
Epigenetic Insights on PARP-1 Activity in Cancer Therapy

Abstract
:Simple Summary
Abstract
1. Introduction
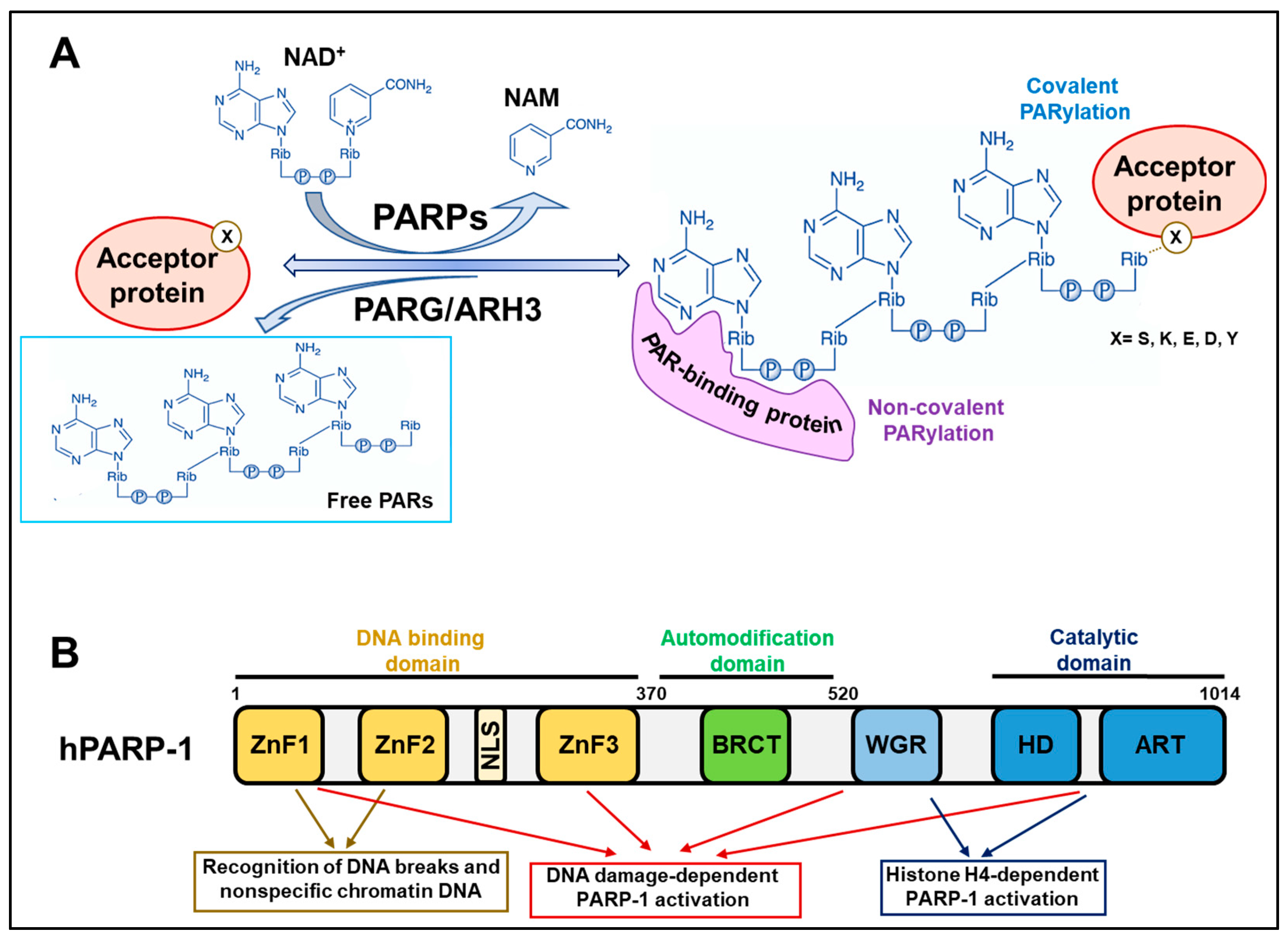
2. Background Knowledge of PARP Reactions
2.1. Covalent PARylation
2.2. Non-Covalent PARylation
3. PARP-1 Activity Shapes Chromatin during DNA Repair and Transcription
4. PARP-1 and DNA Epigenetic Modifications
4.1. DNA Methylation
4.2. DNA Demethylation and Hydroxymethylation
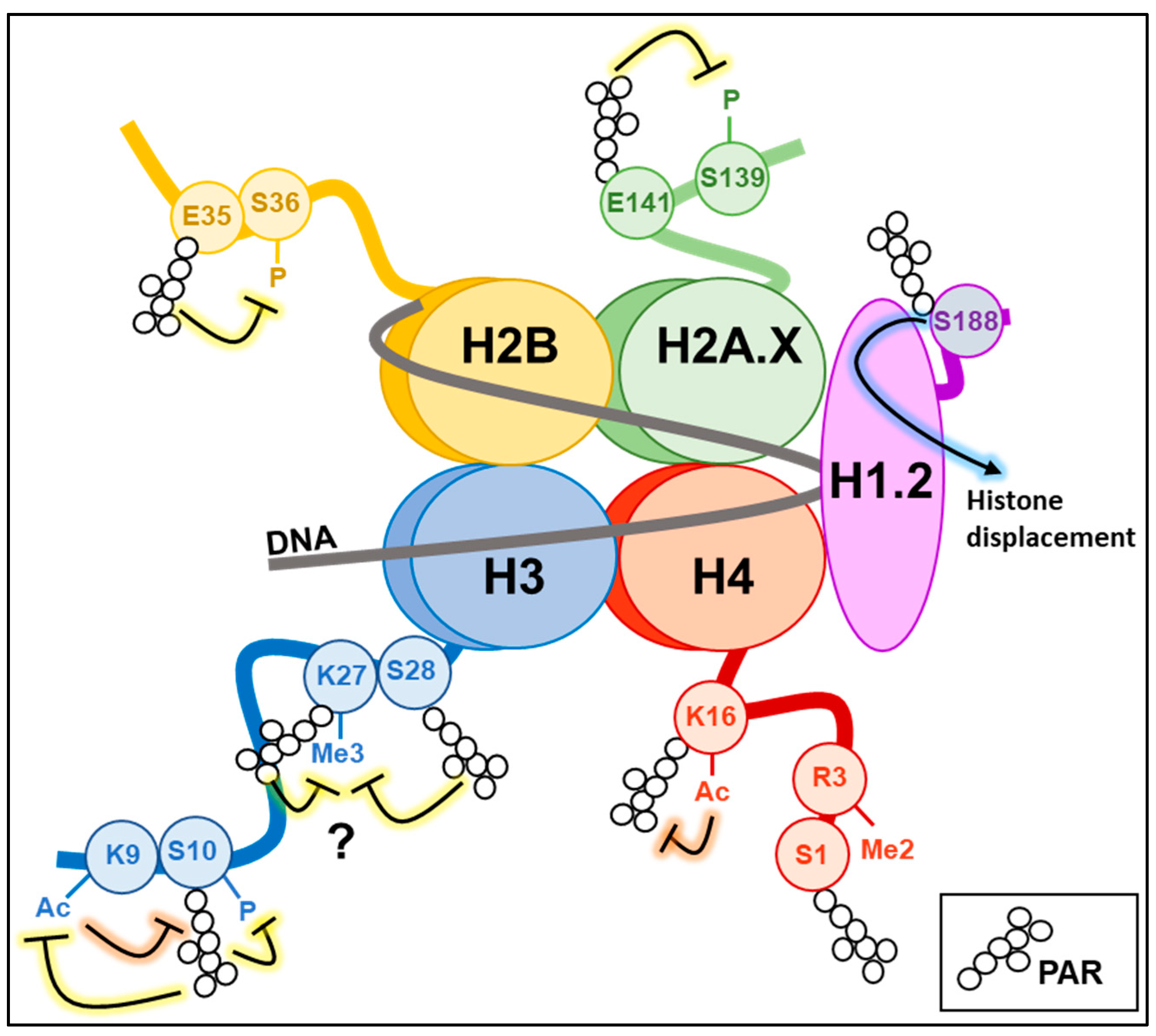
5. PARylation and Histone Modifications
5.1. Histone H1
5.2. Histones H2A/H2B and Their Variants
5.3. Histone H3

{kind=link}
{kind=link}
| Enzyme | Activity | PARylation | Outcome |
|---|---|---|---|
| DNMT1 | DNA methyltransferase | Non-Covalent | Enzyme inhibition [41] |
| TET1/2 | 5mC hydroxylase | Covalent Non-Covalent | Enzyme inhibition [48,49]; recruitment at gene regulatory regions [50] |
| EZH2 | H3K27 methyltransferase | Covalent | Enzyme inhibition [72]; dissociation from PRC2 complex [73] |
| KDM4D | H3K9me3/2 demethylase | Covalent | Recruitment at DNA damage sites [74]; putative enzyme inhibition [76] |
| KDM5A | H3K4me3 demethylase | Non-Covalent | Recruitment at DNA damage sites [67] |
| KDM5B | H3K4me3 demethylase | Covalent | Enzyme inhibition [34]; recruitment at DNA damage sites [75] |
| NSD2 | H3K36 demethylase | Covalent | Enzyme inhibition and impairment of nucleosome binding [77] |
| p300 | Histone acetyltransferase | Covalent | Enzyme activation [78] |
5.4. Histone H4
6. Epigenetic Mechanisms in PARPi-Based Cancer Therapy
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Gibson, B.A.; Kraus, W.L. New Insights into the Molecular and Cellular Functions of Poly(ADP-Ribose) and PARPs. Nat. Rev. Mol. Cell Biol. 2012, 13, 411–424. [Google Scholar] [CrossRef]
- Hassa, P.O.; Hottiger, M.O. The Diverse Biological Roles of Mammalian PARPS, a Small but Powerful Family of Poly-ADP-Ribose Polymerases. Front. Biosci. 2008, 13, 3046–3082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crawford, K.; Bonfiglio, J.J.; Mikoč, A.; Matic, I.; Ahel, I. Specificity of Reversible ADP-Ribosylation and Regulation of Cellular Processes. Crit. Rev. Biochem. Mol. Biol 2018, 53, 64–82. [Google Scholar] [CrossRef] [PubMed]
- Lüscher, B.; Ahel, I.; Altmeyer, M.; Ashworth, A.; Bai, P.; Chang, P.; Cohen, M.; Corda, D.; Dantzer, F.; Daugherty, M.D.; et al. ADP-ribosyltransferases, an update on function and nomenclature. FEBS J. 2021, 289, 7399–7410. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Kraus, W.L. The Expanding Universe of PARP1-Mediated Molecular and Therapeutic Mechanisms. Mol. Cell 2022, 82, 2315–2334. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.N.; Carbone, M.; Mostocotto, C.; Mancone, C.; Tripodi, M.; Maione, R.; Amati, P. Mitochondrial Localization of PARP-1 Requires Interaction with Mitofilin and Is Involved in the Maintenance of Mitochondrial DNA Integrity. J. Biol. Chem. 2009, 284, 31616–31624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific Killing of BRCA2-Deficient Tumours with Inhibitors of Poly(ADP-Ribose) Polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA Repair Defect in BRCA Mutant Cells as a Therapeutic Strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. PARP Inhibitors: Synthetic Lethality in the Clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef] [PubMed]
- Giudice, E.; Gentile, M.; Salutari, V.; Ricci, C.; Musacchio, L.; Carbone, M.V.; Ghizzoni, V.; Camarda, F.; Tronconi, F.; Nero, C.; et al. PARP Inhibitors Resistance: Mechanisms and Perspectives. Cancers 2022, 14, 1420. [Google Scholar] [CrossRef]
- Murai, J.; Huang, S.N.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res 2012, 72, 5588–5599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pommier, Y.; O’Connor, M.J.; de Bono, J. Laying a Trap to Kill Cancer Cells: PARP Inhibitors and Their Mechanisms of Action. Sci. Transl. Med. 2016, 8, 362ps17. [Google Scholar] [CrossRef] [PubMed]
- Pettitt, S.J.; Krastev, D.B.; Brandsma, I.; Dréan, A.; Song, F.; Aleksandrov, R.; Harrell, M.I.; Menon, M.; Brough, R.; Campbell, J.; et al. Genome-Wide and High-Density CRISPR-Cas9 Screens Identify Point Mutations in PARP1 Causing PARP Inhibitor Resistance. Nat. Commun. 2018, 9, 1849. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.K.; Harrell, M.I.; Oza, A.M.; Oaknin, A.; Ray-Coquard, I.; Tinker, A.V.; Helman, E.; Radke, M.R.; Say, C.; Vo, L.-T.; et al. BRCA Reversion Mutations in Circulating Tumor DNA Predict Primary and Acquired Resistance to the PARP Inhibitor Rucaparib in High-Grade Ovarian Carcinoma. Cancer Discov. 2019, 9, 210–219. [Google Scholar] [CrossRef] [Green Version]
- Jaspers, J.E.; Kersbergen, A.; Boon, U.; Sol, W.; van Deemter, L.; Zander, S.A.; Drost, R.; Wientjens, E.; Ji, J.; Aly, A.; et al. Loss of 53BP1 Causes PARP Inhibitor Resistance in Brca1-Mutated Mouse Mammary Tumors. Cancer Discov. 2013, 3, 68–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray Chaudhuri, A.; Nussenzweig, A. The Multifaceted Roles of PARP1 in DNA Repair and Chromatin Remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621. [Google Scholar] [CrossRef]
- Zong, W.; Gong, Y.; Sun, W.; Li, T.; Wang, Z.-Q. PARP1: Liaison of Chromatin Remodeling and Transcription. Cancers 2022, 14, 4162. [Google Scholar] [CrossRef]
- Vyas, S.; Matic, I.; Uchima, L.; Rood, J.; Zaja, R.; Hay, R.T.; Ahel, I.; Chang, P. Family-Wide Analysis of Poly(ADP-Ribose) Polymerase Activity. Nat. Commun. 2014, 5, 4426. [Google Scholar] [CrossRef] [Green Version]
- Messner, S.; Altmeyer, M.; Zhao, H.; Pozivil, A.; Roschitzki, B.; Gehrig, P.; Rutishauser, D.; Huang, D.; Caflisch, A.; Hottiger, M.O. PARP1 ADP-Ribosylates Lysine Residues of the Core Histone Tails. Nucleic Acids Res. 2010, 38, 6350–6362. [Google Scholar] [CrossRef] [Green Version]
- Leslie Pedrioli, D.M.; Leutert, M.; Bilan, V.; Nowak, K.; Gunasekera, K.; Ferrari, E.; Imhof, R.; Malmström, L.; Hottiger, M.O. Comprehensive ADP-Ribosylome Analysis Identifies Tyrosine as an ADP-Ribose Acceptor Site. EMBO Rep. 2018, 19, e45310. [Google Scholar] [CrossRef]
- Bartlett, E.; Bonfiglio, J.J.; Prokhorova, E.; Colby, T.; Zobel, F.; Ahel, I.; Matic, I. Interplay of Histone Marks with Serine ADP-Ribosylation. Cell Rep. 2018, 24, 3488–3502.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suskiewicz, M.J.; Zobel, F.; Ogden, T.E.H.; Fontana, P.; Ariza, A.; Yang, J.-C.; Zhu, K.; Bracken, L.; Hawthorne, W.J.; Ahel, D.; et al. HPF1 Completes the PARP Active Site for DNA Damage-Induced ADP-Ribosylation. Nature 2020, 579, 598–602. [Google Scholar] [CrossRef] [PubMed]
- Prokhorova, E.; Zobel, F.; Smith, R.; Zentout, S.; Gibbs-Seymour, I.; Schützenhofer, K.; Peters, A.; Groslambert, J.; Zorzini, V.; Agnew, T.; et al. Serine-Linked PARP1 Auto-Modification Controls PARP Inhibitor Response. Nat. Commun. 2021, 12, 4055. [Google Scholar] [CrossRef] [PubMed]
- Kamaletdinova, T.; Fanaei-Kahrani, Z.; Wang, Z.-Q. The Enigmatic Function of PARP1: From PARylation Activity to PAR Readers. Cells 2019, 8, 1625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teloni, F.; Altmeyer, M. Readers of Poly(ADP-Ribose): Designed to Be Fit for Purpose. Nucleic Acids Res. 2016, 44, 993–1006. [Google Scholar] [CrossRef] [Green Version]
- Wacker, D.A.; Ruhl, D.D.; Balagamwala, E.H.; Hope, K.M.; Zhang, T.; Kraus, W.L. The DNA Binding and Catalytic Domains of Poly(ADP-Ribose) Polymerase 1 Cooperate in the Regulation of Chromatin Structure and Transcription. Mol. Cell Biol. 2007, 27, 7475–7485. [Google Scholar] [CrossRef] [Green Version]
- Langelier, M.-F.; Pascal, J.M. PARP-1 Mechanism for Coupling DNA Damage Detection to Poly(ADP-Ribose) Synthesis. Curr. Opin. Struct. Biol. 2013, 23, 134–143. [Google Scholar] [CrossRef] [Green Version]
- Muthurajan, U.M.; Hepler, M.R.D.; Hieb, A.R.; Clark, N.J.; Kramer, M.; Yao, T.; Luger, K. Automodification Switches PARP-1 Function from Chromatin Architectural Protein to Histone Chaperone. Proc. Natl. Acad. Sci. USA 2014, 111, 12752–12757. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.Y.; Mauro, S.; Gévry, N.; Lis, J.T.; Kraus, W.L. NAD+-Dependent Modulation of Chromatin Structure and Transcription by Nucleosome Binding Properties of PARP-1. Cell 2004, 119, 803–814. [Google Scholar] [CrossRef] [Green Version]
- Thomas, C.; Ji, Y.; Wu, C.; Datz, H.; Boyle, C.; MacLeod, B.; Patel, S.; Ampofo, M.; Currie, M.; Harbin, J.; et al. Hit and Run versus Long-Term Activation of PARP-1 by Its Different Domains Fine-Tunes Nuclear Processes. Proc. Natl. Acad. Sci. USA 2019, 116, 9941–9946. [Google Scholar] [CrossRef]
- Ciccarone, F.; Zampieri, M.; Caiafa, P. PARP1 Orchestrates Epigenetic Events Setting up Chromatin Domains. Semin. Cell Dev. Biol. 2017, 63, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Langelier, M.-F.; Planck, J.L.; Roy, S.; Pascal, J.M. Crystal Structures of Poly(ADP-Ribose) Polymerase-1 (PARP-1) Zinc Fingers Bound to DNA: Structural and Functional Insights into DNA-Dependent PARP-1 Activity. J. Biol. Chem. 2011, 286, 10690–10701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotova, E.Y.; Hsieh, F.-K.; Chang, H.-W.; Maluchenko, N.V.; Langelier, M.-F.; Pascal, J.M.; Luse, D.S.; Feofanov, A.V.; Studitsky, V.M. Human PARP1 Facilitates Transcription through a Nucleosome and Histone Displacement by Pol II In Vitro. Int. J. Mol. Sci. 2022, 23, 7107. [Google Scholar] [CrossRef]
- Krishnakumar, R.; Kraus, W.L. PARP-1 Regulates Chromatin Structure and Transcription through a KDM5B-Dependent Pathway. Mol. Cell 2010, 39, 736–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenberg, M.V.C.; Bourc’his, D. The Diverse Roles of DNA Methylation in Mammalian Development and Disease. Nat. Rev. Mol. Cell Biol. 2019, 20, 590–607. [Google Scholar] [CrossRef] [PubMed]
- Nocchi, L.; Tomasetti, M.; Amati, M.; Neuzil, J.; Santarelli, L.; Saccucci, F. Thrombomodulin Is Silenced in Malignant Mesothelioma by a Poly(ADP-Ribose) Polymerase-1-Mediated Epigenetic Mechanism. J. Biol. Chem. 2011, 286, 19478–19488. [Google Scholar] [CrossRef] [Green Version]
- Zampieri, M.; Passananti, C.; Calabrese, R.; Perilli, M.; Corbi, N.; De Cave, F.; Guastafierro, T.; Bacalini, M.G.; Reale, A.; Amicosante, G.; et al. Parp1 Localizes within the Dnmt1 Promoter and Protects Its Unmethylated State by Its Enzymatic Activity. PLoS ONE 2009, 4, e4717. [Google Scholar] [CrossRef]
- Zampieri, M.; Guastafierro, T.; Calabrese, R.; Ciccarone, F.; Bacalini, M.G.; Reale, A.; Perilli, M.; Passananti, C.; Caiafa, P. ADP-Ribose Polymers Localized on Ctcf-Parp1-Dnmt1 Complex Prevent Methylation of Ctcf Target Sites. Biochem. J. 2012, 441, 645–652. [Google Scholar] [CrossRef]
- Ciccarone, F.; Valentini, E.; Bacalini, M.G.; Zampieri, M.; Calabrese, R.; Guastafierro, T.; Mariano, G.; Reale, A.; Franceschi, C.; Caiafa, P. Poly(ADP-Ribosyl)Ation Is Involved in the Epigenetic Control of TET1 Gene Transcription. Oncotarget 2014, 5, 10356–10367. [Google Scholar] [CrossRef] [Green Version]
- Caiafa, P.; Guastafierro, T.; Zampieri, M. Epigenetics: Poly(ADP-Ribosyl)Ation of PARP-1 Regulates Genomic Methylation Patterns. FASEB J. 2009, 23, 672–678. [Google Scholar] [CrossRef]
- Reale, A.; Matteis, G.D.; Galleazzi, G.; Zampieri, M.; Caiafa, P. Modulation of DNMT1 Activity by ADP-Ribose Polymers. Oncogene 2005, 24, 13–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, H.; Sifakis, E.G.; Sumida, N.; Millán-Ariño, L.; Scholz, B.A.; Svensson, J.P.; Chen, X.; Ronnegren, A.L.; Mallet de Lima, C.D.; Varnoosfaderani, F.S.; et al. PARP1- and CTCF-Mediated Interactions between Active and Repressed Chromatin at the Lamina Promote Oscillating Transcription. Mol. Cell 2015, 59, 984–997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witcher, M.; Emerson, B.M. Epigenetic Silencing of the P16(INK4a) Tumor Suppressor Is Associated with Loss of CTCF Binding and a Chromatin Boundary. Mol. Cell 2009, 34, 271–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, W.; Ginjala, V.; Pant, V.; Chernukhin, I.; Whitehead, J.; Docquier, F.; Farrar, D.; Tavoosidana, G.; Mukhopadhyay, R.; Kanduri, C.; et al. Poly(ADP-Ribosyl)Ation Regulates CTCF-Dependent Chromatin Insulation. Nat. Genet. 2004, 36, 1105–1110. [Google Scholar] [CrossRef] [Green Version]
- Guastafierro, T.; Cecchinelli, B.; Zampieri, M.; Reale, A.; Riggio, G.; Sthandier, O.; Zupi, G.; Calabrese, L.; Caiafa, P. CCCTC-Binding Factor Activates PARP-1 Affecting DNA Methylation Machinery. J. Biol. Chem. 2008, 283, 21873–21880. [Google Scholar] [CrossRef] [Green Version]
- Nalabothula, N.; Al-jumaily, T.; Eteleeb, A.M.; Flight, R.M.; Xiaorong, S.; Moseley, H.; Rouchka, E.C.; Fondufe-Mittendorf, Y.N. Genome-Wide Profiling of PARP1 Reveals an Interplay with Gene Regulatory Regions and DNA Methylation. PLoS ONE 2015, 10, e0135410. [Google Scholar] [CrossRef]
- Guetg, C.; Scheifele, F.; Rosenthal, F.; Hottiger, M.O.; Santoro, R. Inheritance of Silent RDNA Chromatin Is Mediated by PARP1 via Noncoding RNA. Mol. Cell 2012, 45, 790–800. [Google Scholar] [CrossRef] [Green Version]
- Ciccarone, F.; Valentini, E.; Zampieri, M.; Caiafa, P. 5mC-Hydroxylase Activity Is Influenced by the PARylation of TET1 Enzyme. Oncotarget 2015, 6, 24333–24347. [Google Scholar] [CrossRef] [Green Version]
- Tolić, A.; Ravichandran, M.; Rajić, J.; Đorđević, M.; Đorđević, M.; Dinić, S.; Grdović, N.; Jovanović, J.A.; Mihailović, M.; Nestorović, N.; et al. TET-Mediated DNA Hydroxymethylation Is Negatively Influenced by the PARP-Dependent PARylation. Epigenetics Chromatin 2022, 15, 11. [Google Scholar] [CrossRef]
- Fujiki, K.; Shinoda, A.; Kano, F.; Sato, R.; Shirahige, K.; Murata, M. PPARγ-Induced PARylation Promotes Local DNA Demethylation by Production of 5-Hydroxymethylcytosine. Nat. Commun. 2013, 4, 2262. [Google Scholar] [CrossRef]
- Bannister, A.J.; Kouzarides, T. Regulation of Chromatin by Histone Modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef]
- Ogata, N.; Ueda, K.; Kagamiyama, H.; Hayaishi, O. ADP-Ribosylation of Histone H1. Identification of Glutamic Acid Residues 2, 14, and the COOH-Terminal Lysine Residue as Modification Sites. J. Biol. Chem. 1980, 255, 7616–7620. [Google Scholar] [CrossRef] [PubMed]
- Panzeter, P.L.; Zweifel, B.; Malanga, M.; Waser, S.H.; Richard, M.; Althaus, F.R. Targeting of Histone Tails by Poly(ADP-Ribose). J. Biol. Chem. 1993, 268, 17662–17664. [Google Scholar] [CrossRef]
- Rouleau, M.; Aubin, R.A.; Poirier, G.G. Poly(ADP-Ribosyl)Ated Chromatin Domains: Access Granted. J. Cell. Sci. 2004, 117, 815–825. [Google Scholar] [CrossRef] [Green Version]
- Strickfaden, H.; McDonald, D.; Kruhlak, M.J.; Haince, J.-F.; Th’ng, J.P.H.; Rouleau, M.; Ishibashi, T.; Corry, G.N.; Ausio, J.; Underhill, D.A.; et al. Poly(ADP-Ribosyl)Ation-Dependent Transient Chromatin Decondensation and Histone Displacement Following Laser Microirradiation. J. Biol. Chem. 2016, 291, 1789–1802. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Li, Y.; Tang, M.; Peng, B.; Lu, X.; Yang, Q.; Zhu, Q.; Hou, T.; Li, M.; Liu, C.; et al. Destabilization of Linker Histone H1.2 Is Essential for ATM Activation and DNA Damage Repair. Cell. Res. 2018, 28, 756–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azad, G.K.; Ito, K.; Sailaja, B.S.; Biran, A.; Nissim-Rafinia, M.; Yamada, Y.; Brown, D.T.; Takizawa, T.; Meshorer, E. PARP1-Dependent Eviction of the Linker Histone H1 Mediates Immediate Early Gene Expression during Neuronal Activation. J. Cell Biol. 2018, 217, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Wright, R.H.G.; Castellano, G.; Bonet, J.; Le Dily, F.; Font-Mateu, J.; Ballaré, C.; Nacht, A.S.; Soronellas, D.; Oliva, B.; Beato, M. CDK2-Dependent Activation of PARP-1 Is Required for Hormonal Gene Regulation in Breast Cancer Cells. Genes Dev. 2012, 26, 1972–1983. [Google Scholar] [CrossRef] [Green Version]
- Krishnakumar, R.; Gamble, M.J.; Frizzell, K.M.; Berrocal, J.G.; Kininis, M.; Kraus, W.L. Reciprocal Binding of PARP-1 and Histone H1 at Promoters Specifies Transcriptional Outcomes. Science 2008, 319, 819–821. [Google Scholar] [CrossRef]
- Martinez-Zamudio, R.; Ha, H.C. Histone ADP-Ribosylation Facilitates Gene Transcription by Directly Remodeling Nucleosomes. Mol. Cell Biol. 2012, 32, 2490–2502. [Google Scholar] [CrossRef]
- Yang, G.; Chen, Y.; Wu, J.; Chen, S.-H.; Liu, X.; Singh, A.K.; Yu, X. Poly(ADP-Ribosyl)Ation Mediates Early Phase Histone Eviction at DNA Lesions. Nucleic Acids Res. 2020, 48, 3001–3013. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Camacho, C.V.; Setlem, R.; Ryu, K.W.; Parameswaran, B.; Gupta, R.K.; Kraus, W.L. Functional Interplay between Histone H2B ADP-Ribosylation and Phosphorylation Controls Adipogenesis. Mol. Cell 2020, 79, 934–949.e14. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Bian, C.; Wang, X.; Liu, X.; Ahmad Kassab, M.; Yu, Y.; Yu, X. ADP-Ribosylation of Histone Variant H2AX Promotes Base Excision Repair. EMBO J. 2021, 40, e104542. [Google Scholar] [CrossRef] [PubMed]
- Timinszky, G.; Till, S.; Hassa, P.O.; Hothorn, M.; Kustatscher, G.; Nijmeijer, B.; Colombelli, J.; Altmeyer, M.; Stelzer, E.H.K.; Scheffzek, K.; et al. A Macrodomain-Containing Histone Rearranges Chromatin upon Sensing PARP1 Activation. Nat. Struct. Mol. Biol. 2009, 16, 923–929. [Google Scholar] [CrossRef] [PubMed]
- Ouararhni, K.; Hadj-Slimane, R.; Ait-Si-Ali, S.; Robin, P.; Mietton, F.; Harel-Bellan, A.; Dimitrov, S.; Hamiche, A. The Histone Variant MH2A1.1 Interferes with Transcription by down-Regulating PARP-1 Enzymatic Activity. Genes Dev. 2006, 20, 3324–3336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Ruiz, P.D.; Novikov, L.; Casill, A.D.; Park, J.W.; Gamble, M.J. MacroH2A1.1 and PARP-1 Cooperate to Regulate Transcription by Promoting CBP-Mediated H2B Acetylation. Nat. Struct. Mol. Biol. 2014, 21, 981–989. [Google Scholar] [CrossRef] [Green Version]
- Kumbhar, R.; Sanchez, A.; Perren, J.; Gong, F.; Corujo, D.; Medina, F.; Devanathan, S.K.; Xhemalce, B.; Matouschek, A.; Buschbeck, M.; et al. Poly(ADP-Ribose) Binding and MacroH2A Mediate Recruitment and Functions of KDM5A at DNA Lesions. J. Cell Biol. 2021, 220, e202006149. [Google Scholar] [CrossRef]
- Fuks, F. DNA Methylation and Histone Modifications: Teaming up to Silence Genes. Curr. Opin. Genet. Dev. 2005, 15, 490–495. [Google Scholar] [CrossRef]
- Palazzo, L.; Leidecker, O.; Prokhorova, E.; Dauben, H.; Matic, I.; Ahel, I. Serine Is the Major Residue for ADP-Ribosylation upon DNA Damage. Elife 2018, 7, e34334. [Google Scholar] [CrossRef]
- Larsen, S.C.; Hendriks, I.A.; Lyon, D.; Jensen, L.J.; Nielsen, M.L. Systems-Wide Analysis of Serine ADP-Ribosylation Reveals Widespread Occurrence and Site-Specific Overlap with Phosphorylation. Cell Rep. 2018, 24, 2493–2505.e4. [Google Scholar] [CrossRef]
- Liszczak, G.; Diehl, K.L.; Dann, G.P.; Muir, T.W. Acetylation Blocks DNA Damage-Induced Chromatin ADP-Ribosylation. Nat. Chem. Biol. 2018, 14, 837–840. [Google Scholar] [CrossRef]
- Caruso, L.B.; Martin, K.A.; Lauretti, E.; Hulse, M.; Siciliano, M.; Lupey-Green, L.N.; Abraham, A.; Skorski, T.; Tempera, I. Poly(ADP-Ribose) Polymerase 1, PARP1, Modifies EZH2 and Inhibits EZH2 Histone Methyltransferase Activity after DNA Damage. Oncotarget 2018, 9, 10585–10605. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, H.; Du, Y.; Nakai, K.; Ding, M.; Chang, S.-S.; Hsu, J.L.; Yao, J.; Wei, Y.; Nie, L.; Jiao, S.; et al. EZH2 Contributes to the Response to PARP Inhibitors through Its PARP-Mediated Poly-ADP Ribosylation in Breast Cancer. Oncogene 2018, 37, 208–217. [Google Scholar] [CrossRef] [Green Version]
- Khoury-Haddad, H.; Guttmann-Raviv, N.; Ipenberg, I.; Huggins, D.; Jeyasekharan, A.D.; Ayoub, N. PARP1-Dependent Recruitment of KDM4D Histone Demethylase to DNA Damage Sites Promotes Double-Strand Break Repair. Proc. Natl. Acad. Sci. USA 2014, 111, E728–E737. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Liu, L.; Yang, S.; Song, N.; Zhou, X.; Gao, J.; Yu, N.; Shan, L.; Wang, Q.; Liang, J.; et al. Histone Demethylase KDM5B Is a Key Regulator of Genome Stability. Proc. Natl. Acad. Sci. USA 2014, 111, 7096–7101. [Google Scholar] [CrossRef] [Green Version]
- Le May, N.; Iltis, I.; Amé, J.-C.; Zhovmer, A.; Biard, D.; Egly, J.-M.; Schreiber, V.; Coin, F. Poly (ADP-Ribose) Glycohydrolase Regulates Retinoic Acid Receptor-Mediated Gene Expression. Mol. Cell 2012, 48, 785–798. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; LeDuc, R.D.; Fornelli, L.; Schunter, A.J.; Bennett, R.L.; Kelleher, N.L.; Licht, J.D. Defining the NSD2 Interactome: PARP1 PARylation Reduces NSD2 Histone Methyltransferase Activity and Impedes Chromatin Binding. J. Biol. Chem. 2019, 294, 12459–12471. [Google Scholar] [CrossRef]
- Sobczak, M.; Pitt, A.R.; Spickett, C.M.; Robaszkiewicz, A. PARP1 Co-Regulates EP300-BRG1-Dependent Transcription of Genes Involved in Breast Cancer Cell Proliferation and DNA Repair. Cancers 2019, 11, 1539. [Google Scholar] [CrossRef] [Green Version]
- Shogren-Knaak, M.; Ishii, H.; Sun, J.-M.; Pazin, M.J.; Davie, J.R.; Peterson, C.L. Histone H4-K16 Acetylation Controls Chromatin Structure and Protein Interactions. Science 2006, 311, 844–847. [Google Scholar] [CrossRef] [Green Version]
- Dhar, S.; Gursoy-Yuzugullu, O.; Parasuram, R.; Price, B.D. The Tale of a Tail: Histone H4 Acetylation and the Repair of DNA Breaks. Philos. Trans. R Soc. Lond. B Biol. Sci. 2017, 372, 284. [Google Scholar] [CrossRef]
- Maifrede, S.; Le, B.V.; Nieborowska-Skorska, M.; Golovine, K.; Sullivan-Reed, K.; Dunuwille, W.M.B.; Nacson, J.; Hulse, M.; Keith, K.; Madzo, J.; et al. TET2 and DNMT3A Mutations Exert Divergent Effects on DNA Repair and Sensitivity of Leukemia Cells to PARP Inhibitors. Cancer Res. 2021, 81, 5089–5101. [Google Scholar] [CrossRef]
- Jing, C.-B.; Fu, C.; Prutsch, N.; Wang, M.; He, S.; Look, A.T. Synthetic Lethal Targeting of TET2-Mutant Hematopoietic Stem and Progenitor Cells (HSPCs) with TOP1-Targeted Drugs and PARP1 Inhibitors. Leukemia 2020, 34, 2992–3006. [Google Scholar] [CrossRef]
- Bamezai, S.; Demir, D.; Pulikkottil, A.J.; Ciccarone, F.; Fischbein, E.; Sinha, A.; Borga, C.; Te Kronnie, G.; Meyer, L.-H.; Mohr, F.; et al. TET1 Promotes Growth of T-Cell Acute Lymphoblastic Leukemia and Can Be Antagonized via PARP Inhibition. Leukemia 2021, 35, 389–403. [Google Scholar] [CrossRef]
- Kharat, S.S.; Ding, X.; Swaminathan, D.; Suresh, A.; Singh, M.; Sengodan, S.K.; Burkett, S.; Marks, H.; Pamala, C.; He, Y.; et al. Degradation of 5hmC-Marked Stalled Replication Forks by APE1 Causes Genomic Instability. Sci. Signal. 2020, 13, eaba8091. [Google Scholar] [CrossRef]
- Rondinelli, B.; Gogola, E.; Yücel, H.; Duarte, A.A.; van de Ven, M.; van der Sluijs, R.; Konstantinopoulos, P.A.; Jonkers, J.; Ceccaldi, R.; Rottenberg, S.; et al. EZH2 Promotes Degradation of Stalled Replication Forks by Recruiting MUS81 through Histone H3 Trimethylation. Nat. Cell Biol. 2017, 19, 1371–1378. [Google Scholar] [CrossRef]
- Karakashev, S.; Fukumoto, T.; Zhao, B.; Lin, J.; Wu, S.; Fatkhutdinov, N.; Park, P.-H.; Semenova, G.; Jean, S.; Cadungog, M.G.; et al. EZH2 Inhibition Sensitizes CARM1-High, Homologous Recombination Proficient Ovarian Cancers to PARP Inhibition. Cancer Cell 2020, 37, 157–167.e6. [Google Scholar] [CrossRef] [PubMed]
- Wiegmans, A.P.; Yap, P.-Y.; Ward, A.; Lim, Y.C.; Khanna, K.K. Differences in Expression of Key DNA Damage Repair Genes after Epigenetic-Induced BRCAness Dictate Synthetic Lethality with PARP1 Inhibition. Mol. Cancer Ther. 2015, 14, 2321–2331. [Google Scholar] [CrossRef] [Green Version]
- Ekstrom, T.L.; Pathoulas, N.M.; Huehls, A.M.; Kanakkanthara, A.; Karnitz, L.M. VLX600 Disrupts Homologous Recombination and Synergizes with PARP Inhibitors and Cisplatin by Inhibiting Histone Lysine Demethylases. Mol. Cancer Ther. 2021, 20, 1561–1571. [Google Scholar] [CrossRef]
- Abbotts, R.; Topper, M.J.; Biondi, C.; Fontaine, D.; Goswami, R.; Stojanovic, L.; Choi, E.Y.; McLaughlin, L.; Kogan, A.A.; Xia, L.; et al. DNA Methyltransferase Inhibitors Induce a BRCAness Phenotype That Sensitizes NSCLC to PARP Inhibitor and Ionizing Radiation. Proc. Natl. Acad. Sci. USA 2019, 116, 22609–22618. [Google Scholar] [CrossRef]
- Muvarak, N.E.; Chowdhury, K.; Xia, L.; Robert, C.; Choi, E.Y.; Cai, Y.; Bellani, M.; Zou, Y.; Singh, Z.N.; Duong, V.H.; et al. Enhancing the Cytotoxic Effects of PARP Inhibitors with DNA Demethylating Agents—A Potential Therapy for Cancer. Cancer Cell 2016, 30, 637–650. [Google Scholar] [CrossRef]
- Pulliam, N.; Fang, F.; Ozes, A.R.; Tang, J.; Adewuyi, A.; Keer, H.; Lyons, J.; Baylin, S.B.; Matei, D.; Nakshatri, H.; et al. An Effective Epigenetic-PARP Inhibitor Combination Therapy for Breast and Ovarian Cancers Independent of BRCA Mutations. Clin. Cancer Res. 2018, 24, 3163–3175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giansanti, M.; De Gabrieli, A.; Prete, S.P.; Ottone, T.; Divona, M.D.; Karimi, T.; Ciccarone, F.; Voso, M.T.; Graziani, G.; Faraoni, I. Poly(ADP-Ribose) Polymerase Inhibitors for Arsenic Trioxide-Resistant Acute Promyelocytic Leukemia: Synergistic In Vitro Antitumor Effects with Hypomethylating Agents or High-Dose Vitamin C. J. Pharmacol. Exp. Ther. 2021, 377, 385–397. [Google Scholar] [CrossRef] [PubMed]
- Musheev, M.U.; Schomacher, L.; Basu, A.; Han, D.; Krebs, L.; Scholz, C.; Niehrs, C. Mammalian N1-Adenosine PARylation Is a Reversible DNA Modification. Nat. Commun. 2022, 13, 6138. [Google Scholar] [CrossRef] [PubMed]
- Sterling, J.; Menezes, S.V.; Abbassi, R.H.; Munoz, L. Histone lysine demethylases and their functions in cancer. Int. J. Cancer 2020, 148, 2375–2388. [Google Scholar] [CrossRef]
- Karakashev, S.; Zhu, H.; Yokoyama, Y.; Zhao, B.; Fatkhutdinov, N.; Kossenkov, A.V.; Wilson, A.J.; Simpkins, F.; Speicher, D.; Khabele, D.; et al. BET Bromodomain Inhibition Synergizes with PARP Inhibitor in Epithelial Ovarian Cancer. Cell Rep. 2017, 21, 3398–3405. [Google Scholar] [CrossRef] [Green Version]
- Mio, C.; Gerratana, L.; Bolis, M.; Caponnetto, F.; Zanello, A.; Barbina, M.; Di Loreto, C.; Garattini, E.; Damante, G.; Puglisi, F. BET Proteins Regulate Homologous Recombination-Mediated DNA Repair: BRCAness and Implications for Cancer Therapy. Int. J. Cancer 2019, 144, 755–766. [Google Scholar] [CrossRef]
- Yang, L.; Zhang, Y.; Shan, W.; Hu, Z.; Yuan, J.; Pi, J.; Wang, Y.; Fan, L.; Tang, Z.; Li, C.; et al. Repression of BET Activity Sensitizes Homologous Recombination-Proficient Cancers to PARP Inhibition. Sci. Transl. Med. 2017, 9, eaal1645. [Google Scholar] [CrossRef]


| NCT Identifier (Estimated Study Completion Date) | Cancer Type | Interventions | Phase | Output |
|---|---|---|---|---|
| NCT 02878785 (December 2022) | Acute Myeloid Leukaemia | PARPi: talazoparib DNMTi: decitabine | 1 2 | Dose finding based on tolerability, efficacy, and pharmacodynamic data Efficacy of the selected combination regimen |
| NCT 04846478 (September 2023) | Metastatic castration-resistant prostate cancer | PARPi: talazoparib EZH2i: tazemetostat | 1 | Safety, tolerability, and preliminary clinical activity of drug combination |
| NCT 03742245 (September 2024) | Relapsed/refractory and/or metastatic breast cancer | PARPi: olaparib HDACi: vorinostat | 1 | Safety and preliminary efficacy of drug combination |
| NCT 04355858 (April 2025) | HR+/HER2- endocrine-resistant advanced breast cancer | PARPi: SHR3162 EZH2i: SHR2554 | 2 | Screening valuable treatment cohorts for randomized controlled phase III clinical studies with larger sample size |
| NCT 05071937 (November 2027) | Recurrent ovarian, fallopian tube, or primary peritoneal carcinoma | PARPi: talazoparib BETi: ZEN003696 | 2 | Efficacy of drug combination |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pinton, G.; Boumya, S.; Ciriolo, M.R.; Ciccarone, F. Epigenetic Insights on PARP-1 Activity in Cancer Therapy. Cancers 2023, 15, 6. https://doi.org/10.3390/cancers15010006
Pinton G, Boumya S, Ciriolo MR, Ciccarone F. Epigenetic Insights on PARP-1 Activity in Cancer Therapy. Cancers. 2023; 15(1):6. https://doi.org/10.3390/cancers15010006
Chicago/Turabian StylePinton, Giulia, Sara Boumya, Maria Rosa Ciriolo, and Fabio Ciccarone. 2023. "Epigenetic Insights on PARP-1 Activity in Cancer Therapy" Cancers 15, no. 1: 6. https://doi.org/10.3390/cancers15010006