
CRAC and SK Channels: Their Molecular Mechanisms Associated with Cancer Cell Development

Abstract
:Simple Summary
Abstract
1. Introduction
1.1. Physiological Role of Ca2+
1.2. Ca2+ Signal Transduction in Major Cancer Hallmarks
1.3. Ca2+-Dependent Dysregulation of Proliferation
1.4. Ca2+-Dependent Dysregulation of Cell Survival and Cell Death
1.5. Ca2+-Dependent Dysregulation of Migration and Invasion
1.6. Ca2+-Dependent Dysregulation of Other Cancer Hallmarks
2. Ca2+ Ion Channels and Their Role in Cancer
2.1. CRAC Channels
2.1.1. STIM Proteins
2.1.2. Orai
2.1.3. CRAC Channels and Cholesterol-Rich Regions
2.2. CRAC Channels and Cancer
2.2.1. Cancer Associated with Distinct Expression Levels of CRAC Channel Proteins
2.2.2. Proliferation
2.2.3. Cell Survival and Cell Death
2.2.4. Epithelial–Mesenchymal Transition (EMT), Migration, and Invasion
2.2.5. Hypoxia Linked to CRAC Channel Components in Cancer
2.3. Cancer Associated with Mutations of CRAC Channel Proteins
2.4. Therapeutic Approaches Associated with the Expression of CRAC Channel Components in Cancer
3. The Range of Ca2+-Activated Ion Channels
3.1. Ca2+-Activated K+ Channels
3.2. SK Channels and Cholesterol-Rich Regions
3.3. SK Channels and Cancer
3.3.1. Proliferation
3.3.2. EMT, Migration, and Metastasis
3.4. Therapeutic Approaches Associated with the Expression of SK Channels in Cancer
4. Co-Regulation of Ca2+ and K+ Ion Channels Linked to Cancer Development
4.1. SK3-Orai1 Interplay
4.1.1. The Molecular Determinants of the SK3-Orai1 Interplay
4.1.2. The Interplay of SK3 and Orai1 in Breast, Colon, and Prostate Cancer Cells
5. Perspectives
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 1-EBIO | 1-ethylbenzimidazolinone |
| 5-FU | 5-fluorouracil |
| Å | Angstrom (unit of length equal to 1 × 10−10 m) |
| aa | amino acid |
| Akt | known as protein kinase B or PKB |
| AP1 | activator protein 1 |
| AR | androgen receptor |
| ASIC | acid-sensing channels |
| AMLC | acute myeloid leukemia cells |
| ANO8 | Anoctamin 8 |
| ATPase | adenosine triphosphatase |
| BCL2 | B-cell lymphoma 2 |
| BCLxL | B-cell lymphoma-extra large |
| BID | BH3 Interacting Domain Death Agonist |
| Bim | BCL2-interacting mediator of cell death |
| BK | large conductance, Ca2+-activated potassium channels |
| Ca2+ | calcium ion |
| CAD | Ca2+ release-activated Ca2+ activating domain |
| CAI | carboxyamidotriazole |
| CaM | calmodulin |
| CaMBD | calmodulin-binding domain |
| CaMK | calmodulin kinase |
| CaMMUT | calmodulin mutant |
| cAMP | cyclic adenosine monophosphate |
| Cav1 | Caveolin-1 |
| CC | coiled-coil |
| CDK2/4/6 | cyclin-dependent kinase |
| cEF | canonical EF hand |
| CK2 | casein kinase 2 |
| CLL | chronic lymphocyte leukemia |
| CN | calcineurin |
| COSMIC | catalogue of somatic mutations in cancer |
| COX-2 | cyclooxygenase-2 |
| CRAC | Ca2+ Release-Activated Ca2+ |
| CREB | cAMP-responsive element binding protein |
| CRISPR/Cas9 | clustered regularly interspaced short palindromic repeats/Cas |
| cryo-EM | cryogenic electron microscopy |
| DAG | diacylglycerol |
| DES | Diethylstilbestrol |
| DKO | double knock-out |
| dOrai | Drosophila melanogaster Orai |
| EB | end-binding domain |
| ECM | extracellular matrix |
| EGF | epidermal growth factor |
| EGFR | epidermal growth factor receptor |
| EGTA | ethylene glycol tetraacetic acid |
| EMT | epithelial–mesenchymal transition |
| ER | endoplasmic Reticulum |
| ERα | estrogen receptor-α |
| ERK | extracellular signal-regulated kinase |
| E-Syts | extended-synaptotagmins |
| ETON | extended transmembrane Orai1 N-terminal |
| FAK | focal adhesion kinase |
| FRET | fluorescence resonance energy transfer |
| GBM | glioblastoma multiforme cells |
| GoF | gain of function |
| GPCR | G-protein coupled receptor |
| GRAMD2A | GRAM Domain Containing 2A |
| GSK | GlaxoSmithKline compounds |
| GTPase | guanosine triphosphatase |
| HBL | human diffuse large B-cell lymphoma |
| hEag1 | human ether a-gogo potassium channel 1 |
| HEK | human embryonic kidney |
| HeLa | Henrietta Lacks |
| HIF-1 | hypoxia-inducible factor |
| hOrai1 | human Orai1 |
| Huh-7 | human hepatoma cell line |
| ID | inactivation domain |
| IK | intermediate Ca2+ activated K+ channels |
| IP3 | inositol-triphosphate |
| IP3R | inositol-triphosphate receptor |
| K+ | potassium ion |
| KCa | Ca2+ activated K+ channels |
| KV | voltage-gated potassium channels |
| Kir | inward-rectifier potassium channels |
| KRAS | RAS/ERK pathway |
| La3+ | lanthanum ion |
| LGC | ligand-gated calcium channels |
| LNCaP | lymph node carcinoma of the prostate |
| LoF | loss of function |
| MACC-1 | metastasis-associated in colon cancer-1 |
| MAPK | mitogen-activated protein kinases |
| MßCD | methyl-beta-cyclodextrin |
| MCF-7 | Michigan Cancer Foundation-7 |
| MET | mesenchymal-epithelial transition |
| miRNA | microRNA |
| MLCK | myosin light chain kinase |
| MMPs | matrix metalloproteins |
| nEF | non-canonical EF-hand domain |
| NFAT | nuclear factor of activated T cells |
| NFkB | nuclear factor k-light-chain-enhancer of activated B cells |
| NMR | nuclear magnetic resonance |
| NSCLC | chronic lymphocyte leukemia |
| OASF | Orai—activating small fragment |
| Orai 1–3 | Orai proteins (also as O1–3) |
| PAT | protein acyl transferase |
| PBD | polybasic domain |
| PGE2 | prostaglandin E2 |
| PI3K | phosphoinositide 3-kinases |
| PI4P | phosphatidylinositol 4-phosphate |
| PIP2 | phosphatidylinositol 4,5-bisphosphate |
| PIP3 | phosphatidylinositol-3,4,5-trisphosphat |
| PKA | protein kinase A |
| PKC | protein kinase C |
| PLCε | phospholipase C |
| PM | plasma membrane |
| PMCA | plasma membrane Ca2+ ATPase |
| PYK2 | proline-rich tyrosine kinase 2 |
| Rac1 | Ras-related C3 botulinum toxin substrate 1 |
| Raf | serine/threonine kinase |
| Ras | rat sarcoma virus |
| RASSF4 | Ras Association Domain Family Member 4 |
| Ref | references |
| RNA | ribonucleic acid |
| ROC | receptor-operated channels |
| ROS | reactive oxygen species |
| RTK | growth factor receptor tyrosine kinase |
| SAM | sterile α-motif |
| SARAF | store-operated calcium entry-associated regulatory factor |
| SCID | severe combined immune deficiency |
| SERCA | sarcoplasmic/endoplasmic reticulum calcium ATPase |
| SICE | store-independent calcium entry |
| SigmaR1 | Sigma non-opioid intracellular receptor 1 |
| siRNA | small interfering RNA |
| SK channels | small-conductance Ca2+-activated K+ channels |
| SMase | sphingomyelinase |
| SMOC | second messenger-operated channels |
| SOAP | STIM—Orai association pocket |
| SOAR | STIM—Orai activating region |
| SOC | store operated channel |
| SOCE | store-operated calcium entry |
| SPCA2 | secretory pathway Ca2+-ATPase |
| STIM | stromal interaction molecule |
| STIMATE | STIM Activating Enhancer |
| TM | transmembrane helices |
| TRAM-34 | triarylmethane-34 |
| TRP | transient receptor potential ion channel (C-canonical, M-melastatin, V-vallinoid) |
| VGCC | voltage-gated calcium channels |
| Wnt | Wingless Int-1 |
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prevarskaya, N.; Ouadid-Ahidouch, H.; Skryma, R.; Shuba, Y. Remodelling of Ca2+ transport in cancer: How it contributes to cancer hallmarks? Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130097. [Google Scholar] [CrossRef] [Green Version]
- Fouad, Y.A.; Aanei, C. Revisiting the hallmarks of cancer. Am. J. Cancer Res. 2017, 7, 1016–1036. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prevarskaya, N.; Skryma, R.; Shuba, Y. Ion channels and the hallmarks of cancer. Trends Mol. Med. 2010, 16, 107–121. [Google Scholar] [CrossRef] [PubMed]
- Prevarskaya, N.; Skryma, R.; Shuba, Y. Ion Channels in Cancer: Are Cancer Hallmarks Oncochannelopathies? Physiol. Rev. 2018, 98, 559–621. [Google Scholar] [CrossRef] [Green Version]
- Berridge, M.J.; Bootman, M.D.; Roderick, H.L. Calcium signalling: Dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 2003, 4, 517–529. [Google Scholar] [CrossRef] [Green Version]
- Berridge, M.J.; Bootman, M.D.; Lipp, P. Calcium—A life and death signal. Nature 1998, 395, 645–648. [Google Scholar] [CrossRef]
- Derler, I.; Butorac, C.; Krizova, A.; Stadlbauer, M.; Muik, M.; Fahrner, M.; Frischauf, I.; Romanin, C. Authentic CRAC channel activity requires STIM1 and the conserved portion of the Orai N terminus. J. Biol. Chem. 2018, 293, 1259–1270. [Google Scholar] [CrossRef] [Green Version]
- Parkash, J.; Asotra, K. Calcium wave signaling in cancer cells. Life Sci. 2010, 87, 587–595. [Google Scholar] [CrossRef] [PubMed]
- Skupin, A.; Thurley, K. Calcium signaling: From single channels to pathways. Adv. Exp. Med. Biol. 2012, 740, 531–551. [Google Scholar] [PubMed]
- Yen, M.; Lewis, R.S. Numbers count: How STIM and Orai stoichiometry affect store-operated calcium entry. Cell Calcium 2019, 79, 35–43. [Google Scholar] [CrossRef]
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Marchi, S.; Patergnani, S.; Missiroli, S.; Morciano, G.; Rimessi, A.; Wieckowski, M.R.; Giorgi, C.; Pinton, P. Mitochondrial and endoplasmic reticulum calcium homeostasis and cell death. Cell Calcium 2018, 69, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Catterall, W.A.; Perez-Reyes, E.; Snutch, T.P.; Striessnig, J. International Union of Pharmacology. XLVIII. Nomenclature and structure-function relationships of voltage-gated calcium channels. Pharmacol. Rev. 2005, 57, 411–425. [Google Scholar] [CrossRef]
- Guo, Y.R.; MacKinnon, R. Structure-based membrane dome mechanism for Piezo mechanosensitivity. eLife 2017, 6, e33660. [Google Scholar] [CrossRef]
- Monteith, G.R.; Prevarskaya, N.; Roberts-Thomson, S.J. The calcium–cancer signalling nexus. Nat. Rev. Cancer 2017, 17, 373–380. [Google Scholar] [CrossRef] [Green Version]
- Monteith, G.R.; Davis, F.M.; Roberts-Thomson, S.J. Calcium channels and pumps in cancer: Changes and consequences. J. Biol. Chem. 2012, 287, 31666–31673. [Google Scholar] [CrossRef] [Green Version]
- Berridge, M.J. Calcium signalling remodelling and disease. Biochem. Soc. Trans. 2012, 40, 297–309. [Google Scholar] [CrossRef] [Green Version]
- Sever, R.; Brugge, J.S. Signal transduction in cancer. Cold Spring Harb. Perspect. Med. 2015, 5, a006098. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Wu, T.; Xu, Y.; Dong, Q.; Xiao, J.; Xu, Y.; Li, Q.; Zhang, C.; Gao, J.; Liu, L.; et al. A comprehensive overview of oncogenic pathways in human cancer. Brief. Bioinform. 2020, 21, 957–969. [Google Scholar] [CrossRef] [PubMed]
- Giehl, K. Oncogenic Ras in tumour progression and metastasis. Biol. Chem. 2005, 386, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Divolis, G.; Mavroeidi, P.; Mavrofrydi, O.; Papazafiri, P. Differential effects of calcium on PI3K-Akt and HIF-1α survival pathways. Cell Biol. Toxicol. 2016, 32, 437–449. [Google Scholar] [CrossRef] [PubMed]
- Roderick, H.L.; Cook, S.J. Ca2+ signalling checkpoints in cancer: Remodelling Ca2+ for cancer cell proliferation and survival. Nat. Rev. Cancer 2008, 8, 361–375. [Google Scholar] [CrossRef] [Green Version]
- Nussinov, R.; Wang, G.; Tsai, C.J.; Jang, H.; Lu, S.; Banerjee, A.; Zhang, J.; Gaponenko, V. Calmodulin and PI3K Signaling in KRAS Cancers. Trends Cancer 2017, 3, 214–224. [Google Scholar] [CrossRef] [Green Version]
- Santella, L.; Ercolano, E.; Nusco, G.A. The cell cycle: A new entry in the field of Ca2+ signaling. Cell. Mol. Life Sci. 2005, 62, 2405–2413. [Google Scholar] [CrossRef]
- Wang, Y.Y.; Zhao, R.; Zhe, H. The emerging role of CaMKII in cancer. Oncotarget 2015, 6, 11725–11734. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z. Regulation of cell cycle progression by growth factor-induced cell signaling. Cells 2021, 10, 3327. [Google Scholar] [CrossRef]
- De, A. Wnt/Ca2+ signaling pathway: A brief overview. Acta Biochim. Biophys. Sin. 2011, 43, 745–756. [Google Scholar] [CrossRef]
- Pataki, C.A.; Couchman, J.R.; Brábek, J. Wnt Signaling Cascades and the Roles of Syndecan Proteoglycans. J. Histochem. Cytochem. 2015, 63, 465–480. [Google Scholar] [CrossRef] [PubMed]
- Bruce, J.I.E.; James, A.D. Targeting the Calcium Signalling Machinery in Cancer. Cancers 2020, 12, 2351. [Google Scholar] [CrossRef] [PubMed]
- Haupt, S.; Raghu, D.; Haupt, Y. P53 calls upon CIA (Calcium Induced Apoptosis) to counter stress. Front. Oncol. 2015, 5, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J. The cell-cycle arrest and apoptotic functions of p53 in tumor initiation and progression. Cold Spring Harb. Perspect. Med. 2016, 6, a026104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorrentino, G.; Comel, A.; del Sal, G. p53 orchestrates calcium signaling in vivo. Cell Cycle 2015, 14, 1343–1344. [Google Scholar] [CrossRef] [Green Version]
- Bononi, A.; Bonora, M.; Marchi, S.; Missiroli, S.; Poletti, F.; Giorgi, C.; Pandolfi, P.P.; Pinton, P. Identification of PTEN at the ER and MAMs and its regulation of Ca2+ signaling and apoptosis in a protein phosphatase-dependent manner. Cell Death Differ. 2013, 20, 1631–1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bittremieux, M.; Bultynck, G. p53 and Ca2+ signaling from the endoplasmic reticulum: Partners in anti-cancer therapies. Oncoscience 2015, 2, 233–238. [Google Scholar] [CrossRef]
- Madan, E.; Gogna, R.; Keppler, B.; Pati, U. p53 Increases Intra-Cellular Calcium Release by Transcriptional Regulation of Calcium Channel TRPC6 in GaQ3-Treated Cancer Cells. PLoS ONE 2013, 8, e71016. [Google Scholar] [CrossRef] [Green Version]
- Afzal, M.; Kren, B.T.; Naveed, A.K.; Trembley, J.H.; Ahmed, K. Protein kinase CK2 impact on intracellular calcium homeostasis in prostate cancer. Mol. Cell. Biochem. 2020, 470, 131–143. [Google Scholar] [CrossRef]
- Déliot, N.; Constantin, B. Plasma membrane calcium channels in cancer: Alterations and consequences for cell proliferation and migration. Biochim. Biophys. Acta BBA Biomembr. 2015, 1848, 2512–2522. [Google Scholar] [CrossRef] [Green Version]
- Stewart, T.A.; Yapa, K.T.D.S.; Monteith, G.R. Altered calcium signaling in cancer cells. Biochim. Biophys. Acta BBA Biomembr. 2015, 1848, 2502–2511. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.A.; Hazlehurst, L.A. Role of Calcium Homeostasis in Modulating EMT in Cancer. Biomedicines 2021, 9, 1200. [Google Scholar] [CrossRef] [PubMed]
- Dejos, C.; Gkika, D.; Cantelmo, A.R. The Two-Way Relationship Between Calcium and Metabolism in Cancer. Front. Cell Dev. Biol. 2020, 8, 1251. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A.; Huang, R.Y.Y.J.; Jackson, R.A.A.; Thiery, J.P.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [Green Version]
- Georgakopoulos-Soares, I.; Chartoumpekis, D.V.; Kyriazopoulou, V.; Zaravinos, A. EMT Factors and Metabolic Pathways in Cancer. Front. Oncol. 2020, 10, 499. [Google Scholar] [CrossRef]
- Bronsert, P.; Enderle-Ammour, K.; Bader, M.; Timme, S.; Kuehs, M.; Csanadi, A.; Kayser, G.; Kohler, I.; Bausch, D.; Hoeppner, J.; et al. Cancer cell invasion and EMT marker expression: A three-dimensional study of the human cancer-host interface. J. Pathol. 2014, 234, 410–422. [Google Scholar] [CrossRef]
- Roche, J. The epithelial-to-mesenchymal transition in cancer. Cancers 2018, 10, 79. [Google Scholar] [CrossRef] [Green Version]
- Thiery, J.P.; Sleeman, J.P. Complex networks orchestrate epithelial-mesenchymal transitions. Nat. Rev. Mol. Cell Biol. 2006, 7, 131–142. [Google Scholar] [CrossRef]
- Zhang, Y.; Weinberg, R.A. Epithelial-to-mesenchymal transition in cancer: Complexity and opportunities. Front. Med. 2018, 12, 361–373. [Google Scholar] [CrossRef] [Green Version]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.J.; Nieto, M.A. Epithelial-Mesenchymal Transitions in Development and Disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Adiga, D.; Radhakrishnan, R.; Chakrabarty, S.; Kumar, P.; Kabekkodu, S.P. The Role of Calcium Signaling in Regulation of Epithelial-Mesenchymal Transition. Cells Tissues Organs 2022, 211, 134–156. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Huang, H.; Liao, W.; Ma, H.; Liu, J.; Wang, L.; Huang, N.; Liao, Y.; Liao, W. MACC1 supports human gastric cancer growth under metabolic stress by enhancing the Warburg effect. Oncogene 2015, 34, 2700–2710. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Wang, H.; Huang, H.; Sun, L.; Dong, S.; Huang, N.; Shi, M.; Bin, J.; Liao, Y.; Liao, W. Elevated Orai1 and STIM1 expressions upregulate MACC1 expression to promote tumor cell proliferation, metabolism, migration, and invasion in human gastric cancer. Cancer Lett. 2016, 381, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Goto, H.; Kosako, H.; Tanabe, K.; Yanagida, M.; Sakurai, M.; Amano, M.; Kaibuchi, K.; Inagaki, M. Phosphorylation of Vimentin by Rho-Associated Kinase at a Unique Amino-Terminal Site That Is Specifically Phosphorylated during Cytokinesis. J. Biol. Chem. 1998, 273, 11728–11736. [Google Scholar] [CrossRef] [Green Version]
- Gross, S.; Mallu, P.; Joshi, H.; Schultz, B.; Go, C.; Soboloff, J. Ca2+ as a therapeutic target in cancer. Adv. Cancer Res. 2020, 148, 233–317. [Google Scholar]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia 2015, 3, 83. [Google Scholar] [CrossRef] [Green Version]
- Bertout, J.A.; Patel, S.A.; Simon, M.C. The impact of O2 availability on human cancer. Nat. Rev. Cancer 2008, 8, 967–975. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. Hypoxia-inducible factors: Mediators of cancer progression and targets for cancer therapy. Trends Pharmacol. Sci. 2012, 33, 207–214. [Google Scholar] [CrossRef] [Green Version]
- Girault, A.; Ahidouch, A.; Ouadid-Ahidouch, H. Roles for Ca2+ and K+ channels in cancer cells exposed to the hypoxic tumour microenvironment. Biochim. Biophys. Acta BBA Mol. Cell Res. 2020, 1867, 118644. [Google Scholar] [CrossRef]
- Nielsen, N.; Lindemann, O.; Schwab, A. TRP channels and STIM/ORAI proteins: Sensors and effectors of cancer and stroma cell migration. Br. J. Pharmacol. 2014, 171, 5524–5540. [Google Scholar] [CrossRef] [Green Version]
- Chalmers, S.B.; Monteith, G.R. ORAI channels and cancer. Cell Calcium 2018, 74, 160–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azimi, I.; Milevskiy, M.; Chalmers, S.; Yapa, K.; Robitaille, M.; Henry, C.; Baillie, G.J.; Thompson, E.W.; Roberts-Thomson, S.J.; Monteith, G.R. ORAI1 and ORAI3 in Breast Cancer Molecular Subtypes and the Identification of ORAI3 as a Hypoxia Sensitive Gene and a Regulator of Hypoxia Responses. Cancers 2019, 11, 208. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wan, X.; Kan, H.; Wang, Y.; Yu, F.; Feng, L.; Jin, J.; Zhang, P.; Ma, X. Hypoxia-induced upregulation of Orai1 drives colon cancer invasiveness and angiogenesis. Eur. J. Pharmacol. 2018, 832, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, T.; Wang, Y.; Chen, Z.; Hua, D.; Yao, X.; Ma, X.; Zhang, P. Orai1 is critical for Notch-driven aggressiveness under hypoxic conditions in triple-negative breast cancers. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2018, 1864, 975–986. [Google Scholar] [CrossRef]
- Batra, S.; Alenfall, J. Effect of diverse categories of drugs on human colon tumour cell proliferation. Anticancer Res. 1991, 11, 1221–1224. [Google Scholar]
- Lee, S.C.; Deutsch, C.; Beck, W.T. Comparison of ion channels in multidrug-resistant and -sensitive human leukemic cells. Proc. Natl. Acad. Sci. USA 1988, 85, 2019–2023. [Google Scholar] [CrossRef] [Green Version]
- Pancrazio, J.J.; Tabbara, I.A.; Kim, Y.I. Voltage-activated K+ conductance and cell proliferation in small-cell lung cancer. Anticancer Res. 1993, 13, 1231–1234. [Google Scholar]
- Pancrazio, J.J.; Viglione, M.P.; Tabbara, I.A.; Kim, Y.I. Voltage-dependent ion channels in small-cell lung cancer cells. Cancer Res. 1989, 49, 5901–5906. [Google Scholar]
- Taylor, J.M.; Simpson, R.U. Inhibition of cancer cell growth by calcium channel antagonists in the athymic mouse. Cancer Res. 1992, 52, 2413–2418. [Google Scholar]
- Yamashita, N.; Hamada, H.; Tsuruo, T.; Ogata, E. Enhancement of voltage-gated Na+ channel current associated with multidrug resistance in human leukemia cells. Cancer Res. 1987, 47, 3736–3741. [Google Scholar]
- Buchanan, P.J.; McCloskey, K.D. CaV channels and cancer: Canonical functions indicate benefits of repurposed drugs as cancer therapeutics. Eur. Biophys. J. 2016, 45, 621–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.Y.; Lai, M.D.; Phan, N.N.; Sun, Z.; Lin, Y.C. Meta-Analysis of Public Microarray Datasets Reveals Voltage-Gated Calcium Gene Signatures in Clinical Cancer Patients. PLoS ONE 2015, 10, e0125766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pankratov, Y.; Lalo, U. Calcium permeability of ligand-gated Ca2+ channels. Eur. J. Pharmacol. 2014, 739, 60–73. [Google Scholar] [CrossRef] [PubMed]
- Prakriya, M.; Lewis, R.S. Store-Operated Calcium Channels. Physiol. Rev. 2015, 95, 1383–1436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofmann, T.; Obukhov, A.G.; Schaefer, M.; Harteneck, C.; Gudermann, T.; Schultz, G. Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature 1999, 397, 259–263. [Google Scholar] [CrossRef]
- Venkatachalam, K.; Montell, C. TRP channels. Annu. Rev. Biochem. 2007, 76, 387–417. [Google Scholar] [CrossRef] [Green Version]
- Kellenberger, S.; Schild, L. International Union of Basic and Clinical Pharmacology. XCI. structure, function, and pharmacology of acid-sensing ion channels and the epithelial Na+ channel. Pharmacol. Rev. 2015, 67, 1–35. [Google Scholar] [CrossRef]
- Ranade, S.S.; Syeda, R.; Patapoutian, A. Mechanically Activated Ion Channels. Neuron 2015, 87, 1162–1179. [Google Scholar] [CrossRef] [Green Version]
- Hou, M.F.; Kuo, H.C.; Li, J.H.; Wang, Y.S.; Chang, C.C.; Chen, K.C.; Chen, W.C.; Chiu, C.C.; Yang, S.; Chang, W.C. Orai1/CRACM1 overexpression suppresses cell proliferation via attenuation of the store-operated calcium influx-mediated signalling pathway in A549 lung cancer cells. Biochim. Biophys. Acta BBA Gen. Subj. 2011, 1810, 1278–1284. [Google Scholar] [CrossRef]
- Flourakis, M.; Lehen’kyi, V.; Beck, B.; Raphael, M.; Vandenberghe, M.; Abeele, F.V.; Roudbaraki, M.; Lepage, G.; Mauroy, B.; Romanin, C.; et al. Orai1 contributes to the establishment of an apoptosis-resistant phenotype in prostate cancer cells. Cell Death Dis. 2010, 1, e75. [Google Scholar] [CrossRef] [Green Version]
- Jardin, I.; Diez-Bello, R.; Lopez, J.J.; Redondo, P.C.; Salido, G.M.; Smani, T.; Rosado, J.A. TRPC6 Channels Are Required for Proliferation, Migration and Invasion of Breast Cancer Cell Lines by Modulation of Orai1 and Orai3 Surface Exposure. Cancers 2018, 10, 331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.; Zhang, J.J.; Huang, X.Y. Orai1 and STIM1 are critical for breast tumor cell migration and metastasis. Cancer Cell 2009, 15, 124–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantonero, C.; Sanchez-Collado, J.; Gonzalez-Nunez, M.A.; Salido, G.M.; Lopez, J.J.; Jardin, I.; Rosado, J.A. Store-independent Orai1-mediated Ca2+ entry and cancer. Cell Calcium 2019, 80, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhang, M.; Xu, L.; Lin, D.; Cai, S.; Zou, F. The apoptosis of non-small cell lung cancer induced by cisplatin through modulation of STIM1. Exp. Toxicol. Pathol. 2013, 65, 1073–1081. [Google Scholar] [CrossRef]
- Wang, J.Y.; Sun, J.; Huang, M.Y.; Wang, Y.S.; Hou, M.F.; Sun, Y.; He, H.; Krishna, N.; Chiu, S.-J.; Lin, S.; et al. STIM1 overexpression promotes colorectal cancer progression, cell motility and COX-2 expression. Oncogene 2015, 34, 4358–4367. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Liu, X.; Feng, B.; Liu, N.; Wu, Q.; Han, Y.; Nie, Y.; Wu, K.; Shi, Y.; Fan, D. STIM1, a direct target of microRNA-185, promotes tumor metastasis and is associated with poor prognosis in colorectal cancer. Oncogene 2015, 34, 4808–4820. [Google Scholar] [CrossRef] [Green Version]
- saint Fleur-Lominy, S.; Maus, M.; Vaeth, M.; Lange, I.; Zee, I.; Suh, D.; Liu, C.; Wu, X.; Tikhonova, A.; Aifantis, I.; et al. STIM1 and STIM2 Mediate Cancer-Induced Inflammation in T Cell Acute Lymphoblastic Leukemia. Cell Rep. 2018, 24, 3045–3060.e5. [Google Scholar] [CrossRef]
- Li, G.; Zhang, Z.; Wang, R.; Ma, W.; Yang, Y.; Wei, J.; Wei, Y. Suppression of STIM1 inhibits human glioblastoma cell proliferation and induces G0/G1 phase arrest. J. Exp. Clin. Cancer Res. 2013, 32, 20. [Google Scholar] [CrossRef] [Green Version]
- Debant, M.; Burgos, M.; Hemon, P.; Buscaglia, P.; Fali, T.; Melayah, S.; Goux, N.L.; Vandier, C.; Potier-Cartereau, M.; Pers, J.O.; et al. STIM1 at the plasma membrane as a new target in progressive chronic lymphocytic leukemia. J. Immunother. Cancer 2019, 7, 111. [Google Scholar] [CrossRef] [Green Version]
- Prevarskaya, N.; Zhang, L.; Barritt, G. TRP channels in cancer. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2007, 1772, 937–946. [Google Scholar] [CrossRef] [Green Version]
- Shapovalov, G.; Ritaine, A.; Skryma, R.; Prevarskaya, N. Role of TRP ion channels in cancer and tumorigenesis. Semin. Immunopathol. 2016, 38, 357–369. [Google Scholar] [CrossRef] [PubMed]
- Canales, J.; Morales, D.; Blanco, C.; Rivas, J.; Diaz, N.; Angelopoulos, I.; Cerda, O. A TR(i)P to Cell Migration: New Roles of TRP Channels in Mechanotransduction and Cancer. Front. Physiol. 2019, 10, 757. [Google Scholar] [CrossRef] [PubMed]
- Baba, Y.; Hayashi, K.; Fujii, Y.; Mizushima, A.; Watarai, H.; Wakamori, M.; Numaga, T.; Mori, Y.; Iino, M.; Hikida, M.; et al. Coupling of STIM1 to store-operated Ca2+ entry through its constitutive and inducible movement in the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 2006, 103, 16704–16709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Lu, J.; Xu, P.; Xie, X.; Chen, L.; Xu, T. Mapping the interacting domains of STIM1 and Orai1 in Ca2+ release-activated Ca2+ channel activation. J. Biol. Chem. 2007, 282, 29448–29456. [Google Scholar] [CrossRef] [Green Version]
- Stathopulos, P.B.; Zheng, L.; Li, G.Y.; Plevin, M.J.; Ikura, M. Structural and mechanistic insights into STIM1-mediated initiation of store-operated calcium entry. Cell 2008, 135, 110–122. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.M.; Buchanan, J.; Luik, R.M.; Lewis, R.S. Ca2+ store depletion causes STIM1 to accumulate in ER regions closely associated with the plasma membrane. J. Cell Biol. 2006, 174, 803–813. [Google Scholar] [CrossRef]
- Barr, V.A.; Bernot, K.M.; Srikanth, S.; Gwack, Y.; Balagopalan, L.; Regan, C.K.; Helman, D.J.; Sommers, C.L.; Oh-Hora, M.; Rao, A.; et al. Dynamic movement of the calcium sensor STIM1 and the calcium channel Orai1 in activated T-cells: Puncta and distal caps. Mol. Biol. Cell 2008, 19, 2802–2817. [Google Scholar] [CrossRef]
- Calloway, N.; Vig, M.; Kinet, J.P.; Holowka, D.; Baird, B. Molecular clustering of STIM1 with Orai1/CRACM1 at the plasma membrane depends dynamically on depletion of Ca2+ stores and on electrostatic interactions. Mol. Biol. Cell 2009, 20, 389–399. [Google Scholar] [CrossRef] [Green Version]
- Park, C.Y.; Hoover, P.J.; Mullins, F.M.; Bachhawat, P.; Covington, E.D.; Raunser, S.; Walz, T.; Garcia, K.C.; Dolmetsch, R.E.; Lewis, R.S. STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1. Cell 2009, 136, 876–890. [Google Scholar] [CrossRef] [Green Version]
- Muik, M.; Fahrner, M.; Schindl, R.; Stathopulos, P.; Frischauf, I.; Derler, I.; Plenk, P.; Lackner, B.; Groschner, K.; Ikura, M.; et al. STIM1 couples to ORAI1 via an intramolecular transition into an extended conformation. EMBO J. 2011, 30, 1678–1689. [Google Scholar] [CrossRef]
- Muik, M.; Frischauf, I.; Derler, I.; Fahrner, M.; Bergsmann, J.; Eder, P.; Schindl, R.; Hesch, C.; Polzinger, B.; Fritsch, R.; et al. Dynamic coupling of the putative coiled-coil domain of ORAI1 with STIM1 mediates ORAI1 channel activation. J. Biol. Chem. 2008, 283, 8014–8022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, J.P.; Zeng, W.; Dorwart, M.R.; Choi, Y.J.; Worley, P.F.; Muallem, S. SOAR and the polybasic STIM1 domains gate and regulate Orai channels. Nat. Cell Biol. 2009, 11, 337–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dragoni, S.; Turin, I.; Laforenza, U.; Potenza, D.M.; Bottino, C.; Glasnov, T.N.; Prestia, M.; Ferulli, F.; Saitta, A.; Mosca, A.; et al. Store-operated Ca2+ entry does not control proliferation in primary cultures of human metastatic renal cellular carcinoma. Biomed. Res. Int. 2014, 2014, 739494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jardin, I.; Lopez, J.J.; Salido, G.M.; Rosado, J.A. Store-Operated Ca2+ Entry in Breast Cancer Cells: Remodeling and Functional Role. Int. J. Mol. Sci. 2018, 19, 4053. [Google Scholar] [CrossRef] [Green Version]
- Mignen, O.; Constantin, B.; Potier-Cartereau, M.; Penna, A.; Gautier, M.; Gueguinou, M.; Renaudineau, Y.; Shoji, K.F.; Félix, R.; Bayet, E.; et al. Constitutive calcium entry and cancer: Updated views and insights. Eur. Biophys. J. 2017, 46, 395–413. [Google Scholar] [CrossRef]
- Motiani, R.K.; Hyzinski-Garcia, M.C.; Zhang, X.; Henkel, M.M.; Abdullaev, I.F.; Kuo, Y.H.; Matrougui, K.; Mongin, A.A.; Trebak, M. STIM1 and Orai1 mediate CRAC channel activity and are essential for human glioblastoma invasion. Pflug. Arch. 2013, 465, 1249–1260. [Google Scholar] [CrossRef] [Green Version]
- Rosado, J.A. Introduction: Overview of the Pathophysiological Implications of Store-Operated Calcium Entry in Mammalian Cells. Adv. Exp. Med. Biol. 2017, 993, 391–395. [Google Scholar]
- Stanisz, H.; Saul, S.; Muller, C.S.; Kappl, R.; Niemeyer, B.A.; Vogt, T.; Hoth, M.; Roesch, A.; Bogeski, I. Inverse regulation of melanoma growth and migration by Orai1/STIM2-dependent calcium entry. Pigment Cell Melanoma Res. 2014, 27, 442–453. [Google Scholar] [CrossRef]
- Stanisz, H.; Vultur, A.; Herlyn, M.; Roesch, A.; Bogeski, I. The role of Orai-STIM calcium channels in melanocytes and melanoma. J. Physiol. 2016, 594, 2825–2835. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.L.; Yu, Y.; Roos, J.; Kozak, J.A.; Deerinck, T.J.; Ellisman, M.H.; Stauderman, K.A.; Cahalan, M.D. STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature 2005, 437, 902–905. [Google Scholar] [CrossRef] [Green Version]
- Berna-Erro, A.; Jardin, I.; Salido, G.M.; Rosado, J.A. Role of STIM2 in cell function and physiopathology. J. Physiol. 2017, 595, 3111–3128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butorac, C.; Krizova, A.; Derler, I. Review: Structure and Activation Mechanisms of CRAC Channels. Adv. Exp. Med. Biol. 2020, 1131, 547–604. [Google Scholar] [PubMed]
- Derler, I.; Jardin, I.; Romanin, C. Molecular mechanisms of STIM/Orai communication. Am. J. Physiol. Cell Physiol. 2016, 310, C643–C662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krizova, A.; Maltan, L.; Derler, I. Critical parameters maintaining authentic CRAC channel hallmarks. Eur. Biophys. J. 2019, 48, 425–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parekh, A.B.; Putney, J.W., Jr. Store-operated calcium channels. Physiol. Rev. 2005, 85, 757–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeung, P.S.; Prakriya, M. The exquisitely cooperative nature of Orai1 channel activation. J. Gen. Physiol. 2018, 150, 1352–1355. [Google Scholar] [CrossRef]
- Yeung, P.S.; Yamashita, M.; Prakriya, M. Molecular basis of allosteric Orai1 channel activation by STIM1. J. Physiol. 2019, 598, 1707–1723. [Google Scholar] [CrossRef]
- Liou, J. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr. Biol. 2005, 15, 1235–1241. [Google Scholar] [CrossRef]
- Hou, X.; Pedi, L.; Diver, M.M.; Long, S.B. Crystal structure of the calcium release-activated calcium channel Orai. Science 2012, 338, 1308–1313. [Google Scholar] [CrossRef] [Green Version]
- Prakriya, M.; Feske, S.; Gwack, Y.; Srikanth, S.; Rao, A.; Hogan, P.G. Orai1 is an essential pore subunit of the CRAC channel. Nature 2006, 443, 230–233. [Google Scholar] [CrossRef]
- Feske, S.; Gwack, Y.; Prakriya, M.; Srikanth, S.; Puppel, S.H.; Tanasa, B.; Hogan, P.G.; Lewis, R.S.; Daly, M.; Rao, A. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 2006, 441, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Berna-Erro, A.; Braun, A.; Kraft, R.; Kleinschnitz, C.; Schuhmann, M.K.; Stegner, D.; Wultsch, T.; Eilers, J.; Meuth, S.G.; Stoll, G.; et al. STIM2 regulates capacitive Ca2+ entry in neurons and plays a key role in hypoxic neuronal cell death. Sci. Signal. 2009, 2, ra67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraft, R. STIM and ORAI proteins in the nervous system. Channels 2015, 9, 245–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, R.T.; Manji, S.S.; Parker, N.J.; Hancock, M.S.; van Stekelenburg, L.; Eid, J.P.; Senior, P.V.; Kazenwadel, J.S.; Shandala, T.; Saint, R.; et al. Identification and characterization of the STIM (stromal interaction molecule) gene family: Coding for a novel class of transmembrane proteins. Biochem. J. 2001, 357 Pt 3, 673–685. [Google Scholar] [CrossRef] [PubMed]
- Vashisht, A.; Tanwar, J.; Motiani, R.K. Regulation of proto-oncogene Orai3 by miR18a/b and miR34a. Cell Calcium 2018, 75, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Motiani, R.K.; Abdullaev, I.F.; Trebak, M. A novel native store-operated calcium channel encoded by Orai3: Selective requirement of Orai3 versus Orai1 in estrogen receptor-positive versus estrogen receptor-negative breast cancer cells. J. Biol. Chem. 2010, 285, 19173–19183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motiani, R.K.; Zhang, X.; Harmon, K.E.; Keller, R.S.; Matrougui, K.; Bennett, J.A.; Trebak, M. Orai3 is an estrogen receptor alpha-regulated Ca2+ channel that promotes tumorigenesis. FASEB J. 2013, 27, 63–75. [Google Scholar] [CrossRef] [Green Version]
- Soboloff, J.; Spassova, M.A.; Hewavitharana, T.; He, L.P.; Xu, W.; Johnstone, L.S.; Dziadek, M.A.; Gill, D.L. STIM2 is an inhibitor of STIM1-mediated store-operated Ca2+ Entry. Curr. Biol. 2006, 16, 1465–1470. [Google Scholar] [CrossRef]
- Miederer, A.M.; Alansary, D.; Schwar, G.; Lee, P.H.; Jung, M.; Helms, V.; Niemeyer, B.A. A STIM2 splice variant negatively regulates store-operated calcium entry. Nat. Commun. 2015, 6, 6899. [Google Scholar] [CrossRef] [Green Version]
- Rana, A.; Yen, M.; Sadaghiani, A.M.; Malmersjo, S.; Park, C.Y.; Dolmetsch, R.E.; Lewis, R.S. Alternative splicing converts STIM2 from an activator to an inhibitor of store-operated calcium channels. J. Cell Biol. 2015, 209, 653–669. [Google Scholar] [CrossRef] [Green Version]
- Rosado, J.A.; Diez, R.; Smani, T.; Jardin, I. STIM and Orai1 Variants in Store-Operated Calcium Entry. Front. Pharmacol. 2015, 6, 325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramesh, G.; Jarzembowski, L.; Schwarz, Y.; Poth, V.; Konrad, M.; Knapp, M.L.; Schwär, G.; Lauer, A.A.; Grimm, M.O.W.; Alansary, D.; et al. A short isoform of STIM1 confers frequency-dependent synaptic enhancement. Cell Rep. 2021, 34, 108844. [Google Scholar] [CrossRef]
- Brandman, O.; Liou, J.; Park, W.S.; Meyer, T. STIM2 is a feedback regulator that stabilizes basal cytosolic and endoplasmic reticulum Ca2+ levels. Cell 2007, 131, 1327–1339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subedi, K.P.; Ong, H.L.; Son, G.Y.; Liu, X.; Ambudkar, I.S. STIM2 Induces Activated Conformation of STIM1 to Control Orai1 Function in ER-PM Junctions. Cell Rep. 2018, 23, 522–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wissenbach, U.; Philipp, S.E.; Gross, S.A.; Cavalie, A.; Flockerzi, V. Primary structure, chromosomal localization and expression in immune cells of the murine ORAI and STIM genes. Cell Calcium 2007, 42, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Derler, I.; Plenk, P.; Fahrner, M.; Muik, M.; Jardin, I.; Schindl, R.; Gruber, H.J.; Groschner, K.; Romanin, C. The extended transmembrane Orai1 N-terminal (ETON) region combines binding interface and gate for Orai1 activation by STIM1. J. Biol. Chem. 2013, 288, 29025–29034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNally, B.A.; Somasundaram, A.; Jairaman, A.; Yamashita, M.; Prakriya, M. The C- and N-terminal STIM1 binding sites on Orai1 are required for both trapping and gating CRAC channels. J. Physiol. 2013, 591, 2833–2850. [Google Scholar] [CrossRef]
- Palty, R.; Stanley, C.; Isacoff, E.Y. Critical role for Orai1 C-terminal domain and TM4 in CRAC channel gating. Cell Res. 2015, 25, 963–980. [Google Scholar] [CrossRef]
- Stathopulos, P.B.; Zheng, L.; Ikura, M. Stromal interaction molecule (STIM) 1 and STIM2 calcium sensing regions exhibit distinct unfolding and oligomerization kinetics. J. Biol. Chem. 2009, 284, 728–732. [Google Scholar] [CrossRef] [Green Version]
- Hirve, N.; Rajanikanth, V.; Hogan, P.G.; Gudlur, A. Coiled-Coil Formation Conveys a STIM1 Signal from ER Lumen to Cytoplasm. Cell Rep. 2018, 22, 72–83. [Google Scholar] [CrossRef] [Green Version]
- Gudlur, A.; Zeraik, A.E.; Hirve, N.; Rajanikanth, V.; Bobkov, A.A.; Ma, G.; Zheng, S.; Wang, Y.; Zhou, Y.; Komives, E.A.; et al. Calcium sensing by the STIM1 ER-luminal domain. Nat. Commun. 2018, 9, 4536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, G.; Wei, M.; He, L.; Liu, C.; Wu, B.; Zhang, S.L.; Jing, J.; Liang, X.; Senes, A.; Tan, P.; et al. Inside-out Ca2+ signalling prompted by STIM1 conformational switch. Nat. Commun. 2015, 6, 7826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sallinger, M.; Tiffner, A.; Schmidt, T.; Bonhenry, D.; Waldherr, L.; Frischauf, I.; Lunz, I.; Derler, I.; Schober, R.; Schindl, R. Luminal STIM1 mutants that cause tubular aggregate myopathy promote autophagic processes. Int. J. Mol. Sci. 2020, 21, 4410. [Google Scholar] [CrossRef] [PubMed]
- Schober, R.; Bonhenry, D.; Lunz, V.; Zhu, J.; Krizova, A.; Frischauf, I.; Fahrner, M.; Zhang, M.; Waldherr, L.; Schmidt, T.; et al. Sequential activation of STIM1 links Ca2+ with luminal domain unfolding. Sci. Signal. 2019, 12, eaax3194. [Google Scholar] [CrossRef] [PubMed]
- Mullins, F.M.; Lewis, R.S. The inactivation domain of STIM1 is functionally coupled with the Orai1 pore to enable Ca2+-dependent inactivation. J. Gen. Physiol. 2016, 147, 153–164. [Google Scholar] [CrossRef] [Green Version]
- Derler, I.; Fahrner, M.; Muik, M.; Lackner, B.; Schindl, R.; Groschner, K.; Romanin, C. A Ca2+ release-activated Ca2+ (CRAC) modulatory domain (CMD) within STIM1 mediates fast Ca2(+)-dependent inactivation of ORAI1 channels. J. Biol. Chem. 2009, 284, 24933–24938. [Google Scholar] [CrossRef] [Green Version]
- Hewavitharana, T.; Deng, X.; Soboloff, J.; Gill, D.L. Role of STIM and Orai proteins in the store-operated calcium signaling pathway. Cell Calcium 2007, 42, 173–182. [Google Scholar] [CrossRef]
- Hogan, P.G.; Lewis, R.S.; Rao, A. Molecular basis of calcium signaling in lymphocytes: STIM and ORAI. Annu. Rev. Immunol. 2010, 28, 491–533. [Google Scholar] [CrossRef]
- Rathner, P.; Fahrner, M.; Cerofolini, L.; Grabmayr, H.; Horvath, F.; Krobath, H.; Gupta, A.; Ravera, E.; Fragai, M.; Bechmann, M.; et al. Interhelical interactions within the STIM1 CC1 domain modulate CRAC channel activation. Nat. Chem. Biol. 2021, 17, 196–204. [Google Scholar] [CrossRef]
- van Dorp, S.; Qiu, R.; Choi, U.B.; Wu, M.M.; Yen, M.; Kirmiz, M.; Brunger, A.T.; Lewis, R.S. Conformational dynamics of auto-inhibition in the ER calcium sensor STIM1. eLife 2021, 10, e66194. [Google Scholar] [CrossRef]
- Grabmayr, H.; Romanin, C.; Fahrner, M. STIM Proteins: An Ever-Expanding Family. Int. J. Mol. Sci. 2020, 22, 378. [Google Scholar] [CrossRef] [PubMed]
- Cui, B.; Yang, X.; Li, S.; Lin, Z.; Wang, Z.; Dong, C.; Shen, Y. The inhibitory helix controls the intramolecular conformational switching of the C-terminus of STIM1. PLoS ONE 2013, 8, e74735. [Google Scholar] [CrossRef] [Green Version]
- Ma, G.; He, L.; Liu, S.; Xie, J.; Huang, Z.; Jing, J.; Lee, Y.-T.; Wang, R.; Luo, H.; Han, W.; et al. Optogenetic engineering to probe the molecular choreography of STIM1-mediated cell signaling. Nat. Commun. 2020, 11, 1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muik, M.; Fahrner, M.; Derler, I.; Schindl, R.; Bergsmann, J.; Frischauf, I.; Groschner, K.; Romanin, C. A Cytosolic Homomerization and a Modulatory Domain within STIM1 C Terminus Determine Coupling to ORAI1 Channels. J. Biol. Chem. 2009, 284, 8421–8426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawasaki, T.; Lange, I.; Feske, S. A minimal regulatory domain in the C terminus of STIM1 binds to and activates ORAI1 CRAC channels. Biochem. Biophys. Res. Commun. 2009, 385, 49–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stathopulos, P.B.; Schindl, R.; Fahrner, M.; Zheng, L.; Gasmi-Seabrook, G.M.; Muik, M.; Romanin, C.; Ikura, M. STIM1/Orai1 coiled-coil interplay in the regulation of store-operated calcium entry. Nat. Commun. 2013, 4, 2963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Jin, H.; Cai, X.; Li, S.; Shen, Y. Structural and mechanistic insights into the activation of Stromal interaction molecule 1 (STIM1). Proc. Natl. Acad. Sci. USA 2012, 109, 5657–5662. [Google Scholar] [CrossRef] [Green Version]
- Höglinger, C.; Grabmayr, H.; Maltan, L.; Horvath, F.; Krobath, H.; Muik, M.; Tiffner, A.; Renger, T.; Romanin, C.; Fahrner, M.; et al. Defects in the STIM1 SOARα2 domain affect multiple steps in the CRAC channel activation cascade. Cell. Mol. Life Sci. 2021, 78, 6645–6667. [Google Scholar] [CrossRef]
- Frischauf, I.; Muik, M.; Derler, I.; Bergsmann, J.; Fahrner, M.; Schindl, R.; Groschner, K.; Romanin, C. Molecular determinants of the coupling between STIM1 and Orai channels: Differential activation of Orai1-3 channels by a STIM1 coiled-coil mutant. J. Biol. Chem. 2009, 284, 21696–21706. [Google Scholar] [CrossRef] [Green Version]
- Vig, M.; Peinelt, C.; Beck, A.; Koomoa, D.L.; Rabah, D.; Koblan-Huberson, M.; Kraft, S.; Turner, H.; Fleig, A.; Penner, R.; et al. CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science 2006, 312, 1220–1223. [Google Scholar] [CrossRef] [Green Version]
- Gross, S.A.; Wissenbach, U.; Philipp, S.E.; Freichel, M.; Cavalie, A.; Flockerzi, V. Murine ORAI2 splice variants form functional Ca2+ release-activated Ca2+ (CRAC) channels. J. Biol. Chem. 2007, 282, 19375–19384. [Google Scholar] [CrossRef] [Green Version]
- Gwack, Y.; Srikanth, S.; Feske, S.; Cruz-Guilloty, F.; Oh-Hora, M.; Neems, D.S.; Hogan, P.G.; Rao, A. Biochemical and functional characterization of Orai proteins. J. Biol. Chem. 2007, 282, 16232–16243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dziadek, M.A.; Johnstone, L.S. Biochemical properties and cellular localisation of STIM proteins. Cell Calcium 2007, 42, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Shuttleworth, T.J. Orai3—The ‘exceptional’ Orai? J. Physiol. 2012, 590, 241–257. [Google Scholar] [CrossRef] [PubMed]
- Ji, W.; Xu, P.; Li, Z.; Lu, J.; Liu, L.; Zhan, Y.; Chen, Y.; Hille, B.; Xu, T.; Chen, L. Functional stoichiometry of the unitary calcium-release-activated calcium channel. Proc. Natl. Acad. Sci. USA 2008, 105, 13668–13673. [Google Scholar] [CrossRef] [Green Version]
- Mignen, O.; Thompson, J.L.; Shuttleworth, T.J. Orai1 subunit stoichiometry of the mammalian CRAC channel pore. J. Physiol. 2008, 586, 419–425. [Google Scholar] [CrossRef]
- Penna, A.; Demuro, A.; Yeromin, A.V.; Zhang, S.L.; Safrina, O.; Parker, I.; Cahalan, M.D. The CRAC channel consists of a tetramer formed by Stim-induced dimerization of Orai dimers. Nature 2008, 456, 116–120. [Google Scholar] [CrossRef] [Green Version]
- Srikanth, S.; Yee, M.K.W.; Gwack, Y.; Ribalet, B. The third transmembrane segment of Orai1 protein modulates Ca2+ release-activated Ca2+ (CRAC) channel gating and permeation properties. J. Biol. Chem. 2011, 286, 35318–35328. [Google Scholar] [CrossRef]
- Fahrner, M.; Pandey, S.K.; Muik, M.; Traxler, L.; Butorac, C.; Stadlbauer, M.; Zayats, V.; Krizova, A.; Plenk, P.; Frischauf, I.; et al. Communication between N terminus and loop2 tunes Orai activation. J. Biol. Chem. 2018, 293, 1271–1285. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Cai, X.; Loktionova, N.A.; Wang, X.; Nwokonko, R.M.; Wang, X.; Wang, Y.; Rothberg, B.S.; Trebak, M.; Gill, D.L. The STIM1-binding site nexus remotely controls Orai1 channel gating. Nat. Commun. 2016, 7, 13725. [Google Scholar] [CrossRef] [Green Version]
- Baraniak, J.H.; Zhou, Y.; Nwokonko, R.M.; Jennette, M.R.; Kazzaz, S.A.; Stenson, J.M.; Whitsell, A.L.; Wang, Y.; Trebak, M.; Gill, D.L. Orai channel C-terminal peptides are key modulators of STIM-Orai coupling and calcium signal generation. Cell Rep. 2021, 35, 109322. [Google Scholar] [CrossRef] [PubMed]
- Yen, M.; Lokteva, L.A.; Lewis, R.S. Functional Analysis of Orai1 Concatemers Supports a Hexameric Stoichiometry for the CRAC Channel. Biophys. J. 2016, 111, 1897–1907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korzeniowski, M.K.; Manjarrés, I.M.; Varnai, P.; Balla, T. Activation of STIM1-Orai1 Involves an Intramolecular Switching Mechanism. Sci. Signal. 2010, 3, ra82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butorac, C.; Muik, M.; Derler, I.; Stadlbauer, M.; Lunz, V.; Krizova, A.; Lindinger, S.; Schober, R.; Frischauf, I.; Bhardwaj, R.; et al. A novel STIM1-Orai1 gating interface essential for CRAC channel activation. Cell Calcium 2019, 79, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Derler, I.; Schindl, R.; Fritsch, R.; Romanin, C. Gating and permeation of Orai channels. Front. Biosci. Landmark Ed. 2012, 17, 1304–1322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frischauf, I.; Schindl, R.; Bergsmann, J.; Derler, I.; Fahrner, M.; Muik, M.; Fritsch, R.; Lackner, B.; Groschner, K.; Romanin, C. Cooperativeness of Orai cytosolic domains tunes subtype-specific gating. J. Biol. Chem. 2011, 286, 8577–8584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, X.; Burstein, S.R.; Long, S. Structures reveal opening of the store-operated calcium channel Orai. bioRxiv 2018. [Google Scholar] [CrossRef]
- Hou, X.; Outhwaite, I.R.; Pedi, L.; Long, S.B. Cryo-EM structure of the calcium release-activated calcium channel Orai in an open conformation. eLife 2020, 9, e62772. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wu, G.; Yu, Y.; Chen, X.; Ji, R.; Lu, J.; Li, X.; Zhang, X.; Yang, X.; Shen, Y. Molecular understanding of calcium permeation through the open Orai channel. PLoS Biol. 2019, 17, e3000096. [Google Scholar] [CrossRef]
- Tiffner, A.; Maltan, L.; Weiß, S.; Derler, I. The orai pore opening mechanism. Int. J. Mol. Sci. 2021, 22, 533. [Google Scholar] [CrossRef]
- Yeung, P.S.W.; Yamashita, M.; Ing, C.E.; Pomès, R.; Freymann, D.M.; Prakriya, M. Mapping the functional anatomy of Orai1 transmembrane domains for CRAC channel gating. Proc. Natl. Acad. Sci. USA 2018, 115, E5193–E5202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiffner, A.; Schober, R.; Höglinger, C.; Bonhenry, D.; Pandey, S.; Lunz, V.; Sallinger, M.; Frischauf, I.; Fahrner, M.; Lindinger, S.; et al. CRAC channel opening is determined by a series of Orai1 gating checkpoints in the transmembrane and cytosolic regions. J. Biol. Chem. 2021, 296, 100224. [Google Scholar] [CrossRef]
- Takahashi, Y.; Murakami, M.; Watanabe, H.; Hasegawa, H.; Ohba, T.; Munehisa, Y.; Nobori, K.; Ono, K.; Iijima, T.; Ito, H. Essential role of the N-terminus of murine Orai1 in store-operated Ca2+ entry. Biochem. Biophys. Res. Commun. 2007, 356, 45–52. [Google Scholar] [CrossRef]
- Bergsmann, J.; Derler, I.; Muik, M.; Frischauf, I.; Fahrner, M.; Pollheimer, P.; Schwarzinger, C.; Gruber, H.J.; Groschner, K.; Romanin, C. Molecular Determinants within N Terminus of Orai3 Protein That Control Channel Activation and Gating. J. Biol. Chem. 2011, 286, 31565–31575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiffner, A.; Derler, I. Isoform-specific properties of orai homologues in activation, downstream signaling, physiology and pathophysiology. Int. J. Mol. Sci. 2021, 22, 8020. [Google Scholar] [CrossRef] [PubMed]
- Tiffner, A.; Maltan, L.; Fahrner, M.; Sallinger, M.; Weiß, S.; Grabmayr, H.; Höglinger, C.; Derler, I. Transmembrane Domain 3 (TM3) Governs Orai1 and Orai3 Pore Opening in an Isoform-Specific Manner. Front. Cell Dev. Biol. 2021, 9, 635705. [Google Scholar] [CrossRef] [PubMed]
- Maltan, L.; Andova, A.M.; Derler, I. The Role of Lipids in CRAC Channel Function. Biomolecules 2022, 12, 352. [Google Scholar] [CrossRef] [PubMed]
- Hogan, P.G. The STIM1-ORAI1 microdomain. Cell Calcium 2015, 58, 357–367. [Google Scholar] [CrossRef]
- Cao, X.; Choi, S.; Maleth, J.J.; Park, S.; Ahuja, M.; Muallem, S. The ER/PM microdomain, PI(4,5)P(2) and the regulation of STIM1-Orai1 channel function. Cell Calcium 2015, 58, 342–348. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Ramachandran, S.; Oh-Hora, M.; Rao, A.; Hogan, P.G. Pore architecture of the ORAI1 store-operated calcium channel. Proc. Natl. Acad. Sci. USA 2010, 107, 4896–4901. [Google Scholar] [CrossRef] [Green Version]
- Deng, X.; Wang, Y.; Zhou, Y.; Soboloff, J.; Gill, D.L. STIM and orai: Dynamic intermembrane coupling to control cellular calcium signals. J. Biol. Chem. 2009, 284, 22501–22505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balla, T. Ca2+ and lipid signals hold hands at endoplasmic reticulum-plasma membrane contact sites. J. Physiol. 2018, 596, 2709–2716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gwozdz, T.; Dutko-Gwozdz, J.; Schafer, C.; Bolotina, V.M. Overexpression of Orai1 and STIM1 proteins alters regulation of store-operated Ca2+ entry by endogenous mediators. J. Biol. Chem. 2012, 287, 22865–22872. [Google Scholar] [CrossRef] [Green Version]
- Derler, I.; Jardin, I.; Stathopulos, P.B.; Muik, M.; Fahrner, M.; Zayats, V.; Pandey, S.K.; Poteser, M.; Lacker, B.; Absolonova, M.; et al. Cholesterol modulates Orai1 channel function. Sci. Signal. 2016, 9, ra10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pacheco, J.; Dominguez, L.; Bohorquez-Hernandez, A.; Asanov, A.; Vaca, L. A cholesterol-binding domain in STIM1 modulates STIM1-Orai1 physical and functional interactions. Sci. Rep. 2016, 6, 29634. [Google Scholar] [CrossRef] [Green Version]
- Bohorquez-Hernandez, A.; Gratton, E.; Pacheco, J.; Asanov, A.; Vaca, L. Cholesterol modulates the cellular localization of Orai1 channels and its disposition among membrane domains. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2017, 1862, 1481–1490. [Google Scholar] [CrossRef]
- Carreras-Sureda, A.; Abrami, L.; Ji-Hee, K.; Wang, W.A.; Henry, C.; Frieden, M.; Didier, M.; van der Goot, F.G.; Demaurex, N. S-acylation by ZDHHC20 targets ORAI1 channels to lipid rafts for efficient Ca2+ signaling by Jurkat T cell receptors at the immune synapse. eLife 2021, 10, e72051. [Google Scholar] [CrossRef]
- Heo, W.D.; Inoue, T.; Park, W.S.; Kim, M.L.; Park, B.O.; Wandless, T.J.; Meyer, T. PI(3,4,5)P3 and PI(4,5)P2 lipids target proteins with polybasic clusters to the plasma membrane. Science 2006, 314, 1458–1461. [Google Scholar] [CrossRef]
- Liou, J.; Fivaz, M.; Inoue, T.; Meyer, T. Live-cell imaging reveals sequential oligomerization and local plasma membrane targeting of stromal interaction molecule 1 after Ca2+ store depletion. Proc. Natl. Acad. Sci. USA 2007, 104, 9301–9306. [Google Scholar] [CrossRef] [Green Version]
- Ercan, E.; Momburg, F.; Engel, U.; Temmerman, K.; Nickel, W.; Seedorf, M. A conserved, lipid-mediated sorting mechanism of yeast Ist2 and mammalian STIM proteins to the peripheral ER. Traffic 2009, 10, 1802–1818. [Google Scholar] [CrossRef] [PubMed]
- Collins, S.R.; Meyer, T. Evolutionary origins of STIM1 and STIM2 within ancient Ca2+ signaling systems. Trends Cell Biol. 2011, 21, 202–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chvanov, M.; Walsh, C.M.; Haynes, L.P.; Voronina, S.G.; Lur, G.; Gerasimenko, O.V.; Barraclough, R.; Rudland, P.S.; Petersen, O.H.; Burgoyne, R.D.; et al. ATP depletion induces translocation of STIM1 to puncta and formation of STIM1-ORAI1 clusters: Translocation and re-translocation of STIM1 does not require ATP. Pflug. Arch. 2008, 457, 505–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korzeniowski, M.K.; Popovic, M.A.; Szentpetery, Z.; Varnai, P.; Stojilkovic, S.S.; Balla, T. Dependence of STIM1/Orai1-mediated calcium entry on plasma membrane phosphoinositides. J. Biol. Chem. 2009, 284, 21027–21035. [Google Scholar] [CrossRef] [Green Version]
- Walsh, C.M.; Doherty, M.K.; Tepikin, A.V.; Burgoyne, R.D. Evidence for an interaction between Golli and STIM1 in store-operated calcium entry. Biochem. J. 2010, 430, 453–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maleth, J.; Choi, S.; Muallem, S.; Ahuja, M. Translocation between PI(4,5)P2-poor and PI(4,5)P2-rich microdomains during store depletion determines STIM1 conformation and Orai1 gating. Nat. Commun. 2014, 5, 5843. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.M.; Covington, E.D.; Lewis, R.S. Single-molecule analysis of diffusion and trapping of STIM1 and Orai1 at endoplasmic reticulum-plasma membrane junctions. Mol. Biol. Cell 2014, 25, 3672–3685. [Google Scholar] [CrossRef]
- Calloway, N.; Owens, T.; Corwith, K.; Rodgers, W.; Holowka, D.; Baird, B. Stimulated association of STIM1 and Orai1 is regulated by the balance of PtdIns(4,5)P(2) between distinct membrane pools. J. Cell Sci. 2011, 124 Pt 15, 2602–2610. [Google Scholar] [CrossRef] [Green Version]
- Rosado, J.A.; Sage, S.O. Phosphoinositides are required for store-mediated calcium entry in human platelets. J. Biol. Chem. 2000, 275, 9110–9113. [Google Scholar] [CrossRef]
- Broad, L.M.; Braun, F.J.; Lievremont, J.P.; Bird, G.S.; Kurosaki, T.; Putney, J.W., Jr. Role of the phospholipase C-inositol 1,4,5-trisphosphate pathway in calcium release-activated calcium current and capacitative calcium entry. J. Biol. Chem. 2001, 276, 15945–15952. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Papadopoulos, V. Peripheral-type benzodiazepine receptor function in cholesterol transport. Identification of a putative cholesterol recognition/interaction amino acid sequence and consensus pattern. Endocrinology 1998, 139, 4991–4997. [Google Scholar] [CrossRef]
- Epand, R.M. Cholesterol and the interaction of proteins with membrane domains. Prog. Lipid Res. 2006, 45, 279–294. [Google Scholar] [CrossRef] [PubMed]
- Epand, R.M. Proteins and cholesterol-rich domains. Biochim. Biophys. Acta BBA Biomembr. 2008, 1778, 1576–1582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fantini, J.; Barrantes, F. How cholesterol interacts with membrane proteins: An exploration of cholesterol-binding sites including CRAC, CARC, and tilted domains. Front. Physiol. 2013, 4, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galan, C.; Woodard, G.E.; Dionisio, N.; Salido, G.M.; Rosado, J.A. Lipid rafts modulate the activation but not the maintenance of store-operated Ca2+ entry. Biochim. Biophys. Acta BBA Mol. Cell Res. 2010, 1803, 1083–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Combs, D.J.; Lu, Z. Sphingomyelinase D inhibits store-operated Ca2+ entry in T lymphocytes by suppressing ORAI current. J. Gen. Physiol. 2015, 146, 161–172. [Google Scholar] [CrossRef] [Green Version]
- Hogan, P.G. Sphingomyelin, ORAI1 channels, and cellular Ca2+ signaling. J. Gen. Physiol. 2015, 146, 195–200. [Google Scholar] [CrossRef] [Green Version]
- Kodakandla, G.; West, S.J.; Wang, Q.; Tewari, R.; Zhu, M.X.; Akimzhanov, A.M.; Boehning, D. Dynamic S-acylation of the ER-resident protein stromal interaction molecule 1 (STIM1) is required for store-operated Ca2+ entry. J. Biol. Chem. 2022, 298, 102303. [Google Scholar] [CrossRef]
- Son, A.; Park, S.; Shin, D.M.; Muallem, S. Orai1 and STIM1 in ER/PM junctions: Roles in pancreatic cell function and dysfunction. Am. J. Physiol. Cell Physiol. 2016, 310, C414–C422. [Google Scholar] [CrossRef]
- Giordano, F.; Saheki, Y.; Idevall-Hagren, O.; Colombo, S.F.; Pirruccello, M.; Milosevic, I.; Gracheva, E.O.; Bagriantsev, S.N.; Borgese, N.; De Camilli, P. PI(4,5)P(2)-dependent and Ca2+-regulated ER-PM interactions mediated by the extended synaptotagmins. Cell 2013, 153, 1494–1509. [Google Scholar] [CrossRef] [Green Version]
- Besprozvannaya, M.; Dickson, E.; Li, H.; Ginburg, K.S.; Bers, D.M.; Auwerx, J.; Nunnari, J. GRAM domain proteins specialize functionally distinct ER-PM contact sites in human cells. eLife 2018, 7, e31019. [Google Scholar] [CrossRef]
- Jha, A.; Chung, W.Y.; Vachel, L.; Maleth, J.; Lake, S.; Zhang, G.; Ahuja, M.; Muallem, S. Anoctamin 8 tethers endoplasmic reticulum and plasma membrane for assembly of Ca2+ signaling complexes at the ER/PM compartment. EMBO J. 2019, 38, e101452. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Quintana, A.; Findlay, G.M.; Mettlen, M.; Baust, B.; Jain, M.; Nilsson, R.; Rao, A.; Hogan, P.G. An siRNA screen for NFAT activation identifies septins as coordinators of store-operated Ca2+ entry. Nature 2013, 499, 238–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deb, B.K.; Hasan, G. Regulation of Store-Operated Ca2+ Entry by Septins. Front. Cell Dev. Biol. 2016, 4, 142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katz, Z.B.; Zhang, C.; Quintana, A.; Lillemeier, B.F.; Hogan, P.G. Septins organize endoplasmic reticulum-plasma membrane junctions for STIM1-ORAI1 calcium signalling. Sci. Rep. 2019, 9, 10839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deb, B.K.; Pathak, T.; Hasan, G. Store-independent modulation of Ca2+ entry through Orai by Septin 7. Nat. Commun. 2016, 7, 11751. [Google Scholar] [CrossRef] [Green Version]
- Deb, B.K.; Chakraborty, P.; Gopurappilly, R.; Hasan, G. SEPT7 regulates Ca2+ entry through Orai channels in human neural progenitor cells and neurons. Cell Calcium 2020, 90, 102252. [Google Scholar] [CrossRef]
- De Souza, L.B.; Ong, H.L.; Liu, X.; Ambudkar, I.S. PIP2 and septin control STIM1/Orai1 assembly by regulating cytoskeletal remodeling via a CDC42-WASP/WAVE-ARP2/3 protein complex. Cell Calcium 2021, 99, 102475. [Google Scholar] [CrossRef]
- Srikanth, S.; Jew, M.; Kim, K.D.; Yee, M.K.; Abramson, J.; Gwack, Y. Junctate is a Ca2+-sensing structural component of Orai1 and stromal interaction molecule 1 (STIM1). Proc. Natl. Acad. Sci. USA 2012, 109, 8682–8687. [Google Scholar] [CrossRef]
- Woo, J.S.; Srikanth, S.; Nishi, M.; Ping, P.; Takeshima, H.; Gwack, Y. Junctophilin-4, a component of the endoplasmic reticulum-plasma membrane junctions, regulates Ca2+ dynamics in T cells. Proc. Natl. Acad. Sci. USA 2016, 113, 2762–2767. [Google Scholar] [CrossRef] [Green Version]
- Jing, J.; He, L.; Sun, A.; Quintana, A.; Ding, Y.; Ma, G.; Tan, P.; Liang, X.; Zheng, X.; Chen, L.; et al. Proteomic mapping of ER-PM junctions identifies STIMATE as a regulator of Ca2+ influx. Nat. Cell Biol. 2015, 17, 1339–1347. [Google Scholar] [CrossRef] [Green Version]
- Jha, A.; Ahuja, M.; Maleth, J.; Moreno, C.M.; Yuan, J.P.; Kim, M.S.; Muallem, S. The STIM1 CTID domain determines access of SARAF to SOAR to regulate Orai1 channel function. J. Cell Biol. 2013, 202, 71–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palty, R.; Raveh, A.; Kaminsky, I.; Meller, R.; Reuveny, E. SARAF inactivates the store operated calcium entry machinery to prevent excess calcium refilling. Cell 2012, 149, 425–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, F.; Sun, L.; Machaca, K. Constitutive recycling of the store-operated Ca2+ channel Orai1 and its internalization during meiosis. J. Cell Biol. 2010, 191, 523–535. [Google Scholar] [CrossRef] [Green Version]
- Jiao, H.X.; Mu, Y.P.; Gui, L.X.; Yan, F.R.; Lin, D.C.; Sham, J.S.; Lin, M.J. Increase in caveolae and caveolin-1 expression modulates agonist-induced contraction and store- and receptor-operated Ca2+ entry in pulmonary arteries of pulmonary hypertensive rats. Vascul. Pharmacol. 2016, 84, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Weisleder, N.; Wu, P.; Cai, C.; Chen, J.W. Caveolae/caveolin-1 are important modulators of store-operated calcium entry in Hs578/T breast cancer cells. J. Pharmacol. Sci. 2008, 106, 287–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeh, Y.C.; Parekh, A.B. Distinct structural domains of caveolin-1 independently regulate Ca2+ release-activated Ca2+ channels and Ca2+ microdomain-dependent gene expression. Mol. Cell. Biol. 2015, 35, 1341–1349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pani, B.; Ong, H.L.; Liu, X.; Rauser, K.; Ambudkar, I.S.; Singh, B.B. Lipid Rafts Determine Clustering of STIM1 in Endoplasmic Reticulum-Plasma Membrane Junctions and Regulation of Store-operated Ca2+ Entry (SOCE). J. Biol. Chem. 2008, 283, 17333–17340. [Google Scholar] [CrossRef] [Green Version]
- Alicia, S.; Angélica, Z.; Carlos, S.; Alfonso, S.; Vaca, L. STIM1 converts TRPC1 from a receptor-operated to a store-operated channel: Moving TRPC1 in and out of lipid rafts. Cell Calcium 2008, 44, 479–491. [Google Scholar] [CrossRef]
- Lockwich, T.P.; Liu, X.; Singh, B.B.; Jadlowiec, J.; Weiland, S.; Ambudkar, I.S. Assembly of Trp1 in a Signaling Complex Associated with Caveolin-Scaffolding Lipid Raft Domains. J. Biol. Chem. 2000, 275, 11934–11942. [Google Scholar] [CrossRef] [Green Version]
- Rathor, N.; Chung, H.K.; Wang, S.R.; Wang, J.Y.; Turner, D.J.; Rao, J.N. Caveolin-1 enhances rapid mucosal restitution by activating TRPC1-mediated Ca2+ signaling. Physiol. Rep. 2014, 2, e12193. [Google Scholar]
- Murata, T.; Lin, M.I.; Stan, R.V.; Bauer, P.M.; Yu, J.; Sessa, W.C. Genetic Evidence Supporting Caveolae Microdomain Regulation of Calcium Entry in Endothelial Cells. J. Biol. Chem. 2007, 282, 16631–16643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prakash, Y.S.; Thompson, M.A.; Vaa, B.; Matabdin, I.; Peterson, T.E.; He, T.; Pabelick, C.M. Caveolins and intracellular calcium regulation in human airway smooth muscle. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2007, 293, L1118–L1126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pani, B.; Ong, H.L.; Brazer, S.C.; Liu, X.; Rauser, K.; Singh, B.B.; Ambudkar, I.S. Activation of TRPC1 by STIM1 in ER-PM microdomains involves release of the channel from its scaffold caveolin-1. Proc. Natl. Acad. Sci. USA 2009, 106, 20087–20092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaeth, M.; Kahlfuss, S.; Feske, S. CRAC Channels and Calcium Signaling in T Cell-Mediated Immunity. Trends Immunol. 2020, 41, 878–901. [Google Scholar] [CrossRef]
- Diez-Bello, R.; Jardin, I.; Salido, G.M.; Rosado, J.A. Orai1 and Orai2 mediate store-operated calcium entry that regulates HL60 cell migration and FAK phosphorylation. Biochim. Biophys. Acta BBA Mol. Cell Res. 2017, 1864, 1064–1070. [Google Scholar] [CrossRef]
- Tanwar, J.; Arora, S.; Motiani, R.K. Orai3: Oncochannel with therapeutic potential. Cell Calcium 2020, 90, 102247. [Google Scholar] [CrossRef]
- Chen, Y.T.; Chen, Y.F.; Chiu, W.T.; Liu, K.Y.; Liu, Y.L.; Chang, J.Y.; Chang, H.C.; Shen, M.R. Microtubule-associated histone deacetylase 6 supports the calcium store sensor STIM1 in mediating malignant cell behaviors. Cancer Res. 2013, 73, 4500–4509. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.F.; Chiu, W.T.; Chen, Y.T.; Lin, P.Y.; Huang, H.J.; Chou, C.Y.; Chang, H.C.; Tang, M.J.; Shen, M.R. Calcium store sensor stromal-interaction molecule 1-dependent signaling plays an important role in cervical cancer growth, migration, and angiogenesis. Proc. Natl. Acad. Sci. USA 2011, 108, 15225–15230. [Google Scholar] [CrossRef] [Green Version]
- Ay, A.S.; Benzerdjerb, N.; Sevestre, H.; Ahidouch, A.; Ouadid-Ahidouch, H. Orai3 Constitutes a Native Store-Operated Calcium Entry That Regulates Non Small Cell Lung Adenocarcinoma Cell Proliferation. PLoS ONE 2013, 8, e72889. [Google Scholar] [CrossRef]
- McAndrew, D.; Grice, D.M.; Peters, A.A.; Davis, F.M.; Stewart, T.; Rice, M.; Smart, C.E.; Brown, M.A.; Kenny, P.A.; Roberts-Thomson, S.J.; et al. ORAI1-Mediated Calcium Influx in Lactation and in Breast Cancer. Mol. Cancer Ther. 2011, 10, 448–460. [Google Scholar] [CrossRef] [Green Version]
- Tang, B.D.; Xia, X.; Lv, X.F.; Yu, B.X.; Yuan, J.N.; Mai, X.Y.; Shang, J.Y.; Zhou, J.G.; Liang, S.J.; Pang, R.P. Inhibition of Orai1-mediated Ca 2+ entry enhances chemosensitivity of HepG2 hepatocarcinoma cells to 5-fluorouracil. J. Cell. Mol. Med. 2017, 21, 904–915. [Google Scholar] [CrossRef] [PubMed]
- Faouzi, M.; Kischel, P.; Hague, F.; Ahidouch, A.; Benzerdjeb, N.; Sevestre, H.; Penner, R.; Ouadid-Ahidouch, H. ORAI3 silencing alters cell proliferation and cell cycle progression via c-myc pathway in breast cancer cells. Biochim. Biophys. Acta BBA Mol. Cell Res. 2013, 1833, 752–760. [Google Scholar] [CrossRef] [Green Version]
- Faouzi, M.; Hague, F.; Potier, M.; Ahidouch, A.; Sevestre, H.; Ouadid-Ahidouch, H. Down-regulation of Orai3 arrests cell-cycle progression and induces apoptosis in breast cancer cells but not in normal breast epithelial cells. J. Cell. Physiol. 2011, 226, 542–551. [Google Scholar] [CrossRef] [PubMed]
- Sobradillo, D.; Hernández-Morales, M.; Ubierna, D.; Moyer, M.P.; Núñez, L.; Villalobos, C. A Reciprocal Shift in Transient Receptor Potential Channel 1 (TRPC1) and Stromal Interaction Molecule 2 (STIM2) Contributes to Ca2+ Remodeling and Cancer Hallmarks in Colorectal Carcinoma Cells. J. Biol. Chem. 2014, 289, 28765–28782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, H.; Zhang, H.; Jin, F.; Fang, M.; Huang, M.; Yang, C.S.; Chen, T.; Fu, L.; Pan, Z. Elevated Orai1 expression mediates tumor-promoting intracellular Ca2+ oscillations in human esophageal squamous cell carcinoma. Oncotarget 2014, 5, 3455–3471. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Hughes, J.D.; Rollins, S.; Chen, B.; Perkins, E. Calcium entry via ORAI1 regulates glioblastoma cell proliferation and apoptosis. Exp. Mol. Pathol. 2011, 91, 753–760. [Google Scholar] [CrossRef]
- Yang, N.; Tang, Y.; Wang, F.; Zhang, H.; Xu, D.; Shen, Y.; Sun, S.; Yang, G. Blockade of store-operated Ca2+ entry inhibits hepatocarcinoma cell migration and invasion by regulating focal adhesion turnover. Cancer Lett. 2013, 330, 163–169. [Google Scholar] [CrossRef]
- El Boustany, C.; Bidaux, G.; Enfissi, A.; Delcourt, P.; Prevarskaya, N.; Capiod, T. Capacitative calcium entry and transient receptor potential canonical 6 expression control human hepatoma cell proliferation. Hepatology 2008, 47, 2068–2077. [Google Scholar] [CrossRef]
- Benzerdjeb, N.; Sevestre, H.; Ahidouch, A.; Ouadid-Ahidouch, H. Orai3 is a predictive marker of metastasis and survival in resectable lung adenocarcinoma. Oncotarget 2016, 7, 81588–81597. [Google Scholar] [CrossRef]
- Duffy, S.M.; Ashmole, I.; Smallwood, D.T.; Leyland, M.L.; Bradding, P. Orai/CRACM1 and KCa3.1 ion channels interact in the human lung mast cell plasma membrane. Cell Commun. Signal. 2015, 13, 32. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Ren, Y.; Wang, L.; Zhao, W.; Dong, X.; Pan, J.; Gao, H.; Tian, Y. Orai1 and Stim1 Mediate the Majority of Store-Operated Calcium Entry in Multiple Myeloma and Have Strong Implications for Adverse Prognosis. Cell. Physiol. Biochem. 2018, 48, 2273–2285. [Google Scholar] [CrossRef] [PubMed]
- Yanamandra, N.; Buzzeo, R.W.; Gabriel, M.; Hazlehurst, L.A.; Mari, Y.; Beaupre, D.M.; Cuevas, J. Tipifarnib-Induced Apoptosis in Acute Myeloid Leukemia and Multiple Myeloma Cells Depends on Ca2+ Influx through Plasma Membrane Ca2+ Channels. J. Pharmacol. Exp. Ther. 2011, 337, 636–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, B.; Yuan, C.; Yang, X.L.; Atkin, S.; Xu, S.Z. TRPC Channels and Their Splice Variants Are Essential for Promoting Human Ovarian Cancer Cell Proliferation and Tumorigenesis. Curr. Cancer Drug Targets 2012, 13, 103–116. [Google Scholar] [CrossRef]
- Schmidt, S.; Liu, G.; Liu, G.; Yang, W.; Honisch, S.; Pantelakos, S.; Stournaras, C.; Hönig, A.; Lang, F. Enhanced Orai1 and STIM1 expression as well as store operated Ca2+ entry in therapy resistant ovary carcinoma cells. Oncotarget 2014, 5, 4799–4810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondratska, K.; Kondratskyi, A.; Yassine, M.; Lemonnier, L.; Lepage, G.; Morabito, A.; Skryma, R.; Prevarskaya, N. Orai1 and STIM1 mediate SOCE and contribute to apoptotic resistance of pancreatic adenocarcinoma. Biochim. Biophys. Acta BBA Mol. Cell Res. 2014, 1843, 2263–2269. [Google Scholar] [CrossRef] [Green Version]
- Dubois, C.; Kondratska, K.; Kondratskyi, A.; Morabito, A.; Mesilmany, L.; Farfariello, V.; Toillon, R.A.; Gelus, N.Z.; Laurenge, E.; van den Abeele, F.; et al. ORAI3 silencing alters cell proliferation and promotes mitotic catastrophe and apoptosis in pancreatic adenocarcinoma. Biochim. Biophys. Acta BBA Mol. Cell Res. 2021, 1868, 119023. [Google Scholar] [CrossRef]
- Holzmann, C.; Kilch, T.; Kappel, S.; Armbruster, A.; Jung, V.; Stockle, M.; Bogeski, I.; Schwarz, E.C.; Peinelt, C. ICRAC controls the rapid androgen response in human primary prostate epithelial cells and is altered in prostate cancer. Oncotarget 2013, 4, 2096–2107. [Google Scholar] [CrossRef] [Green Version]
- Perrouin Verbe, M.A.; Bruyere, F.; Rozet, F.; Vandier, C.; Fromont, G. Expression of store-operated channel components in prostate cancer: The prognostic paradox. Hum. Pathol. 2016, 49, 77–82. [Google Scholar] [CrossRef]
- Dubois, C.; van den Abeele, F.; Lehen’kyi, V.; Gkika, D.; Guarmit, B.; Lepage, G.; Slomianny, C.; Borowiec, A.S.; Bidaux, G.; Benahmed, M.; et al. Remodeling of channel-forming ORAI proteins determines an oncogenic switch in prostate cancer. Cancer Cell 2014, 26, 19–32. [Google Scholar] [CrossRef]
- Kappel, S.; Marques, I.J.; Zoni, E.; Stoklosa, P.; Peinelt, C.; Mercader, N.; Kruithof-de Julio, M.; Borgström, A. Store-Operated Ca2+ Entry as a Prostate Cancer Biomarker—A Riddle with Perspectives. Curr. Mol. Biol. Rep. 2017, 3, 208–217. [Google Scholar] [CrossRef] [Green Version]
- Hasna, J.; Hague, F.; Rodat-Despoix, L.; Geerts, D.; Leroy, C.; Tulasne, D.; Ouadid-Ahidouch, H.; Kischel, P. Orai3 calcium channel and resistance to chemotherapy in breast cancer cells: The p53 connection. Cell Death Differ. 2018, 25, 693–707. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Chen, L.; Zhao, P.; Zhou, H.; Zhang, C.; Yu, S.; Lin, Y.; Yang, X. Store-operated Ca2+ entry regulates glioma cell migration and invasion via modulation of Pyk2 phosphorylation. J. Exp. Clin. Cancer Res. 2014, 33, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makena, M.R.; Ko, M.; Dang, D.K.; Rao, R. Epigenetic Modulation of SPCA2 Reverses Epithelial to Mesenchymal Transition in Breast Cancer Cells. Cancers 2021, 13, 259. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.W.; Chen, Y.F.; Chen, Y.T.; Chiu, W.T.; Shen, M.R. The STIM1-Orai1 pathway of store-operated Ca2+ entry controls the checkpoint in cell cycle G1/S transition. Sci. Rep. 2016, 6, 22142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, D.; He, R.; Yu, W.; Li, C.; Cheng, H.; Zhu, B.; Yan, J. ORAI3 contributes to hypoxia-inducible factor 1/2α-sensitive colon cell migration. Physiol. Int. 2021, 108, 221–237. [Google Scholar] [CrossRef] [PubMed]
- Umemura, M.; Baljinnyam, E.; Feske, S.; de Lorenzo, M.S.; Xie, L.H.; Feng, X.; Oda, K.; Makino, A.; Fujita, T.; Yokoyama, U.; et al. Store-Operated Ca2+ Entry (SOCE) Regulates Melanoma Proliferation and Cell Migration. PLoS ONE 2014, 9, e89292. [Google Scholar] [CrossRef]
- Kim, J.H.; Lkhagvadorj, S.; Lee, M.R.; Hwang, K.H.; Chung, H.C.; Jung, J.H.; Cha, S.K.; Eom, M. Orai1 and STIM1 are critical for cell migration and proliferation of clear cell renal cell carcinoma. Biochem. Biophys. Res. Commun. 2014, 448, 76–82. [Google Scholar] [CrossRef]
- Feng, M.; Grice, D.M.; Faddy, H.M.; Nguyen, N.; Leitch, S.; Wang, Y.; Muend, S.; Kenny, P.A.; Sukumar, S.; Roberts-Thomson, S.J.; et al. Store-independent activation of Orai1 by SPCA2 in mammary tumors. Cell 2010, 143, 84–98. [Google Scholar] [CrossRef] [Green Version]
- Tajeddine, N.; Gailly, P. TRPC1 Protein Channel Is Major Regulator of Epidermal Growth Factor Receptor Signaling. J. Biol. Chem. 2012, 287, 16146–16157. [Google Scholar] [CrossRef]
- Wang, L.; Wu, Y.; Lin, L.; Liu, P.; Huang, H.; Liao, W.; Zheng, D.; Zuo, Q.; Sun, L.; Huang, N.; et al. Metastasis-associated in colon cancer-1 upregulation predicts a poor prognosis of gastric cancer, and promotes tumor cell proliferation and invasion. Int. J. Cancer 2013, 133, 1419–1430. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Pan, W.; Liu, S.; Shen, Z.; Xu, Y.; Hu, L. ERK/MAPK signalling pathway and tumorigenesis (Review). Exp. Ther. Med. 2020, 19, 1997–2007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mebratu, Y.; Tesfaigzi, Y. How ERK1/2 activation controls cell proliferation and cell death is subcellular localization the answer? Cell Cycle 2009, 8, 1168–1175. [Google Scholar] [CrossRef] [PubMed]
- Muzny, D.M.; Bainbridge, M.N.; Chang, K.; Dinh, H.H.; Drummond, J.A.; Fowler, G.; Kovar, C.L.; Lewis, L.R.; Morgan, M.B.; Newsham, I.F.; et al. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar]
- Wang, J.Y.; Chen, B.K.; Wang, Y.S.; Tsai, Y.T.; Chen, W.C.; Chang, W.C.; Hou, M.F.; Wu, Y.C.; Chang, W.C. Involvement of store-operated calcium signaling in EGF-mediated COX-2 gene activation in cancer cells. Cell Signal. 2012, 24, 162–169. [Google Scholar] [CrossRef]
- Greenhough, A.; Smartt, H.J.M.; Moore, A.E.; Roberts, H.R.; Williams, A.C.; Paraskeva, C.; Kaidi, A. The COX-2/PGE2 pathway: Key roles in the hallmarks of cancer and adaptation to the tumour microenvironment. Carcinogenesis 2009, 30, 377–386. [Google Scholar] [CrossRef] [Green Version]
- Azimi, I.; Milevskiy, M.J.G.; Kaemmerer, E.; Turner, D.; Yapa, K.T.D.S.; Brown, M.A.; Thompson, E.W.; Roberts-Thomson, S.J.; Monteith, G.R. TRPC1 is a differential regulator of hypoxia-mediated events and Akt signaling in PTEN-deficient breast cancer cells. J. Cell Sci. 2017, 130, 2292–2305. [Google Scholar] [CrossRef] [Green Version]
- Chigurupati, S.; Venkataraman, R.; Barrera, D.; Naganathan, A.; Madan, M.; Paul, L.; Pattisapu, J.V.; Kyriazis, G.A.; Sugaya, K.; Bushnev, S.; et al. Receptor Channel TRPC6 Is a Key Mediator of Notch-Driven Glioblastoma Growth and Invasiveness. Cancer Res. 2010, 70, 418–427. [Google Scholar] [CrossRef] [Green Version]
- Bass, A.J.; Thorsson, V.; Shmulevich, I.; Reynolds, S.M.; Miller, M.; Bernard, B.; Hinoue, T.; Laird, P.W.; Curtis, C.; Shen, H.; et al. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014, 513, 202–209. [Google Scholar]
- Getz, G.; Gabriel, S.B.; Cibulskis, K.; Lander, E.; Sivachenko, A.; Sougnez, C.; Lawrence, M.; Kandoth, C.; Dooling, D.; Fulton, R.; et al. Integrated genomic characterization of endometrial carcinoma. Nature 2013, 497, 67–73. [Google Scholar]
- Frischauf, I.; Litviňuková, M.; Schober, R.; Zayats, V.; Svobodová, B.; Bonhenry, D.; Lunz, V.; Cappello, S.; Tociu, L.; Reha, D.; et al. Transmembrane helix connectivity in Orai1 controls two gates for calcium-dependent transcription. Sci. Signal. 2017, 10, eaao0358. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, M.S.; Sougnez, C.; Lichtenstein, L.; Cibulskis, K.; Lander, E.; Gabriel, S.B.; Getz, G.; Ally, A.; Balasundaram, M.; Birol, I.; et al. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [Google Scholar]
- Tiffner, A.; Derler, I. Molecular choreography and structure of Ca2+ release-activated Ca2+ (CRAC) and KCa2+ channels and their relevance in disease with special focus on cancer. Membranes 2020, 10, 425. [Google Scholar] [CrossRef] [PubMed]
- Srikanth, S.; Gwack, Y. Orai1-NFAT signalling pathway triggered by T cell receptor stimulation. Mol. Cells 2013, 35, 182–194. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.P.; Yuan, J.P.; Zeng, W.; So, I.; Worley, P.F.; Muallem, S. Molecular determinants of fast Ca 2+ -dependent inactivation and gating of the Orai channels. Proc. Natl. Acad. Sci. USA 2009, 106, 14687–14692. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.L.; Cao, Q.; Zhou, K.C.; Feng, Y.J.; Wang, Y.Z. Transient receptor potential channel C3 contributes to the progression of human ovarian cancer. Oncogene 2009, 28, 1320–1328. [Google Scholar] [CrossRef] [Green Version]
- Siaw, W.N.; di Capite, J.; Singaravelu, K.; Parekh, A.B. Sustained activation of the tyrosine kinase syk by antigen in mast cells requires local Ca2+ influx through Ca2+ release-activated Ca2+ channels. J. Biol. Chem. 2008, 283, 31348–31355. [Google Scholar]
- Di Sabatino, A.; Rovedatti, L.; Kaur, R.; Spencer, J.P.; Brown, J.T.; Morisset, V.D.; Biancheri, P.; Leakey, N.A.B.; Wilde, J.I.; Scott, L.; et al. Targeting Gut T Cell Ca 2+ Release-Activated Ca2+ Channels Inhibits T Cell Cytokine Production and T-Box Transcription Factor T-Bet in Inflammatory Bowel Disease. J. Immunol. 2009, 183, 3454–3462. [Google Scholar] [CrossRef] [Green Version]
- Waldherr, L.; Tiffner, A.; Mishra, D.; Sallinger, M.; Schober, R.; Frischauf, I.; Schmidt, T.; Handl, V.; Sagmeister, P.; Köckinger, M.; et al. Blockage of store-operated Ca2+ influx by synta66 is mediated by direct inhibition of the Ca2+ selective orai1 pore. Cancers 2020, 12, 2876. [Google Scholar] [CrossRef]
- Thomas, S.; Quinn, B.A.; Das, S.K.; Dash, R.; Emdad, L.; Dasgupta, S.; Wang, X.Y.; Dent, P.; Reed, J.C.; Pellecchia, M.; et al. Targeting the Bcl-2 family for cancer therapy. Expert Opin. Ther. Targets 2013, 17, 61–75. [Google Scholar] [CrossRef] [Green Version]
- Thomas, L.W.; Lam, C.; Edwards, S.W. Mcl-1; the molecular regulation of protein function. FEBS Lett. 2010, 584, 2981–2989. [Google Scholar] [CrossRef] [Green Version]
- Mignen, O.; Brink, C.; Enfissi, A.; Nadkarni, A.; Shuttleworth, T.J.; Giovannucci, D.R.; Capiod, T. Carboxyamidotriazole-induced inhibition of mitochondrial calcium import blocks capacitative calcium entry and cell proliferation in HEK-293 cells. J. Cell Sci. 2005, 118, 5615–5623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonnefond, M.L.; Florent, R.; Lenoir, S.; Lambert, B.; Abeilard, E.; Giffard, F.; Louis, M.H.; Elie, N.; Briand, M.; Vivien, D.; et al. Inhibition of store-operated channels by carboxyamidotriazole sensitizes ovarian carcinoma cells to anti-BclxL strategies through Mcl-1 down-regulation. Oncotarget 2018, 9, 33896–33911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercer, J.C.; Qi, Q.; Mottram, L.F.; Law, M.; Bruce, D.; Iyer, A.; Morales, J.L.; Yamazaki, H.; Shirao, T.; Peterson, B.R.; et al. Chemico-genetic identification of drebrin as a regulator of calcium responses. Int. J. Biochem. Cell Biol. 2010, 42, 337–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohga, K.; Takezawa, R.; Arakida, Y.; Shimizu, Y.; Ishikawa, J. Characterization of YM-58483/BTP2, a novel store-operated Ca2+ entry blocker, on T cell-mediated immune responses in vivo. Int. Immunopharmacol. 2008, 8, 1787–1792. [Google Scholar] [CrossRef]
- End, D.W.; Smets, G.; Todd, A.V.; Applegate, T.L.; Fuery, C.J.; Angibaud, P.; Venet, M.; Sanz, G.; Poignet, H.; Skrzat, S.; et al. Characterization of the antitumor effects of the selective farnesyl protein transferase inhibitor R115777 in vivo and in vitro. Cancer Res. 2001, 61, 131–137. [Google Scholar]
- Khan, H.Y.; Mpilla, G.B.; Sexton, R.; Viswanadha, S.; Penmetsa, K.V.; Aboukameel, A.; Diab, M.; Kamgar, M.; Al-Hallak, M.N.; Szlaczky, M.; et al. Calcium Release-Activated Calcium (CRAC) Channel Inhibition Suppresses Pancreatic Ductal Adenocarcinoma Cell Proliferation and Patient-Derived Tumor Growth. Cancers 2020, 12, 750. [Google Scholar] [CrossRef] [Green Version]
- Cui, C.; Chang, Y.; Zhang, X.; Choi, S.; Tran, H.; Penmetsa, K.V.; Viswanadha, S.; Fu, L.; Pan, Z. Targeting Orai1-mediated store-operated calcium entry by RP4010 for anti-tumor activity in esophagus squamous cell carcinoma. Cancer Lett. 2018, 432, 169–179. [Google Scholar] [CrossRef]
- Daya, H.A.; Kouba, S.; Ouled-Haddou, H.; Benzerdjeb, N.; Telliez, M.S.; Dayen, C.; Sevestre, H.; Garçon, L.; Hague, F.; Ouadid-Ahidouch, H. Orai3-Mediates Cisplatin-Resistance in Non-Small Cell Lung Cancer Cells by Enriching Cancer Stem Cell Population through PI3K/AKT Pathway. Cancers 2021, 13, 2314. [Google Scholar] [CrossRef]
- Stauderman, K.A. CRAC channels as targets for drug discovery and development. Cell Calcium 2018, 74, 147–159. [Google Scholar] [CrossRef]
- Adelman, J.P. SK channels and calmodulin. Channels 2016, 10, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.S.; Ngo-Anh, T.J.; Bruening-Wright, A.; Maylie, J.; Adelman, J.P. Small conductance Ca2+-activated K+ channels and calmodulin—Cell surface expression and gating. J. Biol. Chem. 2003, 278, 25940–25946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.H.; MacKinnon, R. Activation mechanism of a human SK-calmodulin channel complex elucidated by cryo-EM structures. Science 2018, 360, 508–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, K.; Wang, H.; Yu, J.; Wei, M.; Xiao, Q. New Insights on the Regulation of Ca2+ -Activated Chloride Channel TMEM16A. J. Cell. Physiol. 2017, 232, 707–716. [Google Scholar] [CrossRef]
- Wang, H.; Zou, L.; Ma, K.; Yu, J.; Wu, H.; Wei, M.; Xiao, Q. Cell-specific mechanisms of TMEM16A Ca2+-activated chloride channel in cancer. Mol. Cancer 2017, 16, 152. [Google Scholar] [CrossRef] [PubMed]
- Stocker, M. Ca2+-activated K+ channels: Molecular determinants and function of the SK family. Nat. Rev. Neurosci. 2004, 5, 758–770. [Google Scholar] [CrossRef] [PubMed]
- Gueguinou, M.; Gambade, A.; Felix, R.; Chantome, A.; Fourbon, Y.; Bougnoux, P.; Weber, G.; Potier-Cartereau, M.; Vandier, C. Lipid rafts, KCa/ClCa/Ca2+ channel complexes and EGFR signaling: Novel targets to reduce tumor development by lipids? Biochim. Biophys. Acta BBA Biomembr. 2015, 1848, 2603–2620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berkefeld, H.; Fakler, B.; Schulte, U. Ca2+-activated K+ channels: From protein complexes to function. Physiol. Rev. 2010, 90, 1437–1459. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Liu, Z.; Wang, K. The Ca2+-activated chloride channel ANO1/TMEM16A: An emerging therapeutic target for epithelium-originated diseases? Acta. Pharm. Sin. B 2021, 11, 1412–1433. [Google Scholar] [CrossRef]
- Lallet-Daher, H.; Roudbaraki, M.; Bavencoffe, A.; Mariot, P.; Gackiere, F.; Bidaux, G.; Urbain, R.; Gosset, P.; Delcourt, P.; Fleurisse, L.; et al. Intermediate-conductance Ca2+-activated K+ channels (IKCa1) regulate human prostate cancer cell proliferation through a close control of calcium entry. Oncogene 2009, 28, 1792–1806. [Google Scholar] [CrossRef] [Green Version]
- Chantome, A.; Potier-Cartereau, M.; Clarysse, L.; Fromont, G.; Marionneau-Lambot, S.; Gueguinou, M.; Pagès, J.C.; Collin, C.; Oullier, T.; Girault, V.; et al. Pivotal role of the lipid Raft SK3-Orai1 complex in human cancer cell migration and bone metastases. Cancer Res. 2013, 73, 4852–4861. [Google Scholar] [CrossRef] [Green Version]
- Gueguinou, M.; Crottes, D.; Chantome, A.; Rapetti-Mauss, R.; Potier-Cartereau, M.; Clarysse, L.; Girault, A.; Fourbon, Y.; Jézéquel, P.; Guérin-Charbonnel, C.; et al. The SigmaR1 chaperone drives breast and colorectal cancer cell migration by tuning SK3-dependent Ca2+ homeostasis. Oncogene 2017, 36, 3640–3647. [Google Scholar] [CrossRef] [PubMed]
- Clarysse, L.; Gueguinou, M.; Potier-Cartereau, M.; Vandecasteele, G.; Bougnoux, P.; Chevalier, S.; Chantôme, A.; Vandier, C. cAMP-PKA inhibition of SK3 channel reduced both Ca2+ entry and cancer cell migration by regulation of SK3-Orai1 complex. Pflug. Arch. 2014, 466, 1921–1932. [Google Scholar] [CrossRef] [PubMed]
- Gueguinou, M.; Harnois, T.; Crottes, D.; Uguen, A.; Deliot, N.; Gambade, A.; Chantôme, A.; Haelters, J.P.; Jaffrès, P.A.; Jourdan, M.L.; et al. SK3/TRPC1/Orai1 complex regulates SOCE-dependent colon cancer cell migration: A novel opportunity to modulate anti-EGFR mAb action by the alkyl-lipid Ohmline. Oncotarget 2016, 7, 36168–36184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weaver, A.K.; Bomben, V.C.; Sontheimer, H. Expression and function of calcium-activated potassium channels in human glioma cells. Glia 2006, 54, 223–233. [Google Scholar] [CrossRef] [Green Version]
- Girault, A.; Haelters, J.P.; Potier-Cartereau, M.; Chantome, A.; Jaffres, P.A.; Bougnoux, P.; Joulin, V.; Vandier, C. Targeting SKCa Channels in Cancer: Potential New Therapeutic Approaches. Curr. Med. Chem. 2012, 19, 697–713. [Google Scholar] [CrossRef]
- Klumpp, L.; Sezgin, E.C.; Skardelly, M.; Eckert, F.; Huber, S.M. KCa3.1 channels and glioblastoma: In vitro studies. Curr. Neuropharmacol. 2017, 16, 627–635. [Google Scholar] [CrossRef]
- Potier, M.; Joulin, V.; Roger, S.; Besson, P.; Jourdan, M.L.; Leguennec, J.Y.; Bougnoux, P.; Vandier, C. Identification of SK3 channel as a new mediator of breast cancer cell migration. Mol. Cancer Ther. 2006, 5, 2946–2953. [Google Scholar] [CrossRef] [Green Version]
- Carignani, C.; Roncarati, R.; Rimini, R.; Terstappen, G.C. Pharmacological and molecular characterisation of SK3 channels in the TE671 human medulloblastoma cell line. Brain Res. 2002, 939, 11–18. [Google Scholar] [CrossRef]
- Girault, A.; Haelters, J.P.; Potier-Cartereau, M.; Chantome, A.; Pinault, M.; Marionneau-Lambot, S.; Oullier, T.; Simon, G.; Couthon-Gourvès, H.; Jaffrès, P.A.; et al. New alkyl-lipid blockers of SK3 channels reduce cancer cell migration and occurrence of metastasis. Curr. Cancer Drug Targets 2011, 11, 1111–1125. [Google Scholar] [CrossRef]
- Jelassi, B.; Chantme, A.; Alcaraz-Pérez, F.; Baroja-Mazo, A.; Cayuela, M.L.; Pelegrin, P.; Surprenant, A.; Roger, S. P2X 7 receptor activation enhances SK3 channels- and cystein cathepsin-dependent cancer cells invasiveness. Oncogene 2011, 30, 2108–2122. [Google Scholar] [CrossRef] [Green Version]
- Bery, F.; Cancel, M.; Guéguinou, M.; Potier-Cartereau, M.; Vandier, C.; Chantôme, A.; Guibon, R.; Bruyère, F.; Fromont, G.; Mahéo, K. Zeb1 and sk3 channel are up-regulated in castration-resistant prostate cancer and promote neuroendocrine differentiation. Cancers 2021, 13, 2947. [Google Scholar] [CrossRef] [PubMed]
- Vergara, C.; Latorre, R.; Marrion, N.V.; Adelman, J.P. Calcium-activated potassium channels. Curr. Opin. Neurobiol. 1998, 8, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Stocker, M.; Pedarzani, P. Differential distribution of three Ca2+-activated K+ channel subunits, SK1, SK2, and SK3, in the adult rat central nervous system. Mol. Cell. Neurosci. 2000, 15, 476–493. [Google Scholar] [CrossRef] [PubMed]
- Ishii, T.M.; Silvia, C.; Hirschberg, B.; Bond, C.T.; Adelman, J.P.; Maylie, J. A human intermediate conductance calcium-activated potassium channel. Proc. Natl. Acad. Sci. USA 1997, 94, 11651–11656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, U.S.; Cui, J. BK channel activation: Structural and functional insights. Trends Neurosci. 2010, 33, 415–423. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef]
- Keen, J.E.; Khawaled, R.; Farrens, D.L.; Neelands, T.; Rivard, A.; Bond, C.T.; Janowsky, A.; Fakler, B.; Adelman, J.P.; Maylie, J. Domains Responsible for Constitutive and Ca2+ -Dependent Interactions between Calmodulin and Small Conductance Ca2+ -Activated Potassium Channels. J. Neurosci. 1999, 19, 8830–8838. [Google Scholar] [CrossRef]
- Orfali, R.; Nam, Y.W.; Nguyen, H.M.; Rahman, M.A.; Yang, G.; Cui, M.; Wulff, H.; Zhang, M. Channelopathy-causing mutations in the S(45)A/S(45)B and HA/HB helices of K(Ca)2.3 and K(Ca)3.1 channels alter their apparent Ca2+ sensitivity. Cell Calcium 2022, 102, 102538. [Google Scholar] [CrossRef]
- Herrera, F.E.; Sevrain, C.M.; Jaffrès, P.A.; Couthon, H.; Grélard, A.; Dufourc, E.J.; Chantôme, A.; Potier-Cartereau, M.; Vandier, C.; Bouchet, A.M. Singular interaction between an antimetastatic agent and the lipid bilayer: The ohmline case. ACS Omega 2017, 2, 6361–6370. [Google Scholar] [CrossRef]
- Zhang, M.; Meng, X.Y.; Cui, M.; Pascal, J.M.; Logothetis, D.E.; Zhang, J.F. Selective phosphorylation modulates the PIP 2 sensitivity of the CaM-SK channel complex. Nat. Chem. Biol. 2014, 10, 753–759. [Google Scholar] [CrossRef] [Green Version]
- Levitan, I.; Fang, Y.; Rosenhouse-Dantsker, A.; Romanenko, V. Cholesterol and ion channels. Subcell. Biochem. 2010, 51, 509–549. [Google Scholar]
- Chantome, A.; Girault, A.; Potier, M.; Collin, C.; Vaudin, P.; Pages, J.C.; Vanndier, C.; Joulin, V. KCa2.3 channel-dependent hyperpolarization increases melanoma cell motility. Exp. Cell Res. 2009, 315, 3620–3630. [Google Scholar] [CrossRef]
- Tiffner, A.; Hopl, V.; Schober, R.; Sallinger, M.; Grabmayr, H.; Höglinger, C.; Fahrner, M.; Lunz, V.; Maltan, L.; Frischauf, I.; et al. Orai1 Boosts SK3 Channel Activation. Cancers 2021, 13, 6357. [Google Scholar] [CrossRef]
- Bloch, M.; Ousingsawat, J.; Simon, R.; Schraml, P.; Gasser, T.C.; Mihatsch, M.J.; Kunzelmann, K.; Bubendorf, L. KCNMA1 gene amplification promotes tumor cell proliferation in human prostate cancer. Oncogene 2007, 26, 2525–2534. [Google Scholar] [CrossRef] [Green Version]
- Parihar, A.S.; Coghlan, M.J.; Gopalakrishnan, M.; Shieh, C.C. Effects of intermediate-conductance Ca2+-activated K+ channel modulators on human prostate cancer cell proliferation. Eur. J. Pharmacol. 2003, 471, 157–164. [Google Scholar] [CrossRef]
- Abdullaev, I.F.; Rudkouskaya, A.; Mongin, A.A.; Kuo, Y.H. Calcium-Activated Potassium Channels BK and IK1 Are Functionally Expressed in Human Gliomas but Do Not Regulate Cell Proliferation. PLoS ONE 2010, 5, e12304. [Google Scholar] [CrossRef]
- McFerrin, M.B.; Turner, K.L.; Cuddapah, V.A.; Sontheimer, H. Differential role of IK and BK potassium channels as mediators of intrinsic and extrinsic apoptotic cell death. Am. J. Physiol.-Cell Physiol. 2012, 303, C1070–C1078. [Google Scholar] [CrossRef] [Green Version]
- D’Alessandro, G.; Limatola, C.; Catalano, M. Functional Roles of the Ca2+-activated K+ Channel, KCa3.1, in Brain Tumors. Curr. Neuropharmacol. 2018, 16, 636–643. [Google Scholar] [CrossRef]
- Ohya, S.; Kanatsuka, S.; Hatano, N.; Kito, H.; Matsui, A.; Fujimoto, M.; Matsuba, S.; Niwa, S.; Zhan, P.; Suzuki, T.; et al. Downregulation of the Ca2+-activated K+ channel KCa3.1 by histone deacetylase inhibition in human breast cancer cells. Pharmacol. Res. Perspect. 2016, 4, e00228. [Google Scholar] [CrossRef]
- Sforna, L.; Megaro, A.; Pessia, M.; Franciolini, F.; Catacuzzeno, L. Structure, Gating and Basic Functions of the Ca2+-activated K Channel of Intermediate Conductance. Curr. Neuropharmacol. 2018, 16, 608–617. [Google Scholar] [CrossRef]
- Bulk, E.; Ay, A.S.; Hammadi, M.; Ouadid-Ahidouch, H.; Schelhaas, S.; Hascher, A.; Rohde, C.; Thoennissen, N.H.; Wiewrodt, R.; Schmidt, E.; et al. Epigenetic dysregulation of KCa3.1 channels induces poor prognosis in lung cancer. Int. J. Cancer 2015, 137, 1306–1317. [Google Scholar] [CrossRef]
- Grössinger, E.M.; Weiss, L.; Zierler, S.; Rebhandl, S.; Krenn, P.W.; Hinterseer, E.; Schmölzer, J.; Asslaber, D.; Hainzl, S.; Neureiter, D.; et al. Targeting proliferation of chronic lymphocytic leukemia (CLL) cells through KCa3.1 blockade. Leukemia 2014, 28, 954–958. [Google Scholar] [CrossRef] [Green Version]
- Quast, S.A.; Berger, A.; Buttstädt, N.; Friebel, K.; Schönherr, R.; Eberle, J. General Sensitization of Melanoma Cells for TRAIL-Induced Apoptosis by the Potassium Channel Inhibitor TRAM-34 Depends on Release of SMAC. PLoS ONE 2012, 7, e39290. [Google Scholar] [CrossRef] [Green Version]
- Pillozzi, S.; D’Amico, M.; Bartoli, G.; Gasparoli, L.; Petroni, G.; Crociani, O.; Marzo, T.; Guerriero, A.; Messori, L.; Severi, M.; et al. The combined activation of KCa3.1 and inhibition of Kv11.1/hERG1 currents contribute to overcome Cisplatin resistance in colorectal cancer cells. Br. J. Cancer 2018, 118, 200–212. [Google Scholar] [CrossRef] [Green Version]
- Rabjerg, M.; Oliván-Viguera, A.; Hansen, L.K.; Jensen, L.; Sevelsted-Møller, L.; Walter, S.; Jensen, B.L.; Marcussen, N.; Köhler, R. High Expression of KCa3.1 in Patients with Clear Cell Renal Carcinoma Predicts High Metastatic Risk and Poor Survival. PLoS ONE 2015, 10, e0122992. [Google Scholar] [CrossRef] [Green Version]
- Jiang, S.; Zhu, L.; Yang, J.; Hu, L.; Gu, J.; Xing, X.; Sun, Y.; Zhang, Z. Integrated expression profiling of potassium channels identifys KCNN4 as a prognostic biomarker of pancreatic cancer. Biochem. Biophys. Res. Commun. 2017, 494, 113–119. [Google Scholar] [CrossRef]
- Wen, J.; Lin, B.; Lin, L.; Chen, Y.; Wang, O. KCNN4 is a diagnostic and prognostic biomarker that promotes papillary thyroid cancer progression. Aging 2020, 12, 16437–16456. [Google Scholar] [CrossRef]
- Zhang, P.; Yang, X.; Yin, Q.; Yi, J.; Shen, W.; Zhao, L.; Zhu, Z.; Liu, J. Inhibition of SK4 Potassium Channels Suppresses Cell Proliferation, Migration and the Epithelial-Mesenchymal Transition in Triple-Negative Breast Cancer Cells. PLoS ONE 2016, 11, e0154471. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, S.; Chaigne, J.; Dakik, H.; Fourbon, Y.; Corset, L.; Lecomte, T.; Raoul, W.; Guéguinou, M. SK4 oncochannels regulate calcium entry and promote cell migration in KRAS-mutated colorectal cancer. Cell Calcium 2021, 96, 102384. [Google Scholar] [CrossRef]
- Steudel, F.A.; Mohr, C.J.; Stegen, B.; Nguyen, H.Y.; Barnert, A.; Steinle, M.; Beer-Hammer, S.; Koch, P.; Lo, W.Y.; Schroth, W.; et al. SK4 channels modulate Ca2+ signalling and cell cycle progression in murine breast cancer. Mol. Oncol. 2017, 11, 1172–1188. [Google Scholar] [CrossRef] [Green Version]
- Mohr, C.J.; Gross, D.; Sezgin, E.C.; Steudel, F.A.; Ruth, P.; Huber, S.M.; Lukowski, R. KCa3.1 Channels Confer Radioresistance to Breast Cancer Cells. Cancers 2019, 11, 1285. [Google Scholar] [CrossRef] [Green Version]
- D’Alessandro, G.; Grimaldi, A.; Chece, G.; Porzia, A.; Esposito, V.; Santoro, A.; Salvati, M.; Mainiero, F.; Ragozzino, D.; Di Angelantonio, S.; et al. KCa3.1 channel inhibition sensitizes malignant gliomas to temozolomide treatment. Oncotarget 2016, 7, 30781–30796. [Google Scholar] [CrossRef]
- Robles-Martínez, L.; Garay, E.; Martel-Gallegos, M.G.; Cisneros-Mejorado, A.; Pérez-Montiel, D.; Lara, A.; Arellano, R.O. KCa3.1 Activation via P2y2 Purinergic Receptors Promotes Human Ovarian Cancer Cell (Skov-3) Migration. Sci. Rep. 2017, 7, 4340. [Google Scholar] [CrossRef] [Green Version]
- Figiel, S.; Bery, F.; Chantôme, A.; Fontaine, D.; Pasqualin, C.; Maupoil, V.; Domingo, I.; Guibon, R.; Bruyère, F.; Potier-Cartereau, M.; et al. A Novel Calcium-Mediated EMT Pathway Controlled by Lipids: An Opportunity for Prostate Cancer Adjuvant Therapy. Cancers 2019, 11, 1814. [Google Scholar] [CrossRef] [Green Version]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [Green Version]
- Bauer, C.K.; Schneeberger, P.E.; Kortüm, F.; Altmüller, J.; Santos-Simarro, F.; Baker, L.; Keller-Ramey, J.; White, S.; Campeau, P.M.; Gripp, K.W.; et al. Gain-of-Function Mutations in KCNN3 Encoding the Small-Conductance Ca2+-Activated K+ Channel SK3 Cause Zimmermann-Laband Syndrome. Am. J. Hum. Genet. 2019, 104, 1139–1157. [Google Scholar] [CrossRef]
- Ouadid-Ahidouch, H.; Roudbaraki, M.; Delcourt, P.; Ahidouch, A.; Joury, N.; Prevarskaya, N. Functional and molecular identification of intermediate-conductance Ca2+-activated K+ channels in breast cancer cells: Association with cell cycle progression. Am. J. Physiol.-Cell Physiol. 2004, 287, C125–C134. [Google Scholar] [CrossRef]
- Sundelacruz, S.; Levin, M.; Kaplan, D.L. Role of Membrane Potential in the Regulation of Cell Proliferation and Differentiation. Stem Cell Rev. Rep. 2009, 5, 231–246. [Google Scholar] [CrossRef]
- Wang, Z.H.; Shen, B.; Yao, H.L.; Jia, Y.C.; Ren, J.; Feng, Y.J.; Wang, Y.Z. Blockage of intermediate-conductance-Ca2+-activated K+ channels inhibits progression of human endometrial cancer. Oncogene 2007, 26, 5107–5114. [Google Scholar] [CrossRef] [Green Version]
- D’Alessandro, G.; Catalano, M.; Sciaccaluga, M.; Chece, G.; Cipriani, R.; Rosito, M.; Grimaldi, A.; Lauro, C.; Cantore, G.; Santoro, V.; et al. KCa3.1 channels are involved in the infiltrative behavior of glioblastoma in vivo. Cell Death Dis. 2013, 4, e773. [Google Scholar] [CrossRef] [Green Version]
- Kischel, P.; Girault, A.; Rodat-Despoix, L.; Chamlali, M.; Radoslavova, S.; Abou Daya, H.; Lefebvre, T.; Foulon, A.; Rybarczyk, P.; Hague, F.; et al. Ion Channels: New Actors Playing in Chemotherapeutic Resistance. Cancers 2019, 11, 376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lloyd, K.L.; Cree, I.A.; Savage, R.S. Prediction of resistance to chemotherapy in ovarian cancer: A systematic review. BMC Cancer 2015, 15, 117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Pennisi, A.; Zhan, F.; Sawyer, J.R.; Shaughnessy, J.D.; Yaccoby, S. Establishment and exploitation of hyperdiploid and non-hyperdiploid human myeloma cell lines. Br. J. Haematol. 2007, 138, 802–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, E.L.; Hasegawa, Y.; Shimizu, T.; Okada, Y. IK1 channel activity contributes to cisplatin sensitivity of human epidermoid cancer cells. Am. J. Physiol.-Cell Physiol. 2008, 294, C1398–C1406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, P.; Li, J.; Ye, F.; Fu, W.; Hu, X.; Shao, Z.; Song, C. KCNN4 induces multiple chemoresistance in breast cancer by regulating BCL2A1. Am. J. Cancer Res. 2020, 10, 3302–3315. [Google Scholar]
- Steinestel, K.; Eder, S.; Ehinger, K.; Schneider, J.; Genze, F.; Winkler, E.; Wardelmann, E.; Schrader, A.J.; Steinestel, J. The small conductance calcium-activated potassium channel 3 (SK3) is a molecular target for Edelfosine to reduce the invasive potential of urothelial carcinoma cells. Tumor Biol. 2016, 37, 6275–6283, Erratum in Tumor Biol. 2016, 37, 2773. [Google Scholar] [CrossRef]
- Bery, F.; Figiel, S.; Kouba, S.; Fontaine, D.; Guéguinou, M.; Potier-Cartereau, M.; Vandier, C.; Guibon, R.; Bruyère, F.; Fromont, G.; et al. Hypoxia Promotes Prostate Cancer Aggressiveness by Upregulating EMT-Activator Zeb1 and SK3 Channel Expression. Int. J. Mol. Sci. 2020, 21, 4786. [Google Scholar] [CrossRef]
- Gackiere, F.; Warnier, M.; Katsogiannou, M.; Derouiche, S.; Delcourt, P.; Dewailly, E.; Slomianny, C.; Humez, S.; Prevarskaya, N.; Roudbaraki, M.; et al. Functional coupling between large-conductance potassium channels and Cav3.2 voltage-dependent calcium channels participates in prostate cancer cell growth. Biol. Open 2013, 2, 941–951. [Google Scholar] [CrossRef] [Green Version]
- Hammadi, M.; Chopin, V.; Matifat, F.; Dhennin-Duthille, I.; Chasseraud, M.; Sevestre, H.; Ouadid-Ahidouch, H. Human ether a-gogo K+ channel 1 (hEag1) regulates MDA-MB-231 breast cancer cell migration through Orai1-dependent calcium entry. J. Cell. Physiol. 2012, 227, 3837–3846. [Google Scholar] [CrossRef]
- Peretti, M.; Badaoui, M.; Girault, A.; van Gulick, L.; Mabille, M.P.; Tebbakha, R.; Sevestre, H.; Morjani, H.; Ouadid-Ahidouch, H. Original association of ion transporters mediates the ECM-induced breast cancer cell survival: Kv10.1-Orai1-SPCA2 partnership. Sci. Rep. 2019, 9, 1175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girault, A.; Peretti, M.; Badaoui, M.; Hémon, A.; Morjani, H.; Ouadid-Ahidouch, H. The N and C-termini of SPCA2 regulate differently Kv10.1 function: Role in the collagen 1-induced breast cancer cell survival. Am. J. Cancer Res. 2021, 11, 251–263. [Google Scholar] [PubMed]
- Song, K.; Zhong, X.G.; Xia, X.M.; Huang, J.H.; Fan, Y.F.; Yuan, R.X.; Xue, N.R.; Du, J.; Han, W.X.; Xu, A.M.; et al. Orai1 forms a signal complex with SK3 channel in gallbladder smooth muscle. Biochem. Biophys. Res. Commun. 2015, 466, 456–462. [Google Scholar] [CrossRef] [PubMed]
- Saljic, A.; Muthukumarasamy, K.M.; la Cour, J.M.; Boddum, K.; Grunnet, M.; Berchtold, M.W.; Jespersen, T. Impact of arrhythmogenic calmodulin variants on small conductance Ca2+-activated K+ (SK3) channels. Physiol. Rep. 2019, 7, e14210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willoughby, D.; Everett, K.L.; Halls, M.L.; Pacheco, J.; Skroblin, P.; Vaca, L.; Klussmann, E.; Cooper, D.M.F. Direct binding between Orai1 and AC8 mediates dynamic interplay between Ca2+ and cAMP signaling. Sci. Signal. 2012, 5, ra29. [Google Scholar] [CrossRef]
- Hooper, R.; Zhang, X.; Webster, M.; Go, C.; Kedra, J.; Marchbank, K.; Gill, D.L.; Weeraratna, A.T.; Trebak, M.; Soboloff, J. Novel Protein Kinase C-Mediated Control of Orai1 Function in Invasive Melanoma. Mol. Cell. Biol. 2015, 35, 2790–2798. [Google Scholar] [CrossRef] [Green Version]
- Kawasaki, T.; Ueyama, T.; Lange, I.; Feske, S.; Saito, N. Protein kinase C-induced phosphorylation of Orai1 regulates the intracellular Ca2+ level via the store-operated Ca2+ channel. J. Biol. Chem. 2010, 285, 25720–25730. [Google Scholar] [CrossRef] [Green Version]
- Smaardijk, S.; Chen, J.; Wuytack, F.; Vangheluwe, P. SPCA2 couples Ca2+ influx via Orai1 to Ca2+ uptake into the Golgi/secretory pathway. Tissue Cell 2017, 49, 141–149. [Google Scholar] [CrossRef]
- Nicholson, R.I.; Gee, J.M.W.; Harper, M.E. EGFR and cancer prognosis. Eur. J. Cancer 2001, 37 (Suppl. S4), 9. [Google Scholar] [CrossRef]
- Galizia, G.; Lieto, E.; Ferraraccio, F.; de Vita, F.; Castellano, P.; Orditura, M.; Imperatore, V.; La Mura, A.; La Manna, G.; Pinto, M.; et al. Prognostic significance of epidermal growth factor receptor expression in colon cancer patients undergoing curative surgery. Ann. Surg. Oncol. 2006, 13, 823–835. [Google Scholar] [CrossRef]
- Srivats, S.; Balasuriya, D.; Pasche, M.; Vistal, G.; Edwardson, J.M.; Taylor, C.W.; Murrell-Lagnado, R.D. Sigma1 receptors inhibit store-operated Ca2+ entry by attenuating coupling of STIM1 to Orai1. J. Cell Biol. 2016, 213, 65–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potier, M.; Chantome, A.; Joulin, V.; Girault, A.; Roger, S.; Besson, P.; Jourdan, M.-L.; LeGuennec, J.-Y.; Bougnoux, P.; Vandier, C.; et al. The SK3/KCa2.3 potassium channel is a new cellular target for edelfosine. Br. J. Pharmacol. 2011, 162, 464–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaffres, P.A.; Gajate, C.; Bouchet, A.M.; Couthon-Gourves, H.; Chantome, A.; Potier-Cartereau, M.; Besson, P.; Bougnoux, P.; Mollinedo, F.; Vandier, C.; et al. Alkyl ether lipids, ion channels and lipid raft reorganization in cancer therapy. Pharmacol. Ther. 2016, 165, 114–131. [Google Scholar] [CrossRef] [PubMed]









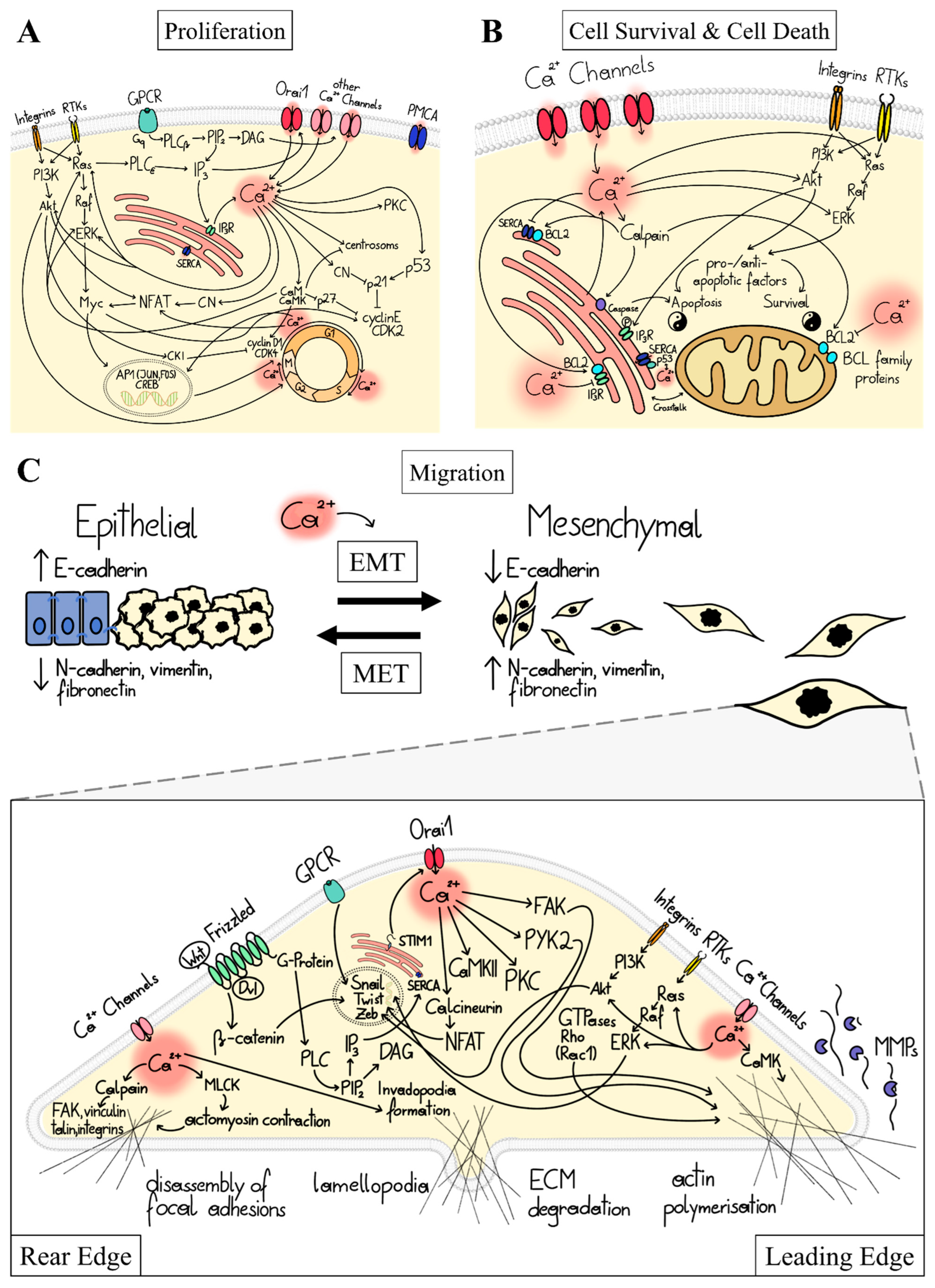{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer type | Critical Proteins | Targeted Signaling Pathways | Affected Cancer Hallmarks | Cell Type | Ref | |
|---|---|---|---|---|---|---|
| Breast | ↑STIM1, ↑Orai1 | small GTPases ↑Ras and ↑Rac -> ↓focal adhesions -> | migration, metastasis | MDA-MB231 | [82] | |
| ↑Orai1 +↑SPCA2 | ↑vimentin ↑Wnt/Ca2+ sig. pathway -> CaMKII -> ß-catenin ->↓Wnt | EMT | MCF-7 | [273] | ||
| ↑Orai3 | ↑ER -> | cell proliferation | [126,127] | |||
| ↑ERK1/2 ->↑Myc -> ↑cell cycle (G1) -> | cell proliferation | [253] | ||||
| ↑PI3K -> ↓p53 -> | apoptosis | [271] | ||||
| hypoxia -> | EMT | MDA-MB-468 | [62] | |||
| STIM2 + Orai1 | (↑?)PAR-2 -> | survival, invasion, cancer prognosis | MCF-7, MDA-MB-231 | [250] | ||
| Cervical | ↑STIM1, ↑Orai1 | ↑cell cycle (G1/S (CDK2, cyclin E)) -> | proliferation | SiHa, HeLa, U2OS | [274] | |
| ↑EGF -> ↑calpain ->↑α-spectrin -> | migration, metastasis | SiHa, CaSki, human patient and mice tissues/cells | [248] | |||
| ↑FAK and ↑Pyk2 ->↓focal adhesions -> | migration, metastasis | |||||
| S and G2/M phases (↓p21, ↑Cdc25C) -> | proliferation | |||||
| VEGF (vascular endothelial growth factor) -> | angiogenesis | |||||
| Colorectal | ↑STIM1 | ↑EGF -> ↑COX-2 -> ↑PGE2 -> | migration, EMT | DDL-1, HT-29, patient samples | [85] | |
| ↑Orai1, Orai3 | hypoxia -> ↑HIF-1/2a -> ↑Orai3 -> | migration | CW-2 | [275] | ||
| ↓STIM2 + ↑TRPC1 + ↑Orai1 + ↑STIM1 | n.d. | proliferation, invasion, survival, apoptosis | HT29 | [254] | ||
| Esophageal | ↑Orai1 (STIM2?) | ↑ERK & Akt -> cell cycle (↓cdc2, ↓cyclin B1, ↓p27) -> | proliferation | KYSE-150, patient and mouse samples | [255] | |
| ↑vimentin, ↑Rac1, ↓E-cadherin -> cytoskeleton -> | migration, invasion | |||||
| Gastric | ↑STIM1, ↑Orai1 | cell cycle (↑cyclin D1, ↓p21) -> | proliferation, metabolism, migration, invasion, metastasis | GS, BGC-803, BGC-823, MGC-803, MKN-28, MKN-45, SGC-7901, nude mice, patient samples | [53] | |
| ↑vimentin, ↑fibronectin, ↑MACC1, ↓E-cadherin -> | migration, metastasis | |||||
| Glioblastoma | ↑STIM1 | cell cycle (G0/G1 phase, ↑cyclin D1, ↑CDK4, ↓p21) -> | proliferation | U251, U87 and U373 | [88] | |
| ↑Orai1 | ↑Pyk2 -> ↓focal adhesion -> ↑vimentin, ↓E-cadherin, ↑N-cadherin (EMT like) -> | migration, invasion | U251/SNB19 | [272] | ||
| Hematologic | AML | ↑Orai1, ↑Orai2 | ↑FAK -> ↓focal adhesions -> | proliferation, migration | HL60 | [245] |
| Orai3 | ↑Ras -> ↑Orai3 -> | cell survival | U937, 8226 | [262] | ||
| MM | ↑STIM1, ↑Orai1 | ↑cell cycle -> | proliferation, apoptosis | KM3, U266 | [261] | |
| CLL | ↑STIM1, ↑Orai1, ↑TRPC1 | n.d. | proliferation, cancer progression | U937, 8226 | [89] | |
| Liver | ↑STIM1 | ↑FAK-Y397 -> ↓focal adhesions -> | migration | HCC-LM3 | [257] | |
| ↑STIM1, ↑Orai1, ↑TRPC6 | cell cycle (↑cyclin D1) -> | proliferation | Huh-7 | [258] | ||
| Lung | ↓Orai1 | ↑EGF -> ↑PI3K/Akt -> cell cycle (G1/S phase; ↑cyclin D) -> | proliferation | A549 | [79] | |
| ↑Orai3/Orai1 | ↑EGF -> PI3K/Akt -> cell cycle (G1/S phase; ↑cyclin D1/E, ↑CDK4 and ↑CDK2) -> | proliferation, cell cycle progression | NCI-H23, NCI-H460, patients | [249] | ||
| Melanoma | ↑STIM1, ↑Orai1 | ↑CaMKII/Raf-1/ERK -> | proliferation, migration, metastasis | SK-Mel-2, C8161, SK-Mel-24, UACC2577, WM3248, | [276] | |
| ↑STIM2, ↑Orai1 | n.d. | migration, invasion | SK-MEL-5, SK-MEL-28, WM3734 | [108] | ||
| ↓STIM2, ↓Orai1 | CREB/β-catenin -> MITF | proliferation | ||||
| Ovarian | ↑STIM1, ↑Orai1 | ↑Akt -> | apoptosis | A2780 | [264] | |
| ↑TRPC1, ↑TRPC3, ↑TRPC4, ↑TRPC6 | RTK? -> | proliferation | SKOV3, ATCC HTB-77 | [263] | ||
| Pancreatic | ↑STIM1, ↑Orai1 | n.d. | apoptosis | Panc1, (ASPC1, BxPC3, MiaPaca2, Capan1) | [265] | |
| ↑Orai3 | ↑cell cycle (G2/M-phase) | proliferation | Panc1, (ASPC1, BxPC3, MiaPaCa2, Capan1) | [266] | ||
| Prostate | ↓Orai1 | ↓AR | apoptosis | LNCaP, DU-145, and PC-3 | [80] | |
| ↑Orai1/Orai3 | cell cycle (G1/S phase; ↑cyclin D) | proliferation | LNCaP | [269] | ||
| ↓SOCE -> | apoptosis | |||||
| Renal | ↑STIM1, ↑Orai1 | n.d. | proliferation, migration | ccRCC, ACHN and Caki1, patient samples | [277] | |
| Cancer Type | Critical Proteins | Targeted Signaling Pathways | Affected Cancer Hallmarks | Cell Type | Ref |
|---|---|---|---|---|---|
| breast | ↑SK3 | n.d. | migration, metastasis | MDA-MB-435s | [327] |
| ↑SK4 | cell cycle (G1, S phases), ↑cdc25C | proliferation | mice | [360] | |
| EGF -> vimentin, snail1 | proliferation, migration, EMT | MCF-7, T47D, MDA-MB-231 and MDA-MB-468, patients | [358] | ||
| ↑SK3, ↑P2X7R | hypoxia ->↑ERK1/2 ->↑Akt (?) | proliferation, invasiveness, migration | MDA-MB-435s | [330] | |
| colorectal | ↑SK4 | ↑Ras/ERK (KRAS)/HIF1a/ROS | migration, invasion, metastasis | HCT116 | [359] |
| glioblastoma | ↑SK4 | ↑cell cycle (G2, M phases) | invasion, proliferation, poor prognosis | T98G, U87MG, GL261, patients | [362] |
| hematologic CCL | ↑SK4 | n.d. | proliferation | patients | [352] |
| melanoma | ↑SK3 | n.d. | migration, metastasis | 518A2, HBL, Bris | [342] |
| ovarian | ↑SK4, P2y2 | n.d. | proliferation, migration, cancer progression | Skov-3, patients | [361,363] |
| pancreatic | ↑SK4 | ↑KRAS -> ↑RAS -> ↑ERK/PI3K | proliferation, poor prognosis | mice, patients | [356] |
| prostate | ↑SK3 (↑ZEB1) | ↑Snail, ↑Slug, ↑Twist | EMT, neuroendocrine differentiation, drug resistance | LNCaP, patients | [331,364] |
| Cancer Type | Critical Proteins | Targeted Signaling Pathways | Affected Cancer Hallmarks | Cell Type | Ref |
|---|---|---|---|---|---|
| Breast | SK3 + ↑Orai1 | cAMP-PKA (↓SK3 activity due to phosphorylation) | migration, metastasis | MDA-MB-435s | [322] |
| SK3 + ↑Orai1 + ↑SigmaR1 | n.d. | migration | MDA-MB-435 | [321] | |
| colon | SK3 + ↑Orai1 + ↑SigmaR1 | n.d. | migration | HCT-116, patients | [321] |
| ↑SK3 + ↑Orai1 + ↑TRPC1 + ↑STIM1 | ↑EGFR -> ↑PI3K -> ↑Akt -> ↑Rac-1 -> ↑Calpain | migration | HCT-116 | [323] | |
| ↑EGFR -> ↑PI3K -> ↑Akt -> STIM1 | migration | ||||
| prostate | SK3 + Orai1 | n.d. | proliferation | LNCaP | [343] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tiffner, A.; Hopl, V.; Derler, I. CRAC and SK Channels: Their Molecular Mechanisms Associated with Cancer Cell Development. Cancers 2023, 15, 101. https://doi.org/10.3390/cancers15010101
Tiffner A, Hopl V, Derler I. CRAC and SK Channels: Their Molecular Mechanisms Associated with Cancer Cell Development. Cancers. 2023; 15(1):101. https://doi.org/10.3390/cancers15010101
Chicago/Turabian StyleTiffner, Adéla, Valentina Hopl, and Isabella Derler. 2023. "CRAC and SK Channels: Their Molecular Mechanisms Associated with Cancer Cell Development" Cancers 15, no. 1: 101. https://doi.org/10.3390/cancers15010101