pigmentosa into the roles of isocitrate dehydrogenases in the Krebs cycle. Nat. Genet. 2008, 40, 1230–1234. [Google Scholar] [CrossRef]
- Pierrache, L.H.M.; Kimchi, A.; Ratnapriya, R.; Roberts, L.; Astuti, G.D.N.; Obolensky, A.; Beryozkin, A.; Tjon-Fo-Sang, M.J.H.; Schuil, J.; Klaver, C.C.W.; et al. Whole-Exome Sequencing Identifies Biallelic IDH3A Variants as a Cause of Retinitis Pigmentosa Accompanied by Pseudocoloboma. Ophthalmology 2017, 124, 992–1003. [Google Scholar] [CrossRef]
- Peter, V.G.; Nikopoulos, K.; Quinodoz, M.; Granse, L.; Farinelli, P.; Superti-Furga, A.; Andréasson, S.; Rivolta, C. A novel missense variant in IDH3A causes autosomal recessive retinitis pigmentosa. Ophthalmic Genet. 2019, 40, 177–181. [Google Scholar] [CrossRef]
- Spiegel, R.; Pines, O.; Ta-Shma, A.; Burak, E.; Shaag, A.; Halvardson, J.; Edvardson, S.; Mahajna, M.; Zenvirt, S.; Saada, A.; et al. Infantile cerebellar-retinal degeneration associated with a mutation in mitochondrial aconitase, ACO2. Am. J. Hum. Genet. 2012, 90, 518–523. [Google Scholar] [CrossRef]
- Aptowitzer, I.; Saada, A.; Faber, J.; Kleid, D.; Elpeleg, O.N. Liver disease in the Ashkenazi-Jewish lipoamide dehydrogenase deficiency. J. Pediatr. Gastroenterol. Nutr. 1997, 24, 599–601. [Google Scholar] [CrossRef]
- Elpeleg, O.N.; Saada, A.B.; Shaag, A.; Glustein, J.Z.; Ruitenbeek, W.; Tein, I.; Halevy, J. Lipoamide dehydrogenase deficiency: A new cause for recurrent myoglobinuria. Muscle Nerve 1997, 20, 238–240. [Google Scholar] [CrossRef]
- Shany, E.; Saada, A.; Landau, D.; Shaag, A.; Hershkovitz, E.; Elpeleg, O.N. Lipoamide dehydrogenase deficiency due to a novel mutation in the interface domain. Biochem. Biophys. Res. Commun. 1999, 262, 163–166. [Google Scholar] [CrossRef]
- Rosenberg, M.J.; Agarwala, R.; Bouffard, G.; Davis, J.; Fiermonte, G.; Hilliard, M.S.; Koch, T.; Kalikin, L.M.; Makalowska, I.; Morton, D.H.; et al. Mutant deoxynucleotide carrier is associated with congenital microcephaly. Nat. Genet. 2002, 32, 175–179. [Google Scholar] [CrossRef]
- Spiegel, R.; Shaag, A.; Edvardson, S.; Mandel, H.; Stepensky, P.; Shalev, S.A.; Horovitz, Y.; Pines, O.; Elpeleg, O. SLC25A19 mutation as a cause of neuropathy and bilateral striatal necrosis. Ann. Neurol. 2009, 66, 419–424. [Google Scholar] [CrossRef]
- Alston, C.L.; Davison, J.E.; Meloni, F.; van der Westhuizen, F.H.; He, L.; Hornig-Do, H.T.; Peet, A.C.; Gissen, P.; Goffrini, P.; Ferrero, I.; et al. Recessive germline SDHA and SDHB mutations causing leukodystrophy and isolated mitochondrial complex II deficiency. J. Med. Genet. 2012, 49, 569–577. [Google Scholar] [CrossRef]
- Jackson, C.B.; Nuoffer, J.M.; Hahn, D.; Prokisch, H.; Haberberger, B.; Gautschi, M.; Häberli, A.; Gallati, S.; Schaller, A. Mutations in SDHD lead to autosomal recessive encephalomyopathy and isolated mitochondrial complex II deficiency. J. Med. Genet. 2014, 51, 170–175. [Google Scholar] [CrossRef]
- Gellera, C.; Uziel, G.; Rimoldi, M.; Zeviani, M.; Laverda, A.; Carrara, F.; DiDonato, S. Fumarase deficiency is an autosomal recessive encephalopathy affecting both the mitochondrial and the cytosolic enzymes. Neurology 1990, 40, 495–499. [Google Scholar] [CrossRef]
- Fattal-Valevski, A.; Eliyahu, H.; Fraenkel, N.D.; Elmaliach, G.; Hausman-Kedem, M.; Shaag, A.; Mandel, D.; Pines, O.; Elpeleg, O. Homozygous mutation, p.Pro304His, in IDH3A, encoding isocitrate dehydrogenase subunit is associated with severe encephalopathy in infancy. Neurogenetics 2017, 18, 57–61. [Google Scholar] [CrossRef]
- Krell, D.; Assoku, M.; Galloway, M.; Mulholland, P.; Tomlinson, I.; Bardella, C. Screen for IDH1, IDH2, IDH3, D2HGDH and L2HGDH mutations in glioblastoma. PLoS ONE 2011, 6, e19868. [Google Scholar] [CrossRef]
- Zeng, L.; Morinibu, A.; Kobayashi, M.; Zhu, Y.; Wang, X.; Goto, Y.; Yeom, C.J.; Zhao, T.; Hirota, K.; Shinomiya, K.; et al. Aberrant IDH3α expression promotes malignant tumor growth by inducing HIF-1-mediated metabolic reprogramming and angiogenesis. Oncogene 2015, 34, 4758–4766. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Wang, Y.; Shi, Z.; Liu, J.; Sun, P.; Hou, X.; Zhang, J.; Zhao, S.; Zhou, B.P.; Mi, J. Metabolic reprogramming of cancer-associated fibroblasts by IDH3α downregulation. Cell Rep. 2015, 10, 1335–1348. [Google Scholar] [CrossRef] [PubMed]
- May, J.L.; Kouri, F.M.; Hurley, L.A.; Liu, J.; Tommasini-Ghelfi, S.; Ji, Y.; Gao, P.; Calvert, A.E.; Lee, A.; Chandel, N.S.; et al. IDH3α regulates one-carbon metabolism in glioblastoma. Sci. Adv. 2019, 5, eaat0456. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Qiao, Y.; Ting, X.; Si, W. Isocitrate dehydrogenase 3A, a rate-limiting enzyme of the TCA cycle, promotes hepatocellular carcinoma migration and invasion through regulation of MTA1, a core component of the NuRD complex. Am. J. Cancer Res. 2020, 10, 3212–3229. [Google Scholar] [PubMed]
- Rohle, D.; Popovici-Muller, J.; Palaskas, N.; Turcan, S.; Grommes, C.; Campos, C.; Tsoi, J.; Clark, O.; Oldrini, B.; Komisopoulou, E.; et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science 2013, 340, 626–630. [Google Scholar] [CrossRef]
- Popovici-Muller, J.; Lemieux, R.M.; Artin, E.; Saunders, J.O.; Salituro, F.G.; Travins, J.; Cianchetta, G.; Cai, Z.; Zhou, D.; Cui, D.; et al. Discovery of AG-120 (Ivosidenib): A First-in-Class Mutant IDH1 Inhibitor for the Treatment of IDH1 Mutant Cancers. ACS Med. Chem. Lett. 2018, 9, 300–305. [Google Scholar] [CrossRef]
- Johannessen, T.A.; Mukherjee, J.; Viswanath, P.; Ohba, S.; Ronen, S.M.; Bjerkvig, R.; Pieper, R.O. Rapid Conversion of Mutant IDH1 from Driver to Passenger in a Model of Human Gliomagenesis. Mol. Cancer Res. 2016, 14, 976–983. [Google Scholar] [CrossRef]
- Tateishi, K.; Wakimoto, H.; Iafrate, A.J.; Tanaka, S.; Loebel, F.; Lelic, N.; Wiederschain, D.; Bedel, O.; Deng, G.; Zhang, B.; et al. Extreme Vulnerability of IDH1 Mutant Cancers to NAD+ Depletion. Cancer Cell 2015, 28, 773–784. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Stein, E.M.; de Botton, S.; Roboz, G.J.; Altman, J.K.; Mims, A.S.; Swords, R.; Collins, R.H.; Mannis, G.N.; Pollyea, D.A.; et al. Durable Remissions with Ivosidenib in IDH1-Mutated Relapsed or Refractory AML. N. Engl. J. Med. 2018, 378, 2386–2398. [Google Scholar] [CrossRef]
- Mellinghoff, I.; Penas-Prado, M.; Peters, K.; Cloughesy, T.; Burris, H.; Maher, E.; Janku, F.; Cote, G.; De La Fuente, M.; Clarke, J.; et al. Actr-31. Phase 1 Study of Ag-881, an Inhibitor of Mutant Idh1 and Idh2: Results from the Recurrent/Progressive Glioma Population. Neuro Oncol. 2018, 20, vi18. [Google Scholar] [CrossRef]
- Yen, K.; Travins, J.; Wang, F.; David, M.D.; Artin, E.; Straley, K.; Padyana, A.; Gross, S.; DeLaBarre, B.; Tobin, E.; et al. AG-221, a First-in-Class Therapy Targeting Acute Myeloid Leukemia Harboring Oncogenic IDH2 Mutations. Cancer Discov. 2017, 7, 478–493. [Google Scholar] [CrossRef]
- Wang, F.; Travins, J.; DeLaBarre, B.; Penard-Lacronique, V.; Schalm, S.; Hansen, E.; Straley, K.; Kernytsky, A.; Liu, W.; Gliser, C.; et al. Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science 2013, 340, 622–626. [Google Scholar] [CrossRef]
- Yang, B.; Zhong, C.; Peng, Y.; Lai, Z.; Ding, J. Molecular mechanisms of “off-on switch” of activities of human IDH1 by tumor-associated mutation R132H. Cell Res. 2010, 20, 1188–1200. [Google Scholar] [CrossRef]
- Stein, E.M. Enasidenib, a targeted inhibitor of mutant IDH2 proteins for treatment of relapsed or refractory acute myeloid leukemia. Future Oncol. 2018, 14, 23–40. [Google Scholar] [CrossRef]
- Burris, H.; Mellinghoff, I.; Maher, E.; Wen, P.; Beeram, M.; Touat, M.; Faris, J.; Azad, N.; Cloughesy, T.; Gore, L.; et al. Abstract PL04-05: The first reported results of AG-120, a first-in-class, potent inhibitor of the IDH1 mutant protein, in a Phase I study of patients with advanced IDH1-mutant solid tumors, including gliomas. Mol. Cancer Ther. 2015, 14, PL04-05. [Google Scholar] [CrossRef]
- Fan, B.; Mellinghoff, I.K.; Wen, P.Y.; Lowery, M.A.; Goyal, L.; Tap, W.D.; Pandya, S.S.; Manyak, E.; Jiang, L.; Liu, G.; et al. Clinical pharmacokinetics and pharmacodynamics of ivosidenib, an oral, targeted inhibitor of mutant IDH1, in patients with advanced solid tumors. Investig. New Drugs 2020, 38, 433–444. [Google Scholar] [CrossRef]
- Yen, K.; Konteatis, Z.; Sui, Z.; Artin, E.; Dang, L.; Straley, K.; Tobin, E.; Campos, C.; Yang, H.; Nagaraja, R.; et al. Abstract B126: AG-881, a brain penetrant, potent, pan-mutant IDH (mIDH) inhibitor for use in mIDH solid and hematologic malignancies. Mol. Cancer Ther. 2018, 17, B126. [Google Scholar] [CrossRef]
- Mellinghoff, I.K.; Bent, M.J.V.D.; Clarke, J.L.; Maher, E.A.; Peters, K.B.; Touat, M.; Groot, J.F.D.; Fuente, M.I.D.L.; Arrillaga-Romany, I.; Wick, W.; et al. INDIGO: A global, randomized, double-blind, phase III study of vorasidenib (VOR.; AG-881) vs placebo in patients (pts) with residual or recurrent grade II glioma with an isocitrate dehydrogenase 1/2 (IDH1/2) mutation. J. Clin. Oncol. 2020, 38, TPS2574. [Google Scholar] [CrossRef]
- Molenaar, R.J.; Botman, D.; Smits, M.A.; Hira, V.V.; van Lith, S.A.; Stap, J.; Henneman, P.; Khurshed, M.; Lenting, K.; Mul, A.N.; et al. Radioprotection of IDH1-Mutated Cancer Cells by the IDH1-Mutant Inhibitor AGI-5198. Cancer Res. 2015, 75, 4790–4802. [Google Scholar] [CrossRef]
- Pusch, S.; Krausert, S.; Fischer, V.; Balss, J.; Ott, M.; Schrimpf, D.; Capper, D.; Sahm, F.; Eisel, J.; Beck, A.C.; et al. Pan-mutant IDH1 inhibitor BAY 1436032 for effective treatment of IDH1 mutant astrocytoma in vivo. Acta Neuropathol. 2017, 133, 629–644. [Google Scholar] [CrossRef]
- Heuser, M.; Palmisiano, N.; Mantzaris, I.; Mims, A.; DiNardo, C.; Silverman, L.R.; Wang, E.S.; Fiedler, W.; Baldus, C.; Schwind, S.; et al. Safety and efficacy of BAY1436032 in IDH1-mutant AML: Phase I study results. Leukemia 2020, 34, 2903–2913. [Google Scholar] [CrossRef]
- Cho, Y.S.; Levell, J.R.; Liu, G.; Caferro, T.; Sutton, J.; Shafer, C.M.; Costales, A.; Manning, J.R.; Zhao, Q.; Sendzik, M.; et al. Discovery and Evaluation of Clinical Candidate IDH305, a Brain Penetrant Mutant IDH1 Inhibitor. ACS Med. Chem. Lett. 2017, 8, 1116–1121. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, C.D.; Hochhaus, A.; Frattini, M.G.; Yee, K.; Zander, T.; Krämer, A.; Chen, X.; Ji, Y.; Parikh, N.S.; Choi, J.; et al. A phase 1 study of IDH305 in patients with IDH1(R132)-mutant acute myeloid leukemia or myelodysplastic syndrome. J. Cancer Res. Clin. Oncol. 2023, 149, 1145–1158. [Google Scholar] [CrossRef] [PubMed]
- Tian, W.; Zhang, W.; Wang, Y.; Jin, R.; Wang, Y.; Guo, H.; Tang, Y.; Yao, X. Recent advances of IDH1 mutant inhibitor in cancer therapy. Front. Pharmacol. 2022, 13, 982424. [Google Scholar] [CrossRef] [PubMed]
- Gatto, L.; Franceschi, E.; Tosoni, A.; Di Nunno, V.; Maggio, I.; Lodi, R.; Brandes, A.A. IDH Inhibitors and Beyond: The Cornerstone of Targeted Glioma Treatment. Mol. Diagn. Ther. 2021, 25, 457–473. [Google Scholar] [CrossRef]
- de la Fuente, M.I.; Colman, H.; Rosenthal, M.; Van Tine, B.A.; Levacic, D.; Walbert, T.; Gan, H.K.; Vieito, M.; Milhem, M.M.; Lipford, K.; et al. Olutasidenib (FT-2102) in patients with relapsed or refractory IDH1-mutant glioma: A multicenter, open-label, phase Ib/II trial. Neuro-oncology 2023, 25, 146–156. [Google Scholar] [CrossRef]
- Okoye-Okafor, U.C.; Bartholdy, B.; Cartier, J.; Gao, E.N.; Pietrak, B.; Rendina, A.R.; Rominger, C.; Quinn, C.; Smallwood, A.; Wiggall, K.J.; et al. New IDH1 mutant inhibitors for treatment of acute myeloid leukemia. Nat. Chem. Biol. 2015, 11, 878–886. [Google Scholar] [CrossRef]
- Natsume, A.; Arakawa, Y.; Narita, Y.; Sugiyama, K.; Hata, N.; Muragaki, Y.; Shinojima, N.; Kumabe, T.; Saito, R.; Motomura, K.; et al. The first-in-human phase I study of a brain-penetrant mutant IDH1 inhibitor DS-1001 in patients with recurrent or progressive IDH1-mutant gliomas. Neuro-oncology 2023, 25, 326–336. [Google Scholar] [CrossRef]
- Dhillon, S. Ivosidenib: First Global Approval. Drugs 2018, 78, 1509–1516. [Google Scholar] [CrossRef]
- Deng, G.; Shen, J.; Yin, M.; McManus, J.; Mathieu, M.; Gee, P.; He, T.; Shi, C.; Bedel, O.; McLean, L.R.; et al. Selective inhibition of mutant isocitrate dehydrogenase 1 (IDH1) via disruption of a metal binding network by an allosteric small molecule. J. Biol. Chem. 2015, 290, 762–774. [Google Scholar] [CrossRef]
- Bello, L.; Giussani, C.; Carrabba, G.; Pluderi, M.; Costa, F.; Bikfalvi, A. Angiogenesis and invasion in gliomas. Cancer Treat. Res. 2004, 117, 263–284. [Google Scholar] [CrossRef]
- Heredia, V.; Mendiola, M.; Ortiz, E.; Bernabéu, D.; Pozo-Kreilinger, J.J.; Miguel, M.; Crespo, R.; Berjón, A.; Martínez-Marín, V.; Redondo, A. AG-120, a novel IDH1 targeted molecule, inhibits invasion and migration of chondrosarcoma cells in vitro. Ann. Oncol. 2017, 28, v538. [Google Scholar] [CrossRef]
- Nicolay, B.; Narayanaswamy, R.; Aguado, E.; Nagaraja, R.; Murtie, J.; Liu, G.; Ishii, Y. EXTH-59. The IDH1 mutant inhibitor AG-120 shows strong inhibition of 2-HG production in an orthotopic IDH1 mutant glioma model in vivo. Neuro-oncology 2017, 19, vi86. [Google Scholar] [CrossRef]
- Yen, K.; Chopra, V.S.; Tobin, E.; Avanzino, B.; Mavrommatis, K.; DiMartino, J.; MacBeth, K.J. Abstract 4956: Functional characterization of the ivosidenib (AG-120) and azacitidine combination in a mutant IDH1 AML cell model. Cancer Res. 2018, 78, 4956. [Google Scholar] [CrossRef]
- Mellinghoff, I.K.; Wen, P.Y.; Taylor, J.W.; Maher, E.A.; Arrillaga-Romany, I.; Peters, K.B.; Le, K.; Tai, F.; Steelman, L.; Cloughesy, T.F. PL3.1 A phase 1, open-label, perioperative study of ivosidenib (AG-120) and vorasidenib (AG-881) in recurrent, IDH1-mutant, low-grade glioma: Results from cohort 1. Neuro Oncol. 2019, 21, iii2. [Google Scholar] [CrossRef]
- Ma, R.; Yun, C.H. Crystal structures of pan-IDH inhibitor AG-881 in complex with mutant human IDH1 and IDH2. Biochem. Biophys. Res. Commun. 2018, 503, 2912–2917. [Google Scholar] [CrossRef]
- Konteatis, Z.; Artin, E.; Nicolay, B.; Straley, K.; Padyana, A.K.; Jin, L.; Chen, Y.; Narayaraswamy, R.; Tong, S.; Wang, F.; et al. Vorasidenib (AG-881): A First-in-Class, Brain-Penetrant Dual Inhibitor of Mutant IDH1 and 2 for Treatment of Glioma. ACS Med. Chem. Lett. 2020, 11, 101–107. [Google Scholar] [CrossRef]
- Golub, D.; Iyengar, N.; Dogra, S.; Wong, T.; Bready, D.; Tang, K.; Modrek, A.S.; Placantonakis, D.G. Mutant Isocitrate Dehydrogenase Inhibitors as Targeted Cancer Therapeutics. Front. Oncol. 2019, 9, 417. [Google Scholar] [CrossRef]
- Kopinja, J.; Sevilla, R.S.; Levitan, D.; Dai, D.; Vanko, A.; Spooner, E.; Ware, C.; Forget, R.; Hu, K.; Kral, A.; et al. A Brain Penetrant Mutant IDH1 Inhibitor Provides In Vivo Survival Benefit. Sci. Rep. 2017, 7, 13853. [Google Scholar] [CrossRef]
- Sulkowski, P.L.; Corso, C.D.; Robinson, N.D.; Scanlon, S.E.; Purshouse, K.R.; Bai, H.; Liu, Y.; Sundaram, R.K.; Hegan, D.C.; Fons, N.R.; et al. 2-Hydroxyglutarate produced by neomorphic IDH mutations suppresses homologous recombination and induces PARP inhibitor sensitivity. Sci. Transl. Med. 2017, 9, aal2463. [Google Scholar] [CrossRef]
- Watts, C.; Savage, J.; Patel, A.; Mant, R.; Wykes, V.; Pohl, U.; Bulbeck, H.; Apps, J.; Sharpe, R.; Thompson, G.; et al. Protocol for the Tessa Jowell BRAIN MATRIX Platform Study. BMJ Open 2022, 12, e067123. [Google Scholar] [CrossRef] [PubMed]
- Lai, A.; Kharbanda, S.; Pope, W.B.; Tran, A.; Solis, O.E.; Peale, F.; Forrest, W.F.; Pujara, K.; Carrillo, J.A.; Pandita, A.; et al. Evidence for sequenced molecular evolution of IDH1 mutant glioblastoma from a distinct cell of origin. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2011, 29, 4482–4490. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.E.; Mazor, T.; Hong, C.; Barnes, M.; Aihara, K.; McLean, C.Y.; Fouse, S.D.; Yamamoto, S.; Ueda, H.; Tatsuno, K.; et al. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science 2014, 343, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Bai, H.; Harmancı, A.S.; Erson-Omay, E.Z.; Li, J.; Coşkun, S.; Simon, M.; Krischek, B.; Özduman, K.; Omay, S.B.; Sorensen, E.A.; et al. Integrated genomic characterization of IDH1-mutant glioma malignant progression. Nat. Genet. 2016, 48, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.; Bunse, L.; Pusch, S.; Sahm, F.; Wiestler, B.; Quandt, J.; Menn, O.; Osswald, M.; Oezen, I.; Ott, M.; et al. A vaccine targeting mutant IDH1 induces antitumour immunity. Nature 2014, 512, 324–327. [Google Scholar] [CrossRef]
- Platten, M.; Bunse, L.; Wick, A.; Bunse, T.; Le Cornet, L.; Harting, I.; Sahm, F.; Sanghvi, K.; Tan, C.L.; Poschke, I.; et al. A vaccine targeting mutant IDH1 in newly diagnosed glioma. Nature 2021, 592, 463–468. [Google Scholar] [CrossRef]
- Wahner, H.C.W.; Träger, M.; Bender, K.; Schweizer, L.; Onken, J.; Senger, C.; Ehret, F.; Budach, V.; Kaul, D. Predicting survival in anaplastic astrocytoma patients in a single-center cohort of 108 patients. Radiat. Oncol. 2020, 15, 282. [Google Scholar] [CrossRef]
- Christians, A.; Adel-Horowski, A.; Banan, R.; Lehmann, U.; Bartels, S.; Behling, F.; Barrantes-Freer, A.; Stadelmann, C.; Rohde, V.; Stockhammer, F.; et al. The prognostic role of IDH mutations in homogeneously treated patients with anaplastic astrocytomas and glioblastomas. Acta Neuropathol. Commun. 2019, 7, 156. [Google Scholar] [CrossRef]
- Huang, J.; Yu, J.; Tu, L.; Huang, N.; Li, H.; Luo, Y. Isocitrate Dehydrogenase Mutations in Glioma: From Basic Discovery to Therapeutics Development. Front. Oncol. 2019, 9, 506. [Google Scholar] [CrossRef]
- Waitkus, M.S.; Diplas, B.H.; Yan, H. Biological Role and Therapeutic Potential of IDH Mutations in Cancer. Cancer Cell 2018, 34, 186–195. [Google Scholar] [CrossRef]
- Doi, A.; Park, I.H.; Wen, B.; Murakami, P.; Aryee, M.J.; Irizarry, R.; Herb, B.; Ladd-Acosta, C.; Rho, J.; Loewer, S.; et al. Differential methylation of tissue- and cancer-specific CpG island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts. Nat. Genet. 2009, 41, 1350–1353. [Google Scholar] [CrossRef]
- Turcan, S.; Rohle, D.; Goenka, A.; Walsh, L.A.; Fang, F.; Yilmaz, E.; Campos, C.; Fabius, A.W.; Lu, C.; Ward, P.S.; et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 2012, 483, 479–483. [Google Scholar] [CrossRef]
- Flavahan, W.A.; Drier, Y.; Liau, B.B.; Gillespie, S.M.; Venteicher, A.S.; Stemmer-Rachamimov, A.O.; Suvà, M.L.; Bernstein, B.E. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature 2016, 529, 110–114. [Google Scholar] [CrossRef]
- Lokker, N.A.; Sullivan, C.M.; Hollenbach, S.J.; Israel, M.A.; Giese, N.A. Platelet-derived growth factor (PDGF) autocrine signaling regulates survival and mitogenic pathways in glioblastoma cells: Evidence that the novel PDGF-C and PDGF-D ligands may play a role in the development of brain tumors. Cancer Res. 2002, 62, 3729–3735. [Google Scholar]
- Turcan, S.; Fabius, A.W.; Borodovsky, A.; Pedraza, A.; Brennan, C.; Huse, J.; Viale, A.; Riggins, G.J.; Chan, T.A. Efficient induction of differentiation and growth inhibition in IDH1 mutant glioma cells by the DNMT Inhibitor Decitabine. Oncotarget 2013, 4, 1729–1736. [Google Scholar] [CrossRef]
- Borodovsky, A.; Salmasi, V.; Turcan, S.; Fabius, A.W.; Baia, G.S.; Eberhart, C.G.; Weingart, J.D.; Gallia, G.L.; Baylin, S.B.; Chan, T.A.; et al. 5-azacytidine reduces methylation, promotes differentiation and induces tumor regression in a patient-derived IDH1 mutant glioma xenograft. Oncotarget 2013, 4, 1737–1747. [Google Scholar] [CrossRef]
- Rastogi, R.P.; Richa; Kumar, A.; Tyagi, M.B.; Sinha, R.P. Molecular mechanisms of ultraviolet radiation-induced DNA damage and repair. J. Nucleic Acids 2010, 2010, 592980. [Google Scholar] [CrossRef]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA damage, repair, and mutagenesis. Environ. Mol. Mutagen. 2017, 58, 235–263. [Google Scholar] [CrossRef]
- Wang, P.; Wu, J.; Ma, S.; Zhang, L.; Yao, J.; Hoadley, K.A.; Wilkerson, M.D.; Perou, C.M.; Guan, K.L.; Ye, D.; et al. Oncometabolite D-2-Hydroxyglutarate Inhibits ALKBH DNA Repair Enzymes and Sensitizes IDH Mutant Cells to Alkylating Agents. Cell Rep. 2015, 13, 2353–2361. [Google Scholar] [CrossRef]
- Chen, F.; Bian, K.; Tang, Q.; Fedeles, B.I.; Singh, V.; Humulock, Z.T.; Essigmann, J.M.; Li, D. Oncometabolites d- and l-2-Hydroxyglutarate Inhibit the AlkB Family DNA Repair Enzymes under Physiological Conditions. Chem. Res. Toxicol. 2017, 30, 1102–1110. [Google Scholar] [CrossRef]
- Morales, J.; Li, L.; Fattah, F.J.; Dong, Y.; Bey, E.A.; Patel, M.; Gao, J.; Boothman, D.A. Review of poly (ADP-ribose) polymerase (PARP) mechanisms of action and rationale for targeting in cancer and other diseases. Crit. Rev. Eukaryot. Gene Expr. 2014, 24, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Tateishi, K.; Higuchi, F.; Miller, J.J.; Koerner, M.V.A.; Lelic, N.; Shankar, G.M.; Tanaka, S.; Fisher, D.E.; Batchelor, T.T.; Iafrate, A.J.; et al. The Alkylating Chemotherapeutic Temozolomide Induces Metabolic Stress in IDH1-Mutant Cancers and Potentiates NAD(+) Depletion-Mediated Cytotoxicity. Cancer Res. 2017, 77, 4102–4115. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.K.; Kizilbash, S.H.; Carlson, B.L.; Mladek, A.C.; Boakye-Agyeman, F.; Bakken, K.K.; Pokorny, J.L.; Schroeder, M.A.; Decker, P.A.; Cen, L.; et al. Delineation of MGMT Hypermethylation as a Biomarker for Veliparib-Mediated Temozolomide-Sensitizing Therapy of Glioblastoma. J. Natl. Cancer Inst. 2016, 108. [Google Scholar] [CrossRef]
- Okamoto, K.; Seimiya, H. Revisiting Telomere Shortening in Cancer. Cells 2019, 8, 107. [Google Scholar] [CrossRef]
- Mukherjee, J.; Johannessen, T.C.; Ohba, S.; Chow, T.T.; Jones, L.; Pandita, A.; Pieper, R.O. Mutant IDH1 Cooperates with ATRX Loss to Drive the Alternative Lengthening of Telomere Phenotype in Glioma. Cancer Res. 2018, 78, 2966–2977. [Google Scholar] [CrossRef]
- Zhang, J.M.; Zou, L. Alternative lengthening of telomeres: From molecular mechanisms to therapeutic outlooks. Cell Biosci. 2020, 10, 30. [Google Scholar] [CrossRef]
- Garten, A.; Schuster, S.; Penke, M.; Gorski, T.; de Giorgis, T.; Kiess, W. Physiological and pathophysiological roles of NAMPT and NAD metabolism. Nat. Rev. Endocrinol. 2015, 11, 535–546. [Google Scholar] [CrossRef]
- Emadi, A.; Jun, S.A.; Tsukamoto, T.; Fathi, A.T.; Minden, M.D.; Dang, C.V. Inhibition of glutaminase selectively suppresses the growth of primary acute myeloid leukemia cells with IDH mutations. Exp. Hematol. 2014, 42, 247–251. [Google Scholar] [CrossRef]
- Elhammali, A.; Ippolito, J.E.; Collins, L.; Crowley, J.; Marasa, J.; Piwnica-Worms, D. A high-throughput fluorimetric assay for 2-hydroxyglutarate identifies Zaprinast as a glutaminase inhibitor. Cancer Discov. 2014, 4, 828–839. [Google Scholar] [CrossRef]
- Matre, P.; Velez, J.; Jacamo, R.; Qi, Y.; Su, X.; Cai, T.; Chan, S.M.; Lodi, A.; Sweeney, S.R.; Ma, H.; et al. Inhibiting glutaminase in acute myeloid leukemia: Metabolic dependency of selected AML subtypes. Oncotarget 2016, 7, 79722–79735. [Google Scholar] [CrossRef]
- Kizilbash, S.H.; McBrayer, S.; Port, J.; Reid, J.M.; Lanza, I.; Allred, J.B.; Chakravarti, A.; Kunos, C.; Adjei, A.A. A phase Ib trial of CB-839 (telaglenastat) in combination with radiation therapy and temozolomide in patients with IDH-mutated diffuse astrocytoma and anaplastic astrocytoma (NCT03528642). J. Clin. Oncol. 2019, 37, TPS2075. [Google Scholar] [CrossRef]
- Garrett, M.; Sperry, J.; Braas, D.; Yan, W.; Le, T.M.; Mottahedeh, J.; Ludwig, K.; Eskin, A.; Qin, Y.; Levy, R.; et al. Metabolic characterization of isocitrate dehydrogenase (IDH) mutant and IDH wildtype gliomaspheres uncovers cell type-specific vulnerabilities. Cancer Metab. 2018, 6, 4. [Google Scholar] [CrossRef]
- Andronesi, O.C.; Arrillaga-Romany, I.C.; Ly, K.I.; Bogner, W.; Ratai, E.M.; Reitz, K.; Iafrate, A.J.; Dietrich, J.; Gerstner, E.R.; Chi, A.S.; et al. Pharmacodynamics of mutant-IDH1 inhibitors in glioma patients probed by in vivo 3D MRS imaging of 2-hydroxyglutarate. Nat. Commun. 2018, 9, 1474. [Google Scholar] [CrossRef]
- McBrayer, S.K.; Mayers, J.R.; DiNatale, G.J.; Shi, D.D.; Khanal, J.; Chakraborty, A.A.; Sarosiek, K.A.; Briggs, K.J.; Robbins, A.K.; Sewastianik, T.; et al. Transaminase Inhibition by 2-Hydroxyglutarate Impairs Glutamate Biosynthesis and Redox Homeostasis in Glioma. Cell 2018, 175, 101–116.e125. [Google Scholar] [CrossRef]
- Sasaki, M.; Knobbe, C.B.; Munger, J.C.; Lind, E.F.; Brenner, D.; Brustle, A.; Harris, I.S.; Holmes, R.; Wakeham, A.; Haight, J.; et al. IDH1(R132H) mutation increases murine haematopoietic progenitors and alters epigenetics. Nature 2012, 488, 656–659. [Google Scholar] [CrossRef]
- Lu, C.; Ward, P.S.; Kapoor, G.S.; Rohle, D.; Turcan, S.; Abdel-Wahab, O.; Edwards, C.R.; Khanin, R.; Figueroa, M.E.; Melnick, A.; et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012, 483, 474–478. [Google Scholar] [CrossRef]
- Saha, S.K.; Parachoniak, C.A.; Ghanta, K.S.; Fitamant, J.; Ross, K.N.; Najem, M.S.; Gurumurthy, S.; Akbay, E.A.; Sia, D.; Cornella, H.; et al. Mutant IDH inhibits HNF-4α to block hepatocyte differentiation and promote biliary cancer. Nature 2014, 513, 110–114. [Google Scholar] [CrossRef]
- Hirata, M.; Sasaki, M.; Cairns, R.A.; Inoue, S.; Puviindran, V.; Li, W.Y.; Snow, B.E.; Jones, L.D.; Wei, Q.; Sato, S.; et al. Mutant IDH is sufficient to initiate enchondromatosis in mice. Proc. Natl. Acad. Sci. USA 2015, 112, 2829–2834. [Google Scholar] [CrossRef]
- Hansen, E.; Quivoron, C.; Straley, K.; Lemieux, R.M.; Popovici-Muller, J.; Sadrzadeh, H.; Fathi, A.T.; Gliser, C.; David, M.; Saada, V.; et al. AG-120, an Oral, Selective, First-in-Class, Potent Inhibitor of Mutant IDH1, Reduces Intracellular 2HG and Induces Cellular Differentiation in TF-1 R132H Cells and Primary Human IDH1 Mutant AML Patient Samples Treated Ex Vivo. Blood 2014, 124, 3734. [Google Scholar] [CrossRef]
- Norsworthy, K.J.; Luo, L.; Hsu, V.; Gudi, R.; Dorff, S.E.; Przepiorka, D.; Deisseroth, A.; Shen, Y.L.; Sheth, C.M.; Charlab, R.; et al. FDA Approval Summary: Ivosidenib for Relapsed or Refractory Acute Myeloid Leukemia with an Isocitrate Dehydrogenase-1 Mutation. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 3205–3209. [Google Scholar] [CrossRef]
- Polychronidou, G.; Karavasilis, V.; Pollack, S.M.; Huang, P.H.; Lee, A.; Jones, R.L. Novel therapeutic approaches in chondrosarcoma. Future Oncol. (Lond. Engl.) 2017, 13, 637–648. [Google Scholar] [CrossRef] [PubMed]
- Zeidner, J.F. Differentiating the Differentiation Syndrome Associated with IDH Inhibitors in AML. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2020, 26, 4174–4176. [Google Scholar] [CrossRef] [PubMed]
- Reyhanoglu, G.; Hughes, B.; King, K.E.; Cambridge, R. Differentiation Syndrome, a Side Effect From the Therapy of Acute Promyelocytic Leukemia. Cureus 2020, 12, e12042. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Drug Name | Target | Mechanism of Action | Advantages | Disadvantages | Current Clinical Trial in Gliomas (s) | Sponsor (s) |
|---|---|---|---|---|---|---|
| AG 120 Ivosidenib (F.D.A. Approved) [169,175,176] | IDH1 R132 C, H, G, S, L | Reversible, allosteric competitive inhibitor | F.D.A. approval was granted in 2018 for relapsed/refractory acute myeloid leukaemia, and in 2019 for newly diagnosed. In 2021, F.D.A received application for the drug to be assessed for cholangiocarcinoma. Currently under investigation in several clinical trials in haematological malignancies. | It is unknown whether it penetrates the BBB, with a 4.1% penetrance in a rat model. | NCT02073994: Phase 1, multicentre, single group assignment, open-label, dose-escalation/safety and clinical activity trial of oral administration for solid tumour, including gliomas. A total of 170 patients to be recruited by 2022. NCT03343197: A phase 1, randomised, multicentre, controlled, open-label, parallel assignment, the perioperative study of AG120 and AG881in patients with non-enhancing IDH1 mutant glioma, both grade II and III. A total of 45 patients estimated. | Agios/Celgene |
| AG 221 Enasidenib (F.D.A. Approved) [174] | IDH2 R140Q, R172K | Allosteric non-competitive inhibitor | F.D.A. approval was granted in 2017 for relapsed/refractory acute myeloid. Currently under investigation in many clinical trials in haematological malignancies. | No information on penetrance through the BBB. | NCT02273739: A phase1/2 multicentre, open-label, dose-escalation trial for solid tumours, including gliomas. The trial was completed with 21 participants. | Agios/Celgene |
| AG 881 [170,177,178] | Pan Inhibitor IDH1&2 | Allosteric non-competitive inhibitor | Penetrance of the BBB in a rat model. Pan inhibitor with an advanced clinical trial design showing good tolerability and safety profile. | NCT02481154: Phase 1, multicentre, open-label, single group assignment, dose-escalation/safety and clinical activity trial of oral administration for gliomas with IDH1 or IDH2 mutation. The trial was completed in 2021. NCT04164901: Phase 3, multicentre, randomised, double-blind, placebo-controlled study of AG-881 in subjects with residual or recurrent grade 2 glioma with an IDH1 or IDH2 mutation. Approximately 366 participants are planned to be randomised 1:1 to receive orally administered Vorasidenib 50 mg QD or placebo. Press release published March 2023. | Agios/Celgene | |
| AGI 5198 [179] | IDH1 R132 C, H | Reversible, allosteric competitive inhibitor to α-KG | Penetrance of BBB in mouse glioma xenografts. | Agios/Celgene | ||
| BAY 1436032 [180,181] | IDH1 R132 C, G, H, L, S | Allosteric non-competitive inhibitor | Penetrance (low): 0.06–0.38 brain to plasma ratio of BBB in a mouse model. | NCT02746081: A phase 1, open-label, non-randomised, multicentre trial of tolerance and pharmacodynamic evaluation in solid tumour with IDH1 mutation. A total of 81 patients. | Bayer | |
| IDH 305 [182,183] | IDH1 R132 C, H | Allosteric non-competitive inhibitor | Penetrance of BBB in murine models. | NCT02381886: A phase 1, single group assignment, open-label trial for advanced malignancies harbouring IDHR132H mutations. A total of 166 patients. Regimen of 75–750 mg twice daily. Complete remission (CR) or CR with incomplete count recovery occurred in 10/37 (27%) patients with AML and 1/4 patients with myelodysplastic syndrome. Adverse events (AEs) suspected to be related to the study drug were reported in 53.7% of patients. Results published March 2023. | Novartis | |
| AGI 6780 [184,185] | IDH2 R140Q | Allosteric non-competitive inhibitor | Unknown penetrance through the BBB. | Agios/Celgene | ||
| FT-2102 [186] | IDH1 R132 C | Competitive inhibitor | Unknown penetrance through the BBB. | NCT03684811: A phase 1b/2, non-randomised, parallel assignment, open-label study of recurrent/progressed glioma plus other tumours with IDH1 mutation. A total of 200 patients estimated. | Forma Hannover Medical School (Germany) | |
| MFK A [184,185] | IDH1 R132 C, H | Unknown | Penetrance through BBB in mouse model shown with brain-to-plasma ratio >1. | Unknown penetrance through the BBB. | Merck | |
| GSK 321 [187] | IDH1 R132 C, H, G | Reversible, allosteric non-competitive inhibitor | Unknown penetrance through the BBB. | GSK | ||
| ML 309 [184,185] | IDH1 R132 H | Reversible inhibitor | Unknown penetrance through the BBB. | |||
| Ds 1001b [188] | IDH1 R132 X | Direct inhibitor | Shown to penetrate the BBB both in human and mouse xenograft models. Designed to penetrate the BBB. | NCT03030066: A phase 1, single group assignment, open-label study. A total of 47 participants. Twice daily oral administration resulted in anti-tumour activity in patients with recurrent/progressive IDH1-mutated glioma. Results published February 2023. | Daichi Sankyo |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Solomou, G.; Finch, A.; Asghar, A.; Bardella, C. Mutant IDH in Gliomas: Role in Cancer and Treatment Options. Cancers 2023, 15, 2883. https://doi.org/10.3390/cancers15112883
Solomou G, Finch A, Asghar A, Bardella C. Mutant IDH in Gliomas: Role in Cancer and Treatment Options. Cancers. 2023; 15(11):2883. https://doi.org/10.3390/cancers15112883
Chicago/Turabian StyleSolomou, Georgios, Alina Finch, Asim Asghar, and Chiara Bardella. 2023. "Mutant IDH in Gliomas: Role in Cancer and Treatment Options" Cancers 15, no. 11: 2883. https://doi.org/10.3390/cancers15112883