
Metastatic Transition of Pancreatic Ductal Cell Adenocarcinoma Is Accompanied by the Emergence of Pro-Invasive Cancer-Associated Fibroblasts


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Single-Cell Data
2.2. Single-Cell RNA Sequencing Processing
2.3. Receptor–Ligand Processing
2.4. Pathway Enrichment and Evolved Levels of Invasability
2.5. Human Tissue Sourcing and Immunostaining
2.6. Accelerated Nanopatterned Stromal Invasion Assay
2.7. Quantification of Invasion Characteristics by Invading Cancer Cells into Stroma
3. Results
3.1. Pancreatic Cancer Transition to Metastasis Is Characterized by the Emergence of Distinct Subclasses of Fibroblasts with Different Phenotypic Roles Supporting Stromal Trespass
3.2. Fibroblasts in Succeeding Metastatic Stages Exhibit Change from Physiological to Pathological Activation
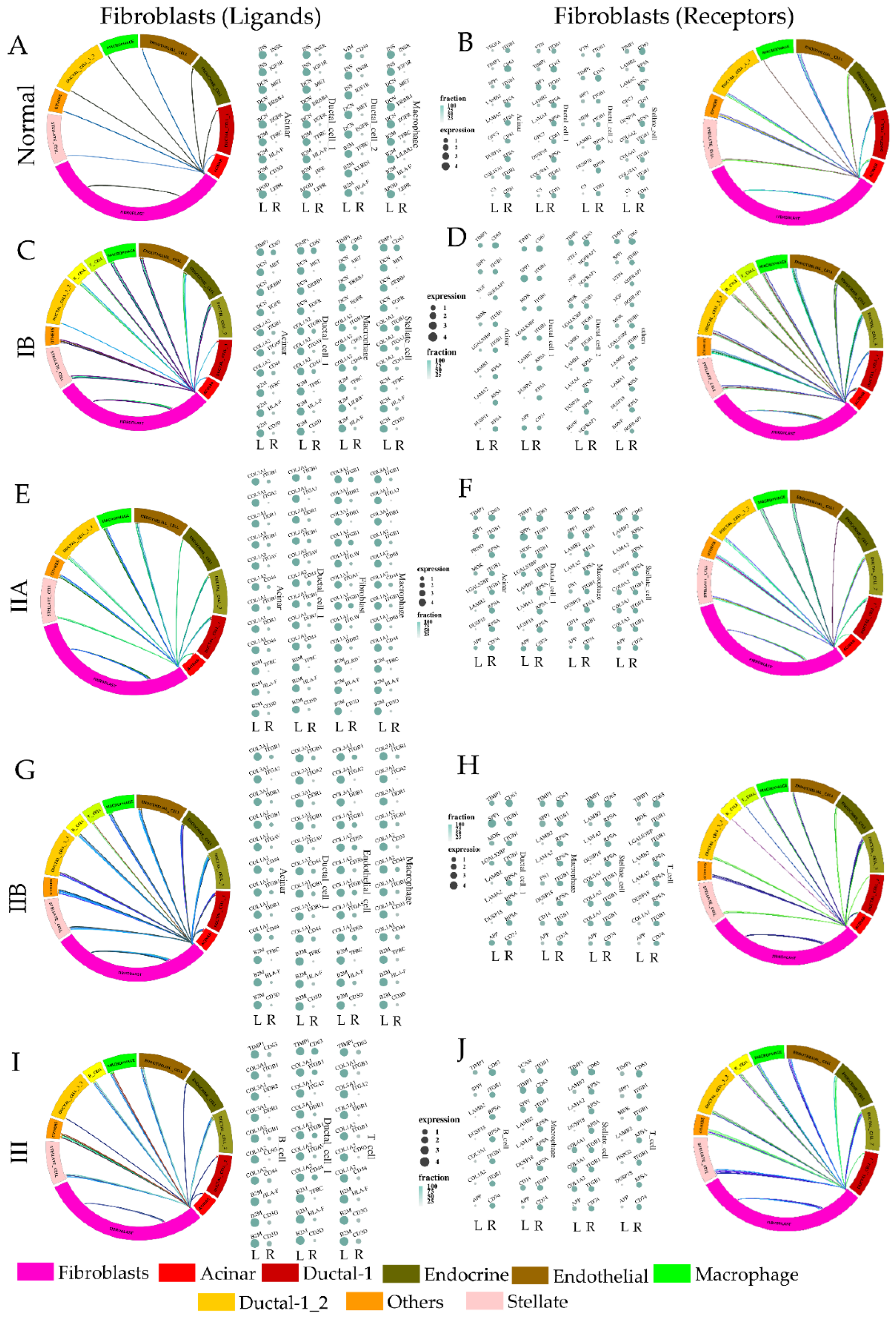
3.3. Paracrine Signaling by Fibroblasts to Other Cell Type Changes Modestly through the Metastatic Cascade
3.4. Transition to Early Metastasis in PDAC Cancer Is Associated with a Dramatic Reduction in Genes Evolved to Resist Invasion
3.5. Stromal Trespass of PDAC Is Associated with Emergence of a Fibroblast Subtype with Evolved Stromal Invasibility Gene Expression
3.6. Genes Characterizing PDAC Fibroblast Subtypes Partly Confer Pro-Invasable and Pro-Resistive Stromal Phenotype
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Suhail, Y.; Cain, M.P.; Vanaja, K.; Kurywchak, P.A.; Levchenko, A.; Kalluri, R.; Kshitiz. Systems biology of cancer metastasis. Cell Syst. 2019, 9, 109–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padmanaban, V.; Krol, I.; Suhail, Y.; Szczerba, B.M.; Aceto, N.; Bader, J.S.; Ewald, A.J. E-cadherin is required for metastasis in multiple models of breast cancer. Nature 2019, 573, 439–444. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.H.; Oh, S.Y.; Lee, H.J.; Kwon, T.G.; Kim, J.W.; Lee, S.T.; Choi, S.Y.; Hong, S.H. Cancer-associated fibroblast subgroups showing differential promoting effect on hnscc progression. Cancers 2021, 13, 654. [Google Scholar] [CrossRef] [PubMed]
- Mizutani, Y.; Kobayashi, H.; Iida, T.; Asai, N.; Masamune, A.; Hara, A.; Esaki, N.; Ushida, K.; Mii, S.; Shiraki, Y.; et al. Meflin-positive cancer-associated fibroblasts inhibit pancreatic carcinogenesis. Cancer Res. 2019, 79, 5367–5381. [Google Scholar] [CrossRef] [Green Version]
- Hwang, R.F.; Moore, T.; Arumugam, T.; Ramachandran, V.; Amos, K.D.; Rivera, A.; Ji, B.; Evans, D.B.; Logsdon, C.D. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 2008, 68, 918–926. [Google Scholar] [CrossRef] [Green Version]
- Gore, J.; Korc, M. Pancreatic cancer stroma: Friend or foe? Cancer Cell 2014, 25, 711–712. [Google Scholar] [CrossRef] [Green Version]
- Kshitiz; Afzal, J.; Maziarz, J.D.; Hamidzadeh, A.; Liang, C.; Erkenbrack, E.M.; Kim, H.N.; Haeger, J.D.; Pfarrer, C.; Hoang, T.; et al. Evolution of placental invasion and cancer metastasis are causally linked. Nat. Ecol. Evol. 2019, 3, 1743–1753. [Google Scholar] [CrossRef]
- Wagner, G.P.; Kshitiz; Dighe, A.; Levchenko, A. The coevolution of placentation and cancer. Annu. Rev. Anim. Biosci. 2021, 10, 259–279. [Google Scholar] [CrossRef]
- Wagner, G.P.; Kshitiz; Levchenko, A. Comments on boddy et al. 2020: Available data suggest positive relationship between placental invasion and malignancy. Evol. Med. Public Health 2020, 2020, 211–214. [Google Scholar] [CrossRef]
- Suhail, Y.; Afzal, J.; Kshitiz. Evolved resistance to placental invasion secondarily confers increased survival in melanoma patients. J. Clin. Med. 2021, 10, 595. [Google Scholar] [CrossRef]
- Reza, K.K.; Dey, S.; Wuethrich, A.; Jing, W.; Behren, A.; Antaw, F.; Wang, Y.; Sina, A.A.; Trau, M. In Situ single cell proteomics reveals circulating tumor cell heterogeneity during treatment. ACS Nano 2021, 15, 11231–11243. [Google Scholar] [CrossRef] [PubMed]
- Atta, L.; Fan, J. Computational challenges and opportunities in spatially resolved transcriptomic data analysis. Nat. Commun. 2021, 12, 5283. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Slowikowski, K.; Zhang, F. Single-cell transcriptomics in cancer: Computational challenges and opportunities. Exp. Mol. Med. 2020, 52, 1452–1465. [Google Scholar] [CrossRef] [PubMed]
- Dong, M.; Thennavan, A.; Urrutia, E.; Li, Y.; Perou, C.M.; Zou, F.; Jiang, Y. Scdc: Bulk gene expression deconvolution by multiple single-cell rna sequencing references. Brief. Bioinform. 2021, 22, 416–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, H.; Liu, Z. A benchmark for rna-seq deconvolution analysis under dynamic testing environments. Genome Biol. 2021, 22, 102. [Google Scholar] [CrossRef] [PubMed]
- Hosein, A.N.; Brekken, R.A.; Maitra, A. Pancreatic cancer stroma: An update on therapeutic targeting strategies. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 487–505. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Sun, B.F.; Chen, C.Y.; Zhou, J.Y.; Chen, Y.S.; Chen, H.; Liu, L.; Huang, D.; Jiang, J.; Cui, G.S.; et al. Single-cell rna-seq highlights intra-tumoral heterogeneity and malignant progression in pancreatic ductal adenocarcinoma. Cell Res. 2019, 29, 725–738. [Google Scholar] [CrossRef]
- Thomas, D.; Radhakrishnan, P. Tumor-stromal crosstalk in pancreatic cancer and tissue fibrosis. Mol. Cancer 2019, 18, 14. [Google Scholar] [CrossRef]
- Rasheed, Z.A.; Matsui, W.; Maitra, A. Pathology of pancreatic stroma in PDAC. In Pancreatic Cancer and Tumor Microenvironment; Grippo, P.J., Munshi, H.G., Eds.; Transworld Research Network: Trivandrum, India, 2012. [Google Scholar]
- Suhail, Y.; Maziarz, J.D.; Novin, A.; Dighe, A.; Afzal, J.; Wagner, G.; Kshitiz. Tracing the cis-regulatory changes underlying the endometrial control of placental invasion. Proc. Natl. Acad. Sci. USA 2022, 119, e2111256119. [Google Scholar] [CrossRef]
- Stuart, T.; Butler, A.; Hoffman, P.; Hafemeister, C.; Papalexi, E.; Mauck, W.M., 3rd; Hao, Y.; Stoeckius, M.; Smibert, P.; Satija, R. Comprehensive integration of single-cell data. Cell 2019, 177, 1888–1902.e21. [Google Scholar] [CrossRef]
- Ramilowski, J.A.; Goldberg, T.; Harshbarger, J.; Kloppmann, E.; Lizio, M.; Satagopam, V.P.; Itoh, M.; Kawaji, H.; Carninci, P.; Rost, B.; et al. A draft network of ligand-receptor-mediated multicellular signalling in human. Nat. Commun. 2015, 6, 7866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korotkevich, G.; Sukhov, V.; Budin, N.; Shpak, B.; Artyomov, M.N.; Sergushichev, A. Fast gene set enrichment analysis. bioRxiv 2021. in preprint. [Google Scholar]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. Clusterprofiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef] [PubMed]
- Jassal, B.; Matthews, L.; Viteri, G.; Gong, C.; Lorente, P.; Fabregat, A.; Sidiropoulos, K.; Cook, J.; Gillespie, M.; Haw, R.; et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2020, 48, D498–D503. [Google Scholar] [CrossRef] [PubMed]
- Ernst, J.; Bar-Joseph, Z. Stem: A tool for the analysis of short time series gene expression data. BMC Bioinform. 2006, 7, 191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, D.W.; Kim, J. The american joint committee on cancer 8th edition staging system for the pancreatic ductal adenocarcinoma: Is it better than the 7th edition? Hepatobiliary Surg. Nutr. 2020, 9, 98–100. [Google Scholar] [CrossRef] [PubMed]
- Louault, K.; Li, R.R.; DeClerck, Y.A. Cancer-associated fibroblasts: Understanding their heterogeneity. Cancers 2020, 12, 3108. [Google Scholar] [CrossRef]
- Werner, S.; Lutzkendorf, J.; Muller, T.; Muller, L.P.; Posern, G. Mrtf-a controls myofibroblastic differentiation of human multipotent stromal cells and their tumour-supporting function in xenograft models. Sci. Rep. 2019, 9, 11725. [Google Scholar] [CrossRef] [Green Version]
- Labernadie, A.; Kato, T.; Brugues, A.; Serra-Picamal, X.; Derzsi, S.; Arwert, E.; Weston, A.; Gonzalez-Tarrago, V.; Elosegui-Artola, A.; Albertazzi, L.; et al. A mechanically active heterotypic e-cadherin/n-cadherin adhesion enables fibroblasts to drive cancer cell invasion. Nat. Cell Biol. 2017, 19, 224–237. [Google Scholar] [CrossRef]
- Pakshir, P.; Alizadehgiashi, M.; Wong, B.; Coelho, N.M.; Chen, X.; Gong, Z.; Shenoy, V.B.; McCulloch, C.A.; Hinz, B. Dynamic fibroblast contractions attract remote macrophages in fibrillar collagen matrix. Nat. Commun. 2019, 10, 1850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davidson, J.; Shen, Z.; Gong, X.; Pollack, J.R. Swi/snf aberrations sensitize pancreatic cancer cells to DNA crosslinking agents. Oncotarget 2018, 9, 9608–9617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.; Zhou, L.; Yang, K.; Iwasawa, K.; Kadekaro, A.L.; Takebe, T.; Andl, T.; Zhang, Y. The beta-catenin/yap signaling axis is a key regulator of melanoma-associated fibroblasts. Signal Transduct. Target Ther. 2019, 4, 63. [Google Scholar] [CrossRef] [Green Version]
- Garcia, P.E.; Scales, M.K.; Allen, B.L.; Pasca di Magliano, M. Pancreatic fibroblast heterogeneity: From development to cancer. Cells 2020, 9, 2464. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Kong, Q.; Wang, J.; Jiang, Y.; Hua, H. Complex roles of camp-pka-creb signaling in cancer. Exp. Hematol. Oncol. 2020, 9, 32. [Google Scholar] [CrossRef]
- Li, G.; Jiang, Q.; Xu, K. Creb family: A significant role in liver fibrosis. Biochimie 2019, 163, 94–100. [Google Scholar] [CrossRef]
- Leung, C.S.; Yeung, T.L.; Yip, K.P.; Pradeep, S.; Balasubramanian, L.; Liu, J.; Wong, K.K.; Mangala, L.S.; Armaiz-Pena, G.N.; Lopez-Berestein, G.; et al. Calcium-dependent fak/creb/tnnc1 signalling mediates the effect of stromal mfap5 on ovarian cancer metastatic potential. Nat. Commun. 2014, 5, 5092. [Google Scholar] [CrossRef]
- Jang, I.; Beningo, K.A. Integrins, cafs and mechanical forces in the progression of cancer. Cancers 2019, 11, 721. [Google Scholar] [CrossRef] [Green Version]
- Ding, J.; Liu, Y.; Lai, Y. Identifying mmp14 and col12a1 as a potential combination of prognostic biomarkers in pancreatic ductal adenocarcinoma using integrated bioinformatics analysis. PeerJ 2020, 8, e10419. [Google Scholar] [CrossRef]
- Weniger, M.; Honselmann, K.C.; Liss, A.S. The extracellular matrix and pancreatic cancer: A complex relationship. Cancers 2018, 10, 316. [Google Scholar] [CrossRef] [Green Version]
- Liu-Chittenden, Y.; Jain, M.; Gaskins, K.; Wang, S.; Merino, M.J.; Kotian, S.; Kumar Gara, S.; Davis, S.; Zhang, L.; Kebebew, E. Rarres2 functions as a tumor suppressor by promoting beta-catenin phosphorylation/degradation and inhibiting p38 phosphorylation in adrenocortical carcinoma. Oncogene 2017, 36, 3541–3552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, W.J.; Zabel, B.A.; Pachynski, R.K. Mechanisms and functions of chemerin in cancer: Potential roles in therapeutic intervention. Front. Immunol. 2018, 9, 2772. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Villalba, A.; Romero, J.P.; Hernandez, S.C.; Vilas-Zornoza, A.; Fortelny, N.; Castro-Labrador, L.; San Martin-Uriz, P.; Lorenzo-Vivas, E.; Garcia-Olloqui, P.; Palacio, M.; et al. Single-cell rna sequencing analysis reveals a crucial role for cthrc1 (collagen triple helix repeat containing 1) cardiac fibroblasts after myocardial infarction. Circulation 2020, 142, 1831–1847. [Google Scholar] [CrossRef] [PubMed]
- Azuma, T.; Witke, W.; Stossel, T.P.; Hartwig, J.H.; Kwiatkowski, D.J. Gelsolin is a downstream effector of rac for fibroblast motility. EMBO J. 1998, 17, 1362–1370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elyada, E.; Bolisetty, M.; Laise, P.; Flynn, W.F.; Courtois, E.T.; Burkhart, R.A.; Teinor, J.A.; Belleau, P.; Biffi, G.; Lucito, M.S.; et al. Cross-species single-cell analysis of pancreatic ductal adenocarcinoma reveals antigen-presenting cancer-associated fibroblasts. Cancer Discov. 2019, 9, 1102–1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crowe, L.A.N.; McLean, M.; Kitson, S.M.; Melchor, E.G.; Patommel, K.; Cao, H.M.; Reilly, J.H.; Leach, W.J.; Rooney, B.P.; Spencer, S.J.; et al. S100a8 & s100a9: Alarmin mediated inflammation in tendinopathy. Sci. Rep. 2019, 9, 1463. [Google Scholar] [PubMed]
- Wang, S.; Song, R.; Wang, Z.; Jing, Z.; Wang, S.; Ma, J. S100a8/a9 in inflammation. Front. Immunol. 2018, 9, 1298. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Jing, Y.; Ding, L.; Zhang, X.; Song, Y.; Chen, S.; Zhao, X.; Huang, X.; Pu, Y.; Wang, Z.; et al. Epiregulin reprograms cancer-associated fibroblasts and facilitates oral squamous cell carcinoma invasion via jak2-stat3 pathway. J. Exp. Clin. Cancer Res. 2019, 38, 274. [Google Scholar] [CrossRef] [Green Version]
- Palmieri, C.; Roberts-Clark, D.; Assadi-Sabet, A.; Coope, R.C.; O’Hare, M.; Sunters, A.; Hanby, A.; Slade, M.J.; Gomm, J.J.; Lam, E.W.; et al. Fibroblast growth factor 7, secreted by breast fibroblasts, is an interleukin-1beta-induced paracrine growth factor for human breast cells. J. Endocrinol. 2003, 177, 65–81. [Google Scholar] [CrossRef] [Green Version]
- Hirakawa, T.; Yashiro, M.; Doi, Y.; Kinoshita, H.; Morisaki, T.; Fukuoka, T.; Hasegawa, T.; Kimura, K.; Amano, R.; Hirakawa, K. Pancreatic fibroblasts stimulate the motility of pancreatic cancer cells through igf1/igf1r signaling under hypoxia. PLoS ONE 2016, 11, e0159912. [Google Scholar] [CrossRef] [Green Version]
- Tao, J.; Yang, G.; Zhou, W.; Qiu, J.; Chen, G.; Luo, W.; Zhao, F.; You, L.; Zheng, L.; Zhang, T.; et al. Targeting hypoxic tumor microenvironment in pancreatic cancer. J. Hematol. Oncol. 2021, 14, 14. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yu, S.; Li, L.; Chen, J.; Quan, M.; Li, Q.; Gao, Y. Klf4-mediated upregulation of cd9 and cd81 suppresses hepatocellular carcinoma development via jnk signaling. Cell Death Dis. 2020, 11, 299. [Google Scholar] [CrossRef] [PubMed]
- Koninger, J.; Giese, N.A.; di Mola, F.F.; Berberat, P.; Giese, T.; Esposito, I.; Bachem, M.G.; Buchler, M.W.; Friess, H. Overexpressed decorin in pancreatic cancer: Potential tumor growth inhibition and attenuation of chemotherapeutic action. Clin. Cancer Res. 2004, 10, 4776–4783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gold, D.V.; Stein, R.; Burton, J.; Goldenberg, D.M. Enhanced expression of cd74 in gastrointestinal cancers and benign tissues. Int. J. Clin. Exp. Pathol. 2010, 4, 1–12. [Google Scholar]
- Zhang, J.F.; Hua, R.; Liu, D.J.; Liu, W.; Huo, Y.M.; Sun, Y.W. Effect of cd74 on the prognosis of patients with resectable pancreatic cancer. Hepatobiliary Pancreat. Dis. Int. 2014, 13, 81–86. [Google Scholar] [CrossRef]
- DiGiacomo, V.; Meruelo, D. Looking into laminin receptor: Critical discussion regarding the non-integrin 37/67-kda laminin receptor/rpsa protein. Biol. Rev. 2016, 91, 288–310. [Google Scholar] [CrossRef] [Green Version]
- Digiacomo, V.; Gando, I.A.; Venticinque, L.; Hurtado, A.; Meruelo, D. The transition of the 37-kda laminin receptor (rpsa) to higher molecular weight species: Sumoylation or artifact? Cell Mol. Biol. Lett. 2015, 20, 571–585. [Google Scholar] [CrossRef] [Green Version]
- Novin, A.; Suhail, Y.; Ajeti, V.; Goyal, R.; Wali, K.; Seck, A.; Jackson, A.; Kshitiz. Diversity in cancer invasion phenotypes indicates specific stroma regulated programs. Hum. Cell 2021, 34, 111–121. [Google Scholar] [CrossRef]
- Griss, J.; Viteri, G.; Sidiropoulos, K.; Nguyen, V.; Fabregat, A.; Hermjakob, H. ReactomeGSA—Efficient multi-omics comparative pathway analysis. Mol. Cell. Proteom. 2020, 19, 2115–2125. [Google Scholar] [CrossRef]
- Ganini, C.; Amelio, I.; Bertolo, R.; Bove, P.; Buonomo, O.C.; Candi, E.; Cipriani, C.; Di Daniele, N.; Juhl, H.; Mauriello, A.; et al. Global mapping of cancers: The cancer genome atlas and beyond. Mol. Oncol. 2021, 15, 2823–2840. [Google Scholar] [CrossRef]
- Karpinsky-Semper, D.; Volmar, C.H.; Brothers, S.P.; Slepak, V.Z. Differential effects of the gbeta5-rgs7 complex on muscarinic m3 receptor-induced Ca2+ influx and release. Mol. Pharmacol. 2014, 85, 758–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, T.; Zhou, Y.; Long, R.; Chen, C.; Zhao, J.; Cui, P.; Guo, M.; Liang, G.; Xu, L. Dusp8 phosphatase: Structure, functions, expression regulation and the role in human diseases. Cell Biosci. 2019, 9, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.; Wang, J.; Fan, G. Nprl2 is an independent prognostic factor of osteosarcoma. Cancer Biomark. 2012, 12, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.Y.; Yu, Q.F.; Shen, W.; Guo, C.M.; Li, Z.; Zhou, X.Y.; Zhou, N.J.; Min, W.P.; Gao, D. Knockdown of elmo3 suppresses growth, invasion and metastasis of colorectal cancer. Int. J. Mol. Sci. 2016, 17, 2119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, G.; Hong, W.; Guo, Y.; Bai, Y.; Chen, B. Molecular mechanism of pancreatic stellate cells activation in chronic pancreatitis and pancreatic cancer. J. Cancer 2020, 11, 1505–1515. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, K. Pancreatic stellate cells: Molecular mechanism of pancreatic fibrosis. J. Gastroenterol. Hepatol. 2008, 23 (Suppl. 1), S119–S121. [Google Scholar] [CrossRef]
- Martinez-Bosch, N.; Vinaixa, J.; Navarro, P. Immune evasion in pancreatic cancer: From mechanisms to therapy. Cancers 2018, 10, 6. [Google Scholar] [CrossRef] [Green Version]
- Zeng, S.; Pottler, M.; Lan, B.; Grutzmann, R.; Pilarsky, C.; Yang, H. Chemoresistance in pancreatic cancer. Int. J. Mol. Sci. 2019, 20, 4504. [Google Scholar] [CrossRef] [Green Version]
- Sarr, A.; Bre, J.; Um, I.H.; Chan, T.H.; Mullen, P.; Harrison, D.J.; Reynolds, P.A. Genome-scale crispr/cas9 screen determines factors modulating sensitivity to protide nuc-1031. Sci. Rep. 2019, 9, 7643. [Google Scholar] [CrossRef]
- He, P.; Yang, J.W.; Yang, V.W.; Bialkowska, A.B. Kruppel-like factor 5, increased in pancreatic ductal adenocarcinoma, promotes proliferation, acinar-to-ductal metaplasia, pancreatic intraepithelial neoplasia, and tumor growth in mice. Gastroenterology 2018, 154, 1494–1508.e13. [Google Scholar] [CrossRef] [Green Version]
- Mori, A.; Moser, C.; Lang, S.A.; Hackl, C.; Gottfried, E.; Kreutz, M.; Schlitt, H.J.; Geissler, E.K.; Stoeltzing, O. Up-regulation of kruppel-like factor 5 in pancreatic cancer is promoted by interleukin-1beta signaling and hypoxia-inducible factor-1alpha. Mol. Cancer Res. 2009, 7, 1390–1398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabacaoglu, D.; Ruess, D.A.; Ai, J.; Algul, H. Nf-kappab/rel transcription factors in pancreatic cancer: Focusing on rela, c-rel, and relb. Cancers 2019, 11, 937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, J.; Li, Y.; Tan, X.; Fu, L. Small heat shock proteins in cancers: Functions and therapeutic potential for cancer therapy. Int. J. Mol. Sci. 2020, 21, 6611. [Google Scholar] [CrossRef] [PubMed]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Hu, C.; Wang, T.; Wang, J.; Zhang, J.; Gao, F.; Ou, Q.; Tian, H.; Jin, C.; Xu, J.; et al. Glia maturation factor beta as a novel biomarker and therapeutic target for hepatocellular carcinoma. Front. Oncol. 2021, 11, 744331. [Google Scholar] [CrossRef]
- Ling, B.; Watt, K.; Banerjee, S.; Newsted, D.; Truesdell, P.; Adams, J.; Sidhu, S.S.; Craig, A.W.B. A novel immunotherapy targeting mmp-14 limits hypoxia, immune suppression and metastasis in triple-negative breast cancer models. Oncotarget 2017, 8, 58372–58385. [Google Scholar] [CrossRef] [Green Version]
- Claesson-Welsh, L. How the matrix metalloproteinase mmp14 contributes to the progression of colorectal cancer. J. Clin. Investig. 2020, 130, 1093–1095. [Google Scholar] [CrossRef]
- Mueller, M.M.; Fusenig, N.E. Friends or foes—Bipolar effects of the tumour stroma in cancer. Nat. Rev. Cancer 2004, 4, 839–849. [Google Scholar] [CrossRef]
- Kwa, M.Q.; Herum, K.M.; Brakebusch, C. Cancer-associated fibroblasts: How do they contribute to metastasis? Clin. Exp. Metastasis 2019, 36, 71–86. [Google Scholar] [CrossRef]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [Green Version]
- Okabe, Y.; Medzhitov, R. Tissue biology perspective on macrophages. Nat. Immunol. 2016, 17, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Colegio, O.R.; Chu, N.Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, L.R.; Borriello, L.; Entenberg, D.; Condeelis, J.S.; Oktay, M.H.; Karagiannis, G.S. The emerging roles of macrophages in cancer metastasis and response to chemotherapy. J. Leukoc. Biol. 2019, 106, 259–274. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, S.; Suhail, Y.; Novin, A.; Perpetua, L.; Kshitiz. Metastatic Transition of Pancreatic Ductal Cell Adenocarcinoma Is Accompanied by the Emergence of Pro-Invasive Cancer-Associated Fibroblasts. Cancers 2022, 14, 2197. https://doi.org/10.3390/cancers14092197
Liu S, Suhail Y, Novin A, Perpetua L, Kshitiz. Metastatic Transition of Pancreatic Ductal Cell Adenocarcinoma Is Accompanied by the Emergence of Pro-Invasive Cancer-Associated Fibroblasts. Cancers. 2022; 14(9):2197. https://doi.org/10.3390/cancers14092197
Chicago/Turabian StyleLiu, Shaofei, Yasir Suhail, Ashkan Novin, Lorrie Perpetua, and Kshitiz. 2022. "Metastatic Transition of Pancreatic Ductal Cell Adenocarcinoma Is Accompanied by the Emergence of Pro-Invasive Cancer-Associated Fibroblasts" Cancers 14, no. 9: 2197. https://doi.org/10.3390/cancers14092197